

Abstract
The mitochondrion relies on compartmentalization of certain enzymes, ions and metabolites for the sake of efficient metabolism. In order to fulfil its activities, a myriad of carriers are properly expressed, targeted and folded in the inner mitochondrial membrane. Among these carriers, the six-transmembrane-helix mitochondrial SLC25 (solute carrier family 25) proteins facilitate transport of solutes with disparate chemical identities across the inner mitochondrial membrane. Although their proper function replenishes building blocks needed for metabolic reactions, dysfunctional SLC25 proteins are involved in pathological states. It is the purpose of the present review to cover the current knowledge on the role of SLC25 transporters in health and disease.
Keywords: disease, mitochondrion, solute carrier family 25 (SLC25), transport
INTRODUCTION
Healthy cells rely on the finely tuned channelling of metabolic substrates and products across their subcellular compartments. To aid this purpose, a myriad of transporters and channels are expressed, correctly folded and properly targeted to their final location. This is how ions and metabolites are funneled across organelle membranes by means of transporters [1]. The mitochondrion is no exception to this, as kilograms of chemical species are exchanged daily in a cycle of metabolic supply and demand with other organelles and the cytoplasm [2]. Although the outer mitochondrial membrane is relatively permeable to solutes up to 5 kDa due the presence of voltage-dependent anion channels, the inner mitochondrial membrane is comparatively impermeable to maintain efficient oxidative phosphorylation [3,4]. To overcome such a physical barrier, 53 members of the mitochondrial inner membrane family of proteins [SLC25 (solute carrier family 25)] facilitate the transport of molecules involved in the urea and citric acid cycles, oxidative phosphorylation, DNA maintenance and iron metabolism among other processes [5–7]. The identity and physiological roles of many of these proteins have been successfully determined by means of ingenious approaches. However, the mechanism by which the transport of its substrates is accomplished is still the subject of a considerable amount of research.
Up to the present time, the only known representative structures of this family correspond to the solved NMR-based structure of the mitochondrial UCP2 (uncoupling protein 2; PDB code 2LCK) and the thoroughly characterized ANT (adenine nucleotide translocator; PDB code 1OKC) [8,9]. Despite their high degree of structural conservation, the chemical identities of the transported species can be considerably different. Discrete residues in a common substrate-binding site determine selectivity [10,11]. In addition, mitochondrial carriers may transport more than one kind of solute as shown in reconstitution experiments [5]. This also implies that the sensitivity to traditional inhibitors may vary among the different members of the SLC25 family [5].
Members of the SLC25 family can also be involved either directly or indirectly in the progression of pathological states. Growing evidence maps SLC25 family members, such as the ANT, in an intricate route by which pro-apoptotic proteins from the Bcl-2 family can elicit cell demise due to oxygen radical signalling [12]. The dual nature of these proteins may equip the cell with an additional tier of regulatory factors by which mitochondrial activity can be altered. The purpose of the present review is therefore to address the physiological and pathological roles of the mitochondrial six-transmembrane-helix transporters in the regulation of cell life and death. For an excellent set of reviews also devoted to the SLC25 family, we refer the reader to [1] and the more recent [7].
THE SLC25 CARRIERS IN TIME
Pioneering studies concerning the mechanisms of oxidative phosphorylation clearly showed that mitochondria play a vital role in the generation of ATP. Antithetically, little was known about the mechanisms underlying transport of molecules between the organelle and the cytosol. The first studies convincingly suggesting the presence of a mitochondrial carrier relied on experiments measuring exchange of 14C-labelled ADP and ATP between the mitochondria and the cytosol [3,13]. These experiments demonstrated the existence of an active ANT embedded in the inner mitochondrial membrane [13,14]. Krämer and Klingenberg [15] found that such an exchange reaction (ADP3−/ATP4−) was dependent on membrane energization. Using a different technique, i.e. the swelling of mitochondria in iso-osmotic solutions, transport systems for phosphate, dicarboxylates, citrate, 2-oxoglutarate and aspartate/glutamate were detected in the late 1960s [16]. These proteins were then purified and reconstituted in the subsequent years (for more details, see [17]). These results suggested that mitochondria express highly selective carriers to fulfil their metabolic needs. A key feature that aided the characterization of such transport systems was the inhibition of such transporters with substrate analogues and key inhibitors such as 2-n-butylmalonate for the case of the malate, citrate and 2-oxoglutarate systems [18].
During the late 1990s the complete sequence of ANT, UCP, PiC (phosphate carrier), citrate, oxoglutarate/malate and carnitine/acylcarnitine carriers were elucidated either by Edman degradation (for ANT and UCP) or specific gene sequencing [6,11,19–22]. These findings were a major breakthrough in mitochondrial transport research as their structural highlights suggested that these proteins belonged to a single family: in all cases, the carriers exhibited a tripartite structure composed of ~ 100 amino acid repeats [23]. Each repeat presented two transmembrane domains and a common signature sequence with the form P-X-D/E-X-X-K/R. Although these findings strongly suggested that proteins from the same family could facilitate many other transport activities, their isolation and structural determination remained a mystery, mainly because these proteins were poorly expressed (see [1]).
However, following the development of massive DNA sequencing techniques as well as bioinformatics tools, the detection of novel carriers from multiple sources was considerably accelerated. Today, researchers estimate the presence of 35 carriers in Saccharomyces cerevisiae, 58 in Arabidopsis thaliana and 53 in humans by means of transport and sequence analysis [24]. As metabolite carriers, these proteins are able to modulate several pathways within the mitochondria, peroxisomes and the cytosol. However, this cellular role indirectly confers on the SLC25 members a dual persona: dysfunctional transport activity of members of this family has been related to pathological states. A direct link between abnormal SLC25 activity and disease has been documented in cases of autism (aspartate/glutamate carrier) [25,26], carnitine deficiency (carnitine/acylcarnitine carrier) [27,28], HHH (hyperornithinaemia, hyperammonaemia and homocitrullinuria) syndrome (ornithine carrier) and the SLC25a38-associated congenital sideroblastic anaemia [27,29,30] (see the Physiological and pathological roles of SLC25 carriers section below).
IDENTIFICATION, PURIFICATION AND ANALYSIS OF MITOCHONDRIAL SLC25 PROTEINS
Early experiments aimed at detecting members of the SLC25 family relied on assays measuring transport of radiolabelled molecules or on the swelling of isolated mitochondria derived from the transport of an osmotically active compound [31,32]. Transport kinetics of radioactive solutes can be measured provided a fast method of transport inhibition is available (i.e. a selective carrier inhibitor). In cases where there is no available inhibitor, transport can be terminated by pelleting the mitochondrial sample out of the medium. This technique is known as centrifugal filtration [33]. Once the reaction has stopped, the presence of the transported molecule can be assayed in the pellet and the supernatant with an appropriate method. As with any other characterization, controls should be included. This involves the measurement of transport of molecules with structural similarities. When measuring transport using the swelling method, addition of an anion will only trigger swelling provided a cation is capable of crossing the inner mitochondrial membrane [31]. Transport of a cation can be mediated by simple diffusion (as in the case of NH4 + salts) or facilitated by ionophores or selective transporters. Once in the mitochondrial matrix, the presence of ammonium salt will induce water-dependent swelling. A marked decrease in the light-scattering properties of the mitochondrial suspension is indicative of a transport process. Some disadvantages of the swelling approach are the presence of more than one transporter with redundant activity (isoforms). In addition, the transport activity monitored in vitro does not necessarily mimic that of the transport activity of a given carrier in vivo [34]. Finally, if the level of expression of a carrier is very low, this would considerably affect the sensitivity of the swelling assay.
As with many integral mitochondrial membrane proteins, purification and reconstitution of operational mitochondrial carriers requires preliminary steps involving isolation of mitochondria and solubilization with suitable detergents. Once in the soluble form, mitochondrial carriers can be reconstituted in liposomes and their activity assayed using appropriate techniques (for authoritative methods, see [35,36]). To avoid contamination with outer membrane proteins, a suspension of purified mitochondria was incubated in hypo-osmotic buffer to produce mitoplasts, i.e. mitochondria devoid of the outer membrane. Once generated, mitoplasts are typically resuspended in non-ionic detergents with a low critical micellar concentration such as Triton (Triton X-100 or Triton X-114). Lipids such as cardiolipin (diphosphatidylglycerol) may be added in the solubilization buffer to enhance purification and activity following reconstitution of certain carriers such as PiC, ANT and the CIC (citrate carrier) [35–37]. Then, the pool of proteins is ultracentrifuged to separate insoluble material. The resulting supernatant is usually applied to a column containing the bone mineral hydroxyapatite. Compared with other mitochondrial proteins, SLC25 proteins usually bind with a relatively low affinity to this mineral. Hence, the first eluting fractions are enriched with these carriers. Additional chromatographic steps involving celite may increase purification yields of a given carrier [37,38]. Finally, variation of solubilization buffer conditions and chromatographic parameters can enhance the selectivity of the SLC25 protein to be purified.
A cleaner approach for measuring the transport activity of a given carrier involves the expression of the protein in Escherichia coli, and its purification and reconstitution in liposomes [39]. The liposome composition should closely mimic the inner mitochondrial membrane where the dimeric lipid cardiolipin can play an important role in the translocation mechanism [35,40,41]. In some cases, sequence comparison between a novel carrier and other carriers of known function can lead to an educated guess of the starting solute to be tested [42]. Unfavourable factors affecting the heterologous expression and reconstitution approach may include incorrect folding due to the lack of post-translational processing [43] or a disparate codon usage [44].
To bypass these common issues, a popular method is the expression of a solute carrier of unknown function in the model organism S. cerevisiae [45–47]. Mutants of this yeast lacking several of the 35 mitochondrial carriers are readily available [48]. Thus phenotype complementation studies can be performed with relative ease [49–51]. The combination of these approaches has led to the identification of the transport activity of dozens of carriers [1,7,52,53]. Despite being highly selective proteins, certain SLC25 carriers can transport solutes with dissimilar chemical moieties. This is the case of SLC25a10 or DIC (dicarboxylate carrier), which is able to transport succinate, phosphate, thiosulfate and malate [54,55]. This finally implies that many carriers are capable of transporting certain solutes in reconstituted systems, but may be responsible for different transport activities in vivo. In addition, recent studies have demonstrated that certain SLC25 proteins can be targeted to other subcellular compartments such as SLC25a17 in humans, which encodes a peroxisomal carrier able to transport CoA, FAD and NAD+ [56].
STRUCTURE OF SLC25 CARRIERS
The SLC25 proteins are nuclear encoded and targeted to the mitochondria by means of a mitochondrial targeting sequence, which may be located anywhere within the six transmembrane helices [57]. PiC appears to present an additional import signal within the N-terminus of the protein, but is dispensable for import [43,58]. Mitochondrial targeting sequences in SLC25 proteins are recognized by the TOM (translocase of the mitochondrial outer membrane) complex in the outer membrane, translocated through the Tom40 pore and inserted in the inner mitochondrial membrane by the carrier translocase Tim22. Once embedded in the inner membrane, carriers are thought to interact with other proteins and lipids, such as respiratory chain components and cardiolipin [57,59,60].
Although there are abundant functional data as to which carriers mediate a given transport activity, the only available 3D structures of solute carriers correspond to ANT (PDB code 1OKC) and UCP2 (PDB code 2LCK) [8,9]. The ANT model from Bos taurus mitochondria shows the carrier in the cytosolic conformation (opening facing the intermembrane space). From this crystal structure, it is possible to define this conformation as a 30 Å (1 Å = 0.1 nm) pit formed by six transmembrane helixes. On the bottom of the void (10 Å), the ANT signature motif RRRMMM is thought to constitute a nucleotide-binding domain (RRR), whereas the tri-methionine sequence has been proposed to form a ‘plug’ directly involved in the pore’s gate. On a proposed translocation model, a patch of positive amino acids located at the carrier entrance in the cytosolic conformation attracts ADP3− at its initial binding site. Additional positive pockets may provide secondary and tertiary binding sites down the ANT cavity. A set of tyrosine residues may provide a scaffold or ladder by which the adenine heterocycle may be transported downwards to the cavity gate. Once correctly bound to its solute, ANT as well as every single carrier is thought to undergo conformational changes within a matrix-orientated salt bridge, facilitating release of the anion [9,61]. For ANT, this conformation is thought to preclude transport of molecules in the opposite direction (ATP transport to the cytoplasm).
As mentioned above, the second SLC25 member with a solved structure at the atomic level is UCP2. A solution NMR method, coupled to a method that uses fragment fitting from the PDB, allowed the determination of the GDP-bound form of UCP2 [8]. Despite relatively low homology with ANT, most hallmarks from the mitochondrial carrier family can be clearly distinguished in this protein (i.e. the tripartite structure). Perhaps the most striking difference between ANT and UCP2 lies in the third repeat. Transmembrane helix 5 appears to be shifted towards the inter-membrane space, whereas the amphipathic helix 3 is tilted around 45°. This deviation appears to have a direct impact on the carrier architecture rendering it in a pore-like configuration.
PHYSIOLOGICAL AND PATHOLOGICAL ROLES OF SLC25 CARRIERS
Mitochondrial SLC25 carriers play a fundamental role during physiological and pathological processes. A schematic diagram illustrating the different carriers and their substrates is shown in Figure 1. Although their proper expression, targeting and transport mechanism is critical for mitochondrial–cytosol crosstalk, dysfunctional or defective carriers can lead to pathological conditions and disease.
Figure 1. Functional roles of mitochondrial six-transmembrane-helix solute carriers.
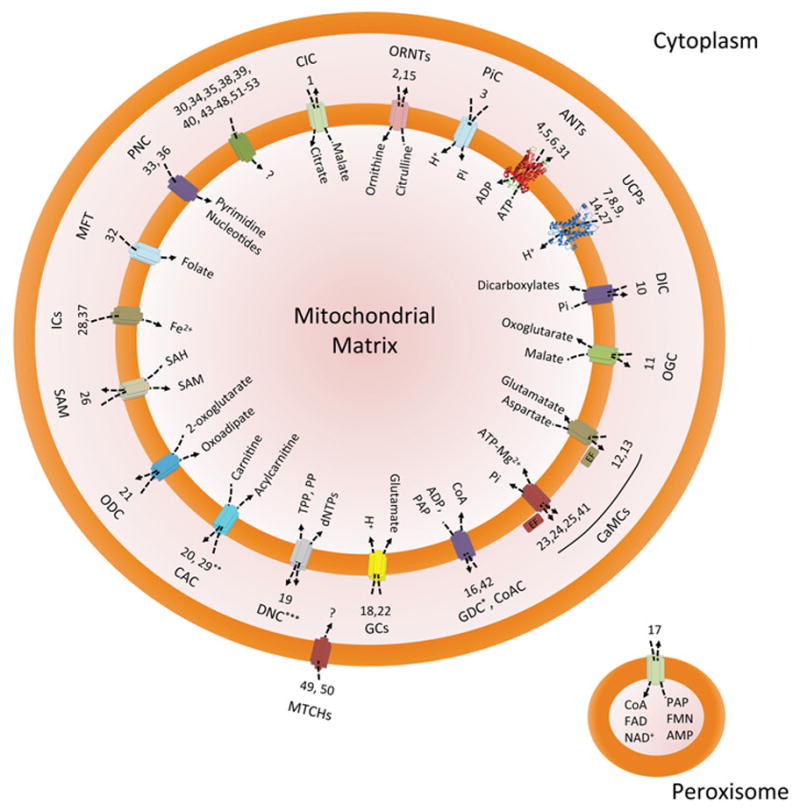
Schematic diagram illustrating the known and unknown activities of the 53 members of this family of proteins. Mitochondrial SLC25 proteins facilitate the transport of diverse chemical species (as summarized in the mitochondrial matrix compartment) by means of uniport, symport and antiport modalities (as described by arrows). The only carriers of known structure are currently SLC25a4 (ANT1) and SLC25a8 (UCP2). Cartoons representing these structures were rendered from available files deposited in the PDB (accession codes 1OKC and 2LCK respectively). Each mitochondrial carrier is identified by acronyms found in the abbreviation list, throughout the text and in previous literature followed by its respective numerical classification (i.e. 1, SLC25a1). Note that SLC25a17 (peroxisome), as well as SLC25a49 and SLC25a50 (outer mitochondrial membrane) are the only human carriers located in different subcellular membranes. *The transport properties of GDC still need to be unequivocally ascertained. **CAC can also exchange ornithine for acylcarnitine. ***The transport activity of DNC is probably reversible.
SLC25a1: citrate (tricarboxylate) carrier
Palmieri, Klingenberg and co-workers [62–64] first purified and reconstituted the CIC of the inner mitochondrial membrane. It was subsequently cloned by Kaplan et al. [19], which confirmed its identity as a SLC25 member. CIC is widely distributed, with its mRNA being detected in the majority of tissues, with highest expression in the liver and kidney [65]. It is a tricarboxylate transporter in that, in addition to citrate, it can also mediate the exchange of malate, isocitrate, aconitate and PEP (phosphoenolpyruvate). The exchange stoichiometry is exactly 1:1, e.g. one citrate out for one malate in, and is thus electroneutral. It can be selectively inhibited by 1,2,3-BTA (1,2,3-benzenetricarboxylate) as well as by phenylglyoxals and PLP (pyridoxal 5′-phosphate). Interestingly, CIC can also be inhibited by anthracyclines such as doxorubicin [66], which may account for the mitochondriotoxic effects of these anticancer agents. CIC has been found to be overexpressed in tumours, and CIC inhibition induces autophagy [67]. Lastly, studies in yeast indicated that, in order to facilitate efficient citrate shuttling, citrate synthase directly interacts with the SLC25a1 carrier [68,69].
CIC is necessary for fatty acid synthesis as it transports citrate out of the mitochondria where it can be cleaved to release acetyl-CoA, the essential starting block for fatty acid and sterol synthesis. The remaining OAA (oxaloacetate) is converted into malate, which then re-enters the mitochondria through CIC in exchange for citrate, and thus the cycle begins anew. From a pathological perspective, starvation [70] and hypothyroidism [71] decrease CIC expression and activity, whereas hyperthyroidism increases activity [72]. Decreased activity is also observed in Type 1, but not Type 2, diabetes [73,74]. Consistent with this, insulin up-regulates CIC expression [75] and its activity appears to be required for efficient glucose-stimulated insulin secretion in β-cells [76]. The SLC25a1 gene is also one of six mitochondrial protein-encoding genes that are deleted in DiGeorge/22q11 deletion syndrome, which causes a variety of cognitive and psychiatric disorders [77,78]. Whether the loss of SLC25a1 protein plays a causative role in the syndrome has yet to be tested and it should be noted that deletion of the carrier gene in yeast yielded no discernible phenotype [79]. CIC has also been reported to be up-regulated in ovarian and colon cancer [80], but again whether this is part of the oncogenic mechanism has yet to be established.
SLC25a2 and SLC25a15: ornithine carriers
There are two genes encoding mitochondrial ornithine carriers: SLC25a2 and SLC25a15 or, somewhat confusingly, ORNT2 and ORNT1 respectively. The first mitochondrial ornithine carrier was identified and cloned in S. cerevisiae [81]. In humans, ORNT1 was the first of the two isoforms to be cloned [30], with the highly homologous ORNT2 being cloned 4 years later [82,83]. ORNT1 is expressed in most tissues, with the highest levels being observed in the liver, pancreas, lung and kidney, whereas ORNT2 was more restricted to the liver, testis, spleen, lung and pancreas [83]. In all cases the level of ORNT2 mRNA was lower than that of ORNT1. Reconstitution of both isoforms demonstrated that they facilitate the import of ornithine into the matrix in exchange for citrulline and H+ out [84]. Transport by both carriers was further enhanced by Pi and dicarboxylates and inhibited by mercurials, PLP and spermine. However, ORNT2 has a lower activity than ORNT1 and also has a lower affinity for ornithine and citrulline [83].
ORNTs are essential for the urea cycle. Ornithine is brought into the matrix through ORNTs where it is converted into citrulline by the addition of carbamoyl phosphate. The citrulline is then exported back out by ORNTs where it can be processed further, ultimately producing urea and ornithine. In addition to ornithine, ORNT1 can also transport the amino acids lysine and arginine, whereas ORNT2 can transport histidine [83]. Therefore ORNTs may represent a major mechanism by which some of the amino acids required for mitochondrial protein synthesis are imported into the mitochondrion.
It has been well established that inactivating mutations in the SLC25a15 gene, i.e. the one that encodes ORNT1, cause HHH syndrome [30], which is characterized by early-onset neurological deficits. This further emphasizes the critical role of this carrier in the urea cycle. Whether there are mutations that cause defective ORNT2, and therefore HHH, has not been documented. Interestingly, overexpression of ORNT2 in fibroblasts from patients with ORNT1 deficiencies can partially rescue the phenotype, indicating that there is some functional redundancy between the two isoforms [82]. Indeed, HHH patients who have an additional gain-of-function mutation in their SLC25a2 (ORNT2-encoding) gene have much milder symptoms than their siblings [85].
SLC25a3: Pi carrier
The mitochondrial PiC transports Pi to the mitochondrial matrix to supply Pi required for ADP phosphorylation. PiC is translated as two isoforms (PiCa and PiCb) as a result of alternative splicing events, which differ in the presence of two exons (3A and 3B) [86–88]. Although the former is expressed in muscle, the latter is expressed ubiquitously. Most of what is known about PiC comes from studies in S. cerevisiae mitochondria.
Mitochondrial PiC deficiency in humans has a lethal outcome. In 2007, the first report describing a homozygous mutation (G72E) in two patients, presenting hypertrophic cardiomyopathy and muscular hypotonia, demonstrated the importance of this carrier for proper oxidative phosphorylation [89]. Studies from biopsies obtained from these patients revealed a non-canonical mitochondrial network with apparently low mitochondrial proliferation. It is possible that the lack of this protein considerably decreases ΔΨ m and this in turn impairs proper import of nuclear-encoded mitochondrial precursor proteins, as seen in model organisms [90].
A potential role for PiC in mitochondria-dependent cell death is also beginning to be understood as recent studies have shown that overexpression of this carrier triggers the intrinsic pathway of apoptosis in many scenarios. Kroemer and co-workers detected the functional interaction between the CMV (cytomegalovirus)-encoded protein vMIA (viral mitochondria-localized inhibitor of apoptosis) and PiC [91]. Once bound, the complex suppresses phosphate transport to the mitochondria thereby inhibiting ATP synthesis. This collapses the phosphorylation potential within the cell, which in turn leads to the characteristic CMV-induced cell rounding. In addition, a role for PiC in mitochondrial permeability transition has also been proposed [92]. This suggests that cell death pathways are conserved in eukaryotic organisms and mitochondrial carriers may indeed play a role in such events.
SLC25a4, SLC25a5, SLC25a6 and SLC25a31: adenine nucleotide translocases
Translocation of adenine nucleotides between the mitochondria and cytosol is among the most characterized of mitochondrial activities. The carriers SLC25a4 (ANT1), SLC25a5 (ANT2), SLC25a6 (ANT3) and SLC25a31 (ANT4) are responsible for this finely tuned task [42]. Their role as the main suppliers of ADP to the mitochondria and ATP to the cytoplasm confers on these proteins metabolic control over many cellular processes. As such, their function is a major determinant of cell fate. This means that ANT expression and transport activity must be finely tuned in order to avoid disturbances in cell homoeostasis. ANT1 is preferentially present in differentiated tissues (i.e. brain, heart and skeletal muscle). ANT2, however, is expressed mainly in proliferating tissues such as tumours. ANT3 is universally expressed in every tissue, and ANT4 is present in liver and testis, although at very low levels when compared with ANT3 [42,93].
Being one of the most expressed proteins in the inner mito-chondrial membrane, ANT can bind respiratory supercomplexes and components of the protein import machinery [59]. In addition, ANT can bind type 2 transglutaminase, a Ca2+ -dependent enzyme involved in post-translational modification of glutamine residues [94]. Studies of this potential interaction demonstrated that transglutaminase, a disulfide isomerase, is crucial in down-regulating ANT1 oligomerization under physiological conditions, thus maintaining its ATP/ADP translocating activity. In cardiac tissue, ANT can also interact with the intermembrane space protein creatine kinase, gaining preferential access to ATP, which is needed to synthesize creatine phosphate in cells with high energetic needs [95].
Pathologically, mutations in the SLC25a4 gene are associated with adPEO (autosomal dominant progressive external ophthalmoplegia). Expression of mutant forms of ANT associated with adPEO reduces ATP/ADP transport [96], and leads to large-scale mtDNA deletions which are characteristic of the disease [97]. ANT deficiency also leads to dilated cardiomyopathy. Mice lacking Slc25a4 display myocardial hypertrophy and ventricular dilation characterized by increased cardiomyocyte apoptosis [98]. The mechanism by which ANT genetic invalidation elicits cell demise may be related to a decrease in the mitochondrial BPC (basal proton conductance) [99]. In mouse muscle mitochondria, the presence of ANT is responsible for 50–66 % of the BPC. Respiratory chain-mediated superoxide production may increase considerably when BPC is poor. Under these conditions, the protonated form of superoxide may cross membranes and further activate pro-apoptotic pathways [99].
If ANT inactivity renders cardiomyocytes prone to disease, it is tempting to hypothesize that ANT overexpression should be cardioprotective. However, cardiomyocytes with elevated levels of ANT are more susceptible to Bax-dependent apoptosis [12]. Bax is thought to mediate translocation of cytochrome c to the cytosol, thus initiating intrinsic apoptosis. The mechanism by which ANT (an inner membrane protein) can facilitate Bax-induced outer membrane permeabilization is not evident; however, this is true even in model organisms [100]. In order to be recruited to the outer membrane in an ANT-dependent pathway, Bax translocation necessitates ROS (reactive oxygen species) signalling as MnTBAP [manganese(III) tetrakis-(4-benzoic acid)porphyrin chloride; a ROS scavenger] efficiently inhibits this process [12].
SLC25a7, SLC25a8, SLC25a9, SLC25a14 and SLC25a27: uncoupling proteins
The substantial family of UCPs includes SLC25a7 (UCP1), SLC25a8 (UCP2), SLC25a9 (UCP3), SLC25a14 (UCP5) and SLC25a27 (UCP4), all of which have been cloned and characterized since their initial discovery by the groups of Nicholls [101] and Klingenberg [102]. With the exception of UCP2, which is ubiquitously expressed [103], UCPs exhibit tissue-specific expression patterns. UCP1 expression is primarily restricted to brown adipose tissue [104], UCP3 is specific to cardiac and skeletal muscle [105], and UCP4 and UCP5 are primarily expressed in the brain [106]. Their regulation is also somewhat different. UCP1 is activated by fatty acids and can be inhibited by purine nucleoside di- and tri-phosphates such as GDP [107]. In contrast, although UCP2 and UCP3 can be also be activated by fatty acids, they are less sensitive to purine inhibition. Less is known about the neuronal UCPs, although both UCP4 and UCP5 have been reported to be GDP-sensitive [108].
UCPs are not involved in basal proton leak across the inner membrane (which is most likely to be through ANT). Instead, they are responsible for inducible increases in proton conductance. However, the mechanism by which UCPs transport protons across the inner membrane is still controversial and may be explained in terms of three different models [107]. In one model, negatively charged fatty acids embedded in UCP facilitate the movement of the positive protons through the transporter. In a second model, protonated fatty acids flip-flop to the matrix where they release the proton and are then transported back to the intermembrane space by UCP. A third model suggests that UCPs are true proton carriers and that fatty acids stimulate transport by relieving tonic inhibition of UCP by purine nucleotides. UCPs are also highly regulated at the protein expression level. For example, cold will rapidly increase levels of all UCPs, as will fatty acids and capsaicin [107]. UCP2 and UCP3 can also be up-regulated by cytokines and thyroid hormone [109,110]. Oxidative stress has been shown to up-regulate UCP5, whereas insulin decreases both UCP4 and UCP5 levels [111,112].
Physiologically, UCP1 is involved in heat production in brown adipose tissue, which is required for non-shivering thermogenesis in neonates, and Ucp1-null mice are more cold sensitive than their wild-type counterparts [113]. Less is known about the physiological role of UCP2 and UCP3. One suggestion is that, by mildly uncoupling oxidative phosphorylation, they reduce ROS production, thereby protecting the cell from these harmful radicals [107]. Another related function is that they may transport fatty acid peroxides out of the mitochondrial matrix, again resulting in a reduction in toxic radicals [114]. Finally, evidence suggests that UCP2 may act as a negative regulator of glucose-stimulated insulin secretion in the pancreas [115]. Still less is known about the physiological contributions of UCP4 and UCP5, although both have been shown to reduce mitochondrial ROS production, much like the other UCPs. In addition, UCP4 interacts with complex II and may regulate the contribution of this complex to oxidative phosphorylation [116].
Pathologically, both pharmacological and genetic inhibition of UCP2 has been shown to reverse the deleterious effects of obesity-and diabetes-induced pancreatic islet dysfunction [115,117]. A recent study has also implicated mutations in the SLC25a7 gene as a cause of diabetic retinopathy [118], indicating that UCP1 may also be present in the retina. Mutations in UCP3-encoding genes have also been correlated with obesity and diabetes [119]. Mutations in the neuronal-specific UCP4-encoding gene have been associated with schizophrenia [120]. However, what effect all of these mutations have on the respective UCP’s expression and function has not been tested. UCPs may also play a role in oncogenesis. UCP2 expression in particular has been reported to be up-regulated in numerous cancers, and its expression is correlated with chemotherapeutic resistance [121]. Gene silencing of UCP2 induced cell death in primary breast cancer cells [122], and pharmacological blockade of UCP2 sensitized leukaemia cells to chemotherapeutic agents [123]. UCP4 and UCP5 are also up-regulated in breast and colon cancer cells respectively [111,124], although a causative role for either of these proteins has yet to be established.
SLC25a10: dicarboxylate carrier
DIC, which is ubiquitously expressed, was first identified in S. cerevisiae [125] and subsequently in mammalian mitochondria [126]. DIC transports malate (and succinate) out of the mitochondrion in exchange for Pi and is therefore electroneutral. It is inhibited by Pi and other phosphate analogues [127] as well as substrate analogues such as alkyl malonates. Inhibition of DIC may also play a role in fatty-acid-mediated uncoupling [128,129]. DIC appears to be similar to CIC in that it interacts with a key component of the TCA (tricarboxylic acid) cycle, malate dehydrogenase in this case [130], presumably to improve functional coupling of the pathway with the shuttle system.
DIC plays a role in gluconeogenesis by acting as an OAA shuttle. OAA is converted into malate by malate dehydrogenase; physical binding of this enzyme to DIC greatly enhances functional coupling. The malate is then transported out by DIC where it is reconverted into OAA. Cytosolic OAA is then converted into PEP by carboxylate kinase. The Pi/malate exchange also provides another source of cytosolic malate to be exchanged for citrate by CIC. Thus DIC could also play a role in fatty acid synthesis [131] and indeed inhibition of DIC prevents fat accumulation during differentiation of 3T3-L1 adipocytes [132]. Another less appreciated physiological role for DIC is that, in addition to transporting dicarboxylates, it is responsible for the import of GSH into the mitochondrial matrix. This trait appears to be shared with the OGC (oxoglutarate carrier; SLC25a11, see below). Overexpression of DIC accelerated mitochondrial GSH accumulation [133], whereas knockdown of the carrier reduced it by ~ 40 % [134]. Thus DIC is important for maintaining the redox status of the mitochondrion.
Little is known about DIC in pathology. Unlike CIC, whose activity and expression are decreased in Type 1 diabetes, the specific activity of DIC is actually increased. However, similar to CIC, its activity remains unchanged in Type 2 diabetes [73]. Prenatal exposure of rats to the pollutant perfluoro-octane sulfonate resulted in the down-regulation of cardiac DIC (along with other carriers) when the pups were weaned [135]. The autosomal recessive disorder ethylmalonic encephalopathy is associated with increased levels of ethyl malonate, which inhibits DIC [136]. How this contributes to its pathology remains to be elucidated.
SLC25a11: oxoglutarate carrier
The OGC, which was first cloned by Walker and Palmeiri in 1990 [11], mediates the export of oxoglutarate in exchange for a dicarboxylate (usually malate). Binding of succinate to the matrix side of the carrier increases the affinity for malate and phenylsuccinate acts as an inhibitor of OGC. PLP, phthalonate and retinoic acid also inhibit OGC [39,137]. It is an important component of the malate–aspartate shuttle. The malate OGC brings in is converted into OAA, which in turn is converted into oxoglutarate by glutamate OAA transaminase, which simultaneously converts glutamate into aspartate. The resultant oxoglutarate is then exported by OGC.
Like DIC, OGC also contributes to the mitochondrial uptake of GSH. Blockade of OGC with phenylsuccinate decreases mitochondrial GSH levels by 40–50 % [138]. OGC also may play a role in glucose-stimulated insulin secretion, as siRNA-mediated knockdown of the carrier reduced insulin release in both β-cells and isolated pancreatic islets [139]. The OGC has also been proposed as the porphryin transporter necessary for mitochondrial import of the precursor porphryin for final conversion to haem. This is on the basis of the finding that OGC inhibition blocks porphryin accumulation and that porphryins can inhibit succinate/oxoglutarate transport by OGC [140].
With regard to pathology, down-regulation of OGC has been reported in horse muscle with recurrent exertional rhabdomyolysis [141]. OGC may also modulate apoptosis. OGC has been reported to bind the anti-apoptotic proteins Bcl-2 and Bcl-XL [142,143], and knockdown of OGC induced apoptosis in mouse insulinoma cells and Caenorhabditis elegans [143]. Moreover, association of Bcl2 with OGC increased mitochondrial GSH accumulation [142], which may account for some of its cytoprotective actions. Antithetically, alcohol intake has been shown to reduce mitochondrial GSH by inhibiting OGC [144].
SLC25a12, SLC25a13, SLCa23, SLC25a24 and SLC25a25: Ca2+-sensitive mitochondrial carriers
Under various physiological and pathological conditions, the ER (endoplasmic reticulum) and mitochondria mediate Ca2+ -dependent metabolic shifts. The Ca2+ cross-talk between the ER and mitochondria can tune the activity of CaMCs (Ca2+ -sensitive mitochondrial carriers) [145]. CaMCs are encoded by the SLC25a12 [AGC1 (aspartate/glutamate carrier 1)], SLC25a13 (AGC2) [145], SLC25a23 [APC2(ATP-Mg/Pi carrier)], SLC25a24 (APC1) and SLC25a25 (APC3) genes [146].
In all cases, CaMCs contain classical octahedral Ca2+ -co-ordination cages (EF hands) in the N-terminal domain, and a highly conserved mitochondrial carrier domain in the C-teminal portion of the protein. The first CaMC to be cloned was a protein of 74 kDa proven to bind calcium and be present in human muscle and brain [147]. Later, this protein was identified by its transport properties as the mitochondrial AGC1 [145]. Its Ca2+ -binding domain is composed of four potential EF hands, three of which may be fully capable of binding the divalent cation. Because AGC1 is predominantly expressed in brain, AGC1 has been implicated in the Ca2+ -induced malate/aspartate/NADH shuttle in neurons and may constitute a protective factor during glutamate excitotoxicity [148]. Mice lacking AGC1 are growth-retarded and display motor co-ordination dysfunctions probably owing to an impaired myelin metabolism triggered by low levels of N-acetylaspartate [149]. In a related case report, AGC1 inactivity in a patient could be associated with a mutation in a conserved glutamine residue. The pathological consequences of this mutation included severe hypotonia, halted psychomotor development and convulsions. As with mice lacking AGC1, the phenotype observed in this patient involves global cerebral hypomyelination [150].
The second CaMC to be described encodes a 675-amino-acid protein sharing 78.3 %identity with AGC1 [151]. As with AGC1, AGC2 mediates proton-coupled glutamate co-transport to the mi-tochondrial matrix in exchange for mitochondrial aspartate [145]. AGC2 is required for nucleotide and protein biosynthesis as well as for the urea cycle by delivering aspartate from mitochondria to the cytosol. Dysfunctional AGC2 induces a metabolic disease characterized by adult-onset type II citrullinaemia and neonatal intrahepatic cholestasis (for authoritative reviews, see [7,27]). AGC2-knockout mice fail to recapitulate human AGC2 deficiency unless glycerol-3-phosphate dehydrogenase is also genetically deleted [152]. This will suppress availability of NADH in the mitochondrial matrix and thus contribute to the deleterious effects of AGC2 deficiency [152].
Fiermonte et al. [146] characterized the function of the SCaMCs (small calcium-binding mitochondrial carriers) APC1, APC2 and APC3. SCaMCs mediate the exchange of adenine nucleotides, in particular ATP-Mg for Pi, thereby promoting the net uptake or efflux of adenine nucleotides into or from the mitochondria (Figure 1). SCaMCs activity possibly results in the F1Fo-ATPase- dependent hydrolysis of ATP and the establishment of a steady ΔΨm required for protein import and the initiation of metabolism in newborn mitochondria [153]. Although APC1 and APC3 are expressed in testis, APC2 can be found mostly in brain and skeletal muscle. APC3 is heavily expressed in lung and brain [146]. SCaMCs play a critical role in the regulation of cell death pathways. During ischaemia/reperfusion, SCaMCs may behave as protective factors by mediating ATP/ADP-induced Ca2+ buffering and facilitating mitochondrial permeability transition pore closure. This in turn protects cells from necrotic cell death triggered by oxidative stress and Ca2+ excess. Conversely, SCaMC overexpression is a typical trait of cancer cells and is thought to represent a negative feedback factor by which transformed and malignant cells can elude death [154].
SLC25a16: Grave’s disease carrier
Grave’s disease is an autoimmune disease of the thyroid that results in hypothyroidism. The GDC (Grave’s disease carrier) SLC25a16 was identified in patients with a disease of the same name due to the presence of antibodies to mitochondrial antigens in serum [155]. This carrier was originally thought to be expressed only in the thyroid. However, tissue analysis of the bovine GDC also revealed expression in the liver, lung, kidney and striated muscle [156]. Very little is known about the function of this carrier. However, deletion of the S. cerevisiae Leu5p (which is 35 %identical to the human protein) results in a 15-fold reduction in mitochondrial CoA levels, suggesting that it is involved in the import of this cofactor [157]. A rescue experiment involving expression of human GDC was able to normalize this phenotype, indicating that the human carrier can also support CoA transport. Whether GDC is indeed a CoA carrier still requires more formal testing.
SLC25a17: the peroxisomal transporter
It is a common notion to assume that mitochondrial carriers (as the name states) are exclusively located in the inner mitochondrial membrane. However, SLC25a17 (along with SLC25a49 and SLC25a50) can be considered an exception to this rule. The existence of human SLC25a17 was first reported in 1998 as a protein displaying 33 % identity with the peroxisomal integral membrane protein CbPMP47 of Candida boidinii [158]. In 2012, Palmieri’s group showed that SLC25a17 was able to transport CoA, FAD, FMN and AMP among other nucleotides almost exclusively by a counter-exchange mechanism in a PLP-sensitive fashion [56]. The SLC25a17 transcript was detected in every tissue; however, its expression was shown to be higher in testis, prostate and ovaries.
In pathological states, SLC25a17 down-regulation (along with many other mitochondrial transcripts) was detected in horses presenting recurrent exertional rhabdomyolysis, a myopathy involving inhibition of muscular relaxation [141]. In that study, the authors proposed that the mitochondrial dysfunction derived from an excess of cytoplasmic calcium triggering ATP depletion could underlie the clinical signs of the disease. A direct correlation between SLC25a17 down-regulation and this pathology remains to be proven.
SLC25a18 and SLC25a22: glutamate carriers
In addition to the AGCs, there are also two dedicated GCs (glutamate carriers) in the inner mitochondrial membrane: SLC25a18 or GC2 and SLC25a22 or GC1. These isoforms are highly homologous, having 63 % identity, and reconstitution experiments demonstrate that both isoforms transport glutamate along with H+ [159]. Thus transport is dependent on the chemical component of the protonmotive force (ΔpH). The main difference between the two isoforms appears to be kinetic, with GC1 having a higher Km and V max than GC2. Agents such as PLP, bathophenanthroline and tannic acid inhibit both equally. There are also tissue differences, with GC1 having a much higher expression level in most tissues, especially in the liver, pancreas and spleen [159].
The primary physiological role of the GCs is to import glutamate into the mitochondrial matrix where it is the substrate of the mitochondrial-restricted glutamate dehydrogenase. This converts glutamate into oxoglutarate and NH4 +, which then enters the urea cycle. The proposed model is that glutamate dehydrogenase produces glutamate from oxoglutarate and NH4 +, which is then exported out of the mitochondria by the GC1 acting in reverse mode. The cytosolic glutamate then stimulates vesicle mobilization [160]. Like CIC, GC1 is critical for glucose-stimulated insulin secretion in pancreatic β-cells, as knockdown of GC1 attenuated the ability of glucose to induce insulin release [161]. As seen in vitro, these results have been recapitulated in vivo in glutamate-dehydrogenase-deficient mice [162], indicating the importance of this pathway for insulin secretion. This would certainly explain the considerable levels of GC1 expression in the pancreas.
Pathologically, a missense mutation in the SLC25a22 gene (P206L) has been identified in autosomal recessive neonatal myoclonic epilepsy, and fibroblasts from these patients exhibited defective glutamate transport and oxidation [163]. SLC25a22 mutations have also been linked to early infantile epileptic encephalopathy [164].
SLC25a19: deoxynucleotide carrier
As the name suggests, the DNC (deoxynucleotide carrier) is thought to be one of the main routes of import for deoxynucleotides into the mitochondrion (along with SLC25a33, which is pyrimidine dNTP selective, see below). Cloning and reconstitution of the human carrier revealed that the DNC preferentially transported all four dNDPs rather than dNTPs in exchange for either ATP or ADP [165]. This transport was completely inhibited by PLP and partially by bongkrekic acid and atractyloside. Tissue analysis of the mRNA and protein demonstrated a near ubiquitous expression pattern, with the highest protein levels seen in the kidney and lung [165].
The presumed function of the DNC is the import of dNDPs into the matrix where they can be converted into dNTPs and therefore be used for the replication of the mtDNA. An additional function as a ThPP (thiamine pyrophosphate) transporter has also been inferred from Slc25a19-knockout mice, which exhibited a profound reduction in mitochondrial ThPP levels to the point of being undetectable [166]. It has even been proposed that this is the main function of the DNC rather than deoxynucleotide transport [167].
A mutation in SLC25a19 was found to be associated with Amish microcephaly, and reconstitution of the mutant carrier demonstrated reduced dNTP transport activity [168]. Interestingly, cells isolated from patients also exhibited marked reductions in mitochondrial ThPP [166]. The DNC was originally though to be involved in the mtDNA depletion caused by antiviral nucleosides, i.e. it transported the drugs into the matrix where they inhibit DNA polymerase-γ. Indeed, transgenic mice overexpressing the DNC specifically in the heart were more sensitive to nucleoside-induced mtDNA deletion, mitochondrial damage and cardiomyopathy than their non-transgenic counterparts [169]. In contrast with this study, Lam et al. [170] showed that neither overexpression nor depletion of the DNC affected nucleoside drug uptake and mtDNA depletion. It is possible that in this case the pyrmidine nucleotide carrier SLC25a33 (and potentially SLC25a36) was an alternative route these drugs took into the mitochondrion.
SLC25a20 and SLC25a29: carnitine/acylcarnitine carriers
There have been two carnitine/acylcarnitine transporters identified to date. The first to be cloned and characterized in depth were the CACs (carnitine/acylcarnitine carriers) of rat and S. cerevisiae [22,46]. CAC transports L-carnitine over D-carnitine in an acyl-chain-length-dependent manner. Acetylcarnitine and propionylcarnitine are transported with high affinity, whereas medium- and long-chain fatty acid esters of carnitine are poor substrates for S. cerevisiae CAC. The rat CAC (SLC25a20) mediates a similar activity, but with an affinity for medium-and long-chain esters of carnitine higher than for short-chain carnitines and L-carnitine [171]. Both the reconstituted carrier from liver mitochondria as well as the recombinant CAC require a countersubstrate for activity and catalyse translocation through a Ping Pong mechanism [22,28,38,171,172]. The CAC can be inhibited by maleimides, mercurials and sulfobetaines. A second CAC isoform has been more recently cloned and termed the CACL (CAC-like protein) or SLC25a29 [173]. Although this protein shares only modest identity with the original CAC, it too exchanges carnitine for palmitoylcarnitine. The CAC is expressed in all tissues, but has especially high levels in liver and cardiac muscle [174]. In contrast, the CACL is somewhat more restricted, being primarily expressed in the heart, brain, liver and kidney [173].
The CAC is essential for β-oxidation. Cytosolic fatty acids are transferred to carnitine by CPT1 (carnitine palmitoyltransferase-1) in the outer membrane. The resultant acylcarnitine is then imported into the matrix by the CAC in exchange for carnitine (and is hence electroneutral). The fatty acids are then transferred off the carnitine and on to CoA by CPT2, where they can now enter the β-oxidation pathway. Whether the CACL is of similar importance has yet to be ascertained. In this regard, many nonsense, frameshift and splicing mutations in the SLC25a20 gene have been identified in patients with CAC deficiency disease [174–177]. To date, 35 different mutations of the CAC have been detected in carnitine carrier-deficient patients [28]. Usually, disease-triggering mutations affect residues of the carrier signature motif. CAC activity deficiency can also be a consequence of an elongation in the C-terminal portion of the protein [174]. In any case, impaired β-oxidation derived from defective CAC results in decreased carnitine/acycarnitine transport. The clinical consequences of such alteration may involve hypoglycaemia, hyperammonaemia, cardiomyopathy, liver failure and encephalopathy [177]. Such findings underscore the essential requirement of the CAC for effective β-oxidation to occur. Moreover, they suggest that CACL cannot compensate for the lack of CAC and that there is very little functional redundancy between the two carriers. To date no inactivating mutations in the SLC25a29 gene have been identified.
Physical inactivity leads to a down-regulation of SLC25a20, whereas exercise up-regulates it [178]. Less is known regarding the regulation of CACL, but its expression is enhanced by starvation [173] and exercise [179]. Diabetes is also associated with CAC down-regulation [180]. Anti-hyperlipidaemic drugs, such as statins and fibrates, up-regulate expression of the CAC [181]. This would be expected to enhance β-oxidation and therefore may contribute to the lipid-lowering effects of these agents.
SLC25a21: oxoadipate carrier
The mitochondrial ODC (oxodicarboxylate carrier), also known as the oxoadipate carrier, mediates the import of 2-oxoadipate into the mitochondrial matrix in exchange for 2-oxoglutarate out. Walker, Palmieri and co-workers first characterized two isoforms of this transporter in yeast [45]. This same group then went on to clone the human and rat orthologues [182], although in these cases only a single isoform was identified. The human and rat ODCs are ubiquitously expressed both at the mRNA and protein levels, with very little variation between tissues. Reconstitution of the human ODC in liposomes revealed a PLP- and bathophenanthroline-sensitive oxoadipate/oxoglutarate counter-exchange mechanism as its primary function [182]. The protein can also transport glutarate and aminoadipate and, to a lesser extent, citrate. A recently identified adipose tissue-restricted splice variant of the ODC, termed ODC-AS, possesses a 29-amino-acid replacement on the protein C-terminus [183].
Oxoadipate is the primary product of lysine and tryptophan catabolism, which occurs in the cytosol. The ODC then brings the oxoadipate into the mitochondria, in exchange for oxoglutarate, where it is converted into acetyl-CoA and enters the TCA cycle. The transport of oxoglutarate into the cytosol is necessary for the first step of lysine breakdown, which is catabolized by lysine-oxoglutarate reductase. Interestingly, siRNA knockdown of the ODC-AS splice variant prevented adipocyte differentiation and lipid accumulation, presumably by decreasing the available acetyl-CoA for fatty acid synthesis. Pathologically, Palmieri and co-workers proposed that a defect in the ODC may contribute to oxoadipate acidaemia, a neurological disorder that is characterized by excessive amounts of oxoadipate and aminoadipate [182]. Deletion of the 14q13.3 region of chromosome 14, which contains the SLC25a21 gene, has also been reported in lung cancer [184], and down-regulation of this gene is associated with diabetic retinopathy in rats [185].
SLC25a26: S-adenosylmethionine carrier
SAM (S-adenosylmethionine) is required for several mito-chondrial processes, including DNA, RNA, protein and sterol methylation, and the synthesis of biotin. In liver cells, 30 % of the total cellular SAM is mitochondrial. However, mitochondria do not possess the machinery to make SAM de novo and must import it. Wagner and co-workers first characterized a mitochondrial SAM transporter and found that it was not dependent on ΔΨm and could be inhibited by sinefungin and SAH (S-adenosylhomocysteine), but not methionine or adenosine [186]. Subsequent cloning of the yeast and human genes by Palmieri’s group revealed an SLC25 family member carrier that imported SAM in exchange for SAH [50,187], although the yeast carrier may have additional uniport activity. The mRNA for SLC25a26 is, for the most part, ubiquitously expressed in human tissues, although very low levels are found in the spleen and the highest levels are found in the testes [187]. Mitochondrial SAM levels are markedly reduced in Down’s syndrome, despite an increase in SAM carrier mRNA [188]. This is suggestive of a defect in the SAM carrier, although this has yet to be proven. What role the carrier has or its dysfunction may play in other diseases has yet to be elucidated.
SLC25a28 and SLC25a37: iron carriers
There are two iron carriers in the inner membrane of the mitochondrion, specifically SLC25a28 and SLC25a37, otherwise known as mitoferrin-2 and mitoferrin-1 respectively. The import of iron into the mitochondrial matrix is required for the formation of iron–sulfur clusters by the Isc/Nfs/Isd/frataxin system. These clusters are then incorporated into many mitochondrial enzymes, including succinate dehydrogenase and aconitase, or are exported out of the mitochondrion. Iron is also needed for the last step of haem biosynthesis, which occurs in the mitochondrial matrix. A mitochondrial iron carrier was proposed to exist, although it took some time to identify and clone. Eventually, Larsson’s laboratory cloned the human and mouse orthologue of two previously identi-fied yeast iron carriers, Mrs3 and Mrs4 [189,190], and found it to be ubiquitously expressed in all tissues, especially striated muscle and the liver. This carrier was designated SLC25a28. A second iron transporter, SLC25a37 or mitoferrin-1, was subsequently cloned and appeared to be in most tissues, but with preferential expression in haemopoietic cells and the brain [191,192].
Both transporters appear to be critical for iron import. Knockdown of either SLC25a28 or SLC25a37 reduced iron incorporation into haem and iron–sulfur cluster synthesis in both erythroid and non-erythroid cells [193]. Interestingly, knockdown of SLC25a28 had a greater effect on haem synthesis, whereas SLC25a37 depletion had greater effect on iron–sulfur cluster formation. Mouse erythroblasts lacking SLC25a37 had significantly reduced iron incorporation into haem and underwent maturation arrest [191]. The import of iron by this transporter is enhanced by an interaction with the ABC (ATP-binding cassette) transporter Abcb10, whose ablation mimics the effects of SLC25a37 deletion [194]. Iron import and haem biosynthesis are efficiently coupled by SLC25a37 and ferrochelatase interaction [195]. What role, if any, these transporters or mutant versions thereof play in disease pathogenesis remains unknown.
SLC25a32: folate carrier
The MFT (mitochondrial folate transporter) carries folate into the mitochondrial matrix. The existence of MFT was first propopsed by Horne et al. [196], who demonstrated that mitochondrial folate import exhibited the characteristics of carrier-mediated transport and could be inhibited by methotrexate. Titus and Moran [197] eventually cloned the human transporter where it was evident that it belonged to the SLC25 family. MFT tissue distribution and transport mechanism are still unknown. However, folate uptake is neither Δ pH- nor ΔΨm-dependent [196].
The MFT appears to be essential for mitochondrial synthesis of glycine, where folate is required for the conversion of serine into glycine by serine hydroxymethyltransferase. Inactivation of the MFT’s activity or MFT deletion renders cells glycine auxotrophs [197,198]. Mitochondrial folate is also required for the glycine cleavage system, which produces the formylmethionine necessary for mitochondrial translation initiation. Interestingly, yeast lacking the mitochondrial FAD transporter Flx1 could be rescued by expressing MFT, suggesting that MFT may also transport FAD across the inner membrane [199].
In disease, chronic alcohol exposure has been shown to reduce mitochondrial folate uptake due to reduced SLC25a32 mRNA and protein levels. In addition, alcohol was shown to reduce the activity of the SLC25a32 promoter [200]. MFT is also down-regulated by chronic kidney disease [201]. Consequences of MFT down-regulation include enhanced sensitivity of cells to oxidative stress-induced mitochondrial dysfunction and apoptosis [202]. Finally, polymorphisms in the SLC25a32 gene were reported to correlate with the variability of methotrexate therapy efficacy in arthritic patients [203], and may be predictive of altered vitamin B12 metabolism in patients with cardiovascular disease [204].
SLC25a33 and SLC25a36: pyrimidine nucleotide carriers
The presence of a specific transport mechanism for pyrimidine nucleotides in mitochondria was first revealed when a mitochondrial fraction possessing dCTP transport activity was reconstituted in liposomes [205]. Moreover, this activity could be blocked by dCDP, dGTP and dATP. Importantly, pyrimidine nucleotide uptake was not affected when the DNC was either overexpressed or knocked down, indicating that the carrier was distinct from the DNC [170]. The yeast orthologue of the PNC (pyrimidine nucleotide carrier) was subsequently cloned and it was found that it was specific for CTP, TTP and UTP, as well as the corresponding di- and mono-phosphates [206]. In that work, the authors proposed the PNC transport mechanism as an exchange with one nucleotide in for one nucleotide out. The transport could be inhibited by tannic acid and Bromocresol Purple, and partially by bongkrekic acid. Inactivation of the gene in yeast resulted in total loss of mitochondrial DNA.
Using this information, the mouse pyrimidine nucleotide carrier PNC1 (SLC25a33) was cloned [207] and found to be expressed primarily in the liver and testis with lower expression in the heart and brain. Its preference was for UTP followed by TTP and CTP and knockdown of the PNC reduced UTP import into the mitochondrion. Since then a second putative PNC has been identified, termed SLC25a36 [192]. Whether this has PNC activity remains to be established.
From a disease perspective, overexpression of the PNC1 was found to increase cell size and proliferation and suppress ROS production in breast cancer cells, whereas depletion had the opposite effect [207,208]. Thus overexpression of PNC1 may play a role in oncogenesis. Along these lines SLC25a36 expression is also up-regulated in cervical cancer [209].
SLC25a38: ??? carrier
SLC25a38 is somewhat unique in that it has no transport function ascribed to it. Mutations in the SLC25a38 gene are believed to be responsible for congenital sideroblastic anaemia [210,211]. Patients with this mutation exhibit elevated serum ferritin levels and immature erythroid progenitors that contain iron deposits. In zebrafish, knockdown of erythroid cell-restricted SLC25a38 caused defective erythropoiesis coupled with reduced haemoglobin levels [211]. S. cerevisiae cells lacking the SLC25a38 homologue also exhibited reduced haem biosynthesis. It has therefore been proposed that SLC25a38 is a glycine transporter, as conversion of glycine into 5-aminolaevulinic acid in the mitochondrial matrix is the first step in haem biosynthesis. However, whether this SLC25 protein is indeed the mitochondrial glycine carrier remains to be thoroughly tested.
SLC25a42: CoA carrier
The SLC25a42 gene was first identified by Haitina et al. [192]. It was then cloned and reconstituted in liposomes by Palmieri’s laboratory [5], establishing that the protein catalysed the exchange of CoA for adenine and deoxyadenine nucleotides. In addition, PAP (adenine 3′,5′-diphosphate) produced in the mitochondria from CoA was shown also to be transported by SLC25a42. Therefore this carrier can facilitate the import of CoA into the matrix in exchange for PAP or adenine nucleotides. SLC25a42 activity can be inhibited by a variety of SLC25 family inhibitors, such as bongkrekic acid, tannic acid and PLP [5].
SLC25a43–SLC25a53
Little is known about the most recently discovered members of the SLC25 family. Accordingly, literature on transport activities for these carriers remains scarce. A more comprehensive search results in some interesting points regarding these relatively unknown proteins. In a GWAS (genome-wide association study), SLC25a43 was proposed to play a role in Paget’s disease of bone [212]. In another GWAS set, two different groups determined a role for SLC25a46 as susceptibility loci for atopic dermatitis in Japanese and Chinese populations respectively [213,214]. The mechanism by which both carriers induce their pathogenesis still waits to be elucidated.
In a further GWAS, single-nucleotide polymorphisms were associated between SLC25a48 and Parkinson’s disease in an Ashkenazi Jewish population [215]. Again, since this carrier activity is unknown, it is difficult to envisage what the protein–disease relationship is in this case.
By using microarray profiling and in silico analyses, a group found a putative thyroid-response element in the promoter region of the SLC25a45 gene [216]. Consequently, the mRNA of this carrier is positively regulated during hyperthyroidism. In that work, the authors suggest that thyroid hormone-mediated mitochondrial biogenesis may be a result of the presence of thyroid-responsive elements in the vicinity of the transcription start site of proteins such as SLC25a45. As with SLC25a46, the transport characteristics of SLC25a45 remain obscure.
A decreased level of expression of SLC25a47 in hepatocellular carcinoma has been implicated in the reluctance of such aggressive cancer to undergo cell death [217]. Upon overexpression of SLC25a47, HEK (human embryonic kidney)-293T cells underwent ΔΨm dissipation and collapse of ATP synthesis. The mechanism by which SLC25a47 overexpression triggers cell death may depend on its UCP-like activity [217]. Although no direct UCP activity determination was performed in one study [217], in another, SLC25a47 overexpression in yeast resulted in a slight increase in state 4 (non-phosphorylating) respiration of isolated mitochondria [218]. These increases were not sensitive to GDP and modulation by fatty acids was not tested. Although we cannot rule out the possibility that SLC25a47 may have promiscuous UCP-like activity [219], its transport characteristics still have to be properly ascertained.
Perhaps one of the most intriguing new findings in the mitochondrial carrier field is the fact that SLC25a49 and SLC25a50, also known as MTCH1 (mitochondrial carrier homologue 1) and MTCH2, are targeted to the outer mitochondrial membrane [220–222]. Although these proteins share an identity of only 48 %, they are both are involved in apoptotic death, albeit by different mechanisms. SLC25a49, also known as presenilin 1-associated protein, encodes a protein known to induce apoptosis independent of the Bcl-2 proteins Bax and Bak [221]. Alternatively spliced transcript variants encoding more than one isoform have been detected ubiquitously. Carrodeguas’ group defined two pro-apoptotic domains comprising residues 65–112 and residues 113–168. Strikingly, mitochondria-targeting peptides composed of each domain separately or a fusion of both segments were shown to induce ΔΨm dissipation and cell death in HEK-293T and HeLa cells [220,223]. SLC25a50 encodes a six-transmembrane-helix 30 kDa protein thought to be an outer membrane receptor of pro-apoptotic Bid and possibly Bax [222]. SLC25a50 is a protein associated with the neuronal regulation of body weight [224]. SLC25a50 possesses the three repeats of a mitochondrial carrier; however, it has a shorter transmembrane 1 helix. As with ANT, this protein presents a single substrate-binding site in the central cavity of the carrier. This site, however, has unusual characteristics leading to the suggestion that SLC25a50 may not be a transporter, or may be recognizing a substrate in a non-typical way [222]. At the present time, there are no reports to our knowledge about any particular phenotype for SLC25a51, SLC25a52 and SLC25a53.
CONCLUDING REMARKS
Solute carriers are indispensable for transport of essential molecules across the inner mitochondrial membrane. Their proper activity is required for many biochemical pathways and for cellular homoeostasis. Consequently, their dysfunction probably leads to pathological outcomes. This explains why mutations affecting the activity of SLC25 proteins drive a wide variety of human pathologies.
Despite the considerable progress that has been achieved in SLC25 research in previous decades, many questions still remain in order to reinforce current hypotheses in the field. For example, is it possible to solve any SLC25 crystal structures in the ‘m’ conformation? What is the molecular mechanism by which Ca2+ binding induces translocation in CaMCs? How was the ancestral activity of mitochondrial carrier homologues altered to perform their putative ‘receptor-like’ activity in the outer mitochondrial membrane?
Future experiments are required to ascertain means of treatment for SLC25-derived diseases. Accordingly, it is equally important to understand how mitochondrial carriers govern the mitochondria, the cells and living systems in general.
Acknowledgments
We thank Kyle S. McCommis and Kurt D. Marshall for helpful comments about the present review. We apologize to the colleagues whose papers we could not cite due to space limitations.
FUNDING
Work in Dr Baines’ laboratory is supported by the National Institutes of Health [grant number HL094404] and the American Heart Association [grant number 13POST14060013 (to M.G.-A.)].
Abbreviations used
- adPEO
autosomal dominant progressive external ophthalmoplegia
- AGC
aspartate/glutamate carrier
- ANT
adenine nucleotide translocator
- APC
ATP-Mg/Pi carrier
- BPC
basal proton conductance
- CAC
carnitine/acylcarnitine carrier
- CACL
CAC-like protein
- CaMC
Ca2+ -sensitive mitochondrial carrier
- CIC
citrate carrier
- CMV
cytomegalovirus
- CPT
carnitine palmitoyltransferase
- DIC
dicarboxylate carrier
- DNC
deoxynucleotide carrier
- ER
endoplasmic reticulum
- GC
glutamate carrier
- GDC
Grave’s disease carrier
- GWAS
genome-wide association study
- HEK
human embryonic kidney
- HHH
hyperornithinaemia, hyperammonaemia and homocitrullinuria
- MFT
mitochondrial folate transporter
- MTCH
mitochondrial carrier homologue
- OAA
oxaloacetate
- ODC
oxodicarboxylate carrier
- OGC
oxoglutarate carrier
- PAP
adenine 3′,5′-diphosphate
- PEP
phosphoenolpyruvate
- PiC
phosphate carrier
- PLP
pyridoxal 5′-phosphate
- PNC
pyridine nucleotide carrier
- ROS
reactive oxygen species
- SAH
S-adenosylhomocysteine
- SAM
S-adenosylmethionine
- SCaMC
small calcium-binding mitochondrial carrier
- SLC25
solute carrier family 25
- TCA
tricarboxylic acid
- ThPP
thiamine pyrophosphate
- UCP
uncoupling protein
References
- 1.Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflügers Arch. 2004;447:689–709. doi: 10.1007/s00424-003-1099-7. [DOI] [PubMed] [Google Scholar]
- 2.Dahout-Gonzalez C, Nury H, Trézéguet V, Lauquin GJM, Pebay-Peyroula E, Brandolin G. Molecular, functional, and pathological aspects of the mitochondrial ADP/ATP carrier. Physiology. 2006;21:242–249. doi: 10.1152/physiol.00005.2006. [DOI] [PubMed] [Google Scholar]
- 3.Pfaff E, Klingenberg M, Heldt HW. Unspecific permeation and specific exchange of adenine nucleotides in liver mitochondria. Biochim Biophys Acta. 1965;104:312–315. doi: 10.1016/0304-4165(65)90258-8. [DOI] [PubMed] [Google Scholar]
- 4.Pfaff E, Klingenberg M, Ritt E, Vogell W. Correlation of the unspecific permeable mitochondrial space with the “intermembrane space”. Eur J Biochem. 1968;5:222–232. doi: 10.1111/j.1432-1033.1968.tb00361.x. [DOI] [PubMed] [Google Scholar]
- 5.Fiermonte G, Paradies E, Todisco S, Marobbio CMT, Palmieri F. A novel member of solute carrier family 25 (SLC25A42) is a transporter of coenzyme A and adenosine 3′,5′-diphosphate in human mitochondria. J Biol Chem. 2009;284:18152–18159. doi: 10.1074/jbc.M109.014118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aquila H, Misra D, Eulitz M, Klingenberg M. Complete amino acid sequence of the ADP/ATP carrier from beef heart mitochondria. Hoppe-Seyler’s Z Physiol Chem. 1982;363:345–349. [PubMed] [Google Scholar]
- 7.Palmieri F. The mitochondrial transporter family SLC25: identification, properties and physiopathology. Mol Aspects Med. 2013;34:465–484. doi: 10.1016/j.mam.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Berardi MJ, Shih WM, Harrison SC, Chou JJ. Mitochondrial uncoupling protein 2 structure determined by NMR molecular fragment searching. Nature. 2011;476:109–113. doi: 10.1038/nature10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trézéguet V, Lauquin GJM, Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426:39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- 10.Kunji ER, Robinson AJ. Coupling of proton and substrate translocation in the transport cycle of mitochondrial carriers. Curr Opin Struct Biol. 2010;20:440–447. doi: 10.1016/j.sbi.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 11.Runswick MJ, Walker JE, Bisaccia F, Iacobazzi V, Palmieri F. Sequence of the bovine 2-oxoglutarate/malate carrier protein: structural relationship to other mitochondrial transport proteins. Biochemistry. 1990;29:11033–11040. doi: 10.1021/bi00502a004. [DOI] [PubMed] [Google Scholar]
- 12.Baines CP, Molkentin JD. Adenine nucleotide translocase-1 induces cardiomyocyte death through upregulation of the pro-apoptotic protein Bax. J Mol Cell Cardiol. 2009;46:969–977. doi: 10.1016/j.yjmcc.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heldt HW, Jacobs H, Klingenberg M. Endogenous ADP of mitochondria, an early phosphate acceptor of oxidative phosphorylation as disclosed by kinetic studies with 14C labelled ADP and ATP and with atractyloside. Biochem Biophys Res Commun. 1965;18:174–179. doi: 10.1016/0006-291x(65)90736-9. [DOI] [PubMed] [Google Scholar]
- 14.Duee ED, Vignais PV. Exchange between extra- and intramitochondrial adenine nucleotides. Biochim Biophys Acta. 1965;107:184–188. doi: 10.1016/0304-4165(65)90419-8. [DOI] [PubMed] [Google Scholar]
- 15.Krämer R, Klingenberg M. Modulation of the reconstituted adenine nucleotide exchange by membrane potential. Biochemistry. 1980;19:556–560. doi: 10.1021/bi00544a025. [DOI] [PubMed] [Google Scholar]
- 16.Chappell JB. Systems used for the transport of substrates into mitochondria. Br Med Bull. 1968;24:150–157. doi: 10.1093/oxfordjournals.bmb.a070618. [DOI] [PubMed] [Google Scholar]
- 17.Palmieri F, Indiveri C, Bisaccia F, Krämer R. Functional properties of purified and reconstituted mitochondrial metabolite carriers. J Bioenerg Biomembr. 1993;25:525–535. doi: 10.1007/BF01108409. [DOI] [PubMed] [Google Scholar]
- 18.Robinson BH, Chappell JB. The inhibition of malate, tricarboxylate and oxoglutarate entry into mitochondria by 2-n-butylmalonate. Biochem Biophys Res Commun. 1967;28:249–255. doi: 10.1016/0006-291x(67)90437-8. [DOI] [PubMed] [Google Scholar]
- 19.Kaplan RS, Mayor JA, Wood DO. The mitochondrial tricarboxylate transport protein. cDNA cloning, primary structure, and comparison with other mitochondrial transport proteins. J Biol Chem. 1993;268:13682–13690. [PubMed] [Google Scholar]
- 20.Aquila H, Link TA, Klingenberg M. The uncoupling protein from brown fat mitochondria is related to the mitochondrial ADP/ATP carrier. Analysis of sequence homologies and of folding of the protein in the membrane. EMBO J. 1985;4:2369–2376. doi: 10.1002/j.1460-2075.1985.tb03941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Runswick MJ, Powell SJ, Nyren P, Walker JE. Sequence of the bovine mitochondrial phosphate carrier protein: structural relationship to ADP/ATP translocase and the brown fat mitochondria uncoupling protein. EMBO J. 1987;6:1367–1373. doi: 10.1002/j.1460-2075.1987.tb02377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Indiveri C, Iacobazzi V, Giangregorio N, Palmieri F. The mitochondrial carnitine carrier protein: cDNA cloning, primary structure and comparison with other mitochondrial transport proteins. Biochem J. 1997;321:713–719. doi: 10.1042/bj3210713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walker JE, Runswick MJ. The mitochondrial transport protein superfamily. J Bioenerg Biomembr. 1993;25:435–446. doi: 10.1007/BF01108401. [DOI] [PubMed] [Google Scholar]
- 24.Palmieri F, Pierri CL. Structure and function of mitochondrial carriers: role of the transmembrane helix P and G residues in the gating and transport mechanism. FEBS Lett. 2010;584:1931–1939. doi: 10.1016/j.febslet.2009.10.063. [DOI] [PubMed] [Google Scholar]
- 25.Silverman JM, Buxbaum JD, Ramoz N, Schmeidler J, Reichenberg A, Hollander E, Angelo G, Smith CJ, Kryzak LA. Autism-related routines and rituals associated with a mitochondrial aspartate/glutamate carrier SLC25A12 polymorphism. Am J Med Genet, Part B. 2008;147:408–410. doi: 10.1002/ajmg.b.30614. [DOI] [PubMed] [Google Scholar]
- 26.Lepagnol-Bestel AM, Maussion G, Boda B, Cardona A, Iwayama Y, Delezoide AL, Moalic JM, Muller D, Dean B, Yoshikawa T, et al. SLC25A12 expression is associated with neurite outgrowth and is upregulated in the prefrontal cortex of autistic subjects. Mol Psychiatry. 2008;13:385–397. doi: 10.1038/sj.mp.4002120. [DOI] [PubMed] [Google Scholar]
- 27.Palmieri F. Diseases caused by defects of mitochondrial carriers: a review. Biochim Biophys Acta. 2008;1777:564–578. doi: 10.1016/j.bbabio.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 28.Indiveri C, Iacobazzi V, Tonazzi A, Giangregorio N, Infantino V, Convertini P, Console L, Palmieri F. The mitochondrial carnitine/acylcarnitine carrier: function, structure and physiopathology. Mol Aspects Med. 2011;32:223–233. doi: 10.1016/j.mam.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 29.Palmieri F, Pierri CL. Mitochondrial metabolite transport. Essays Biochem. 2010;47:37–52. doi: 10.1042/bse0470037. [DOI] [PubMed] [Google Scholar]
- 30.Camacho JA, Obie C, Biery B, Goodman BK, Hu CA, Almashanu S, Steel G, Casey R, Lambert M, Mitchell GA, et al. Hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome is caused by mutations in a gene encoding a mitochondrial ornithine transporter. Nat Genet. 1999;22:151–158. doi: 10.1038/9658. [DOI] [PubMed] [Google Scholar]
- 31.Guerin B, Guérin M, Klingenberg M. Differential inhibition of phosphate efflux and influx and a possible discrimination between an inner and outer location of the phosphate carrier in mitochondria. FEBS Lett. 1970;10:265–268. doi: 10.1016/0014-5793(70)80644-5. [DOI] [PubMed] [Google Scholar]
- 32.Palmieri F, Klingenberg M. Direct methods for measuring metabolite transport and distribution in mitochondria. Methods Enzymol. 1979;56:279–301. doi: 10.1016/0076-6879(79)56029-7. [DOI] [PubMed] [Google Scholar]
- 33.Klingenberg M. The ADP and ATP transport in mitochondria and its carrier. Biochim Biophys Acta. 2008;1778:1978–2021. doi: 10.1016/j.bbamem.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 34.Agrimi G, Russo A, Pierri CL, Palmieri F. The peroxisomal NAD + carrier of Arabidopsis thaliana transports coenzyme A and its derivatives. J Bioenerg Biomembr. 2012;44:333–340. doi: 10.1007/s10863-012-9445-0. [DOI] [PubMed] [Google Scholar]
- 35.Palmieri F, Indiveri C, Bisaccia F, Iacobazzi V. Mitochondrial metabolite carrier proteins: purification, reconstitution, and transport studies. Methods Enzymol. 1995;260:349–369. doi: 10.1016/0076-6879(95)60150-3. [DOI] [PubMed] [Google Scholar]
- 36.Klingenberg M, Winkler E, Huang S. ADP/ATP carrier and uncoupling protein. Methods Enzymol. 1995;260:369–389. doi: 10.1016/0076-6879(95)60151-1. [DOI] [PubMed] [Google Scholar]
- 37.Krämer R, Palmieri F. Molecular aspects of isolated and reconstituted carrier proteins from animal mitochondria. Biochim Biophys Acta. 1989;974:1–23. doi: 10.1016/s0005-2728(89)80160-4. [DOI] [PubMed] [Google Scholar]
- 38.Indiveri C, Iacobazzi V, Giangregorio N, Palmieri F. Bacterial overexpression, purification, and reconstitution of the carnitine/acylcarnitine carrier from rat liver mitochondria. Biochem Biophys Res Commun. 1998;249:589–594. doi: 10.1006/bbrc.1998.9197. [DOI] [PubMed] [Google Scholar]
- 39.Fiermonte G, Walker JE, Palmieri F. Abundant bacterial expression and reconstitution of an intrinsic membrane-transport protein from bovine mitochondria. Biochem J. 1993;294:293–299. doi: 10.1042/bj2940293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klingenberg M. Cardiolipin and mitochondrial carriers. Biochim Biophys Acta. 2009;1788:2048–2058. doi: 10.1016/j.bbamem.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 41.Beyer K, Klingenberg M. ADP/ATP carrier protein from beef heart mitochondria has high amounts of tightly bound cardiolipin, as revealed by 31P nuclear magnetic resonance. Biochemistry. 1985;24:3821–3826. doi: 10.1021/bi00336a001. [DOI] [PubMed] [Google Scholar]
- 42.Dolce V, Scarcia P, Iacopetta D, Palmieri F. A fourth ADP/ATP carrier isoform in man: identification, bacterial expression, functional characterization and tissue distribution. FEBS Lett. 2005;579:633–637. doi: 10.1016/j.febslet.2004.12.034. [DOI] [PubMed] [Google Scholar]
- 43.Zara V, Palmieri F, Mahlke K, Pfanner N. The cleavable presequence is not essential for import and assembly of the phosphate carrier of mammalian mitochondria but enhances the specificity and efficiency of import. J Biol Chem. 1992;267:12077–12081. [PubMed] [Google Scholar]
- 44.Heimpel S, Basset G, Odoy S, Klingenberg M. Expression of the mitochondrial ADP/ATP carrier in Escherichia coli Renaturation, reconstitution, and the effect of mutations on 10 positive residues. J Biol Chem. 2001;276:11499–11506. doi: 10.1074/jbc.M010586200. [DOI] [PubMed] [Google Scholar]
- 45.Palmieri L, Agrimi G, Runswick MJ, Fearnley IM, Palmieri F, Walker JE. Identification in Saccharomyces cerevisiae of two isoforms of a novel mitochondrial transporter for 2-oxoadipate and 2-oxoglutarate. J Biol Chem. 2001;276:1916–1922. doi: 10.1074/jbc.M004332200. [DOI] [PubMed] [Google Scholar]
- 46.Palmieri L, Lasorsa FM, Iacobazzi V, Runswick MJ, Palmieri F, Walker JE. Identification of the mitochondrial carnitine carrier in Saccharomyces cerevisiae. FEBS Lett. 1999;462:472–476. doi: 10.1016/s0014-5793(99)01555-0. [DOI] [PubMed] [Google Scholar]
- 47.Castegna A, Scarcia P, Agrimi G, Palmieri L, Rottensteiner H, Spera I, Germinario L, Palmieri F. Identification and functional characterization of a novel mitochondrial carrier for citrate and oxoglutarate in Saccharomyces cerevisiae. J Biol Chem. 2010;285:17359–17370. doi: 10.1074/jbc.M109.097188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roussel D, Harding M, Runswick MJ, Walker JE, Brand MD. Does any yeast mitochondrial carrier have a native uncoupling protein function? J Bioenerg Biomembr. 2002;34:165–176. doi: 10.1023/a:1016027302232. [DOI] [PubMed] [Google Scholar]
- 49.Palmieri L, Rottensteiner H, Girzalsky W, Scarcia P, Palmieri F, Erdmann R. Identification and functional reconstitution of the yeast peroxisomal adenine nucleotide transporter. EMBO J. 2001;20:5049–5059. doi: 10.1093/emboj/20.18.5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marobbio CM, Agrimi G, Lasorsa FM, Palmieri F. Identification and functional reconstitution of yeast mitochondrial carrier for S-adenosylmethionine. EMBO J. 2003;22:5975–5982. doi: 10.1093/emboj/cdg574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoyos ME, Palmieri L, Wertin T, Arrigoni R, Polacco JC, Palmieri F. Identification of a mitochondrial transporter for basic amino acids in Arabidopsis thaliana by functional reconstitution into liposomes and complementation in yeast. Plant J. 2003;33:1027–1035. doi: 10.1046/j.1365-313x.2003.01685.x. [DOI] [PubMed] [Google Scholar]
- 52.Palmieri F, Agrimi G, Blanco E, Castegna A, Di Noia MA, Iacobazzi V, Lasorsa FM, Marobbio CM, Palmieri L, Scarcia P, et al. Identification of mitochondrial carriers in Saccharomyces cerevisiae by transport assay of reconstituted recombinant proteins. Biochim Biophys Acta. 2006;1757:1249–1262. doi: 10.1016/j.bbabio.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 53.Palmieri F, Pierri CL, De Grassi A, Nunes-Nesi A, Fernie AR. Evolution, structure and function of mitochondrial carriers: a review with new insights. Plant J. 2011;66:161–181. doi: 10.1111/j.1365-313X.2011.04516.x. [DOI] [PubMed] [Google Scholar]
- 54.Palmieri L, Palmieri F, Runswick MJ, Walker JE. Identification by bacterial expression and functional reconstitution of the yeast genomic sequence encoding the mitochondrial dicarboxylate carrier protein. FEBS Lett. 1996;399:299–302. doi: 10.1016/s0014-5793(96)01350-6. [DOI] [PubMed] [Google Scholar]
- 55.Palmieri L, Runswick MJ, Fiermonte G, Walker JE, Palmieri F. Yeast mitochondrial carriers: bacterial expression, biochemical identification and metabolic significance. J Bioenerg Biomembr. 2000;32:67–77. doi: 10.1023/a:1005564429242. [DOI] [PubMed] [Google Scholar]
- 56.Agrimi G, Russo A, Scarcia P, Palmieri F. The human gene SLC25A17 encodes a peroxisomal transporter of coenzyme A, FAD and NAD + Biochem J. 2012;443:241–247. doi: 10.1042/BJ20111420. [DOI] [PubMed] [Google Scholar]
- 57.Zara V, Ferramosca A, Robitaille-Foucher P, Palmieri F, Young JC. Mitochondrial carrier protein biogenesis: role of the chaperones Hsc70 and Hsp90. Biochem J. 2009;419:369–375. doi: 10.1042/BJ20082270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dietmeier K, Zara V, Palmisano A, Palmieri F, Voos W, Schlossmann J, Moczko M, Kispal G, Pfanner N. Targeting and translocation of the phosphate carrier/p32 to the inner membrane of yeast mitochondria. J Biol Chem. 1993;268:25958–25964. [PubMed] [Google Scholar]
- 59.Claypool SM, Oktay Y, Boontheung P, Loo JA, Koehler CM. Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J Cell Biol. 2008;182:937–950. doi: 10.1083/jcb.200801152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Claypool SM. Cardiolipin, a critical determinant of mitochondrial carrier protein assembly and function. Biochim Biophys Acta. 2009;1788:2059–2068. doi: 10.1016/j.bbamem.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nury H, Dahout-Gonzalez C, Trézéguet V, Lauquin GJM, Brandolin G, Pebay-Peyroula E. Relations between structure and function of the mitochondrial ADP/ATP carrier. Annu Rev Biochem. 2006;75:713–741. doi: 10.1146/annurev.biochem.75.103004.142747. [DOI] [PubMed] [Google Scholar]
- 62.Stipani I, Palmieri F. Purification of the active mitochondrial tricarboxylate carrier by hydroxylapatite chromatography. FEBS Lett. 1983;161:269–274. doi: 10.1016/0014-5793(83)81023-0. [DOI] [PubMed] [Google Scholar]
- 63.Stipani I, Krämer R, Palmieri F, Klingenberg M. Citrate transport in liposomes reconstituted with Triton extracts from mitochondria. Biochem Biophys Res Commun. 1980;97:1206–1214. doi: 10.1016/0006-291x(80)91503-x. [DOI] [PubMed] [Google Scholar]
- 64.Bisaccia F, De Palma A, Palmieri F. Identification and purification of the tricarboxylate carrier from rat liver mitochondria. Biochim Biophys Acta. 1989;977:171–176. doi: 10.1016/s0005-2728(89)80068-4. [DOI] [PubMed] [Google Scholar]
- 65.Huizing M, Ruitenbeek W, van den Heuvel LP, Dolce V, Iacobazzi V, Smeitink JA, Palmieri F, Trijbels JM. Human mitochondrial transmembrane metabolite carriers: tissue distribution and its implication for mitochondrial disorders. J Bioenerg Biomembr. 1998;30:277–284. doi: 10.1023/a:1020501021222. [DOI] [PubMed] [Google Scholar]
- 66.Stipani I, Capalbo MI, Zara V. Effect of anthracycline antibiotics on the reconstituted mitochondrial tricarboxylate carrier. Biochem Biophys Res Commun. 1989;164:1281–1287. doi: 10.1016/0006-291x(89)91808-1. [DOI] [PubMed] [Google Scholar]
- 67.Catalina-Rodriguez O, Kolukula VK, Tomita Y, Preet A, Palmieri F, Wellstein A, Byers S, Giaccia AJ, Glasgow E, Albanese C, et al. The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget. 2012;3:1220–1235. doi: 10.18632/oncotarget.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grigorenko EV, Small WC, Persson LO, Srere PA. Citrate synthase 1 interacts with the citrate transporter of yeast mitochondria. J Mol Recognit. 1990;3:215–219. doi: 10.1002/jmr.300030508. [DOI] [PubMed] [Google Scholar]
- 69.Sandor A, Johnson JH, Srere PA. Cooperation between enzyme and transporter in the inner mitochondrial membrane of yeast. Requirement for mitochondrial citrate synthase for citrate and malate transport in Saccharomyces cerevisiae. J Biol Chem. 1994;269:29609–29612. [PubMed] [Google Scholar]
- 70.Zara V, Gnoni GV. Effect of starvation on the activity of the mitochondrial tricarboxylate carrier. Biochim Biophys Acta. 1995;1239:33–38. doi: 10.1016/0005-2736(95)00125-m. [DOI] [PubMed] [Google Scholar]
- 71.Giudetti AM, Leo M, Siculella L, Gnoni GV. Hypothyroidism down-regulates mitochondrial citrate carrier activity and expression in rat liver. Biochim Biophys Acta. 2006;1761:484–491. doi: 10.1016/j.bbalip.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 72.Paradies G, Ruggiero FM. Enhanced activity of the tricarboxylate carrier and modification of lipids in hepatic mitochondria from hyperthyroid rats. Arch Biochem Biophys. 1990;278:425–430. doi: 10.1016/0003-9861(90)90280-c. [DOI] [PubMed] [Google Scholar]
- 73.Kaplan RS, Mayor JA, Blackwell R, Wilson GL, Schaffer SW. Functional levels of mitochondrial anion transport proteins in non-insulin-dependent diabetes mellitus. Mol Cell Biochem. 1991;107:79–86. doi: 10.1007/BF02424578. [DOI] [PubMed] [Google Scholar]
- 74.Damiano F, Mercuri E, Stanca E, Gnoni GV, Siculella L. Streptozotocin-induced diabetes affects in rat liver citrate carrier gene expression by transcriptional and posttranscriptional mechanisms. Int J Biochem Cell Biol. 2011;43:1621–1629. doi: 10.1016/j.biocel.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 75.Infantino V, Iacobazzi V, De Santis F, Mastrapasqua M, Palmieri F. Transcription of the mitochondrial citrate carrier gene: role of SREBP-1, upregulation by insulin and downregulation by PUFA. Biochem Biophys Res Commun. 2007;356:249–254. doi: 10.1016/j.bbrc.2007.02.114. [DOI] [PubMed] [Google Scholar]
- 76.Joseph JW, Jensen MV, Ilkayeva O, Palmieri F, Alarcon C, Rhodes CJ, Newgard CB. The mitochondrial citrate/isocitrate carrier plays a regulatory role in glucose-stimulated insulin secretion. J Biol Chem. 2006;281:35624–35632. doi: 10.1074/jbc.M602606200. [DOI] [PubMed] [Google Scholar]
- 77.Maynard TM, Meechan DW, Dudevoir ML, Gopalakrishna D, Peters AZ, Heindel CC, Sugimoto TJ, Wu Y, Lieberman JA, Lamantia AS. Mitochondrial localization and function of a subset of 22q11 deletion syndrome candidate genes. Mol Cell Neurosci. 2008;39:439–451. doi: 10.1016/j.mcn.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Heisterkamp N, Mulder MP, Langeveld A, ten Hoeve J, Wang Z, Roe BA, Groffen J. Localization of the human mitochondrial citrate transporter protein gene to chromosome 22Q11 in the DiGeorge syndrome critical region. Genomics. 1995;29:451–456. doi: 10.1006/geno.1995.9982. [DOI] [PubMed] [Google Scholar]
- 79.Kaplan RS, Mayor JA, Kakhniashvili D, Gremse DA, Wood DO, Nelson DR. Deletion of the nuclear gene encoding the mitochondrial citrate transport protein from Saccharomyces cerevisiae. Biochem Biophys Res Commun. 1996;226:657–662. doi: 10.1006/bbrc.1996.1411. [DOI] [PubMed] [Google Scholar]
- 80.Cheng L, Lu W, Kulkarni B, Pejovic T, Yan X, Chiang JH, Hood L, Odunsi K, Lin B. Analysis of chemotherapy response programs in ovarian cancers by the next-generation sequencing technologies. Gynecol Oncol. 2010;117:159–169. doi: 10.1016/j.ygyno.2010.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Palmieri L, De Marco V, Iacobazzi V, Palmieri F, Runswick MJ, Walker JE. Identification of the yeast ARG-11 gene as a mitochondrial ornithine carrier involved in arginine biosynthesis. FEBS Lett. 1997;410:447–451. doi: 10.1016/s0014-5793(97)00630-3. [DOI] [PubMed] [Google Scholar]
- 82.Camacho JA, Rioseco-Camacho N, Andrade D, Porter J, Kong J. Cloning and characterization of human ORNT2: a second mitochondrial ornithine transporter that can rescue a defective ORNT1 in patients with the hyperornithinemia-hyperammonemia-homocitrullinuria syndrome, a urea cycle disorder. Mol Genet Metab. 2003;79:257–271. doi: 10.1016/s1096-7192(03)00105-7. [DOI] [PubMed] [Google Scholar]
- 83.Fiermonte G, Dolce V, David L, Santorelli FM, Dionisi-Vici C, Palmieri F, Walker JE. The mitochondrial ornithine transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J Biol Chem. 2003;278:32778–32783. doi: 10.1074/jbc.M302317200. [DOI] [PubMed] [Google Scholar]
- 84.Indiveri C, Tonazzi A, Stipani I, Palmieri F. The purified and reconstituted ornithine/citrulline carrier from rat liver mitochondria: electrical nature and coupling of the exchange reaction with H + translocation. Biochem J. 1997;327:349–355. doi: 10.1042/bj3270349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Camacho JA, Mardach R, Rioseco-Camacho N, Ruiz-Pesini E, Derbeneva O, Andrade D, Zaldivar F, Qu Y, Cederbaum SD. Clinical and functional characterization of a human ORNT1 mutation (T32R) in the hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome. Pediatr Res. 2006;60:423–429. doi: 10.1203/01.pdr.0000238301.25938.f5. [DOI] [PubMed] [Google Scholar]
- 86.Fiermonte G, Dolce V, Palmieri F. Expression in Escherichia coli, functional characterization, and tissue distribution of isoforms A and B of the phosphate carrier from bovine mitochondria. J Biol Chem. 1998;273:22782–22787. doi: 10.1074/jbc.273.35.22782. [DOI] [PubMed] [Google Scholar]
- 87.Dolce V, Fiermonte G, Palmieri F. Tissue-specific expression of the two isoforms of the mitochondrial phosphate carrier in bovine tissues. FEBS Lett. 1996;399:95–98. doi: 10.1016/s0014-5793(96)01294-x. [DOI] [PubMed] [Google Scholar]
- 88.Dolce V, Iacobazzi V, Palmieri F, Walker JE. The sequences of human and bovine genes of the phosphate carrier from mitochondria contain evidence of alternatively spliced forms. J Biol Chem. 1994;269:10451–10460. [PubMed] [Google Scholar]
- 89.Mayr JA, Zimmermann FA, Horváth R, Schneider HC, Schoser B, Holinski-Feder E, Czermin B, Freisinger P, Sperl W. Deficiency of the mitochondrial phosphate carrier presenting as myopathy and cardiomyopathy in a family with three affected children. Neuromuscul Disord. 2011;21:803–808. doi: 10.1016/j.nmd.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 90.Zara V, Dietmeier K, Palmisano A, Vozza A, Rassow J, Palmieri F, Pfanner N. Yeast mitochondria lacking the phosphate carrier/p32 are blocked in phosphate transport but can import preproteins after regeneration of a membrane potential. Mol Cell Biol. 1996;16:6524–6531. doi: 10.1128/mcb.16.11.6524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Poncet D, Pauleau AL, Szabadkai G, Vozza A, Scholz SR, Le Bras M, Brière JJ, Jalil A, Le Moigne R, Brenner C, et al. Cytopathic effects of the cytomegalovirus-encoded apoptosis inhibitory protein vMIA. J Cell Biol. 2006;174:985–996. doi: 10.1083/jcb.200604069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Leung AWC, Varanyuwatana P, Halestrap AP. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J Biol Chem. 2008;283:26312–26323. doi: 10.1074/jbc.M805235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gallerne C, Touat Z, Chen ZX, Martel C, Mayola E, Sharaf el dein O, Buron N, Le Bras M, Jacotot E, Borgne-Sanchez A, et al. The fourth isoform of the adenine nucleotide translocator inhibits mitochondrial apoptosis in cancer cells. Int J Biochem Cell Biol. 2010;42:623–629. doi: 10.1016/j.biocel.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 94.Malorni W, Farrace MG, Matarrese P, Tinari A, Ciarlo L, Mousavi-Shafaei P, D’Eletto M, Di Giacomo G, Melino G, Palmieri L, et al. The adenine nucleotide translocator 1 acts as a type 2 transglutaminase substrate: implications for mitochondrial-dependent apoptosis. Cell Death Differ. 2009;16:1480–1492. doi: 10.1038/cdd.2009.100. [DOI] [PubMed] [Google Scholar]
- 95.Brdiczka DG, Zorov DB, Sheu SS. Mitochondrial contact sites: their role in energy metabolism and apoptosis. Biochim Biophys Acta. 2006;1762:148–163. doi: 10.1016/j.bbadis.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 96.Kawamata H, Tiranti V, Magrane J, Chinopoulos C, Manfredi G. adPEO mutations in ANT1 impair ADP-ATP translocation in muscle mitochondria. Hum Mol Genet. 2011;20:2964–2974. doi: 10.1093/hmg/ddr200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.El-Khoury R, Sainsard-Chanet A. Suppression of mitochondrial DNA instability of autosomal dominant forms of progressive external ophthalmoplegia-associated ANT1 mutations in Podospora anserina. Genetics. 2009;183:861–871. doi: 10.1534/genetics.109.107813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Narula N, Zaragoza MV, Sengupta PP, Li P, Haider N, Verjans J, Waymire K, Vannan M, Wallace DC. Adenine nucleotide translocase 1 deficiency results in dilated cardiomyopathy with defects in myocardial mechanics, histopathological alterations, and activation of apoptosis. JACC Cardiovasc Imaging. 2011;4:1–10. doi: 10.1016/j.jcmg.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Brand MD, Pakay JL, Ocloo A, Kokoszka J, Wallace DC, Brookes PS, Cornwall EJ. The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem J. 2005;392:353–362. doi: 10.1042/BJ20050890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pereira C, Camougrand N, Manon S, Sousa MJ, Côrte-Real M. ADP/ATP carrier is required for mitochondrial outer membrane permeabilization and cytochrome c release in yeast apoptosis. Mol Microbiol. 2007;66:571–582. doi: 10.1111/j.1365-2958.2007.05926.x. [DOI] [PubMed] [Google Scholar]
- 101.Nicholls DG, Locke RM. Thermogenic mechanisms in brown fat. Physiol Rev. 1984;64:1–64. doi: 10.1152/physrev.1984.64.1.1. [DOI] [PubMed] [Google Scholar]
- 102.Klingenberg M, Echtay KS. Uncoupling proteins: the issues from a biochemist point of view. Biochim Biophys Acta. 2001;1504:128–143. doi: 10.1016/s0005-2728(00)00242-5. [DOI] [PubMed] [Google Scholar]
- 103.Fleury C, Neverova M, Collins S, Raimbault S, Champigny O, Levi-Meyrueis C, Bouillaud F, Seldin MF, Surwit RS, Ricquier D, et al. Uncoupling protein-2: a novel gene linked to obesity and hyperinsulinemia. Nat Genet. 1997;15:269–272. doi: 10.1038/ng0397-269. [DOI] [PubMed] [Google Scholar]
- 104.Bouillaud F, Ricquier D, Thibault J, Weissenbach J. Molecular approach to thermogenesis in brown adipose tissue: cDNA cloning of the mitochondrial uncoupling protein. Proc Natl Acad Sci USA. 1985;82:445–448. doi: 10.1073/pnas.82.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Boss O, Samec S, Paoloni-Giacobino A, Rossier C, Dulloo A, Seydoux J, Muzzin P, Giacobino JP. Uncoupling protein-3: a new member of the mitochondrial carrier family with tissue-specific expression. FEBS Lett. 1997;408:39–42. doi: 10.1016/s0014-5793(97)00384-0. [DOI] [PubMed] [Google Scholar]
- 106.Yu XX, Mao W, Zhong A, Schow P, Brush J, Sherwood SW, Adams SH, Pan G. Characterization of novel UCP5/BMCP1 isoforms and differential regulation of UCP4 and UCP5 expression through dietary or temperature manipulation. FASEB J. 2000;14:1611–1618. doi: 10.1096/fj.14.11.1611. [DOI] [PubMed] [Google Scholar]
- 107.Divakaruni AS, Brand MD. The regulation and physiology of mitochondrial proton leak. Physiology. 2011;26:192–205. doi: 10.1152/physiol.00046.2010. [DOI] [PubMed] [Google Scholar]
- 108.Xia C, Liu JZ, Xu Y. Effects of GDP on the activity and expression of mitochondrial uncoupling proteins in rat brain in vitro. Sheng Li Xue Bao. 2008;60:492–496. [PubMed] [Google Scholar]
- 109.Busquets S, Miniero DV, Sanchis D, Cappello AR, Alvarez B, Curcio R, Ricquier D, Ludovico A, Lopez-Soriano FJ, Daddabbo L, et al. In the rat, tumor necrosis factor α administration results in an increase in both UCP2 and UCP3 mRNAs in skeletal muscle: a possible mechanism for cytokine-induced thermogenesis? FEBS Lett. 1998;440:348–350. doi: 10.1016/s0014-5793(98)01485-9. [DOI] [PubMed] [Google Scholar]
- 110.Lanni A, Moreno M, Lombardi A, Goglia F. Thyroid hormone and uncoupling proteins. FEBS Lett. 2003;543:5–10. doi: 10.1016/s0014-5793(03)00320-x. [DOI] [PubMed] [Google Scholar]
- 111.Santandreu FM, Valle A, Fernandez de Mattos S, Roca P, Oliver J. Hydrogen peroxide regulates the mitochondrial content of uncoupling protein 5 in colon cancer cells. Cell Physiol Biochem. 2009;24:379–390. doi: 10.1159/000257430. [DOI] [PubMed] [Google Scholar]
- 112.Yonezawa T, Haga S, Kobayashi Y, Katoh K, Obara Y. Saturated fatty acids stimulate and insulin suppresses BMCP1 expression in bovine mammary epithelial cells. Biochem Biophys Res Commun. 2009;390:915–919. doi: 10.1016/j.bbrc.2009.10.077. [DOI] [PubMed] [Google Scholar]
- 113.Enerback S, Jacobsson, Simpson EM, Guerra C, Yamashita H, Harper ME, Kozak LP. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature. 1997;387:90–94. doi: 10.1038/387090a0. [DOI] [PubMed] [Google Scholar]
- 114.Goglia F, Skulachev VP. A function for novel uncoupling proteins: antioxidant defense of mitochondrial matrix by translocating fatty acid peroxides from the inner to the outer membrane leaflet. FASEB J. 2003;17:1585–1591. doi: 10.1096/fj.03-0159hyp. [DOI] [PubMed] [Google Scholar]
- 115.Zhang CY, Baffy G, Perret P, Krauss S, Peroni O, Grujic D, Hagen T, Vidal-Puig AJ, Boss O, Kim YB, et al. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, β cell dysfunction, and type 2 diabetes. Cell. 2001;105:745–755. doi: 10.1016/s0092-8674(01)00378-6. [DOI] [PubMed] [Google Scholar]
- 116.Ho PW, Ho JW, Tse HM, So DH, Yiu DC, Liu HF, Chan KH, Kung MH, Ramsden DB, Ho SL. Uncoupling protein-4 (UCP4) increases ATP supply by interacting with mitochondrial complex II in neuroblastoma cells. PLoS ONE. 2012;7:e32810. doi: 10.1371/journal.pone.0032810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang CY, Parton LE, Ye CP, Krauss S, Shen R, Lin CT, Porco JAJ, Lowell BB. Genipin inhibits UCP2-mediated proton leak and acutely reverses obesity- and high glucose-induced β cell dysfunction in isolated pancreatic islets. Cell Metab. 2006;3:417–427. doi: 10.1016/j.cmet.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 118.Brondani LA, de Souza BM, Duarte GCK, Kliemann LM, Esteves JF, Marcon AS, Gross JL, Canani LH, Crispim D. The UCP1-3826A/G polymorphism is associated with diabetic retinopathy and increased UCP1 and MnSOD2 gene expression in human retina. Invest Ophthalmol Visual Sci. 2012;53:7449–7457. doi: 10.1167/iovs.12-10660. [DOI] [PubMed] [Google Scholar]
- 119.de Luis DA, Aller R, Izaola O, Sagrado MG, Conde R, Primo D, de la Fuente B, Ovalle HF, Mambrilla MR. Relationship of –55C/T polymorphism of uncoupling protein 3 (UCP3) gene with metabolic syndrome by ATP III classification. J Clin Lab Anal. 2012;26:272–278. doi: 10.1002/jcla.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mouaffak F, Kebir O, Bellon A, Gourevitch R, Tordjman S, Viala A, Millet B, Jaafari N, Olie JP, Krebs MO. Association of an UCP4 (SLC25A27) haplotype with ultra-resistant schizophrenia. Pharmacogenomics. 2011;12:185–193. doi: 10.2217/pgs.10.179. [DOI] [PubMed] [Google Scholar]
- 121.Dalla Pozza E, Fiorini C, Dando I, Menegazzi M, Sgarbossa A, Costanzo C, Palmieri M, Donadelli M. Role of mitochondrial uncoupling protein 2 in cancer cell resistance to gemcitabine. Biochim Biophys Acta. 2012;1823:1856–1863. doi: 10.1016/j.bbamcr.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 122.Sayeed A, Harper JA, Meng Z, Stuart JA, Luciani G, Jekabsons MB, Chen LC, Roussel D, Bennington JL, Brindle KM, et al. Negative regulation of UCP2 by TGFβ signaling characterizes low and intermediate-grade primary breast cancer. Cell Death Dis. 2010;1:e53. doi: 10.1038/cddis.2010.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mailloux RJ, Adjeitey CN, Harper ME. Genipin-induced inhibition of uncoupling protein-2 sensitizes drug-resistant cancer cells to cytotoxic agents. PLoS ONE. 2010;5:e13289. doi: 10.1371/journal.pone.0013289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gonidi M, Athanassiadou AM, Patsouris E, Tsipis A, Dimopoulos S, Kyriakidou V, Chelidonis G, Athanassiadou P. Mitochondrial UCP4 and Bcl-2 expression in imprints of breast carcinomas: relationship with DNA ploidy and classical prognostic factors. Pathol Res Pract. 2011;207:377–382. doi: 10.1016/j.prp.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 125.Palmieri L, Palmieri F, Runswick MJ, Walker JE. Identification by bacterial expression and functional reconstitution of the yeast genomic sequence encoding the mitochondrial dicarboxylate carrier protein. FEBS Lett. 1996;399:299–302. doi: 10.1016/s0014-5793(96)01350-6. [DOI] [PubMed] [Google Scholar]
- 126.Fiermonte G, Palmieri L, Dolce V, Lasorsa FM, Palmieri F, Runswick MJ, Walker JE. The sequence, bacterial expression, and functional reconstitution of the rat mitochondrial dicarboxylate transporter cloned via distant homologs in yeast and Caenorhabditis elegans. J Biol Chem. 1998;273:24754–24759. doi: 10.1074/jbc.273.38.24754. [DOI] [PubMed] [Google Scholar]
- 127.Johnson RN, Chappell JB. The inhibition of mitochondrial dicarboxylate transport by inorganic phosphate, some phosphate esters and some phosphonate compounds. Biochem J. 1974;138:171–175. doi: 10.1042/bj1380171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wojtczak L, Wieckowski MR. The mechanisms of fatty acid-induced proton permeability of the inner mitochondrial membrane. J Bioenerg Biomembr. 1999;31:447–455. doi: 10.1023/a:1005444322823. [DOI] [PubMed] [Google Scholar]
- 129.Ventura FV, Ruiter J, Ijlst L, de Almeida IT, Wanders RJ. Differential inhibitory effect of long-chain acyl-CoA esters on succinate and glutamate transport into rat liver mitochondria and its possible implications for long-chain fatty acid oxidation defects. Mol Genet Metab. 2005;86:344–352. doi: 10.1016/j.ymgme.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 130.Lancar-Benba J, Foucher B, Saint-Macary M. Purification of the rat-liver mitochondrial dicarboxylate carrier by affinity chromatography on immobilized malate dehydrogenase. Biochim Biophys Acta. 1994;1190:213–216. doi: 10.1016/0005-2736(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 131.Mizuarai S, Miki S, Araki H, Takahashi K, Kotani H. Identification of dicarboxylate carrier Slc25a10 as malate transporter in de novo fatty acid synthesis. J Biol Chem. 2005;280:32434–32441. doi: 10.1074/jbc.M503152200. [DOI] [PubMed] [Google Scholar]
- 132.Kajimoto K, Terada H, Baba Y, Shinohara Y. Essential role of citrate export from mitochondria at early differentiation stage of 3T3-L1 cells for their effective differentiation into fat cells, as revealed by studies using specific inhibitors of mitochondrial di- and tricarboxylate carriers. Mol Genet Metab. 2005;85:46–53. doi: 10.1016/j.ymgme.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 133.Lash LH, Putt DA, Matherly LH. Protection of NRK-52E cells, a rat renal proximal tubular cell line, from chemical-induced apoptosis by overexpression of a mitochondrial glutathione transporter. J Pharmacol Exp Ther. 2002;303:476–486. doi: 10.1124/jpet.102.040220. [DOI] [PubMed] [Google Scholar]
- 134.Kamga CK, Zhang SX, Wang Y. Dicarboxylate carrier-mediated glutathione transport is essential for reactive oxygen species homeostasis and normal respiration in rat brain mitochondria. Am J Physiol Cell Physiol. 2010;299:C497–C505. doi: 10.1152/ajpcell.00058.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Xia W, Wan Y, Li YY, Zeng H, Lv Z, Li G, Wei Z, Xu SQ. PFOS prenatal exposure induce mitochondrial injury and gene expression change in hearts of weaned SD rats. Toxicology. 2011;282:23–29. doi: 10.1016/j.tox.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 136.Amaral AU, Cecatto C, Busanello EN, Ribeiro CA, Melo DR, Leipnitz G, Castillo RF, Wajner M. Ethylmalonic acid impairs brain mitochondrial succinate and malate transport. Mol Genet Metab. 2012;105:84–90. doi: 10.1016/j.ymgme.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 137.Cione E, Pingitore A, Perri M, Genchi G. Influence of all-trans-retinoic acid on oxoglutarate carrier via retinoylation reaction. Biochim Biophys Acta. 2009;1791:3–7. doi: 10.1016/j.bbalip.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 138.Zhong Q, Putt DA, Xu F, Lash LH. Hepatic mitochondrial transport of glutathione: studies in isolated rat liver mitochondria and H4IIE rat hepatoma cells. Arch Biochem Biophys. 2008;474:119–127. doi: 10.1016/j.abb.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Odegaard ML, Joseph JW, Jensen MV, Lu D, Ilkayeva O, Ronnebaum SM, Becker TC, Newgard CB. The mitochondrial 2-oxoglutarate carrier is part of a metabolic pathway that mediates glucose- and glutamine-stimulated insulin secretion. J Biol Chem. 2010;285:16530–16537. doi: 10.1074/jbc.M109.092593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kabe Y, Ohmori M, Shinouchi K, Tsuboi Y, Hirao S, Azuma M, Watanabe H, Okura I, Handa H. Porphyrin accumulation in mitochondria is mediated by 2-oxoglutarate carrier. J Biol Chem. 2006;281:31729–31735. doi: 10.1074/jbc.M604729200. [DOI] [PubMed] [Google Scholar]
- 141.Barrey E, Jayr L, Mucher E, Gospodnetic S, Joly F, Benech P, Alibert O, Gidrol X, Mata X, Vaiman A, et al. Transcriptome analysis of muscle in horses suffering from recurrent exertional rhabdomyolysis revealed energetic pathway alterations and disruption in the cytosolic calcium regulation. Anim Genet. 2012;43:271–281. doi: 10.1111/j.1365-2052.2011.02246.x. [DOI] [PubMed] [Google Scholar]
- 142.Wilkins HM, Marquardt K, Lash LH, Linseman DA. Bcl-2 is a novel interacting partner for the 2-oxoglutarate carrier and a key regulator of mitochondrial glutathione. Free Radical Biol Med. 2012;52:410–419. doi: 10.1016/j.freeradbiomed.2011.10.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gallo M, Park D, Luciani DS, Kida K, Palmieri F, Blacque OE, Johnson JD, Riddle DL. MISC-1/OGC links mitochondrial metabolism, apoptosis and insulin secretion. PLoS ONE. 2011;6:e17827. doi: 10.1371/journal.pone.0017827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Coll O, Colell A, Garcia-Ruiz C, Kaplowitz N, Fernandez-Checa JC. Sensitivity of the 2-oxoglutarate carrier to alcohol intake contributes to mitochondrial glutathione depletion. Hepatology. 2003;38:692–702. doi: 10.1053/jhep.2003.50351. [DOI] [PubMed] [Google Scholar]
- 145.Palmieri L, Pardo B, Lasorsa FM, del Arco A, Kobayashi K, Iijima M, Runswick MJ, Walker JE, Saheki T, Satrústegui J, et al. Citrin and aralar1 are Ca2 + -stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001;20:5060–5069. doi: 10.1093/emboj/20.18.5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fiermonte G, De Leonardis F, Todisco S, Palmieri L, Lasorsa FM, Palmieri F. Identification of the mitochondrial ATP-Mg/Pi transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution. J Biol Chem. 2004;279:30722–30730. doi: 10.1074/jbc.M400445200. [DOI] [PubMed] [Google Scholar]
- 147.del Arco A, Satrústegui J. Molecular cloning of Aralar, a new member of the mitochondrial carrier superfamily that binds calcium and is present in human muscle and brain. J Biol Chem. 1998;273:23327–23334. doi: 10.1074/jbc.273.36.23327. [DOI] [PubMed] [Google Scholar]
- 148.Satrústegui J, Contreras L, Ramos M, Marmol P, Del Arco A, Saheki T, Pardo B. Role of aralar, the mitochondrial transporter of aspartate-glutamate, in brain N-acetylaspartate formation and Ca2 + signaling in neuronal mitochondria. J Neurosci Res. 2007;85:3359–3366. doi: 10.1002/jnr.21299. [DOI] [PubMed] [Google Scholar]
- 149.Contreras L, Gomez-Puertas PJ, Iijima M, Kobayashi K, Saheki T, Satrústegui J. Ca 2 + activation kinetics of the two aspartate-glutamate mitochondrial carriers, aralar and citrin: role in the heart malate-aspartate NADH shuttle. J Biol Chem. 2007;282:7098–7106. doi: 10.1074/jbc.M610491200. [DOI] [PubMed] [Google Scholar]
- 150.Wibom R, Lasorsa FM, Töhönen V, Barbaro M, Sterky FH, Kucinski T, Naess K, Jonsson M, Pierri CL, Palmieri F, et al. AGC1 deficiency associated with global cerebral hypomyelination. N Engl J Med. 2009;361:489–495. doi: 10.1056/NEJMoa0900591. [DOI] [PubMed] [Google Scholar]
- 151.Kobayashi K, Sinasac DS, Iijima M, Boright AP, Begum L, Lee JR, Yasuda T, Ikeda S, Hirano R, Terazono H, et al. The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat Genet. 1999;22:159–163. doi: 10.1038/9667. [DOI] [PubMed] [Google Scholar]
- 152.Saheki T, Iijima M, Li MX, Kobayashi K, Horiuchi M, Ushikai M, Okumura F, Meng XJ, Inoue I, Tajima A, et al. Citrin/mitochondrial glycerol-3-phosphate dehydrogenase double knock-out mice recapitulate features of human citrin deficiency. J Biol Chem. 2007;282:25041–25052. doi: 10.1074/jbc.M702031200. [DOI] [PubMed] [Google Scholar]
- 153.Traba J, Satrústegui J, Del Arco A. Adenine nucleotide transporters in organelles: novel genes and functions. Cell Mol Life Sci. 2011;68:1183–1206. doi: 10.1007/s00018-010-0612-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Traba J, del Arco A, Duchen MR, Szabadkai G, Satrústegui J. SCaMC-1 promotes cancer cell survival by desensitizing mitochondrial permeability transition via ATP/ADP-mediated matrix Ca2 + buffering. Cell Death Differ. 2012;19:650–660. doi: 10.1038/cdd.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zarrilli R, Oates EL, McBride OW, Lerman MI, Chan JY, Santisteban P, Ursini MV, Notkins AL, Kohn LD. Sequence and chromosomal assignment of a novel cDNA identified by immunoscreening of a thyroid expression library: similarity to a family of mitochondrial solute carrier proteins. Mol Endocrinol. 1989;3:1498–1505. doi: 10.1210/mend-3-9-1498. [DOI] [PubMed] [Google Scholar]
- 156.Fiermonte G, Runswick MJ, Walker JE, Palmieri F. Sequence and pattern of expression of a bovine homologue of a human mitochondrial transport protein associated with Grave’s disease. DNA Seq. 1992;3:71–78. doi: 10.3109/10425179209033999. [DOI] [PubMed] [Google Scholar]
- 157.Prohl C, Pelzer W, Diekert K, Kmita H, Bedekovics T, Kispal G, Lill R. The yeast mitochondrial carrier Leu5p and its human homologue Graves’ disease protein are required for accumulation of coenzyme A in the matrix. Mol Cell Biol. 2001;21:1089–1097. doi: 10.1128/MCB.21.4.1089-1097.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wylin T, Baes M, Brees C, Mannaerts GP, Fransen M, Van Veldhoven PP. Identification and characterization of human PMP34, a protein closely related to the peroxisomal integral membrane protein PMP47 of Candida boidinii. Eur J Biochem. 1998;258:332–338. doi: 10.1046/j.1432-1327.1998.2580332.x. [DOI] [PubMed] [Google Scholar]
- 159.Fiermonte G, Palmieri L, Todisco S, Agrimi G, Palmieri F, Walker JE. Identification of the mitochondrial glutamate transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J Biol Chem. 2002;277:19289–19294. doi: 10.1074/jbc.M201572200. [DOI] [PubMed] [Google Scholar]
- 160.Maechler P, Wollheim CB. Mitochondrial glutamate acts as a messenger in glucose-induced insulin exocytosis. Nature. 1999;402:685–689. doi: 10.1038/45280. [DOI] [PubMed] [Google Scholar]
- 161.Casimir M, Lasorsa FM, Rubi B, Caille D, Palmieri F, Meda P, Maechler P. Mitochondrial glutamate carrier GC1 as a newly identified player in the control of glucose-stimulated insulin secretion. J Biol Chem. 2009;284:25004–25014. doi: 10.1074/jbc.M109.015495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Carobbio S, Frigerio F, Rubi B, Vetterli L, Bloksgaard M, Gjinovci A, Pournourmohammadi S, Herrera PL, Reith W, Mandrup S, et al. Deletion of glutamate dehydrogenase in β-cells abolishes part of the insulin secretory response not required for glucose homeostasis. J Biol Chem. 2009;284:921–929. doi: 10.1074/jbc.M806295200. [DOI] [PubMed] [Google Scholar]
- 163.Molinari F, Raas-Rothschild A, Rio M, Fiermonte G, Encha-Razavi F, Palmieri L, Palmieri F, Ben-Neriah Z, Kadhom N, Vekemans M, et al. Impaired mitochondrial glutamate transport in autosomal recessive neonatal myoclonic epilepsy. Am J Hum Genet. 2005;76:334–339. doi: 10.1086/427564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Molinari F, Kaminska A, Fiermonte G, Boddaert N, Raas-Rothschild A, Plouin P, Palmieri L, Brunelle F, Palmieri F, Dulac O, et al. Mutations in the mitochondrial glutamate carrier SLC25A22 in neonatal epileptic encephalopathy with suppression bursts. Clin Genet. 2009;76:188–194. doi: 10.1111/j.1399-0004.2009.01236.x. [DOI] [PubMed] [Google Scholar]
- 165.Dolce V, Fiermonte G, Runswick MJ, Palmieri F, Walker JE. The human mitochondrial deoxynucleotide carrier and its role in the toxicity of nucleoside antivirals. Proc Natl Acad Sci USA. 2001;98:2284–2288. doi: 10.1073/pnas.031430998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lindhurst MJ, Fiermonte G, Song S, Struys E, De Leonardis F, Schwartzberg PL, Chen A, Castegna A, Verhoeven N, Mathews CK, et al. Knockout of Slc25a19 causes mitochondrial thiamine pyrophosphate depletion, embryonic lethality, CNS malformations, and anemia. Proc Natl Acad Sci USA. 2006;103:15927–15932. doi: 10.1073/pnas.0607661103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kang J, Samuels DC. The evidence that the DNC (SLC25A19) is not the mitochondrial deoxyribonucleotide carrier. Mitochondrion. 2008;8:103–108. doi: 10.1016/j.mito.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 168.Rosenberg MJ, Agarwala R, Bouffard G, Davis J, Fiermonte G, Hilliard MS, Koch T, Kalikin LM, Makalowska I, Morton DH, et al. Mutant deoxynucleotide carrier is associated with congenital microcephaly. Nat Genet. 2002;32:175–179. doi: 10.1038/ng948. [DOI] [PubMed] [Google Scholar]
- 169.Lewis W, Haase CP, Miller YK, Ferguson B, Stuart T, Ludaway T, McNaught J, Russ R, Steltzer J, Santoianni R, et al. Transgenic expression of the deoxynucleotide carrier causes mitochondrial damage that is enhanced by NRTIs for AIDS. Lab Invest. 2005;85:972–981. doi: 10.1038/labinvest.3700301. [DOI] [PubMed] [Google Scholar]
- 170.Lam W, Chen C, Ruan S, Leung CH, Cheng YC. Expression of deoxynucleotide carrier is not associated with the mitochondrial DNA depletion caused by anti-HIV dideoxynucleoside analogs and mitochondrial dNTP uptake. Mol Pharmacol. 2005;67:408–416. doi: 10.1124/mol.104.007120. [DOI] [PubMed] [Google Scholar]
- 171.Indiveri C, Tonazzi A, Palmieri F. Identification and purification of the carnitine carrier from rat liver mitochondria. Biochim Biophys Acta. 1990;1020:81–86. doi: 10.1016/0005-2728(90)90096-m. [DOI] [PubMed] [Google Scholar]
- 172.Indiveri C, Tonazzi A, Palmieri F. The reconstituted carnitine carrier from rat liver mitochondria: evidence for a transport mechanism different from that of the other mitochondrial translocators. Biochim Biophys Acta. 1994;1189:65–73. doi: 10.1016/0005-2736(94)90281-x. [DOI] [PubMed] [Google Scholar]
- 173.Sekoguchi E, Sato N, Yasui A, Fukada S, Nimura Y, Aburatani H, Ikeda K, Matsuura A. A novel mitochondrial carnitine-acylcarnitine translocase induced by partial hepatectomy and fasting. J Biol Chem. 2003;278:38796–38802. doi: 10.1074/jbc.M306372200. [DOI] [PubMed] [Google Scholar]
- 174.Huizing M, Iacobazzi V, Ijlst L, Savelkoul P, Ruitenbeek W, van den Heuvel L, Indiveri C, Smeitink J, Trijbels F, Wanders R, et al. Cloning of the human carnitine-acylcarnitine carrier cDNA and identification of the molecular defect in a patient. Am J Hum Genet. 1997;61:1239–1245. doi: 10.1086/301628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Costa C, Costa JM, Nuoffer JM, Slama A, Boutron A, Saudubray JM, Legrand A, Brivet M. Identification of the molecular defect in a severe case of carnitine-acylcarnitine carrier deficiency. J Inherit Metab Dis. 1999;22:267–270. doi: 10.1023/a:1005590223680. [DOI] [PubMed] [Google Scholar]
- 176.Ruiter JP, Oostheim W, Niezen-Koning KE, Palmieri F, Wanders RJ. Identification of a missense mutation in a patient with lethal carnitine acyl-carnitine carrier deficiency. Adv Exp Med Biol. 1999;466:347–351. doi: 10.1007/0-306-46818-2_40. [DOI] [PubMed] [Google Scholar]
- 177.Iacobazzi V, Invernizzi F, Baratta S, Pons R, Chung W, Garavaglia B, Dionisi-Vici C, Ribes A, Parini R, Huertas MD, et al. Molecular and functional analysis of SLC25A20 mutations causing carnitine-acylcarnitine translocase deficiency. Hum Mutat. 2004;24:312–320. doi: 10.1002/humu.20085. [DOI] [PubMed] [Google Scholar]
- 178.Lammers G, Poelkens F, van Duijnhoven NT, Pardoel EM, Hoenderop JG, Thijssen DH, Hopman MT. Expression of genes involved in fatty acid transport and insulin signaling is altered by physical inactivity and exercise training in human skeletal muscle. Am J Physiol Endocrinol Metab. 2012;303:E1245–E1251. doi: 10.1152/ajpendo.00356.2012. [DOI] [PubMed] [Google Scholar]
- 179.McGivney BA, McGettigan PA, Browne JA, Evans ACO, Fonseca RG, Loftus BJ, Lohan A, MacHugh DE, Murphy BA, Katz LM, et al. Characterization of the equine skeletal muscle transcriptome identifies novel functional responses to exercise training. BMC Genomics. 2010;11:398. doi: 10.1186/1471-2164-11-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Di Noia MA, Van Driesche S, Palmieri F, Yang LM, Quan S, Goodman AI, Abraham NG. Heme oxygenase-1 enhances renal mitochondrial transport carriers and cytochrome c oxidase activity in experimental diabetes. J Biol Chem. 2006;281:15687–15693. doi: 10.1074/jbc.M510595200. [DOI] [PubMed] [Google Scholar]
- 181.Iacobazzi V, Convertini P, Infantino V, Scarcia P, Todisco S, Palmieri F. Statins, fibrates and retinoic acid upregulate mitochondrial acylcarnitine carrier gene expression. Biochem Biophys Res Commun. 2009;388:643–647. doi: 10.1016/j.bbrc.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 182.Fiermonte G, Dolce V, Palmieri L, Ventura M, Runswick MJ, Palmieri F, Walker JE. Identification of the human mitochondrial oxodicarboxylate carrier. Bacterial expression, reconstitution, functional characterization, tissue distribution, and chromosomal location. J Biol Chem. 2001;276:8225–8230. doi: 10.1074/jbc.M009607200. [DOI] [PubMed] [Google Scholar]
- 183.Niimi M, Tao L, Lin SH, Yin J, Wu X, Fukui H, Kambayashi J, Ye J, Sun B. Involvement of an alternatively spliced mitochondrial oxodicarboxylate carrier in adipogenesis in 3T3-L1 cells. J Biomed Sci. 2009;16:92. doi: 10.1186/1423-0127-16-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Harris T, Pan Q, Sironi J, Lutz D, Tian J, Sapkar J, Perez-Soler R, Keller S, Locker J. Both gene amplification and allelic loss occur at 14q13.3 in lung cancer. Clin Cancer Res. 2011;17:690–699. doi: 10.1158/1078-0432.CCR-10-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhong Q, Kowluru RA. Diabetic retinopathy and damage to mitochondrial structure and transport machinery. Invest Ophthalmol Visual Sci. 2011;52:8739–8746. doi: 10.1167/iovs.11-8045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Horne DW, Holloway RS, Wagner C. Transport of S-adenosylmethionine in isolated rat liver mitochondria. Arch Biochem Biophys. 1997;343:201–206. doi: 10.1006/abbi.1997.0167. [DOI] [PubMed] [Google Scholar]
- 187.Agrimi G, Di Noia MA, Marobbio CM, Fiermonte G, Lasorsa FM, Palmieri F. Identification of the human mitochondrial S-adenosylmethionine transporter: bacterial expression, reconstitution, functional characterization and tissue distribution. Biochem J. 2004;379:183–190. doi: 10.1042/BJ20031664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Infantino V, Castegna A, Iacobazzi F, Spera I, Scala I, Andria G, Iacobazzi V. Impairment of methyl cycle affects mitochondrial methyl availability and glutathione level in Down’s syndrome. Mol Genet Metab. 2011;102:378–382. doi: 10.1016/j.ymgme.2010.11.166. [DOI] [PubMed] [Google Scholar]
- 189.Li FY, Nikali K, Gregan J, Leibiger I, Leibiger B, Schweyen R, Larsson C, Suomalainen A. Characterization of a novel human putative mitochondrial transporter homologous to the yeast mitochondrial RNA splicing proteins 3 and 4. FEBS Lett. 2001;494:79–84. doi: 10.1016/s0014-5793(01)02319-5. [DOI] [PubMed] [Google Scholar]
- 190.Li FY, Leibiger B, Leibiger I, Larsson C. Characterization of a putative murine mitochondrial transporter homology of hMRS3/4. Mamm Genome. 2002;13:20–23. doi: 10.1007/s00335-001-2094-y. [DOI] [PubMed] [Google Scholar]
- 191.Shaw GC, Cope JJ, Li L, Corson K, Hersey C, Ackermann GE, Gwynn B, Lambert AJ, Wingert RA, Traver D, et al. Mitoferrin is essential for erythroid iron assimilation. Nature. 2006;440:96–100. doi: 10.1038/nature04512. [DOI] [PubMed] [Google Scholar]
- 192.Haitina T, Lindblom J, Renstrom T, Fredriksson R. Fourteen novel human members of mitochondrial solute carrier family 25 (SLC25) widely expressed in the central nervous system. Genomics. 2006;88:779–790. doi: 10.1016/j.ygeno.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 193.Paradkar PN, Zumbrennen KB, Paw BH, Ward DM, Kaplan J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol Cell Biol. 2009;29:1007–1016. doi: 10.1128/MCB.01685-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Chen W, Paradkar PN, Li L, Pierce EL, Langer NB, Takahashi-Makise N, Hyde BB, Shirihai OS, Ward DM, Kaplan J, et al. Abcb10 physically interacts with mitoferrin-1 (Slc25a37) to enhance its stability and function in the erythroid mitochondria. Proc Natl Acad Sci USA. 2009;106:16263–16268. doi: 10.1073/pnas.0904519106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Chen W, Dailey HA, Paw BH. Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis. Blood. 2010;116:628–630. doi: 10.1182/blood-2009-12-259614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Horne DW, Holloway RS, Said HM. Uptake of 5-formyltetrahydrofolate in isolated rat liver mitochondria is carrier-mediated. J Nutr. 1992;122:2204–2209. doi: 10.1093/jn/122.11.2204. [DOI] [PubMed] [Google Scholar]
- 197.Titus SA, Moran RG. Retrovirally mediated complementation of the glyB phenotype. Cloning of a human gene encoding the carrier for entry of folates into mitochondria. J Biol Chem. 2000;275:36811–36817. doi: 10.1074/jbc.M005163200. [DOI] [PubMed] [Google Scholar]
- 198.McCarthy EA, Titus SA, Taylor SM, Jackson-Cook C, Moran RG. A mutation inactivating the mitochondrial inner membrane folate transporter creates a glycine requirement for survival of Chinese hamster cells. J Biol Chem. 2004;279:33829–33836. doi: 10.1074/jbc.M403677200. [DOI] [PubMed] [Google Scholar]
- 199.Spaan AN, Ijlst L, van Roermund CW, Wijburg FA, Wanders RJ, Waterham HR. Identification of the human mitochondrial FAD transporter and its potential role in multiple acyl-CoA dehydrogenase deficiency. Mol Genet Metab. 2005;86:441–447. doi: 10.1016/j.ymgme.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 200.Biswas A, Senthilkumar N, Said HM. Effect of chronic alcohol exposure on folate uptake by liver mitochondria. Am J Physiol Cell Physiol. 2012;302:C203–C209. doi: 10.1152/ajpcell.00283.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bukhari FJ, Moradi H, Gollapudi P, Ju Kim H, Vaziri ND, Said HM. Effect of chronic kidney disease on the expression of thiamin and folic acid transporters. Nephrol Dial Transplant. 2011;26:2137–2144. doi: 10.1093/ndt/gfq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ye YL, Chan YT, Liu HC, Lu HT, Huang RF. Depleted folate pool and dysfunctional mitochondria associated with defective mitochondrial folate proteins sensitize Chinese ovary cell mutants to tert-butylhydroperoxide-induced oxidative stress and apoptosis. J Nutr Biochem. 2010;21:793–800. doi: 10.1016/j.jnutbio.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 203.Becker ML, van Haandel L, Gaedigk R, Thomas B, Hoeltzel MF, Lasky A, Dai H, Stobaugh J, Leeder JS. Red blood cell folate concentrations and polyglutamate distribution in juvenile arthritis: predictors of folate variability. Pharmacogenet Genomics. 2012;22:236–246. doi: 10.1097/FPC.0b013e3283500202. [DOI] [PubMed] [Google Scholar]
- 204.Wernimont SM, Clark AG, Stover PJ, Wells MT, Litonjua AA, Weiss ST, Gaziano JM, Vokonas PS, Tucker KL, Cassano PA. Folate network genetic variation predicts cardiovascular disease risk in non-Hispanic white males. J Nutr. 2012;142:1272–1279. doi: 10.3945/jn.111.157180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Bridges EG, Jiang Z, Cheng YC. Characterization of a dCTP transport activity reconstituted from human mitochondria. J Biol Chem. 1999;274:4620–4625. doi: 10.1074/jbc.274.8.4620. [DOI] [PubMed] [Google Scholar]
- 206.Marobbio CM, Di Noia MA, Palmieri F. Identification of a mitochondrial transporter for pyrimidine nucleotides in Saccharomyces cerevisiae: bacterial expression, reconstitution and functional characterization. Biochem J. 2006;393:441–446. doi: 10.1042/BJ20051284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Floyd S, Favre C, Lasorsa FM, Leahy M, Trigiante G, Stroebel P, Marx A, Loughran G, O’Callaghan K, Marobbio CM, et al. The insulin-like growth factor-I-mTOR signaling pathway induces the mitochondrial pyrimidine nucleotide carrier to promote cell growth. Mol Biol Cell. 2007;18:3545–3555. doi: 10.1091/mbc.E06-12-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Favre C, Zhdanov A, Leahy M, Papkovsky D, O’Connor R. Mitochondrial pyrimidine nucleotide carrier (PNC1) regulates mitochondrial biogenesis and the invasive phenotype of cancer cells. Oncogene. 2010;29:3964–3976. doi: 10.1038/onc.2010.146. [DOI] [PubMed] [Google Scholar]
- 209.Wilting SM, de Wilde J, Meijer CJ, Berkhof J, Yi Y, van Wieringen WN, Braakhuis BJ, Meijer GA, Ylstra B, Snijders PJ, et al. Integrated genomic and transcriptional profiling identifies chromosomal loci with altered gene expression in cervical cancer. Genes Chromosome Cancer. 2008;47:890–905. doi: 10.1002/gcc.20590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Bergmann AK, Campagna DR, McLoughlin EM, Agarwal S, Fleming MD, Bottomley SS, Neufeld EJ. Systematic molecular genetic analysis of congenital sideroblastic anemia: evidence for genetic heterogeneity and identification of novel mutations. Pediatr Blood Cancer. 2010;54:273–278. doi: 10.1002/pbc.22244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Guernsey DL, Jiang H, Campagna DR, Evans SC, Ferguson M, Kellogg MD, Lachance M, Matsuoka M, Nightingale M, Rideout A, et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat Genet. 2009;41:651–653. doi: 10.1038/ng.359. [DOI] [PubMed] [Google Scholar]
- 212.Albagha OM, Wani SE, Visconti MR, Alonso N, Goodman K, Brandi ML, Cundy T, Chung PYJ, Dargie R, Devogelaer JP, et al. Genome-wide association identifies three new susceptibility loci for Paget’s disease of bone. Nat Genet. 2011;43:685–689. doi: 10.1038/ng.845. [DOI] [PubMed] [Google Scholar]
- 213.Hirota T, Takahashi A, Kubo M, Tsunoda T, Tomita K, Sakashita M, Yamada T, Fujieda S, Tanaka S, Doi S, et al. Genome-wide association study identifies eight new susceptibility loci for atopic dermatitis in the Japanese population. Nat Genet. 2012;44:1222–1226. doi: 10.1038/ng.2438. [DOI] [PubMed] [Google Scholar]
- 214.Sun LD, Xiao FL, Li Y, Zhou WM, Tang HY, Tang XF, Zhang H, Schaarschmidt H, Zuo XB, Foelster-Holst R, et al. Genome-wide association study identifies two new susceptibility loci for atopic dermatitis in the Chinese Han population. Nat Genet. 2011;43:690–694. doi: 10.1038/ng.851. [DOI] [PubMed] [Google Scholar]
- 215.Liu X, Cheng R, Verbitsky M, Kisselev S, Browne A, Mejia-Sanatana H, Louis ED, Cote LJ, Andrews H, Waters C, et al. Genome-wide association study identifies candidate genes for Parkinson’s disease in an Ashkenazi Jewish population. BMC Med Genet. 2011;12:104. doi: 10.1186/1471-2350-12-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Paquette MA, Dong H, Gagné R, Williams A, Malowany M, Wade MG, Yauk CL. Thyroid hormone-regulated gene expression in juvenile mouse liver: identification of thyroid response elements using microarray profiling and in silico analyses. BMC Genomics. 2011;12:634. doi: 10.1186/1471-2164-12-634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Tan MGK, Ooi LLPJ, Aw SE, Hui KM. Cloning and identification of hepatocellular carcinoma down-regulated mitochondrial carrier protein, a novel liver-specific uncoupling protein. J Biol Chem. 2004;279:45235–45244. doi: 10.1074/jbc.M403683200. [DOI] [PubMed] [Google Scholar]
- 218.Jin X, Yang YD, Chen K, Lv ZY, Zheng L, Liu YP, Chen SH, Yu CH, Jiang XY, Zhang CY, et al. HDMCP uncouples yeast mitochondrial respiration and alleviates steatosis in L02 and hepG2 cells by decreasing ATP and H2O2 levels: a novel mechanism for NAFLD. J Hepatol. 2009;50:1019–1028. doi: 10.1016/j.jhep.2008.10.034. [DOI] [PubMed] [Google Scholar]
- 219.Luevano-Martínez LA. Uncoupling proteins (UCP) in unicellular eukaryotes: true UCPs or UCP1-like acting proteins? FEBS Lett. 2012;586:1073–1078. doi: 10.1016/j.febslet.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 220.Lamarca V, Marzo I, Sanz-Clemente A, Carrodeguas JA. Exposure of any of two proapoptotic domains of presenilin 1-associated protein/mitochondrial carrier homolog 1 on the surface of mitochondria is sufficient for induction of apoptosis in a Bax/Bak-independent manner. Eur J Cell Biol. 2008;87:325–334. doi: 10.1016/j.ejcb.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 221.Mao G, Tan J, Gao W, Shi Y, Cui MZ, Xu X. Both the N-terminal fragment and the protein-protein interaction domain (PDZ domain) are required for the pro-apoptotic activity of presenilin-associated protein PSAP. Biochim Biophys Acta. 2008;1780:696–708. doi: 10.1016/j.bbagen.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Robinson AJ, Kunji ER, Gross A. Mitochondrial carrier homolog 2 (MTCH2): the recruitment and evolution of a mitochondrial carrier protein to a critical player in apoptosis. Exp Cell Res. 2012;318:1316–1323. doi: 10.1016/j.yexcr.2012.01.026. [DOI] [PubMed] [Google Scholar]
- 223.Lamarca V, Sanz-Clemente A, Pérez-Pé R, Martínez-Lorenzo MJ, Halaihel N, Muniesa P, Carrodeguas JA. Two isoforms of PSAP/MTCH1 share two proapoptotic domains and multiple internal signals for import into the mitochondrial outer membrane. Am J Physiol Cell Physiol. 2007;293:C1347–C1361. doi: 10.1152/ajpcell.00431.2006. [DOI] [PubMed] [Google Scholar]
- 224.Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, Heid IM, Berndt SI, Elliott AL, Jackson AU, Lamina C, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]