

Abstract
Sulphonylurea drugs are the therapy of choice for treating neonatal diabetes (ND) caused by mutations in the ATP-sensitive K+ channel (KATP channel). We investigated the interactions between MgATP, MgADP, and the sulphonylurea gliclazide with KATP channels expressed in Xenopus oocytes. In the absence of MgATP, gliclazide block was similar for wild-type channels and those carrying the Kir6.2 ND mutations R210C, G334D, I296L, and V59M. Gliclazide abolished the stimulatory effect of MgATP on all channels. Conversely, high MgATP concentrations reduced the gliclazide concentration, producing a half-maximal block of G334D and R201C channels and suggesting a mutual antagonism between nucleotide and gliclazide binding. The maximal extent of high-affinity gliclazide block of wild-type channels was increased by MgATP, but this effect was smaller for ND channels; channels that were least sensitive to ATP inhibition showed the smallest increase in sulphonylurea block. Consequently, G334D and I296L channels were not fully blocked, even at physiological MgATP concentrations (1 mmol/L). Glibenclamide block was also reduced in β-cells expressing Kir6.2-V59M channels. These data help to explain why patients with some mutations (e.g., G334D, I296L) are insensitive to sulphonylurea therapy, why higher drug concentrations are needed to treat ND than type 2 diabetes, and why patients with severe ND mutations are less prone to drug-induced hypoglycemia.
Neonatal diabetes (ND) usually presents within the first 6 months of life (1). It is caused by mutations in a number of genes, but those in the ATP-sensitive K+ channel (KATP channel) subunits SUR1 and Kir6.2 are of special significance because most patients whose diabetes is caused by these mutations can be treated with sulphonylurea drugs (2,3). The KATP channel plays a critical role in insulin secretion because it controls the resting membrane potential of the pancreatic β-cell (4). When KATP channels are open, as happens when glucose levels are low, the membrane is hyperpolarized, calcium influx is switched off, and insulin secretion is inhibited. Closure of KATP channels in response to glucose metabolism leads to membrane depolarization, opening of voltage-gated calcium channels, and a rise in intracellular calcium concentration that triggers insulin release. Mutations that cause ND impair the ability of the channel to close in response to metabolically generated ATP and thus prevent depolarization and insulin secretion (1,5–8).
Intracellular adenine nucleotides influence KATP channel activity in multiple ways. Binding of ATP (or ADP) to Kir6.2 results in channel closure (9). Interaction of MgATP or MgADP with the two nucleotide-binding sites (NBS1, NBS2) of SUR1 stimulates channel activity by increasing the channel open probability (PO), thereby masking the inhibitory effect of ATP and indirectly reducing ATP block (10–13). It is believed that this interaction is mediated by MgADP occupancy of NBS2 and that MgATP is first hydrolyzed to MgADP (14). MgATP interaction with SUR1 also reduces ATP inhibition at Kir6.2 by most likely allosterically reducing ATP binding (15,16).
In the absence of nucleotides, sulphonylureas bound to SUR1 act as partial agonists and produce a maximum block of ∼50–80% (Fig. 1B) (17,18). Like ATP, they reduce the mean open time and burst duration of the channel and increase the frequency and duration of the interburst closures (19,20). Sulphonylureas also decrease Mg-nucleotide binding to SUR1 and, consequently, abolish channel stimulation (Fig. 1A) (17). This unmasks the inhibitory effect of ATP at Kir6.2, which adds to the sulphonylurea block and produces an apparent increase in the inhibitory effect of the drug (Fig. 1B).
FIG. 1.

The mechanism of sulphonylurea block of KATP channels. A: Schematic of the interaction of Mg-nucleotides with Kir6.2 and SUR1 subunits in the absence (left) and presence (right) of sulphonylureas for wild-type channels and channels carrying an ATP-insensitive mutation (G334) in Kir6.2. Nucleotides reduce the PO of wild-type channels by binding to Kir6.2 and increase PO by interacting with SUR1 (top left). Sulphonylureas inhibit channel activity by binding to SUR1. They also displace MgADP from SUR1, and consequently, ATP block at Kir6.2 is not balanced by MgATP activation at SUR1 (top right). Channels with a mutation that abolishes (G334D) ATP inhibition at Kir6.2 are activated but not blocked by ADP or MgADP (bottom left). Sulphonylureas still inhibit channel activity. They also displace MgADP from SUR1, abolishing activation; however, the total block is less because of the lack of ATP inhibition at Kir6.2 (bottom right). B: Concentration-response curves for gliclazide block of macroscopic KATP currents in excised patches in the absence (○; n = 4–8) and presence (●; n = 7–12) of 100 μmol/L MgADP. The lines are the best fit of Eq. 2 to the mean data, with IC50(1) = 50 nmol/L, IC50(2) = 3 mmol/L, h1 = h2 = 1, and a = 0.4 (0 µmol/L MgADP), or to Eq. 1, with IC50 = 204 nmol/L, h = 1.13, and a = 0.065 (100 µmol/L MgADP). Data are taken from Gribble and Ashcroft (23) and Proks et al. (33). SU, sulphonylurea.
Sulphonylurea block, therefore, is predicted to depend on both cell metabolism and the ATP sensitivity of the KATP channel. By impairing the latter, ND mutations are expected to reduce the efficacy of sulphonylurea block (Fig. 1A). We explored the effect of a range of ND mutations on sulphonylurea block of the KATP channel. These results explain why patients with some mutations respond to sulphonylurea therapy and why those with other mutations do not.
RESEARCH DESIGN AND METHODS
Oocyte studies.
For heterologous expression, we used human Kir6.2 (Genbank NM000525 with E23 and I377) and rat SUR1 (Genbank L40624). Site-directed mutagenesis, preparation of mRNA, and isolation and injection of Xenopus laevis oocytes with mRNA have been described previously (21). Macroscopic currents were recorded from giant inside-out patches at 20–22°C and −60 mV, filtered at 5 kHz, and digitized at 20 kHz. The pipette solution contained (mmol/L) 140 KCl, 1.2 MgCl2, 2.6 CaCl2, and 10 HEPES (pH 7.4 with KOH). The standard internal (bath) solution contained (mmol/L) 107 KCl, 1 CaCl2, 2 MgCl2, 10 EGTA, and 10 HEPES (pH 7.2 with KOH) and either MgATP, MgADP, or gliclazide. The Mg-free solution contained (mmol/L) 107 KCl, 1 K2SO4, 10 EGTA, and 10 HEPES (pH 7.2 with KOH) and either K2ATP, K2ADP, or gliclazide.
The relationship between nucleotide or drug concentration and KATP current inhibition was fit with
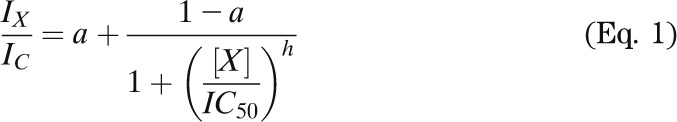 |
where IX is the steady-state KATP current in the presence of the test nucleotide or drug concentration [X], IC is the current in nucleotide-free (or drug-free) solution obtained by averaging the current before and after application, IC50 is the nucleotide (or drug) concentration at which the inhibition is half maximal, h is the Hill coefficient, and a is the fraction of KATP current remaining at gliclazide concentrations that saturate the high-affinity binding site on SUR1 (a = 0 for ATP concentration-inhibition relations).
For noise analysis, the macroscopic mean current (I) and variance (σ) were determined from 1 s data segments. Control data were recorded immediately before gliclazide application, and test data were recorded once a steady-state condition was reached. PO and N (number of active channels in the patch) were calculated as described previously (16).
β-Cell studies.
Mice expressing the Kir6.2-V59M mutation specifically in their pancreatic β-cells (ib-V59M mice) were generated by crossing mice carrying a floxed Kir6.2-V59M gene (8) with RIP-Cre-ER mice (22). Mice were maintained as described previously (8). Tamoxifen (0.4 mL of 20 mg/mL in corn oil) was injected at 12 weeks of age to induce Kir6.2-V59M gene expression. Three days later when blood glucose concentration was >20 mmol/L, islets were isolated and dissociated into single β-cells, and cells were cultured for 1–3 days as previously described (8). Control mice were injected with corn oil alone. All experiments were conducted in accordance with the U.K. Animals (Scientific Procedures) Act 1986 and University of Oxford ethical guidelines.
Whole-cell KATP currents were recorded from isolated β-cells using the standard whole-cell configuration. β-Cells were distinguished from α-cells by their size and (in the case of mutant β-cells) their larger steady-state whole-cell KATP currents. The pipette solution contained (mmol/L) 107 KCl, 1 CaCl2, 1 MgCl2, 10 EGTA, 10 HEPES, and 0.3 ATP (pH 7.2 with KOH). The extracellular solution contained (mmol/L) 138 NaCl, 5.6 KCl, 1.2 MgCl2, 2.6 CaCl2, and 10 HEPES (pH 7.4 with NaOH). A 100 mmol/L stock solution of glibenclamide was made in DMSO and diluted in extracellular solution as required. Concentration-response curves in Fig. 1B (open circles) and Fig. 7D were fit with
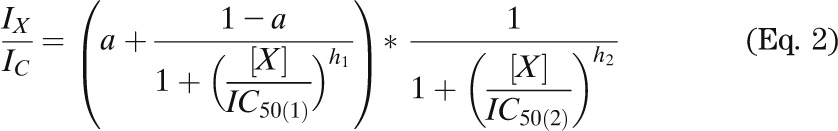 |
where IX and IC are the current amplitudes in the presence and absence of the drug, respectively; IC50(1) and IC50(2) are the concentrations ([X]) at which inhibition is half maximal for the high-affinity and low-affinity sites, respectively; h1 and h2 are slope factors; and a is the fraction of unblocked current at the high-affinity site at saturating drug concentration. For fitting, h2 was fixed at 1.0.
Statistical analysis.
All values are presented as mean ± SEM. Statistical significance was determined by Student t test.
RESULTS
To explore the effects of sulphonylureas on KATP channels containing ND mutations, we used the sulphonylurea gliclazide. Gliclazide is technically advantageous because its effects are readily reversible, and when measured in inside-out patches, its concentration-inhibition relationship exhibits a clearly defined separation between block at the high-affinity (IC50 ∼5 nmol/L) site on SUR1 and block at the low-affinity (IC50 ∼3 mmol/L) site on Kir6.2 (Fig. 1B) (23). We confined our studies to high-affinity block at gliclazide concentrations ≤100 μmol/L because only this is of clinical importance (Fig. 1B) (24).
We also restricted our analysis to mutations in Kir6.2 because these are associated with the most severe reductions in ATP inhibition (7,25–30), and some are known to confer a significantly reduced block by sulphonylureas (28,30,31). We selected four Kir6.2 ND mutations, all of which reduce the ability of ATP to block the channel. Two (R201C and G334D) are located in the putative ATP-binding site (25,26), and two (V59M and I296L) reduce ATP inhibition indirectly by impairing channel gating (26,27). We refer to channels containing these mutations by the name of the mutation (e.g., Kir6.2-R201C/SUR1 is R201C).
Effects of gliclazide on ATP block of mutant channels.
We first examined the effect of gliclazide on ATP block of R201C, V59M, and I296L channels heterologously expressed in Xenopus oocytes. We could not study G334D channels because they are not inhibited by ATP (25,28).
Figure 2 shows KATP currents and ATP concentration-inhibition relationships for wild-type and R201C channels measured in inside-out patches in the presence and absence of Mg2+ or gliclazide. The mutation increased the IC50 of ATP from 7 to 98 μmol/L in the absence of Mg2+ and from 16 μmol/L to 2 mmol/L in the presence of Mg2+ (Table 1). These values are similar to those reported previously for R201C channels expressed in Xenopus oocytes (21,26).
FIG. 2.

Effect of gliclazide on ATP block of wild-type and R201C KATP channels. A–D: Representative Kir6.2/SUR1 (A and C) and Kir6.2-R201C/SUR1 (B and D) currents recorded at −60 mV in the absence (A and B) and presence (C and D) of Mg2+. Gliclazide and ATP were added as indicated by the bars. The dotted line indicates the zero current level. E and F: ATP concentration-inhibition curves for Kir6.2/SUR1 (E) and Kir6.2-R201C/SUR1 (F) channels measured in the absence of both gliclazide (SU) and Mg2+ (□) and in the absence of gliclazide but in the presence of 2 mmol/L Mg2+ (○), 30 μmol/L gliclazide without Mg2+ (■), and 30 μmol/L gliclazide plus 2 mmol/L Mg2+ (●). The lines are the best fit of Eq. 1 to the mean data with the following parameters: Kir6.2/SUR1 (E) IC50 = 6.8 μmol/L, h = 1.0 (□); IC50 = 4 μmol/L, h = 1.0 (■); IC50 = 16 μmol/L, h = 1.0 (○); and IC50 = 4 μmol/L, h = 0.97 (●) and Kir6.2-R201C/SUR1 (F): IC50 = 98 μmol/L, h = 1.4 (□); IC50 = 2.0 mmol/L, h = 1.1 (○); IC50 = 97 μmol/L, h = 0.87 (■); and IC50 = 107 μmol/L, h = 0.90 (●). The dotted line is the concentration-inhibition curve for Kir6.2/SUR1 channels in the absence of Mg2+ and gliclazide (Fig. 2E) (□). SU, sulfonylurea; WT, wild-type.
TABLE 1.
ATP block of wild-type and mutant KATP channels
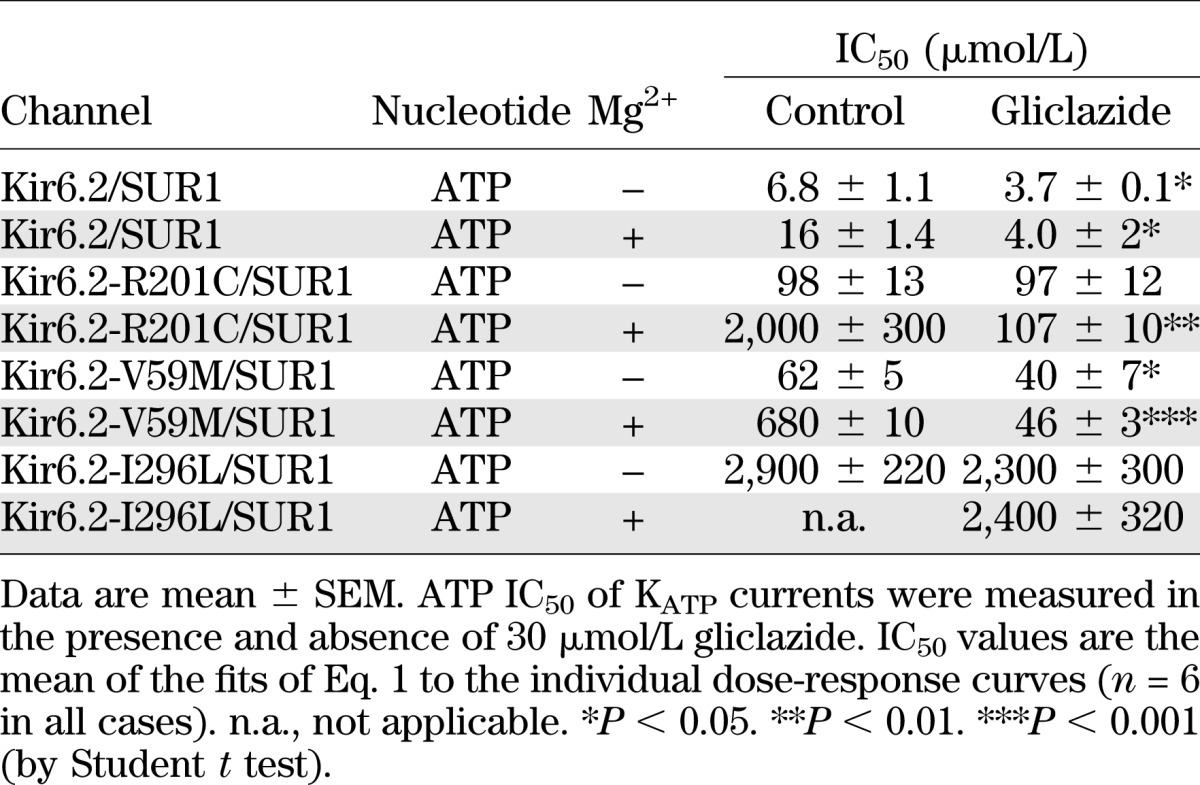
In the absence of Mg2+, gliclazide caused a small, but significant increase in ATP block of wild-type channels (Fig. 2A and E and Table 1). One possible explanation for this finding is that ATP binding to NBS1 of SUR1 (in an Mg-independent manner) reduces channel inhibition and that this effect is reversed by gliclazide. Gliclazide did not alter the IC50 for ATP inhibition of R201C channels in the absence of Mg2+ (Fig. 2F and Table 1), suggesting that the mutation might impair the ability of ATP binding to SUR1-NBS1 to cause channel closure (and because this is absent, gliclazide has no effect). Why gliclazide reduces the slope of the ATP concentration-inhibition curve is unclear.
Addition of Mg2+ reduced ATP block of both wild-type and R201C channels, but the effect was much greater for the mutant channels (Fig. 2E and F), as shown previously (21). In the presence of gliclazide, the MgATP and ATP concentration-inhibition relationships of R201C channels were identical (Fig. 2F and Table 1), suggesting that gliclazide abolishes channel activation by MgATP at SUR1 as it does for wild-type channels.
We next examined the effect of gliclazide on V59M and I296L channels. In the absence of Mg2+, gliclazide enhanced ATP block of V59M channels (Fig. 3A and B and Table 1), suggesting that, as proposed for wild-type channels, ATP binding to SUR1-NBS1 might decrease block at Kir6.2-V59M and that gliclazide abolishes this effect. The ATP sensitivity of V59M and I296L channels were markedly reduced by Mg2+, which is expected if MgATP interaction with SUR1 increases PO and reduces ATP block at Kir6.2. The latter probably makes the greatest contribution because the intrinsic PO (i.e., in the absence of nucleotide binding) of these channels is extremely high (>0.8) and cannot be increased much more. In the presence of gliclazide, MgATP inhibition of both channels was similar to that in the absence of both drug and Mg2+, which is consistent with gliclazide preventing MgATP activation.
FIG. 3.
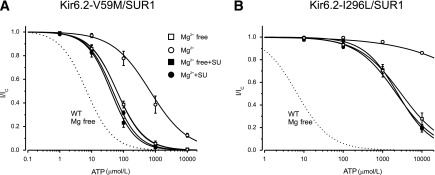
Effect of gliclazide on ATP block of mutant KATP channels with impaired gating. ATP concentration-inhibition curves for Kir6.2-V59M/SUR1 (A) and Kir6.2-I296L/SUR1 (B) channels were measured in the absence of both gliclazide and Mg2+ (□) and in the absence of gliclazide but the presence of 2 mmol/L Mg2+ (○), 30 μmol/L gliclazide without Mg2+ (■), and 30 μmol/L gliclazide plus 2 mmol/L Mg2+ (●). The dotted line is the concentration-inhibition curve for Kir6.2/SUR1 channels in the absence of Mg2+ and gliclazide (Fig. 2E, □). The lines are the best fit of Eq. 1 to the mean data with the following parameters: Kir6.2-V59M/SUR1 (A): IC50 = 62 μmol/L, h = 1.0 (□); IC50 = 510 μmol/L, h = 0.7 (○); IC50 = 40 μmol/L, h = 1.1 (■); and IC50 = 46 μmol/L, h = 1.1 (●) and Kir6.2-I296L/SUR1 (B): IC50 = 2.8 mmol/L, h = 0.76 (□); IC50 = 2.3 mmol/L, h = 0.86 (■); and IC50 = 2.4 mmol/L, h = 1.0 (●). The line through the open circles was drawn by hand. SU, sulfonylurea; WT, wild-type.
Modification of high-affinity gliclazide block by MgATP.
We next compared the effect of MgATP on gliclazide block of wild-type and mutant KATP channels. To enable a quantitative comparison, we used an ATP concentration that produces an approximately half-maximal block at Kir6.2. This was obtained from the ATP concentration-inhibition relationship in the absence of Mg2+ (Table 1). We used 15 μmol/L for wild-type, 100 μmol/L for R201C and V59M, and 3 mmol/L for I296L channels. For G334D channels, we used 1 mmol/L MgATP because this concentration produced near-maximal activation (16), and in the present study, higher concentrations of ATP (without Mg2+) sometimes caused a slight block.
In the absence of MgATP, wild-type, R201C, and G334D channels exhibited similar gliclazide concentration-inhibition curves. The IC50 lay between 50 and 70 nmol/L, and inhibition reached a plateau at 50–60% of maximum (Fig. 4), indicating that there is significant channel activity (i.e., PO is not 0) even when drug-binding sites are fully occupied. These results are similar to previous findings for sulphonylurea block of wild-type (23) and G334D (28) channels in the absence of intracellular nucleotides and indicate that neither the R201C nor the G334D mutation alters the direct block of the channel by gliclazide.
FIG. 4.

ATP modulation of gliclazide block of wild-type KATP channels and mutant channels with impaired ATP binding to Kir6.2. A–C: Gliclazide concentration-inhibition relations for wild-type and mutant channels in the absence (○) and presence (●) of MgATP. Currents are expressed relative to those in the absence of gliclazide. The MgATP concentration was 15 μmol/L for Kir6.2/SUR1, 1 mmol/L for Kir6.2-G334D/SUR1, and 100 μmol/L for Kir6.2-R201C/SUR1, respectively. The lines are the best fit of Eq. 1 to the mean data with the following parameters: Kir6.2/SUR1 (A): IC50 = 67 nmol/L, h = 1.3, a = 0.45 (○) and IC50 = 71 nmol/L, h = 1.0, a = 0.20 (●); Kir6.2-G334D/SUR1 (B): IC50 = 67 nmol/L, h = 1.1, a = 0.39 (○) and IC50 = 213 nmol/L, h = 1.0, a = 0.21 (●); and Kir6.2-R201C/SUR1 (C): IC50 = 49 nmol/L, h = 1.2, a = 0.48 (○) and IC50 = 190 nmol/L, h = 1.2, a = 0.27 (●). n = 6 in all experiments.
In contrast, gliclazide inhibited V59M channels very poorly and was almost without effect on I296L channels (Fig. 5A and B). These mutations impaired KATP channel gating, increasing the intrinsic PO from 0.36 ± 0.06 (n = 6) for wild-type channels to 0.77 ± 0.02 (n = 6) for V59M and 0.85 ± 0.01 (n = 6) for I296L channels. Previous reports indicated that an increased PO is correlated with reduced sulphonylurea sensitivity (30,32,33). We simulated the effect of PO on gliclazide block with a Monod-Wyman-Changeux model (see Fig. 5C and D legend for details). This model predicts that as PO is increased, the extent of block is reduced, and the IC50 increases. For a PO of 0.77, the maximal predicted block was 22%, which agrees well with that measured for V59M channels (20%). A similar correlation between measured and predicted values was found for I126L channels. Thus, in the absence of nucleotides, the PO largely determines the maximal extent of sulphonylurea block.
FIG. 5.

ATP modulation of gliclazide block of mutant KATP channels with impaired gating. A and B: Gliclazide concentration-inhibition relations for Kir6.2-V59M/SUR1 (n = 6) and Kir6.2-I296L/SUR1 (n = 6) channels in the absence (○) and presence (●) of MgATP. The MgATP concentration was 100 μmol/L for Kir6.2-V59M/SUR1 and 3 mmol/L for Kir6.2-I296L/SUR1. The lines are the best fit of Eq. 1 to the mean data with the following parameters: Kir6.2-V59M/SUR1 (A): IC50 = 200 nmol/L, h = 0.92, a = 0.79 (○) and IC50 = 140 nmol/L, h = 0.88, a = 0.39 (●) and Kir6.2-I296L/SUR1 (B): IC50 = 930 nmol/L, h = 1.5, a = 0.95 (○) and IC50 = 1,200 nmol/L, h = 0.98, a = 0.41 (●). C and D: Simulation of the dependence of high-affinity gliclazide block on PO in nucleotide-free solutions with a Monod-Wyman-Changeux model. C: Fractional block of KATP current, as a function of gliclazide concentration, for different values of PO. Currents are expressed relative to those in the absence of gliclazide. The lines are drawn to the following: (1 + F + E0) × (1 + [S]/Kd,O)4/ [(1 + F) × (1 + [S]/Kd,O)4 + E0 × (1 + [S]/Kd,C)4], where F = 0.16 is the equilibrium gating constant for the fast intraburst transitions (from maximal PO of 0.86 when E0 = 0), E0 = [1 − PO × (1 + F)] / PO is the equilibrium gating constant for the slow interburst transitions, [S] is the gliclazide concentration, and Kd,O = 70 nmol/L and Kd,C = 50 nmol/L are the dissociation constants for gliclazide binding to the open and closed states, respectively. D: Monod-Wyman-Changeux model for gliclazide-dependent gating of KATP channels used to derive the current traces in C. This model is the simplest mechanism of concerted gating, which has previously been shown to occur in KATP channels (48–50). KC, sulphonylurea binding constant for the closed state; KO, sulphonylurea binding constant for the open state; S, sulphonylurea; t, proportionality factor reflecting the change in the equilibrium gating constant E0 when sulphonylurea is bound to the channel (t = KO/KC).
In the presence of MgATP, the maximal extent of high-affinity gliclazide block of all channels was enhanced (Figs. 4 and 5). The current reached a plateau at a level that was ∼50% of that in the absence of MgATP. This finding is expected because we used an ATP concentration close to the IC50. Of note, however, is that the ATP concentration required to produce this additional block was substantially greater for mutant channels (e.g., 3 mmol/L MgATP for I296L compared with 15 μmol/L MgATP for wild-type channels).
It has been previously proposed that sulphonylureas abolish the stimulatory effects of Mg-nucleotides on wild-type channels and thus unmask ATP inhibition at Kir6.2, leading to an apparent enhancement of sulphonylurea block (Fig. 1A) (17,33). The present results show the same is true for V59M, I296L, and R201C channels. However, because these channels show less inhibition at Kir6.2, the ability of nucleotides to enhance sulphonylurea block is correspondingly reduced, explaining why much more ATP is required to produce the same maximal block.
Figure 6 plots the current in the presence of gliclazide as a fraction of that in the absence of both drug and nucleotide, showing that with the exception of G334D channels (which are activated) and R201C channels (which are unaltered), MgATP inhibits both wild-type and ND channels. These differences are dictated by the extent to which MgATP inhibits channel activity at Kir6.2. Wild-type channels, which are the most ATP sensitive, show the greatest block. In contrast, G334D channels, which show no block at Kir6.2 (25), are activated by MgATP (16).
FIG. 6.

ATP modulation of gliclazide block of wild-type and mutant KATP channels. A–E: Gliclazide concentration-inhibition relations for wild-type and mutant channels in the absence (○) and presence (●) of MgATP. Currents are expressed relative to those in the absence of both MgATP and gliclazide. MgATP concentrations were 15 μmol/L (A, Kir6.2/SUR1), 100 μmol/L (B, Kir6.2-R201C/SUR1), 1 mmol/L (C, Kir6.2-G334D/SUR1), 100 μmol/L (D, Kir6.2-V59M/SUR1), and 3 mmol/L (E, Kir6.2-I296L/SUR1). The lines are the best fit of Eq. 1 to the mean data with the following parameters: Kir6.2/SUR1 (A): IC50 = 67 nmol/L, h = 1.3, a = 0.45 (○) and IC50 = 71 nmol/L, h = 1.0, a = 0.10 (●); Kir6.2-R201C/SUR1 (B): IC50 = 67 nmol/L, h = 1.1, a = 0.39 (○) and IC50 = 190 nmol/L, h = 1.2, a = 0.25 (●); Kir6.2-G334D/SUR1 (C): IC50 = 140 nmol/L, h = 0.88, a = 0.31 (○) and IC50 = 213 nmol/L, h = 1.0, a = 0.39 (●); Kir6.2-V59M/SUR1 (D): IC50 = 200 nmol/L, h = 0.92, a = 0.79 (○) and IC50 = 49 nmol/L, h = 1.2, a = 0.48 (●); and Kir6.2-I296L/SUR1 (E): IC50 = 930 nmol/L, h = 1.5, a = 0.95 (○) and IC50 = 1,200 nmol/L, h = 0.98, a = 0.38 (●). n = 6 in all experiments. Note that in the absence of gliclazide Kir6.2-G334D/SUR1 currents are greater in the presence of ATP than in the absence of ATP because the G334D mutation abolishes the inhibitory effect of ATP at Kir6.2, leaving only the stimulatory effect at SUR1 (C). In contrast, currents are smaller in the presence of MgATP for all other channels because both inhibition and activation are present. F and G: Current remaining in the presence of 100 μmol/L MgATP or 1 mmol/L MgATP in the absence (open bars) and presence (solid bars) of 30 μmol/L gliclazide for wild-type channels and KATP channels carrying ND mutations. The current is expressed as a fraction of that in drug- and nucleotide-free solution. n = 6 in all experiments. WT, wild-type.
The addition of gliclazide results in further inhibition, the extent of which varies with the ATP sensitivity of the channel. Independent of the molecular mechanism of action of the mutation, channels that are inhibited most strongly by MgATP are also most strongly blocked by gliclazide in the presence of the nucleotide (Fig. 6F and G). For example, R201C and V59M channels are blocked by approximately the same amount (∼70%) because the magnitude of ATP inhibition is similar, even though the mechanism by which the mutation affects ATP inhibition differs.
In the case of G334D channels, which are activated by MgATP but not blocked by ATP, gliclazide removes channel activation by 1 mmol/L MgATP but has no further effect. Because the channel is not blocked by ATP, the maximal block is the same whether or not MgATP is present (Fig. 6C). It is also substantially smaller than that found for wild-type channels in 1 mmol/L MgATP but no gliclazide (Fig. 6G). This finding explains why patients with this mutation fail to respond to sulphonylurea therapy (28).
Mg-nucleotides influence gliclazide binding.
The IC50 for gliclazide block of G334D and R201C channels increased approximately threefold when MgATP was added (Fig. 4B and C). The most likely explanation for this finding is that MgATP binding to the NBSs of SUR1 causes partial displacement of gliclazide from its binding site. Indeed, nucleotide-induced displacement of glibenclamide binding to SUR1 has been reported (34,35). An alternative idea is that MgATP leads to the generation of phosphatidylinositol 4,5-bisphosphate (PIP2), which either impairs gliclazide binding directly or by increasing PO (36).
To distinguish between these possibilities, we measured the effect of various agents on the magnitude of G334D currents in the presence of 100 nmol/L gliclazide (Fig. 7A). At this concentration, the difference in block in the presence (22%) and absence (37%) of 1 mmol/L MgATP is evident (Fig. 4B). Wortmanin (10 µmol/L), which prevents PIP2 production through inhibition of phosphatidylinositol kinases (37), did not affect block by 1 mmol/L ATP (Fig. 7A), suggesting that PIP2 is not involved. The addition of 1 mmol/L MgADP, which should not stimulate PIP2 production, produced an even greater suppression of block than MgATP. Taken together, the data favor the idea that the reduced IC50 is attributable to nucleotide binding to the NBSs of SUR1, which produces a conformational change that displaces gliclazide binding. This effect requires Mg2+ because it is abolished in Mg-free solution (Fig. 7A). Because MgADP is more effective that MgATP, we postulate that MgADP binding to SUR1 displaces gliclazide binding and that MgATP must be converted to MgADP to be effective.
FIG. 7.
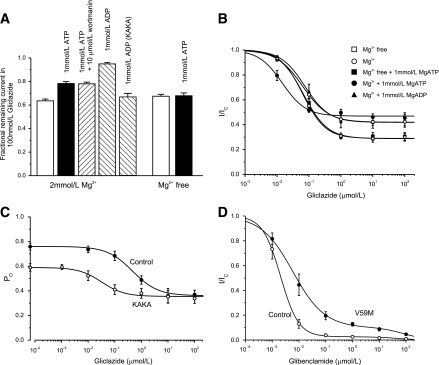
Reduction of gliclazide block by nucleotides. A: Mean ± SEM current remaining in the presence of 100 nmol/L gliclazide and nucleotides (n = 6) for Kir6.2-G334D/SUR1 or Kir6.2-G334D/SUR1-KAKA (KAKA) channels. B: Fractional block of Kir6.2-G334D/SUR1-KAKA currents as a function of gliclazide concentration in the absence (○; n = 6) and presence (●; n = 6) of 1 mmol/L MgATP or 1 mmol/L MgADP (▲; n = 6) and in the absence (□; n = 6) and presence (■; n = 6) of 1 mmol/L ATP (Mg-free solution). The lines are the best fit of Eq. 1 to the mean data with the following parameters: IC50 = 63 nmol/L, h = 1.1, a = 0.42 (○); IC50 = 15 nmol/L, h = 1.2, a = 0.46 (●); IC50 = 75 nmol/L, h = 1.2, a = 0.42 (▲); IC50 = 64 nmol/L, h = 1.2, a = 0.29 (□); and IC50 = 67 nmol/L, h = 1.2, a = 0.29 (■). A and B: Experiments were performed in the inside-out patch configuration. C: Dependence of single-channel PO on gliclazide concentration for Kir6.2-G334D/SUR1 channels (control [●]; n = 6) and Kir6.2-G334D/SUR1-KAKA channels (KAKA [○]; n = 6) measured in cell-attached patches. The lines are the best fit of the following to the mean data: 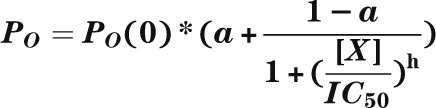 , where PO(0) is the single-channel PO in the absence of gliclazide, [X] is the gliclazide concentration, IC50 is the gliclazide concentration at which the inhibition is half maximal, h is the Hill coefficient, and a is the fractional PO at gliclazide concentrations that saturate the high-affinity SUR1 site. PO(0) = 0.76, IC50 = 440 nmol/L, h = 0.92, a = 0.47 (●) and PO(0) = 0.59, IC50 = 30 nmol/L, h = 1.0, a = 0.60 (●). D: Mean ± SEM relationship between glibenclamide and the whole-cell KATP current measured in β-cells from control mice (○; n = 6 cells, three mice) and mice in which the Kir6.2-V59M mutation was induced (V59M [●]; n = 6 cells, three mice). Current (I) is expressed relative to that in drug-free solution (IC). The curves are the best fit of Eq. 2 to the mean data with the following parameters: IC50(1) = 2 nmol/L, h1 = 1.3, IC50(2) = 45 μmol/L, h2 = 1, a = 0.03 (control [○]) and IC50(1) = 6 nmol/L, h1 = 0.74, IC50(2) = 80 μmol/L, h2 = 1, a = 0.1 (V59M [●]).
, where PO(0) is the single-channel PO in the absence of gliclazide, [X] is the gliclazide concentration, IC50 is the gliclazide concentration at which the inhibition is half maximal, h is the Hill coefficient, and a is the fractional PO at gliclazide concentrations that saturate the high-affinity SUR1 site. PO(0) = 0.76, IC50 = 440 nmol/L, h = 0.92, a = 0.47 (●) and PO(0) = 0.59, IC50 = 30 nmol/L, h = 1.0, a = 0.60 (●). D: Mean ± SEM relationship between glibenclamide and the whole-cell KATP current measured in β-cells from control mice (○; n = 6 cells, three mice) and mice in which the Kir6.2-V59M mutation was induced (V59M [●]; n = 6 cells, three mice). Current (I) is expressed relative to that in drug-free solution (IC). The curves are the best fit of Eq. 2 to the mean data with the following parameters: IC50(1) = 2 nmol/L, h1 = 1.3, IC50(2) = 45 μmol/L, h2 = 1, a = 0.03 (control [○]) and IC50(1) = 6 nmol/L, h1 = 0.74, IC50(2) = 80 μmol/L, h2 = 1, a = 0.1 (V59M [●]).
If the reduction in IC50 arises because MgADP interacts with SUR1 to decrease gliclazide binding, it should be abolished by mutations that impair MgADP binding at SUR1. Mutation of the Walker A lysine in either NBD1 or NBD2 of SUR1 markedly reduces the ability of MgADP (or MgATP) to stimulate wild-type channels (10) and impairs MgADP binding (38). We therefore simultaneously mutated the Walker A lysine in both NBD1 and NBD2 of SUR1 to alanine (SUR1-KAKA) and coexpressed SUR1-KAKA with Kir6.2-G334D. As Fig. 7A shows, the KAKA mutation prevented MgADP from reducing gliclazide block.
In the absence of nucleotide (but presence of Mg2+), the gliclazide concentration-inhibition curve for Kir6.2-G334D/SUR1-KAKA channels was similar to that of wild-type channels (Fig. 7B), indicating that the KAKA mutations do not affect drug binding. There was no effect of 1 mmol/L MgADP on either the IC50 or the maximal extent of gliclazide block, which is expected if MgADP binding is abolished by the KAKA mutation. The data are consistent, therefore, with the idea that MgADP can displace gliclazide binding (and vice versa).
Surprisingly, in the presence of 1 mmol/L MgATP, the IC50 was reduced from 63 to 15 nmol/L. Because the KA mutations impair MgATP hydrolysis but not binding (39), this finding suggests that MgATP binding to SUR1-KAKA increases gliclazide binding, which is in contrast to Kir6.2-G334D/SUR1 channels where MgATP binding decreases gliclazide binding to SUR1 (Fig. 4B).
Figure 7C shows the relationship between gliclazide concentration and PO for Kir6.2-G334D/SUR1 and Kir6.2-G334D/SUR1-KAKA channels in cell-attached patches, where channel activity is determined by the balance between the stimulatory and inhibitory effects of intracellular ligands. In the absence of gliclazide, PO was 0.76 ± 0.03 (n = 6) for Kir6.2-G334D/SUR1 and 0.59 ± 0.02 (n = 6) for Kir6.2-G334D/SUR1-KAKA channels. We assume that the latter represents the intrinsic PO because this channel is largely insensitive to either ATP block or MgATP activation. The greater PO of Kir6.2-G334D/SUR1 channels must, therefore, be produced by MgATP hydrolysis at NBS2. Of note, the increase in PO is quite small: only ∼25% of total. The maximal block of Kir6.2-G334D/SUR1 and Kir6.2-G334D/SUR1-KAKA channels was only 50 and 30%, respectively, because they are insensitive to ATP inhibition, and thus, only the direct block by gliclazide was observed following abolition of channel activation by the drug.
The IC50 for gliclazide block of Kir6.2-G334D/SUR1 channels in cell-attached patches was 440 nmol/L (Fig. 7C). This is approximately sixfold greater than that measured in the presence of 1 mmol/L MgATP (in excised patches) and can be attributed to the higher PO (0.76 in the cell-attached patch vs. <0.4 in the inside-out patch). The IC50 for gliclazide block of Kir6.2-G334D/SUR1-KAKA channels (40 nmol/L) was substantially smaller, consistent with what was observed in excised patches (Fig. 7B).
High-affinity block of V59M channels in pancreatic β-cells is not complete.
Patients with severe ND mutations are treated with much higher doses (∼10-fold) of sulphonylureas than those with type 2 diabetes, yet they rarely experience hypoglycemia (2,40–42). A possible explanation is that therapeutic concentrations of sulphonylureas do not completely block β-cell KATP channels so that excessive insulin secretion is prevented. To explore this hypothesis, we tested the effect of glibenclamide, which is commonly used to treat ND (2,40), on whole-cell KATP currents in β-cells isolated from mice expressing a β-cell–specific Kir6.2-V59M mutation (ib-V59M mice) (8).
Figure 7D shows the relationship between glibenclamide concentration and whole-cell KATP current in control and ib-V59M β-cells. Both datasets were best fit by the sum of a high-affinity and a low-affinity inhibitory site. Wild-type currents were almost fully blocked by 100 nmol/L glibenclamide, with an IC50 for high-affinity block of 2.1 ± 0.2 nmol/L (n = 6 patches, three mice). Occupation of the high-affinity site produced 97 ± 1% inhibition. In contrast, the IC50 for high-affinity block of ib-V59M β-cells was 7.5 ± 3.5 nmol/L (n = 6 patches, three mice), and 10 ± 1% of current remained at drug concentrations that saturated the high-affinity site. Thus, there was a significant increase in the IC50 for channel inhibition by glibenclamide, as was observed for gliclazide block of V59M channels in Xenopus oocytes (Fig. 5A). Furthermore, in agreement with our hypothesis, a clear pedestal of current was observed for mutant channels, and there was only a small reduction in block between glibenclamide concentrations of 100 nmol/L and 10 μmol/L.
DISCUSSION
These data demonstrate that MgATP increases high-affinity block of ND channels by sulphonylureas, as it does for wild-type channels (17,33). However, because mutant channels are less sensitive to ATP inhibition, much higher ATP concentrations are needed to produce the same degree of enhancement. As a consequence, some ND channels are not fully blocked by the drug, even at physiological ATP concentrations, which explains why diabetes cannot be managed by sulphonylureas in patients with certain ND mutations and why hypoglycemia is less common in patients with severe ND mutations than in patients with type 2 diabetes.
Effects of Mg-nucleotides on gliclazide inhibition of mutant KATP channels.
The present results are consistent with a mutual antagonism between binding of gliclazide and Mg-nucleotides to SUR1. At low sulphonylurea concentrations, suppression of gliclazide binding by physiological (mmol/L) MgATP concentrations increases the IC50 for high-affinity gliclazide block of KATP currents. Conversely, at high drug concentrations, gliclazide displaces Mg nucleotide binding to NBS2 of SUR1, abolishing the ability of the nucleotide to stimulate channel activity (10–13) and reduce ATP inhibition at Kir6.2 (11,15), which results in an apparent enhancement of the maximal sulphonylurea block at the high-affinity site on SUR1. The magnitude of this increased block is determined by the extent of ATP inhibition at Kir6.2 (as measured in Mg-free solution). This phenomenon predicts that the magnitude of the enhanced block depends on the metabolic state of the cell being smaller when metabolism and ATP levels are low (although whether ATP levels fall sufficiently under physiological conditions for this to occur is unclear). This phenomenon also accounts for the reduced gliclazide block of ND channels, which have impaired ATP sensitivity.
As the present data show, ND channels that have an intermediate reduction in ATP sensitivity (IC50 ∼100 µmol/L in Mg-free solutions), such as R201C and V59M channels, are almost fully blocked by sulphonylureas at physiological ATP concentrations. Those that are poorly blocked (IC50 >1 mmol/L in Mg-free solutions), such as I296L, or not inhibited at all, such as G334D, show little or no enhancement of sulphonylurea block, and sulphonylureas can never effectively inhibit these channels. Although most of our experiments were performed with homomeric KATP channels heterologously expressed in Xenopus oocytes, a similar reduction in sulphonylurea block was observed in β-cells of mice hemizygously expressing V59M channels.
Clinical relevance.
These results resolve the conundrum of why G334D channels show normal block by sulphonylureas in excised patches in the absence of nucleotides but patients are unresponsive to drug therapy (28). Under physiological conditions (i.e., in the presence of intracellular adenine nucleotides), G334D channels are poorly blocked by sulphonylureas. The IC50 is increased, and the maximal block is less. The present data demonstrate that any mutation that dramatically reduces the sensitivity of the channel to ATP might also impair sulphonylurea block sufficiently to prevent sulphonylurea control of glucose homeostasis in patients with these mutations; this might also account for why patients with the I296L mutation cannot transfer to drug therapy (2).
In the presence of physiological levels of MgATP, ND mutations that decrease ATP binding produce an increase in the IC50 for both gliclazide and glibenclamide block (Figs. 4 and 7) (28) and might explain why higher doses of drug are required to treat ND than to treat type 2 diabetes. In addition, the fact that KATP currents are predicted to be larger in mutant β-cells might be a factor.
KATP channels are largely blocked at resting blood glucose concentrations (5–7 mmol/L) (43–46), so β-cells sit poised on the cusp of secretion, and only a small reduction in PO is needed to trigger electrical activity and insulin secretion (45). We found that 2 μmol/L glibenclamide blocked KATP currents in ib-V59M β-cells to the same extent as ∼7 mmol/L glucose in wild-type mice (44–46). Thus, it appears that the drug restores PO to the level found for wild-type channels at resting blood glucose levels. Consequently, metabolic amplifying pathways, which couple glucose metabolism to insulin secretion downstream of KATP channel closure-mediated Ca2+ entry into the β-cell, are enabled (47), explaining why insulin secretion is meal dependent in ND patients (2) as it is in nondiabetic individuals.
For channels containing severe ND mutations, maximal inhibition at the high-affinity site is incomplete even at physiological ATP levels, which might explain why human patients with these mutations rarely experience hypoglycemia (2,40–42): Because channel activity is never completely abolished, insulin secretion is limited. The present data show that increasing glibenclamide has little effect on the extent of block over a wide range of concentrations; however, this will apply only to those Kir6.2 mutations that show pronounced ATP insensitivity. Kir6.2 mutations that reduce ATP sensitivity only slightly will be completely blocked by glibenclamide at physiological ATP levels, and patients with these mutations will be more susceptible to hypoglycemia.
ACKNOWLEDGMENTS
This study was supported by the Wellcome Trust (grant 089795/Z/09/Z), the Royal Society, and the European Union (LSHM-CT-2006-518153 and 332620). F.M.A. holds a Royal Society Wolfson Research Merit Award.
No potential conflicts of interest relevant to this article were reported.
P.P. performed the data analysis and modeling. P.P. and H.d.W. performed the experiments. P.P., H.d.W., and F.M.A. wrote and reviewed the manuscript. P.P. and F.M.A. designed the experiments. P.P. and F.M.A. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
See accompanying commentary, p. 3666.
REFERENCES
- 1.Hattersley AT, Ashcroft FM. Activating mutations in Kir6.2 and neonatal diabetes: new clinical syndromes, new scientific insights, and new therapy. Diabetes 2005;54:2503–2513 [DOI] [PubMed] [Google Scholar]
- 2.Pearson ER, Flechtner I, Njølstad PR, et al. Neonatal Diabetes International Collaborative Group Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med 2006;355:467–477 [DOI] [PubMed] [Google Scholar]
- 3.Rafiq M, Flanagan SE, Patch AM, Shields BM, Ellard S, Hattersley AT, Neonatal Diabetes International Collaborative Group Effective treatment with oral sulfonylureas in patients with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care 2008;31:204–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic beta-cell. Prog Biophys Mol Biol 1989;54:87–143 [DOI] [PubMed] [Google Scholar]
- 5.Gloyn AL, Pearson ER, Antcliff JF, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med 2004;350:1838–1849 [DOI] [PubMed] [Google Scholar]
- 6.Babenko AP, Polak M, Cavé H, et al. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med 2006;355:456–466 [DOI] [PubMed] [Google Scholar]
- 7.McTaggart JS, Clark RH, Ashcroft FM. The role of the KATP channel in glucose homeostasis in health and disease: more than meets the islet. J Physiol 2010;588:3201–3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Girard CA, Wunderlich FT, Shimomura K, et al. Expression of an activating mutation in the gene encoding the KATP channel subunit Kir6.2 in mouse pancreatic beta cells recapitulates neonatal diabetes. J Clin Invest 2009;119:80–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature 1997;387:179–183 [DOI] [PubMed] [Google Scholar]
- 10.Gribble FM, Tucker SJ, Ashcroft FM. The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide. EMBO J 1997;16:1145–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shyng S, Ferrigni T, Nichols CG. Regulation of KATP channel activity by diazoxide and MgADP. Distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. J Gen Physiol 1997;110:643–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nichols CG, Shyng SL, Nestorowicz A, et al. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science 1996;272:1785–1787 [DOI] [PubMed] [Google Scholar]
- 13.Gribble FM, Tucker SJ, Haug T, Ashcroft FM. MgATP activates the beta cell KATP channel by interaction with its SUR1 subunit. Proc Natl Acad Sci U S A 1998;95:7185–7190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zingman LV, Alekseev AE, Bienengraeber M, et al. Signaling in channel/enzyme multimers: ATPase transitions in SUR module gate ATP-sensitive K+ conductance. Neuron 2001;31:233–245 [DOI] [PubMed] [Google Scholar]
- 15.Abraham MR, Selivanov VA, Hodgson DM, et al. Coupling of cell energetics with membrane metabolic sensing. Integrative signaling through creatine kinase phosphotransfer disrupted by M-CK gene knock-out. J Biol Chem 2002;277:24427–24434 [DOI] [PubMed] [Google Scholar]
- 16.Proks P, de Wet H, Ashcroft FM. Activation of the K(ATP) channel by Mg-nucleotide interaction with SUR1. J Gen Physiol 2010;136:389–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gribble FM, Tucker SJ, Ashcroft FM. The interaction of nucleotides with the tolbutamide block of cloned ATP-sensitive K+ channel currents expressed in Xenopus oocytes: a reinterpretation. J Physiol 1997;504:35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gribble FM, Reimann F. Sulphonylurea action revisited: the post-cloning era. Diabetologia 2003;46:875–891 [DOI] [PubMed] [Google Scholar]
- 19.Gillis KD, Gee WM, Hammoud A, McDaniel ML, Falke LC, Misler S. Effects of sulfonamides on a metabolite-regulated ATPi-sensitive K+ channel in rat pancreatic B-cells. Am J Physiol 1989;257:C1119–C1127 [DOI] [PubMed] [Google Scholar]
- 20.Barrett-Jolley R, Davies NW. Kinetic analysis of the inhibitory effect of glibenclamide on KATP channels of mammalian skeletal muscle. J Membr Biol 1997;155:257–262 [DOI] [PubMed] [Google Scholar]
- 21.Proks P, Girard C, Ashcroft FM. Functional effects of KCNJ11 mutations causing neonatal diabetes: enhanced activation by MgATP. Hum Mol Genet 2005;14:2717–2726 [DOI] [PubMed] [Google Scholar]
- 22.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004;429:41–46 [DOI] [PubMed] [Google Scholar]
- 23.Gribble FM, Ashcroft FM. Differential sensitivity of beta-cell and extrapancreatic K(ATP) channels to gliclazide. Diabetologia 1999;42:845–848 [DOI] [PubMed] [Google Scholar]
- 24.Abdelmoneim AS, Hasenbank SE, Seubert JM, Brocks DR, Light PE, Simpson SH. Variations in tissue selectivity amongst insulin secretagogues: a systematic review. Diabetes Obes Metab 2012;14:130–138 [DOI] [PubMed] [Google Scholar]
- 25.Drain P, Li LH, Wang JK. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc Natl Acad Sci U S A 1998;95:13953–13958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Proks P, Antcliff JF, Lippiat J, Gloyn AL, Hattersley AT, Ashcroft FM. Molecular basis of Kir6.2 mutations associated with neonatal diabetes or neonatal diabetes plus neurological features. Proc Natl Acad Sci U S A 2004;101:17539–17544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Proks P, Girard C, Haider S, et al. A gating mutation at the internal mouth of the Kir6.2 pore is associated with DEND syndrome. EMBO Rep 2005;6:470–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masia R, Koster JC, Tumini S, et al. An ATP-binding mutation (G334D) in KCNJ11 is associated with a sulfonylurea-insensitive form of developmental delay, epilepsy, and neonatal diabetes. Diabetes 2007;56:328–336 [DOI] [PubMed] [Google Scholar]
- 29.Koster JC, Cadario F, Peruzzi C, Colombo C, Nichols CG, Barbetti F. The G53D mutation in Kir6.2 (KCNJ11) is associated with neonatal diabetes and motor dysfunction in adulthood that is improved with sulfonylurea therapy. J Clin Endocrinol Metab 2008;93:1054–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koster JC, Remedi MS, Dao C, Nichols CG. ATP and sulfonylurea sensitivity of mutant ATP-sensitive K+ channels in neonatal diabetes: implications for pharmacogenomic therapy. Diabetes 2005;54:2645–2654 [DOI] [PubMed] [Google Scholar]
- 31.Ashcroft FM. New uses for old drugs: neonatal diabetes and sulphonylureas. Cell Metab 2010;11:179–181 [DOI] [PubMed] [Google Scholar]
- 32.Reimann F, Tucker SJ, Proks P, Ashcroft FM. Involvement of the n-terminus of Kir6.2 in coupling to the sulphonylurea receptor. J Physiol 1999;518:325–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Proks P, Reimann F, Green N, Gribble F, Ashcroft FM. Sulfonylurea stimulation of insulin secretion. Diabetes 2002;51(Suppl. 3):S368–S376 [DOI] [PubMed] [Google Scholar]
- 34.Hambrock A, Löffler-Walz C, Quast U. Glibenclamide binding to sulphonylurea receptor subtypes: dependence on adenine nucleotides. Br J Pharmacol 2002;136:995–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ortiz D, Voyvodic P, Gossack L, Quast U, Bryan J. Two neonatal diabetes mutations on transmembrane helix 15 of SUR1 increase affinity for ATP and ADP at nucleotide binding domain 2. J Biol Chem 2012;287:17985–17995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krauter T, Ruppersberg JP, Baukrowitz T. Phospholipids as modulators of K(ATP) channels: distinct mechanisms for control of sensitivity to sulphonylureas, K(+) channel openers, and ATP. Mol Pharmacol 2001;59:1086–1093 [PubMed] [Google Scholar]
- 37.Nakanishi S, Catt KJ, Balla T. A wortmannin-sensitive phosphatidylinositol 4-kinase that regulates hormone-sensitive pools of inositolphospholipids. Proc Natl Acad Sci U S A 1995;92:5317–5321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsuo M, Kimura Y, Ueda KK. KATP channel interaction with adenine nucleotides. J Mol Cell Cardiol 2005;38:907–916 [DOI] [PubMed] [Google Scholar]
- 39.de Wet H, Mikhailov MV, Fotinou C, et al. Studies of the ATPase activity of the ABC protein SUR1. FEBS J 2007;274:3532–3544 [DOI] [PubMed] [Google Scholar]
- 40.Sagen JV, Raeder H, Hathout E, et al. Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient characteristics and initial response to sulfonylurea therapy. Diabetes 2004;53:2713–2718 [DOI] [PubMed] [Google Scholar]
- 41.Codner E, Flanagan SE, Ugarte F, et al. Sulfonylurea treatment in young children with neonatal diabetes: dealing with hyperglycemia, hypoglycemia, and sick days. Diabetes Care 2007;30:e28–e29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones AG, Hattersley AT. Reevaluation of a case of type 1 diabetes mellitus diagnosed before 6 months of age. Nat Rev Endocrinol 2010;6:347–351 [DOI] [PubMed] [Google Scholar]
- 43.Misler S, Falke LC, Gillis K, McDaniel ML. A metabolite-regulated potassium channel in rat pancreatic B cells. Proc Natl Acad Sci U S A 1986;83:7119–7123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ashcroft FM, Ashcroft SJ, Harrison DE. Properties of single potassium channels modulated by glucose in rat pancreatic beta-cells. J Physiol 1988;400:501–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Göpel S, Kanno T, Barg S, Galvanovskis J, Rorsman P. Voltage-gated and resting membrane currents recorded from B-cells in intact mouse pancreatic islets. J Physiol 1999;521:717–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sakura H, Ashcroft SJH, Terauchi Y, Kadowaki T, Ashcroft FM. Glucose modulation of ATP-sensitive K-currents in wild-type, homozygous and heterozygous glucokinase knock-out mice. Diabetologia 1998;41:654–659 [DOI] [PubMed] [Google Scholar]
- 47.Henquin JC. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia 2009;52:739–751 [DOI] [PubMed] [Google Scholar]
- 48.Drain P, Geng X, Li L. Concerted gating mechanism underlying KATP channel inhibition by ATP. Biophys J 2004;86:2101–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Craig TJ, Ashcroft FM, Proks P. How ATP inhibits the open K(ATP) channel. J Gen Physiol 2008;132:131–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Babenko AP. A novel ABCC8 (SUR1)-dependent mechanism of metabolism-excitation uncoupling. J Biol Chem 2008;283:8778–8782 [DOI] [PMC free article] [PubMed] [Google Scholar]