

Abstract
Oncogenic activation resulting in hyperproliferative lesions within the colonic mucosa has been identified in putative precancerous lesions, aberrant crypt foci (ACF). KRAS and BRAF mutation status was determined in 172 ACF identified in the colorectum of screening subjects by in situ high-definition, magnifying chromoendoscopy. Lesions were stratified according to histology (serrated vs. distended) as previously described [1]. Due to their limiting size, however, it was not technically feasible to examine downstream signaling consequences of these oncogenic mutations. We have combined ultraviolet-infrared (UV/IR) microdissection with an ultrasensitive nanofluidic proteomic immunoassay (NIA) to enable accurate quantification of post-translational modifications to mitogen-activated protein kinase (MAPK) in total protein lysates isolated from hyperproliferative crypts and adjacent normal mucosa. Using this approach, levels of singly and dually (activated) phosphorylated isoforms of extracellular receptor kinase(ERK)-1 and -2 were quantified in samples containing as little as 16-ng of total protein recovered from <200 cells. ERK activation is responsible for observed hyperplasia found in these early lesions, but is not directly dependent on KRAS and/or BRAF mutation status. This study describes the novel use of a sensitive nanofluidic platform to measure oncogene-driven proteomic changes in diminutive lesions and highlights the advantage of this approach over classical immunohistochemistry-based analyses.
Keywords: Aberrant crypt foci, ERK1/2, oncogene activation, nanofluidic proteomic immunoassay, colorectal cancer
1 Introduction
Despite increased awareness of the importance of early detection of colorectal cancer (CRC) by the use of screening colonoscopy, CRC remains the second leading cause of cancer-related deaths in Western countries [2]. Important to successful CRC prevention is the identification of surrogate markers that may indicate a colonic mucosa at elevated risk for cancer. Aberrant crypt foci (ACF) are generally considered the earliest identifiable macroscopic lesion within the colonic mucosa that may be associated with risk of future neoplastic development[3]. We and others have shown that ACF commonly harbor activating mutations to several key proto-oncogenes, including KRAS and BRAF, thereby justifying their molecular characterization [1, 4]. The validity of ACF as a surrogate marker was supported by recent studies that showed that the mean number and prevalence of ACF is significantly correlated with the incidence of adenomas and CRC [5, 6].
While significant advances have been made in our understanding of the molecular events that accompany the progression of normal colonic mucosa to adenocarcinomas, the precise role of ACF within the adenoma-carcinoma sequence has not yet been conclusively established. Identifying genetic and molecular abnormalities that may contribute to their formation and expansion within the colonic mucosa are necessary for establishing their utility as surrogate cancer markers. As we have previously reported [1], ACF from the distal colorectum almost always show histological features of hyperplasia, and these lesions often harbor somatic mutations to key proto-oncogenes, including BRAF and KRAS. Because of their limiting sample size, however, molecular characterization of ACF has proven to be technically challenging beyond the available routine histological and immunohistological analyses.
Recently, an ultra-sensitive platform for protein quantification has been developed that enables capillary-based nanofluidic isoelectric focusing and antibody-based chemilluminescence analyses on only nanogram quantities of total protein[7]. This nanofluidic proteomic immunoassay (NIA) has been used primarily to examine changes in signaling activity in response to drug treatment [8, 9]. To date, however, these applications have relied primarily on whole tissue or cell culture lysates[10]. Since it is impossible to generate a sufficient concentration of protein lysate from small (< 1mm2) biopsy specimens for traditional IEF/Western blotting, we developed a new methodology combining the use of tissue microdissection with nanofluidics to study protein modifications in limiting numbers of cells.
In the following study, we demonstrate the use of NIA to examine the downstream consequences of RAS and RAF activation in ultraviolet-infrared (UV/IR) captured ACF removed from normal human colonic mucosa during screening colonoscopy. We have examined the phosphorylation states of ERK1 and ERK2 in UV/IR microdissected ACF and adjacent normal colonic mucosa. The significance of this experimental approach is that we can quantitatively determine the levels of individual ERK isoforms within the cellular context of oncogenic mutations.
2 Materials and Methods
2.1 Subject selection
All patients included in this study underwent a total colonoscopy at the University of Connecticut Health Center (UCHC) at John Dempsey Hospital (JDH) in accordance with Institutional policies. All patients who met the Amsterdam criteria for familial adenomatous polyposis (FAP) or hereditary non-polyposis CRC (HNPCC) were excluded from this study. This study was performed following Institutional Review Board approval and after receiving written informed consent from the subjects.
2.2 ACF collection and characterization
ACF were identified and isolated from grossly normal-appearing colonic mucosa by biopsy in situ during high-resolution, close-focus magnifying chromoendoscopy as previously described [1]. Briefly, the distal 20-cm of the colorectum (encompassing parts of the rectum and sigmoid colon) were stained with 40 mL of 0.2% indigo carmine. ACF were visualized and photographed using an Olympus close-focus colonoscope (XCF-Q160ALE; Olympus Corp., Center Valley, PA) capable of 60x magnification over a longer visualizing distance (100 mm). A finding was accepted as an ACF if, under magnification, two or more crypts had increased lumen diameter, thick crypt walls or abnormally shaped lumens. Biopsies of individual ACF and normal mucosa were immediately embedded in optimum cutting temperature (OCT) freezing medium, flash-frozen, and stored at −80°C.
Histological analyses were performed on coded hematoxylin and eosin (H&E) stained sections using light microscopy by T.V.R, a board-certified human pathologist who was blinded to clinical and molecular findings, according to previously described characteristics [1]. Briefly, hyperplastic ACF are characterized by the same criteria applied to hyperplastic polyps [11]. Serrated hyperplastic ACF are defined by their stellate luminal shape (serrated on oblique or cross-section) and the prominent component of columnar cells with microvesicular cytoplasm. Distended hyperplastic ACF lack crypt serration and have prominent goblet cell populations. In addition, hyperplastic ACF frequently have tufting of the surface epithelium. H&E slides were visualized using a BX60 upright bright-field microscope with 10x occulars/20x UPlanFl objectives (Olympus), a CoolSNAP imager and RSImage software, v.1.9.2 (RoperScientific, Ottobrunn, Germany)
2.3 Laser capture microdissection, DNA extraction and mutation analyses for BRAFV600E and KRAS codons 12/13
Laser capture microdissection, DNA extraction and subsequent analyses for mutations to BRAF and KRAS were performed as previously described [1, 11]. Briefly, frozen serial sections of ACF were prepared at 5- to 7-μm thickness on glass slides and microdissection was performed on these sections using the Veritas microdissection instrument (Applied Biosystems, Inc., Foster City, CA). Whenever possible, cells from the adjacent normal mucosa (ANM) directly abutting the aberrant crypts were collected separately by laser-capture. On average, approximately 3,000 cells were collected from each sample.
DNA (25 ng) was extracted from micro-dissected aberrant crypts and ANM using the PicoPure DNA Extraction Kit (Applied Biosystems). Mutations were detected by amplifying a 189-bp fragment of the BRAF gene spanning codon 600 using the following primers: forward, 5′-CCTAAACTCTTCATAATGCTTGCTC-3′ and reverse, 5′-CCACAAAATGGATCCAGACA-3′. Similarly, a 205-bp fragment of KRAS spanning codons 12 and 13 was amplified using the following primers: forward, 5′-GTACTGGTGGAGTATTTGAT-3′ and reverse, 5′-TCTATTGTTGGATCATATTC-3′. Amplified products were confirmed by gel electrophoresis and enzymatically purified with 2 μL of EXO SAP-IT (U.S. Biochemical Corp., Cleveland, OH). 2.5-μL aliquots of product were sequenced using the forward primer with an ABI BigDye TerV3.1 Cycle Sequencing kit on an ABI 9700 thermocycler (Applied Biosystems). Fifteen microliters of ethanol-precipitated reaction product was sequenced by capillary electrophoresis using a 3100-avant Genetic Analyzer (Applied Biosystems). The data were analyzed using ABI DNA Sequencing Analysis Software (ver. 3.7). Positive controls for BRAF (human colon carcinoma sample with a known mutation at codon 600) and KRAS (SW480 cells) and a negative control (placental DNA; Sigma, St. Louis, MO) were used in these analyses.
2.4 Immunofluorescence of ERK1/2
Immunofluorescence (IF) staining for total ERK expression was performed on a total of 45 biopsies of either ACF or adjacent normal mucosa. IF was performed on 7-μm sections from fresh-frozen OCT sections prepared on glass slides. Slides were baked for 10 min at 70°C, then fixed in 4% paraformaldehyde for 30 min, and washed in PBS. Membranes were permeabilized using 0.25% Triton-X100 for 20 min and washed in PBS. Tissues were blocked with PowerBlock (Biogenex, San Ramon, CA) for 10 min, then incubated in primary rabbit anti-human ERK1/2 antibody(1:200, Novus) and mouse anti-human beta-catenin (1:1000, Sigma) overnight at 4°C. All antibodies were diluted in 10% normal goat serum and 1% bovine serum albumin. Samples were then washed with PBS and incubated in secondary goat anti-rabbit Alexa-568 antibody (1:200, Invitrogen) and goat anti-mouse Alexa 488 antibody (1:200, Invitrogen) for 1 hour at room temperature (RT). Slides were washed in PBS and incubated with DAPI (2 min, RT; 1:10,000) for nuclear visualization. Beta-catenin is used as a membrane marker for crypt visualization. Slides were read using a Zeiss Axioplan2 upright fluorescence microscope with a 10x occular and 20x Plan-APOCHROMAT objective (Zeiss, Oberkochen, Germany) using fluorescent filters: 568 nm: red, ERK1/2 (Exposure time = 200 ms); 488 nm: green, beta-catenin (82.5 ms); 355 nm: blue, DAPI (15 ms). Images were acquired using a Zeiss AxioCam MRc and Zeiss AxioVision Rel. 4.8 software.
2.5 UV/IR microdissection for protein extraction
Tissues that were immediately frozen and stored at −80°C in OCT freezing medium were sectioned using a Leica CM1900 Cryostat. Twelve-μm sections were collected on Arcturus PEN membrane glass slides (Applied Biosystems). Slides were then shaken for 30 sec in 100% EtOH followed by 30 sec in 100% Xylene, air-dried and stored in a sealed container containing desiccant. Aberrant crypts excluding adjacent stroma were collected (total tissue area collected/sample ≥ 2.00 mm2) on Arcturus CapSure Macro LCM caps (Applied Biosystems) using a Veritas microdissection instrument. During the course of our methods development, it became obvious that the use of IR capture microdissection compromised the quality of the tissue transfer to the cap. Thus we switched to the use of combined UV-cutting and IR-transfer to collect tissue samples for subsequent protein analysis. The same technique was applied to collect adjacent normal crypts from these samples, as well as from normal biopsy specimens obtained independently from the same patient, when available.
2.6 Tissue lysis and MicroBCA assay
Caps with collected tissue were stored at -80°C for less than 24 hours. Tissues were then lysed directly on the cap surface using 10 μL of lysis buffer [Bicine/CHAPS lysis buffer (ProteinSimple, Santa Clara, CA) containing 80 mM NaCl, 1x DMSO Inhibitors mix (ProteinSimple), and 1x Aqueous Inhibitor mix (ProteinSimple). Sample handling was performed using siliconized tips and tubes whenever possible to reduce sample retention. Caps were maintained on ice for 10 min, quickly spun 6,600 rpm, and returned to ice for 10 minutes. Tubes were then spun at 10,000 × g for 10 min at 4°C to clear cellular debris. Supernatants were then transferred to low retention tubes and stored at −80°C (Fig. 1). Prior to freezing, a 0.5 μL aliquot of protein lysate was removed for determination of protein concentration using the microBCA assay (Thermo Fisher Scientific, Inc., Rockford, IL). Reactions and concentration standard curves (Bovine Serum Albumin, 2.0 mg/mL – 0.0625 mg/mL) were prepared in 96-well PCR plates. BCA reactions were incubated for 45 min at 37°C and subsequently measured using a NanoDrop spectrophotometer, performed in triplicate.
Figure 1. Experimental approach.
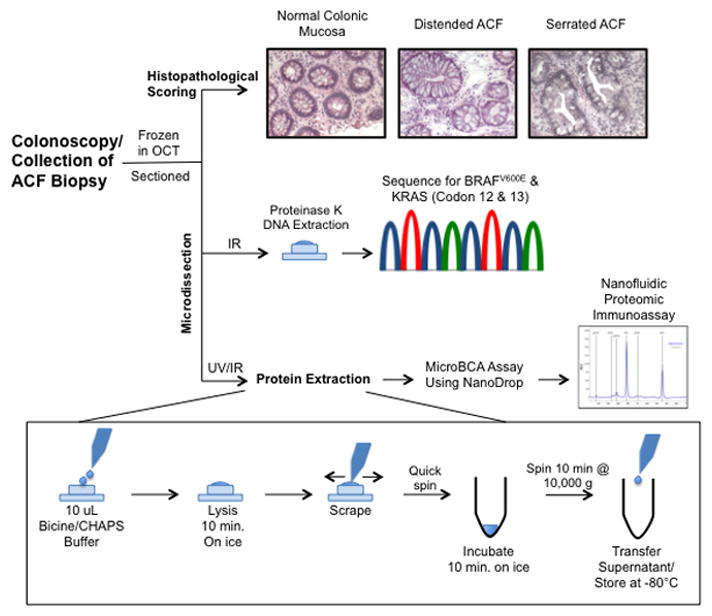
The combined use of UV/IR microdissection with NIA increases the informative potential of size-limited human biopsy specimens. The use of fresh-frozen tissues, cryosectioning and microdissection enables the acquisition of histological, genetic and proteomic data. Histopathological scoring shows H&E staining of representative normal colonic mucosa, distended and serrated hyperplastic ACF (all images shown at 200x magnification).
2.7 Nanofluidic proteomic immunoassay
Nanofluidic proteomic immunoassays for ERK1/2 were performed using the NanoPro CB100 (ProteinSimple), [7, 8]. Protein samples were diluted to 0.16 mg/mL in lysis buffer and mixed at a ratio of 1:4 with ampholyte premix G2 (pH range: 5–8) spiked with fluorescent pI standards: 4.9, 6, 6.4, 7, 7.3 (ProteinSimple). Assay optimization, using phospho-ERK1/2 specific, ERK1-specific and pan-ERK1/2 antibodies on HeLa control lysates (ProteinSimple) identified specific pI values (range: ±0.059) for each ERK isoform. Each sample was run with 6 replicates. The lower limit of detection was approximately 0.03 mg/mL total protein. Luminescent signal was measured by the NanoPro CB100 and data analysis was carried out using Compass Software v.1.8.1(ProteinSimple).
2. 8 Statistical analysis
To compare the group means of phosphorylation levels, a repeated measures general linear mixed model (GLMM) analysis was performed, using the MIXED procedure in SAS (ver. 9.2). Type 3 F-test statistics were used to test the overall hypothesis of equal group means among the five groups (BRAF, KRAS, wild-type for BRAF and KRAS, adjacent normal and normal). The group mean and standard error of the mean were estimated by an LSMEANS statement. When F-tests showed a significant difference among the five groups, adjusted pair-wise comparisons were further performed to identify pairs having significant differences using Tukey’s HSD procedure to control the experiment-wise error rate. We used a two-sided alpha level of significance of 0.05.
3 Results
3.1 KRAS and BRAF mutational status in ACF
172 distal colon ACF were stratified according to their molecular genotype into the following three categories: KRAS, BRAF, or wild-type for both loci (referred to as WT). ACF were independently classified as hyperplastic-serrated or hyperplastic-distended (Fig. 1). Approximately half (51%) of distended ACF exhibit a missense mutation in the KRAS gene at either codon 12 or 13 (Table 1). Interestingly, the presence of a BRAF mutation in distended ACF is rare (3%) and 31% of distended ACF are WT for both KRAS and BRAF. These results correlate with our previously reported frequencies for distended crypts (KRAS: 42%; BRAF: 3%; WT 55%)[1]. In comparison, a large percentage of serrated ACF harbor a BRAFV600E mutation (36%) consistent with our previous results[1]. However, the frequency of KRAS mutations in serrated ACF (40%) is greater than we previously reported (19%)[1]. In addition, the frequency of specific KRAS missense mutations, including G12A, G12C, G12D, G12V, and G13M, occur at equivalent frequencies between serrated and distended ACF, with G12D being the predominant mutation (Table 1).
Table 1.
BRAF and KRAS mutation frequency in hyperplastic ACF.
| Mutation Status | Mutation | Hyperplastic ACF
|
|
|---|---|---|---|
| Distended (%)(n = 67) | Serrated (%)(n = 105) | ||
|
| |||
| Wild-Type | N/A | 31 (46.27) | 22 (20.95) |
|
| |||
| KRAS | G12A | 1 (1.49) | 0 |
| G12C | 4 (5.97) | 2 (1.90) | |
| G12D | 16 (23.88) | 26 (24.76) | |
| G12V | 12 (17.91) | 16 (15.24) | |
| G13M | 1 (1.49) | 0 | |
|
| |||
| ALL | 34 (50.75) | 44 (41.90) | |
|
| |||
| BRAF | V600E | 2 (2.99) | 39 (37.14) |
|
| |||
| TOTAL ACF = 172 | |||
3.2 MAPK activation in ACF
Based on our findings of a distinct ACF histology that is related to the underlying oncogenic alterations, we hypothesized that KRAS or BRAF mutations may be associated with differential ERK-1/2 activation. In order to test this hypothesis, a pan-ERK1/2 antibody was examined in frozen tissue sections. As shown in Fig. 1, ACF displayed a modestly elevated nuclear accumulation of ERK1/2 compared to adjacent NCM. However, immunostaining was highly variable across the panel of ACF that were tested, compromising our ability to quantify the staining intensity. Thus to more accurately assess the potential differential activation of ERK signaling in these hyperplastic lesions, we developed a new method combining the high cellular resolution of tissue microdissection with the sensitivity afforded by a nanofluidic proteomic immunoassay. Our initial approach was to optimize the microdissection method to maximize protein yield. It was determined that the use of UV/IR microdissection followed by an on-cap protein extraction in limited sample buffer was essential (Fig. 2). Furthermore, we determined that the lower limit of detection is 12 ng of total protein for the NIA analysis, requiring at least 1 mm2 of captured tissue from 12-μm sections.
Figure 2. ERK1/2 analysis in human colonic mucosa.
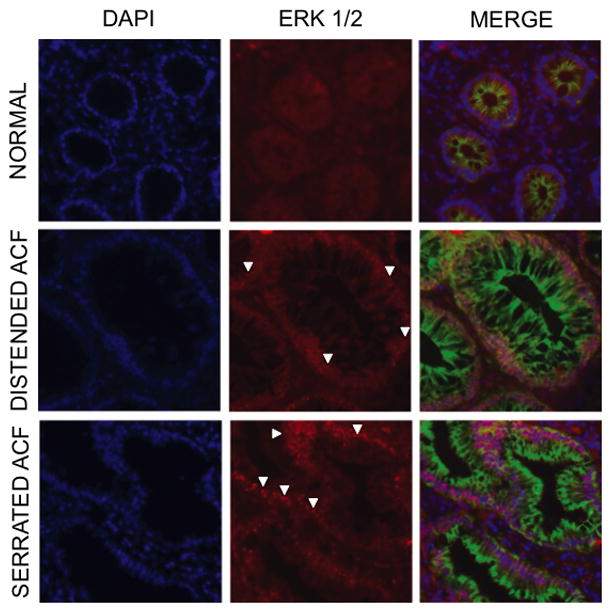
Immunofluorescence analysis of ERK1/2 was determined in frozen tissue sections as described under Materials and methods. ACF show evidence of nuclear accumulation of ERK1/2 (white arrowheads) suggesting its activation compared to normal colonocytes (200x). Blue, DAPI nuclear stain (1:10,000); Green, 1° mouse mAb anti-human β-catenin for membrane visualization (Sigma, 1:1,000), 2° Alexa 488 goat anti-mouse IgG (Invitrogen, 1:200). Red, 1° rabbit pAb anti-human ERK1/2 (Novus, 1:200), 2° Alexa 568 goat anti-rabbit IgG (Invitrogen, 1:200). Rabbit IgG Isotype control shows no positive staining (data not shown).
Using this approach, the relative concentrations of unphosphorylated ERK-1 and -2, singly phosphorylated ERK-1 and -2 (pERK1/2), and the activated dually phosphorylated ERK-1 and -2 (ppERK1/2) isoforms were measured in ACF. HeLa cell lysates were used as controls to determine the relative pIs of the individual isoforms: ERK1 = 5.78 ± 0.059; pERK1 = 5.42 ± 0.059; ppERK1 = 5.10 ± 0.059; ERK2 = 6.50 ± 0.059; pERK2 = 6.03 ± 0.059; ppERK2 = 5.55 ± 0.059. As shown in Fig. 3, colon crypts isolated from adjacent normal mucosa (N=8) showed no significant differences in the extent of ERK activation when compared to colonocytes prepared from independent biopsy specimens histologically confirmed as normal mucosa (N=6).
Figure 3. ERK activation state in normal colonocytes.
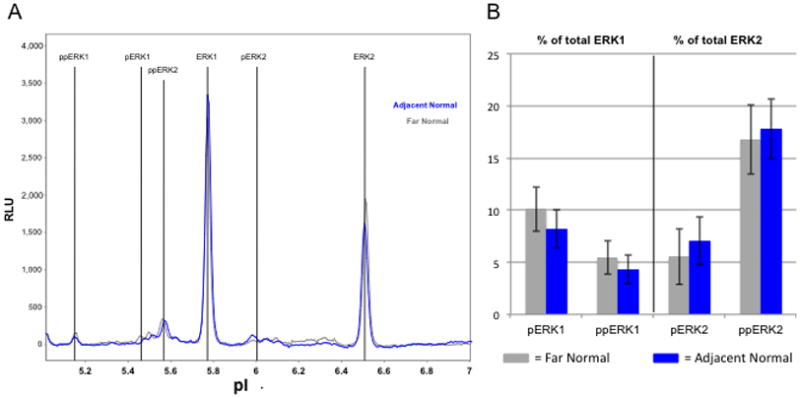
Microdissected colonocytes were prepared from frozen sections as described under Materials and methods. Normal-appearing crypts were microdissected from distant normal biopsies (gray) and from adjacent normal areas within ACF biopsies (blue) and subjected to NIA analyses. A) Representative electrophoretograms of ERK1/2 isoforms. B) Electrophoretograms were quantified for ERK1/2 phosphoforms using Compass software to measure the area under the curve. There are no significant differences in ERK activation between normal crypts according to their proximity to hyperplastic crypts. Error bars represent one standard deviation of the mean.
While a total of 172 ACF were used for the mutational analysis, a subset of 32 samples were sectioned and analyzed for ERK activation status using the NIA. The subset was chosen so as to maximize the use of the most recently obtained human tissues, thus minimizing the extent of protein degradation during sample storage. Of the 32 specimens that were selected, 20 were determined to provide a sufficient quantity of protein for the NIA analysis. Ultimately, colonocytes were micro-dissected from four ACF biopsies that were wild-type for BRAF and KRAS mutations, nine ACF positive for KRAS codon 12 or 13 mutation, and seven ACF positive for the BRAFV600E mutation. Samples were analyzed for ERK activation signatures and compared to colonocytes isolated from adjacent normal mucosa (Fig. 4). Relative concentrations of activated ERK 2 are significantly elevated in all ACF compared to normal crypts. However, only KRAS and WT ACF have significantly increased ERK1 activation. Distal colon ACF with constitutively activated KRAS activate ERK1/2 to the greatest extent, suggesting that these mutations are a more potent activator of the MAPK signaling cascade compared to the BRAFV600E mutation.
Figure 4. ERK activation state in aberrant crypt foci.

Microdissected colonocytes were prepared from frozen sections as described under Materials and methods. Nanofluidic proteomic immunoassays for ERK1/2 result in distinct electrophoretogram signatures (A,C,E) for ACF when compared with adjacent normal crypts (purple). Signatures shown represent three ACF genotypes: WT for BRAF/KRAS (A, green), KRAS mutant (C, blue), and BRAF mutant (E, red). Areas under the curve for each ERK1 and ERK2 phosphoform are quantified as a percent of the total ERK1 or ERK2 (B,D,F). Only WT for BRAF/KRAS ACF (B) and KRAS mutant ACF (D) have significantly elevated levels of pERK1 and ppERK1, while BRAF mutant ACF (F) have only slightly elevated (not significant) pERK1 and ppERK1 levels. All ACF significantly activate ppERK2. **: Significant using Tukey’s HSD procedure, two-sided alpha level of significance of 0.05. Error bars represent one standard deviation of the mean.
4 Discussion
The ‘classical pathway’ of colon carcinogenesis describes the step-wise formation of CRC resulting from the accumulation of genetic mutations, specifically those occurring within the APC, KRAS, and p53 genes[12, 13]. An alternative pathway that has been refined over the past decade results in a spectrum of hyperplastic lesions, ranging from hyperplastic polyps (HP) to sessile serrated adenomas (SSA)[14, 15]. The molecular features of the HPs vary with respect to their colonic location. For example, HPs and SSAs occurring in the proximal colon tend to have a higher frequency of BRAF mutations, microsatellite instability (MSI) and CpG island methylator phenotype (CIMP) in comparison to similar hyperplastic lesions found in the distal colon[16, 17]. In addition, these lesions generally lack APC mutations [18].
We believe that a subset of ACF described in the present study may represent precursor lesions to this alternative pathway. We show that mutations to either KRAS or BRAF are associated with the development of early hyperplasia within the distal colon. Consistent with the molecular aberrations that accompany this alternative pathway [17], we have also identified MSI and hypermethylation of Ras-association domain family 1a (RASSF1a) in 24% of hyperplastic ACF [19]. Because of their limiting sample size, however, it has not been technically feasible to combine genetic and proteomic analyses within the same specimen. The novel methods described in this report have begun to establish a robust approach that minimizes the amount of sample required per assay (Fig. 1), thus directly increasing the informative potential of a single biopsy specimen. By using this technique, it may ultimately be possible to understand the biological outcome of these early colonic lesions.
The equivalent distribution of KRAS and BRAF mutations within the serrated ACF observed in the present study, where we have greatly increased our sample size, differs somewhat from our previous findings [1]. While KRAS mutations occur at a greater frequency in serrated ACF than previously reported (41.9 vs. 18.8%), we have confirmed the rare occurrence of BRAF mutations in hyperplastic-distended crypts [1]. Although we still cannot determine with certainty whether these lesions occur in sequence or represent parallel pathways, the similar frequency of BRAF and KRAS mutations in serrated ACF and the infrequent occurrence of BRAF mutations in distended ACF raises the interesting possibility that distended lesions precede serrated ones. It is possible that the acquisition of a BRAF mutation favors its rapid growth into a serrated histological phenotype. On the other hand, KRAS-mutated distended crypts may activate several key inhibitory pathways, such as RASSF1/2, that may impede their progression to the serrated phenotype [20]. This mechanism may be bypassed in the event of a BRAF mutation [20]. Understanding the downstream signaling consequences of these oncogenic mutations will shed new insights into the relationship between serrated and distended ACF and their potential role within the alternative pathway to carcinogenesis.
Using our NIA approach, we have been able for the first time to evaluate the extent of ERK activation in biopsy specimens obtained from the human colon. Our results show that both KRAS and BRAF mutations result in elevated levels of the activated, dually-phosphorylated ERK2 isoform (Fig. 4). Interestingly, ACF that are wild-type for BRAF and KRAS also demonstrate ERK2 activation, suggesting that other mechanisms can stimulate this proliferative pathway, possibly including members of the EGFR family or other mutations within the Ras-Raf-Mek pathway that have not yet been uncovered. A relatively high frequency of ErbB2/3 and NRAS mutations in human CRC were recently described; their status in ACF, however, has not been established[21]. Nevertheless, our results demonstrate the importance of ERK2 activation in stimulating mucosal hyperplasia, especially since directly adjacent normal crypts do not appear to have increased ERK activation (Fig. 3 & 4). The high level of ERK2 activation compared to ERK1 is not surprising, given its known role in mediating cell proliferation and its typically higher level of expression[22, 23]. Interestingly, only WT and KRAS-mutant ACF showed a significant activation of ERK1, while BRAF-positive ACF demonstrated only a modest (< 2-fold) increase. It has been suggested that activated ERK1 may actually act as a competitive inhibitor of ERK2, thereby blocking its activity[23, 24]. These functional distinctions among the ERK isoforms and their differential activation in ACF of varying histologies again support the idea that the formation of distended crypts may precede the appearance of serrated ACF. Of course, determining the activation status of the ERK isoforms within these very small epithelial lesions would not have been possible using conventional immunohistochemistry, and further underscores the advantages of our novel experimental approach.
An additional consideration is the potential role of oncogene-induced senescence in determining the biological outcome of ACF. Several recent mouse genetic studies have examined the effects of these early oncogenic alterations within the intestinal mucosa. Carragher et. al[25] showed that LSL-BRAFV600E x AhcreERT+/o mice with an activating mutation of the BRAF oncogene develop serrated hyperplasia within three days after tamoxifen activation of the Cre recombinase. The mice developed small intestinal tumors by twelve weeks following BRAF activation, but this only occurred with a concurrent inactivation of p16INK4a via promoter methylation. Similarly, Bennecke et. al[26] showed that LSL-KRASG12D x villin-Cre mice develop serrated, hyperplastic colon crypts accompanied by increased p16INK4a expression. After crossing these mice with Ink4a/Arf−/− mice, which are deficient for p16 expression, > 50% of these mice developed tumors resembling traditional serrated adenomas by twelve weeks of age. These studies in mice highlight the important role of p16-mediated senescence in reducing the transformative potential of these oncogenic mutations[27]. In the present study using IF analysis, although p16 was consistently expressed within the colonic mucosa, its levels were not significantly different between hyperplastic and normal crypts (data not shown). However, it is possible that the activation state of p16 is differentially affected by BRAF or KRAS mutations. It has been shown that p16 is phosphorylated at four different serine residues, two of which may have different effects on its activation state; i.e.) Ser8 (inactivating)[28] and Ser152(activating)[29]. In future studies, we will develop NIA-based assays to examine these post-translational modifications of p16 to overcome the inherent limitations of IF analysis.
In summary, we describe a new methodology that will increase the informative potential of extremely small human biopsy specimens. By combining precise tissue microdissection of early hyperplastic lesions with subsequent proteomic analysis, it is now possible to examine post-translational modifications to specific signaling proteins within the context of underlying genetic and histological changes. Our results highlight the advantages of this approach over conventional immunohistological methods by focusing on two ERK isoforms and their relative activation states. Combining UV/IR microdissection with the NIA assay has made it possible to obtain quantitative proteomic information within the context of histological changes and upstream genetic abnormalities.
Acknowledgments
We thank Dr. J. Grady for his assistance with the statistical analyses and Dr. T.V. Rajan for providing a detailed histological analysis of ACF. We further acknowledge the State of Connecticut Department of Public Health, Biomedical Research Application #2012-0913 and the National Institutes of Health (1RO1CA159976) for funding these studies.
Abbreviations
- ACF
aberrant crypt foci
- ANM
adjacent normal mucosa
- CIMP
CpG island methylator phenotype
- CRC
colorectal cancer. ERK1/2, extracellular receptor kinase 1/2
- GLMM
general linear mixed model
- HP
hyperplastic polyp
- IF
immunofluorescence
- MAPK
mitogen activated protein kinase
- MSI
microsatellite instability
- NIA
nanofluidic proteomic immunoassay
- OCT
optimum cutting temperature medium
- pERK1/2
singly phosphorylated extracellular receptor kinase1/2
- ppERK1/2
dually phosphorylated extracellular receptor kinase1/2
- RASSF
ras-association domain family
- RT
room temperature
- SSA
sessile serrated adenoma
- UV/IR
ultraviolet-infrared
- WT
wild-type
Footnotes
The authors have declared no conflict of interest
References
- 1.Rosenberg DW, Yang S, Pleau DC, Greenspan EJ, et al. Mutations in BRAF and KRAS differentially distinguish serrated versus non-serrated hyperplastic aberrant crypt foci in humans. Cancer research. 2007;67:3551–3554. doi: 10.1158/0008-5472.CAN-07-0343. [DOI] [PubMed] [Google Scholar]
- 2.Rougier P, Mitry E. Epidemiology, treatment and chemoprevention in colorectal cancer. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO. 2003;14(Suppl 2):ii3–5. doi: 10.1093/annonc/mdg722. [DOI] [PubMed] [Google Scholar]
- 3.Yokota T, Sugano K, Kondo H, Saito D, et al. Detection of aberrant crypt foci by magnifying colonoscopy. Gastrointestinal endoscopy. 1997;46:61–65. doi: 10.1016/s0016-5107(97)70212-8. [DOI] [PubMed] [Google Scholar]
- 4.Pretlow TP, Pretlow TG. Mutant KRAS in aberrant crypt foci (ACF): initiation of colorectal cancer? Biochimica et biophysica acta. 2005;1756:83–96. doi: 10.1016/j.bbcan.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 5.Sakai E, Takahashi H, Kato S, Uchiyama T, et al. Investigation of the prevalence and number of aberrant crypt foci associated with human colorectal neoplasm. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive. Oncology. 2011;20:1918–1924. doi: 10.1158/1055-9965.EPI-11-0104. [DOI] [PubMed] [Google Scholar]
- 6.Anderson JC, Swede H, Rustagi T, Protiva P, et al. Aberrant crypt foci as predictors of colorectal neoplasia on repeat colonoscopy. Cancer causes & control: CCC. 2012;23:355–361. doi: 10.1007/s10552-011-9884-7. [DOI] [PubMed] [Google Scholar]
- 7.O’Neill RA, Bhamidipati A, Bi X, Deb-Basu D, et al. Isoelectric focusing technology quantifies protein signaling in 25 cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:16153–16158. doi: 10.1073/pnas.0607973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan AC, Deb-Basu D, Orban MW, Gotlib JR, et al. Nanofluidic proteomic assay for serial analysis of oncoprotein activation in clinical specimens. Nature medicine. 2009;15:566–571. doi: 10.1038/nm.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kedei N, Telek A, Czap A, Lubart ES, et al. The synthetic bryostatin analog Merle 23 dissects distinct mechanisms of bryostatin activity in the LNCaP human prostate cancer cell line. Biochemical pharmacology. 2011;81:1296–1308. doi: 10.1016/j.bcp.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garber K. Beyond sequencing: new diagnostic tests turn to pathways. Journal of the National Cancer Institute. 2011;103:290–292. doi: 10.1093/jnci/djr042. [DOI] [PubMed] [Google Scholar]
- 11.Yang S, Farraye FA, Mack C, Posnik O, O’Brien MJ. BRAF and KRAS Mutations in hyperplastic polyps and serrated adenomas of the colorectum: relationship to histology and CpG island methylation status. The American journal of surgical pathology. 2004;28:1452–1459. doi: 10.1097/01.pas.0000141404.56839.6a. [DOI] [PubMed] [Google Scholar]
- 12.Fearon ER. Molecular genetics of colorectal cancer. Annual review of pathology. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 13.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, et al. Genetic alterations during colorectal-tumor development. The New England journal of medicine. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 14.Longacre TA, Fenoglio-Preiser CM. Mixed hyperplastic adenomatous polyps/serrated adenomas. A distinct form of colorectal neoplasia. The American journal of surgical pathology. 1990;14:524–537. doi: 10.1097/00000478-199006000-00003. [DOI] [PubMed] [Google Scholar]
- 15.O’Brien MJ. Hyperplastic and serrated polyps of the colorectum. Gastroenterol Clin North Am. 2007;36:947–968. viii. doi: 10.1016/j.gtc.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 16.Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology. 2010;138:2088–2100. doi: 10.1053/j.gastro.2009.12.066. [DOI] [PubMed] [Google Scholar]
- 17.Weisenberger DJ, Siegmund KD, Campan M, Young J, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nature genetics. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 18.Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007;50:113–130. doi: 10.1111/j.1365-2559.2006.02549.x. [DOI] [PubMed] [Google Scholar]
- 19.Greenspan EJ, Cyr JL, Pleau DC, Levine J, et al. Microsatellite instability in aberrant crypt foci from patients without concurrent colon cancer. Carcinogenesis. 2007;28:769–776. doi: 10.1093/carcin/bgl209. [DOI] [PubMed] [Google Scholar]
- 20.van der Weyden L, Adams DJ. The Ras-association domain family (RASSF) members and their role in human tumourigenesis. Biochimica et biophysica acta. 2007;1776:58–85. doi: 10.1016/j.bbcan.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lloyd AC. Distinct functions for ERKs? J Biol. 2006;5:13. doi: 10.1186/jbiol46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vantaggiato C, Formentini I, Bondanza A, Bonini C, et al. ERK1 and ERK2 mitogen-activated protein kinases affect Ras-dependent cell signaling differentially. J Biol. 2006;5:14. doi: 10.1186/jbiol38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roskoski R., Jr ERK1/2 MAP kinases: structure function regulation. Pharmacological research: the official journal of the Italian Pharmacological Society. 2012;66:105–143. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 25.Carragher LA, Snell KR, Giblett SM, Aldridge VS, et al. V600EBraf induces gastrointestinal crypt senescence and promotes tumour progression through enhanced CpG methylation of p16INK4a. EMBO molecular medicine. 2010;2:458–471. doi: 10.1002/emmm.201000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bennecke M, Kriegl L, Bajbouj M, Retzlaff K, et al. Ink4a/Arf and oncogene-induced senescence prevent tumor progression during alternative colorectal tumorigenesis. Cancer cell. 2010;18:135–146. doi: 10.1016/j.ccr.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 27.Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–233. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 28.Guo Y, Yuan C, Weghorst CM, Li J. IKKbeta specifically binds to P16 and phosphorylates Ser8 of P16. Biochemical and biophysical research communications. 2010;393:504–508. doi: 10.1016/j.bbrc.2010.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gump J, Stokoe D, McCormick F. Phosphorylation of p16INK4A correlates with Cdk4 association. The Journal of biological chemistry. 2003;278:6619–6622. doi: 10.1074/jbc.C200622200. [DOI] [PubMed] [Google Scholar]