

SUMMARY
Macroautophagy is a bulk clearance mechanism in which the double-membraned phagophore grows and engulfs cytosolic material. In yeast, the phagophore nucleates from a cluster of 20-30 nm diameter Atg9-containing vesicles located at a multiprotein assembly known as the preautophagosomal structure (PAS). The crystal structure of a 2:2:2 complex of the earliest-acting PAS proteins, Atg17, Atg29, and Atg31, was solved at 3.05 Å resolution. Atg17 is crescent-shaped with a 10 nm radius of curvature. Dimerization of the Atg17-Atg31-Atg29 complex is critical for both PAS formation and autophagy and each dimer contains two separate and complete crescents. Upon induction of autophagy, Atg17-Atg31-Atg29 assembles with Atg1 and Atg13, which in turn initiates the formation of the phagophore. The C-terminal EAT domain of Atg1 was shown to sense membrane curvature, dimerize, and tether lipid vesicles. These data suggest a structural mechanism for the organization of Atg9 vesicles into the early phagophore.
INTRODUCTION
Macroautophagy (hereafter, “autophagy”) is a bulk clearance mechanism essential for cell survival during starvation and for the removal of damaged organelles and harmful protein aggregates. In its general features, the autophagy process is conserved from yeast to mammals (Mizushima et al., 2008; Nakatogawa et al., 2009). Autophagy proceeds by the expansion of a unique double-membraned structure known as the phagophore or isolation membrane, which engulfs cytosolosic components including, organelles, pathogens, protein aggregates, and other substrates (Nakatogawa et al., 2009; Rubinsztein et al., 2012). Following closure of the double membrane, the entire structure fuses with the vacuole (in yeast) or lysosome, and its contents are degraded.
The physical basis for the shaping of the phagophore and its maturation into the autophagosome is largely unknown. Autophagosome biogenesis appears not to directly involve the classical coats of vesicle trafficking or the cytoskeleton. Autophagy does use other multipurpose membrane trafficking factors, including SNAREs and SNARE-interacting proteins (Moreau et al., 2011; Nair et al., 2011). The primary factors responsible for phagophore construction are a set of ~35 Atg proteins that are almost exclusively dedicated to autophagosome biogenesis (Mizushima et al., 2011). In yeast, most of the Atg proteins, with the exception of Atg17, Atg29, and Atg31, also participate in the mechanistically related process of cytoplasm-to-vacuole targeting (Cvt) pathway, which is a constitutively active pathway for the transport of specific cargoes to the vacuole via double-membraned carriers.
In yeast, phagophore and Cvt vesicle biogenesis is initiated at the preautophagosomal structure (PAS), also known as the phagophore assembly site (Suzuki and Ohsumi, 2010). The PAS is defined by fluorescence microscopy as a bright punctum that is adjacent to the vacuole and positive for the Cvt cargo aminopeptidase I (Ape1) and Atg proteins. Yeast cells contain a single PAS under both normal and starvation conditions. As visualized by electron microscopy, the membrane component of the PAS consists of a single cluster of vesicles and tubules that are positive for the only integral membrane protein of autophagy, Atg9 (Mari et al., 2010). Many of the tubules visualized in EM of thin sections probably correspond to membrane sheets. The vesicles have a mean diameter of ~30 nm (Mari et al., 2010). The tubules and sheets are similar in diameter to the vesicles in their shortest dimension (Mari et al., 2010), and several-fold larger in their long dimension, and derive from SNARE-dependent fusion of the 30 nm vesicles (Nair et al., 2011). A mean of three 30 nm vesicles, are involved in nucleating one phagophore (Yamamoto et al., 2012). It is not known what drives the clustering of the tubulovesicular structures, or what directs their fusion into a membrane sheet.
The mechanism of ordered assembly of the PAS responsible for starvation-induced autophagy has been established (Kawamata et al., 2008), with Atg17 (Suzuki et al., 2007), Atg29 (Kawamata et al., 2005), and Atg31 (Kabeya et al., 2007) being the first autophagy proteins to coalesce into the PAS. Indeed, loss of Atg17 leads to a significant inhibition of autophagy and the disruption of PAS localization for almost every other autophagy factor (Kabeya et al., 2005; Suzuki et al., 2007). Atg17, Atg29, and Atg31 have been reconstituted by co-expression into a stable multiprotein complex (Kabeya et al., 2005; Kabeya et al., 2009) (hereafter, Atg17-Atg31-Atg29). In parallel and independent of Atg17, the Cvt protein, Atg11, is the most upstream factor in the Cvt PAS. Both Atg11 and Atg17 are large proteins that are predicted to be almost entirely helical, and lack any obvious functional motifs. The autophagy and Cvt pathways converge at the next step in PAS assembly, the recruitment of Atg13, which binds directly to the N-terminal region of Atg17 (Kabeya et al., 2005). Atg13 is a major locus for the regulation of autophagy in response to starvation. Under fed conditions, the protein kinase Tor phosphorylates the C-terminus of Atg13, inactivating it with respect to autophagy. Upon starvation, Atg13 becomes hypophosphorylated and assembles with the protein kinase Atg1, at the PAS via the Atg13-Atg17 interaction. This leads to the formation of the full Atg1 complex, comprising Atg1-Atg13-Atg17-Atg31-Atg29, and to the activation of autophagosome formation. Atg1 has a particularly critical role in autophagy (Kamada et al., 2000; Mizushima, 2010). The N-terminal protein kinase domain of Atg1 seems to be important later in the pathway, and may be involved in triggering the release of Atg proteins from the mature autophagosome (Cheong et al., 2008). It is the C-terminal domain (CTD) of Atg1 that appears to have a structural role in the PAS that is critical very early in phagophore initiation (Cheong et al., 2008).
One reason why the mechanism of autophagosome biogenesis has been so opaque is that several of the key protein complexes involved- notably the autophagy-specific class III phosphatidylinositol (PI) 3-kinase complex, the PI(3)P-sensing Atg2-Atg18 complex, and the Atg1 complex- are large and biochemically intractable. The versions of these complexes from commonly studied organisms have thus far resisted crystallization. Working with the orthologous complex from Lachancea thermotolerans, we have determined the structure of the Atg17-Atg31-Atg29 portion of the Atg1 complex. The complex is a dimer of trimers, and contains two independent crescent-shaped units. The radius of curvature of the crescents is ~10 nm, similar to that of 30 nm vesicles and to the most highly curved portions of the rim of the phagophore. The interplay between the two Atg17 crescents appears to be important for function as engineered Atg17 monomers do not form the PAS, leading to a complete loss in autophagic funtion. The Atg17-Atg31-Atg29 complex does not bind to synthetic protein-free liposomes. However, the dimeric C-terminal domain of Atg1 (which we christen the “EAT” domain) was found to be a potent and selective ligand for highly curved liposomes, and to tether them in vitro. Taken together, these data suggest a model for how the Atg1 complex templates the fusion of small Atg9 vesicles into membrane tubules and sheets at the earliest step in autophagosome formation.
RESULTS
Structure determination of Atg17-Atg31-Atg29
A stable ternary complex of Atg17-Atg31-Atg29 was formed by co-expression of all three proteins in Escherichia coli. To facilitate crystallization, limited proteolysis was carried out on the full-length complex from Saccharomyces cerevisiae, which revealed that the C-terminal half of Atg29 was sensitive to proteolytic cleavage and unnecessary for complex formation. Atg29 was therefore truncated after residue 88 for all crystallization constructs. Crystallization trials with the S. cerevisiae ternary complex were unsuccessful, so crystallization of Atg17-Atg31-Atg29 from closely related budding yeasts was pursued. We initially crystallized the ternary complex from Kluyveromyces lactis, however these crystals were poorly ordered and not of sufficient quality for data collection. Comparative sequence analysis of budding yeasts (Figure S1-S2) led us to screen Atg17-Atg31-Atg29 from L. thermotolerans, which produced well-ordered diffraction quality crystals of the ternary complex.
The structure of the L. thermotolerans ternary complex was solved and refined to 3.05 Å resolution (Figure 1A, Table S1). The structural model encompasses virtually all of Atg17 and Atg31. Atg17 in particular is well-ordered almost throughout its entirety (Figure 1B). With the exception of two residues at the C-terminus, and the loop corresponding to residues 181-193 connecting helices α2 and α3, all of Atg17 is included in the refined structure. In contrast, with the exception of the N-terminal β-strand, most of the density for Atg29 was of poor quality and lacked side-chain definition. This part of Atg29 comprises a three-helix bundle (Figure 1C). The mobility of Atg29, beyond the first β-strand, is such that the sequence register could only be assigned for the first helix, and side-chains could not be placed at all. Density for Atg31 was superior to that for the Atg29 helical bundle, but with the exception of the C-terminal helix, it was not as well defined as for Atg17. Atg31 contains only one well-ordered native Met residue. In order to validate the sequence assignment of the β-sheet region of Atg31, two Leu residues (Leu87 and Leu101) were mutated to Met (Figure S3), and the resulting SeMet SAD synthesis confirmed the correctness of the original Atg31 sequence assignment.
Figure 1.
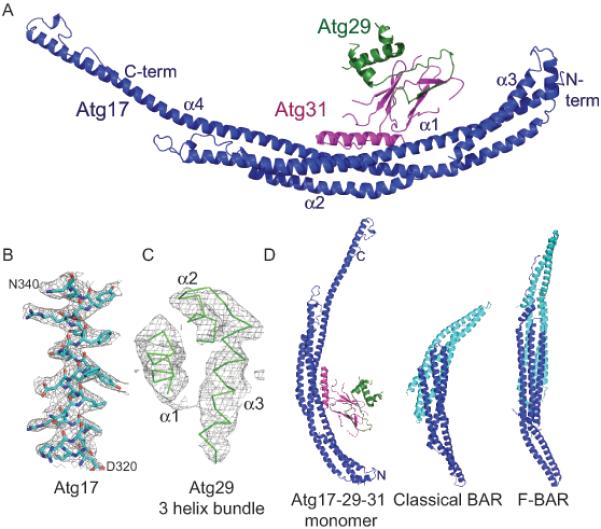
Crystal structure of the Atg17-Atg31-Atg29 complex
(A) Ribbon model for the monomer structure of Atg17 (blue), Atg29 (green) and Atg31 (magenta). The N and C termini and 4 α helices of Atg17 are labeled.
(B) Representative density from a phase-extended solvent-modified Selenium SAD map calculated at 3.1 Å, contoured at 1.2 σ, overlaid on the refined model of Atg17 residues 320-340.
(C) The same map as shown in (B), overlaid on the refined model of the 3 helix bundle of Atg29, illustrating the high mobility of Atg29 relative to the rest of the structure.
(D) Ribbon model comparison of Atg17-Atg31-Atg29 with a comparison to the F-BAR domain of FCHo (PDB ID 2V0O) and the BAR domain Arfaptin (PDB ID 1I49). One BAR monomer is colored blue and the other cyan. See also Figures S1-3 and Table S1.
The Atg17 monomer is a crescent
The Atg17 monomer has a helical fold and a striking crescent shape (Figure 1D), which is suggestive of a function in binding curved membranes. With respect to these general features, Atg17 is similar to the tublular vesicle-binding BAR domains (Frost et al., 2009; Peter et al., 2004). This is remarkable in that there is no sequence homology between the subunits of this complex and any of the BAR domain proteins, and a crescent-shaped fold had not been predicted for Atg17. In contrast to Atg17, BAR domains must dimerize to attain their functional crescent shape (Frost et al., 2009; Peter et al., 2004). The curvature of the BAR crescent is dictated by the angle of intersection of the two subunits in the BAR dimer. Atg17 is made up of 4 helices, which form a well-packed coiled-coil. The longest single helix, α4, stretches the entire length of the monomer, for 156 residues, with a tip-to-tip distance of 194 Å (Figure 1A). The radius of curvature of the crescent is approximately 100 Å, consistent with the radii of the vesicles observed to cluster in the PAS (Mari et al., 2010). The dimensions of the Atg17 monomer are comparable to those of a complete F-BAR dimer (Figure 1D). The finding that the crescent shape of Atg17 is formed from a single molecule represents a fundamental difference between Atg17 and the BAR domain family. A key consequence of the crescent being contained entirely within the monomer is that each Atg17 crescent is asymmetric with respect to the two tips, in contrast to the two-fold symmetry of BAR domain homodimers.
A point mutation of Atg17 Cys24 to Arg has been shown to reduce the affinity of Atg13 for Atg17 (Kabeya et al., 2005) suggesting that this residue makes up part of the binding site for Atg13. This positions Atg13 at the N-terminal tip of Atg17. The Atg17 monomer lacks distinctive electrostatic features, aside from an electronegative patch near the Atg13 binding site and an electropositive path on the convex side of the crescent (Figure 2A-C). Atg17 contains three well-conserved surfaces including an extensive dimerization interface (corresponding to dimer 1, as discussed below), the Atg31 binding site, and the Atg13 binding site (Figure 2 D-F).
Figure 2.
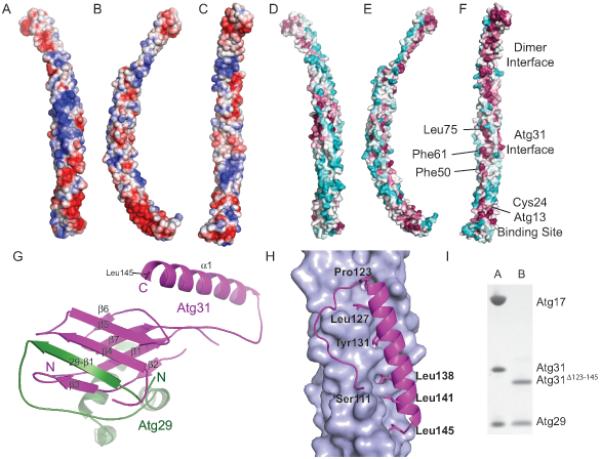
The Atg29-31 subcomplex
(A-C) Surface of the Atg17 monomer colored according to electrostatic potential, with saturating blue and red at ± 3 kT/e; (A) Top view (convex side); (B) Side view; (C) Bottom view (concave side).
(D-F) Conserved surface residues of the Atg17 monomer, with the surface colored according to the sequence conservation among budding yeasts in a gradient from cyan (highly variable) to magenta (most conserved). (D) Top view (convex side); (E) Side view; (F) Bottom view (concave side). Regions important for dimerization, Atg31 and Atg13 binding are indicated.
(G) Detailed view of the interaction between Atg31 (magenta) and Atg29 (green). The seven β strands and one α helix from Atg31 are labeled as well as the N and C-termini. The N-termini and β strand from Atg29 are also labeled.
(H) Detailed view of the 4 helix bundle interaction between Atg31 shown in magenta as a cartoon and Atg17 shown in light blue as a surface.
(I) SDS-PAGE gel illustrating that the truncation of Atg31 after residue 122 blocks binding of Atg17. Column A: Purified complex of Atg17-Atg31-Atg29. Column B: Purified complex from cells expressing Atg17, Atg29, and Atg31Δ123-145. These complexes include the crystallized Atg29 C-terminal truncation constructs. See also Figure S4.
Atg29-Atg31 has a novel fold and is partially mobile relative to Atg17
Atg31 is made up of a β-sheet sandwich followed by a single C-terminal α-helix. Atg31 binds Atg29 through the β sandwich, with the N-terminus of Atg29 making up one of the β-strands of the sandwich (Figure 2G). Indeed, the N-terminal β-strand strand is the only well-ordered portion of Atg29. A search of the Dali server yielded no significant hits, thus we conclude that the Atg29-Atg31 beta sandwich represents a novel protein fold.
Atg31 bridges the ternary complex by binding both Atg17 and Atg29. There are no direct contacts between Atg17 and Atg29. Atg31 binds to Atg17 through its C-terminal helix, which forms a four-helix bundle with three of the helices from Atg17 (Figure 2H). The remainder of the Atg29-Atg31 subcomplex is anchored to the main body of the crescent through ~700 Å2 of mostly polar interactions. This positions Atg29-Atg31 in the center of the Atg17 crescent presenting a steric block for potential protein or vesicle interaction. Modeling suggests that pivoting of the main Atg29-Atg31 unit about the start of the β7-α1 loop, near Atg31 residues 111-112, would remove the steric block. Consistent with the modeling, deletion of the α1 helix (Atg31Δ123-145) in the context of co-expression of Atg17, 29, and 31 in E. coli leads to formation of a stable Atg29-Atg31 subcomplex (Figure 2I) that has no measurable interaction with Atg17 and leads to a loss of autophagic function in vivo (Figure S4). These observations suggest that one role of Atg29-Atg31 could be to regulate the ability of the crescent to bind curved vesicles.
Atg17-Atg31-Atg29 is a rod-like double crescent in solution
The version of Atg17-Atg31-Atg29 complex used in crystallization has a molecular mass equivalent to a 2:2:2 complex in solution as determined by size exclusion chromatography (SEC) and light scattering (Figure 3A, Table S2). This observation is consistent with previous ultracentrifugation studies of full length S. cerevisiae Atg17-Atg31-Atg29 (Kabeya et al., 2009). The crystal lattice reveals several potential dimers. Calculation of the buried surface area of each potential dimer interaction surface with PISA (Krissinel and Henrick, 2007) revealed three candidates that had the most extensive buried surface area (Figure 3B). To determine which, if any, of these potential dimers corresponded to the quaternary structure in solution, we measured small angle X-ray scattering (SAXS) of Atg17-Atg31-Atg29 in solution. SAXS data were recorded on the Atg17-Atg31-Atg29 complex at concentrations of 0.5, 0.8, and 2.3 mg ml−1 (Figures 3C). Because of the rod-like character of the potential dimers, we analyzed the radius of gyration of the cross section (Rc) of the particle in solution rather than using the standard Guinier analysis for determining the radius of gyration which is not applicable to rod shaped samples. The Rc can be determined by plotting ln(qI(q)) versus q2 (Koch et al., 2003). Consistent with the rod-like particle approximation, the ln(qI(q)) vs. q2 data yielded a linear fit to the Rc region of the plot (Figure 3C). Rc was 16.3 ± 0.2 Å at all three concentrations tested, confirming the quality of the SAXS data, the absence of concentration-dependent aggregation in the sample, and the rod-like nature of the Atg17-Atg31-Atg29 particle (Figure 3C inset).
Figure 3.
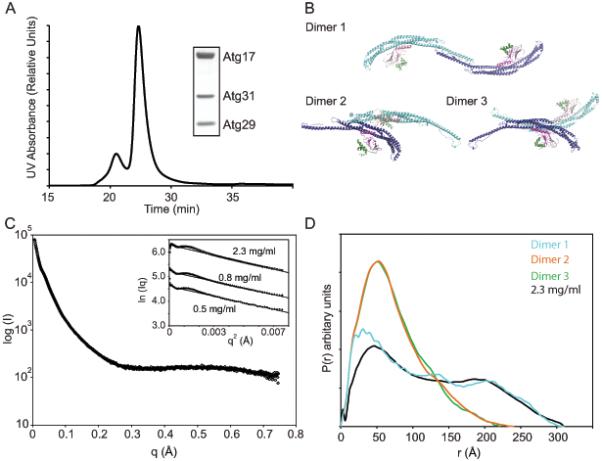
Structure of the Atg17-Atg31-Atg29 dimer in solution
(A) Light scattering experiments were performed on the crystallized construct of Atg17-Atg31-Atg29 containing a C-terminal truncation of Atg29.
(B) Three potential Atg17-Atg31-Atg29 dimers resulting from the crystal lattice are shown and are numbered according to their buried surface area content with dimer 1 containing the highest buried surface area.
(C) SAXS data were recorded on 2.3 mg ml−1 Atg17-Atg31-Atg29. Rc plots for SAXS data recorded at 0.5, 0.8 and 2.3 mg ml−1 are shown as an inset. The lines represent the fit of the linear region used to determine the Rc.
(D) P(r) functions calculated from the experimental scattering data and the three possible dimers structures.
See also Tables S2-3.
To further probe the solution structure of the 2:2:2 Atg17-Atg31-Atg29 complex in solution, the P(r) distribution was calculated and compared to the theoretical P(r) for each potential Atg17-Atg31-Atg29 dimer. The optimal value of Dmax used to obtain P(r) was 310 Å, which is similar to the Dmax of dimer 1, 343 Å, and dimer 2, 310 Å, but not with the value for dimer 3 (Table S3). The real space Rg of Atg17-Atg31-Atg29 calculated from the P(r) is 101.6 ± 1.2 Å. This is in excellent agreement with Rg = 102.7 Å computed from the coordinates of dimer 1, but not consistent with Rg = 61.0 Å for dimer 2 or Rg = 60.8 Å dimer 3. Moreover, the experimentally determined P(r) function is in excellent agreement with the P(r) function computed from the coordinates of dimer 1 (Figure 3D), while this is not so for dimer 2 or 3. These data confirm that dimer 1 represents the correct Atg17-Atg31-Atg29 assembly in solution.
Engineering of stable Atg17-Atg31-Atg29 monomers
Each of the two monomers of Atg17 forms its own intact crescent structure, yet in solution Atg17-Atg31-Atg29 assembles as a 2:2:2 complex. The interactions that hold together the dimer are mediated entirely through the C-teminus of Atg17, with no involvement of Atg29 and Atg31. Thus, the two crescents fold independently of one another, except that the C-terminal portion of helix α4 is stabilized by its partner in the dimer (Figure 4A). Dimerization positions the two molecules of Atg17 so that the crescents oppose one another with the binding sites for Atg13 located at the distal tips of each dimer. The dimer interface of Atg17 is extensive with the burial of 2400 Å2 and an overlapping length of 71 Å in the last 46 amino acids of helix α4. The bulk of the interactions are formed through a series of conserved hydrophobic residues, including Leu355, Ile358, Leu 359, Leu366, and Ile369 which point towards the helix from the opposing monomer (Figure 4B and 4C). To further confirm the importance of this helix for dimer formation and to produce a stable monomeric Atg17-Atg31-Atg29 complex to test the importance of dimer formation for Atg17-Atg31-Atg29 function, deletions of the C-terminal helix termed Δ1, Δ2, and Δ3 were expressed and purified. Atg17 Δ1, Δ2, and Δ3 were predominantly monomeric as tested by light scattering (Figure 4D and Table S2). Each Atg17 truncation construct retained its ability to interact with Atg29 and Atg31, demonstrating that the portion of helix α4 necessary for dimer formation is not essential for the stability of the core Atg17 monomer (Figure 4E).
Figure 4.
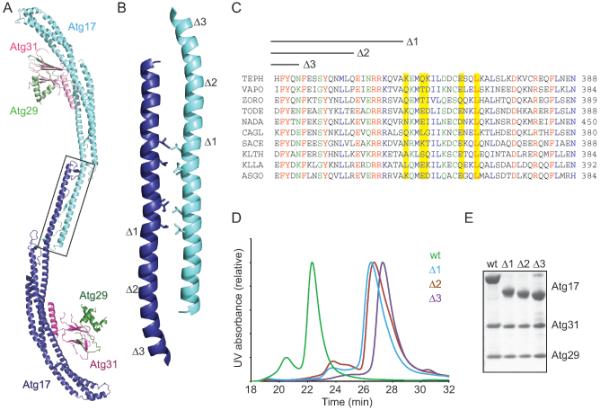
Deconstruction of the Atg17-Atg31-Atg29 dimer
(A) Ribbon model of the Atg17-Atg31-Atg29 dimer. The Atg17 dimer interface is highlighted by a black box.
(B) An enlarged view of the Atg17 dimerization interface as is highlighted in A. Hydrophobic residues forming the bulk of the interaction are shown as stick representations.
(C) Sequence alignment for the region of the Atg17 dimer shown in B. The hydrophobic residues which form the bulk of the interface are highlighted in yellow. The end of each truncated Atg17 sequence (Δ1, Δ2, and Δ3) used to confirm the dimer are highlighted above the alignment. Residues are colored as follows: red, completely conserved, blue, well conserved strongly similar residues, green, well conserved weakly similar residues. Abbreviations used: TEPH: Tetrapisispora phaffii, VAPO: Vanderwaltozyma polyspora, ZORO: Zygosaccharomyces rouxii, TODE: Torulaspora delbrueckii, NADA: Naumovozyma dairenensis, CAGL: Candida glabrata, SACE: Saccharomyces cerevisiae, KLTH: Kluyveromyces thermotolerans, KLLA: Kluyveromyces lactis, AsGO: Ashbya gossypii.
(D) Size exclusion chromatography for Atg17-Atg31-Atg29 and the Δ1, Δ2, and Δ3 dimerization site truncations using a WTC-030S5. This column was in line with a Wyatt Dawn Heleos II instrument and the molecular weight of each peak was confirmed by light scattering.
(E) SDS-PAGE of the purified Atg17-Atg31-Atg29, and the Δ1, Δ2, and Δ3 C-terminal truncations of Atg17. These complexes all include the crystallized Atg29 C-terminal truncation constructs.
Atg17 dimerization is essential for PAS formation and autophagy
To test the function of the Atg17 dimer in vivo, we monitored the localization of Atg17-GFP and constructs encoding the engineered monomeric ATG17Δ1-GFP, ATG17Δ2-GFP and ATG17Δ3-GFP in an atg17Δ S. cerevisiae background. ATG17-GFP cells display bright perivacuolar puncta, corresponding to the PAS in both SMD and SMD with rapamycin treatment (Figure 5A). Cells expressing any of the three monomeric ATG17-GFP constructs were almost entirely devoid of perivacuolar puncta (Figures 5A and B). As Atg17 is one of the earliest organizers of the autophagy specific PAS, we monitored the localization of the downstream autophagy protein Atg8. Most Atg proteins, including Atg8, are localized to the PAS for both Cvt and autophagy. As such, we monitored the localization of Atg8 in an atg11Δ atg17Δ strain to prevent Cvt specific localization of Atg8 at the PAS. In agreement with the loss of localization of Atg17 at the PAS, Atg8 localization at the autophagy specific PAS is completely ablated for all three monomeric Atg17 constructs (Figure 5C).
Figure 5.
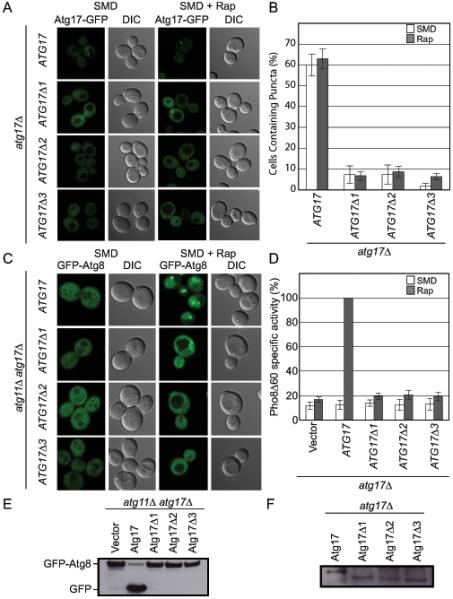
Atg17 dimerization is essential for PAS formation and autophagy
(A) Representative microscopy of ATG17-GFP, ATG17Δ1-GFP, ATG17Δ2-GFP, and ATG17Δ3-GFP in the absence (left) and presence (right) of rapamycin treatment.
(B) Quantification of the microscopy from part (A). A total of 3 trials with 100 cells counted per trial.
(C) Representative microscopy images of atg11Δ atg17Δ GFP-ATG8 cells transformed with ATG17, ATG17Δ1, ATG17Δ2, and ATG17Δ3 in the absence (left) and presence (right) of rapamycin treatment are shown.
(D) Pho8Δ60 assay to monitor autophagy was performed in the absence (white) and presence (grey) of rapamycin treatment. Samples were normalized to the activity of Atg17 in rapamycin treated cells.
(E) GFP-Atg8 processing assay as monitored by western blot against GFP. The GFP-Atg8 and GFP bands are labeled.
(F) Western blot against GFP to monitor the expression of Atg17-GFP.
Error bars in (B) and (D) represent the standard deviation of triplicate experiments.
See also Table S4.
To test the effect of expressing monomeric ATG17 on autophagy function we used a common autophagy reporter assay, based on the engineered cargo Pho8Δ60, which utilizes an engineered alkaline phosphatase that is only active upon targeting to the vacuole by autophagy (Klionsky, 2007). The monomeric ATG17 constructs display a complete loss in autophagy activity (Figure 5D). These results were further confirmed using a second commonly used autophagy reporter assay, the GFP-Atg8 processing assay (Klionsky et al., 2007). GFP-Atg8 is targeted to the vacuole during autophagy and Cvt where Atg8 is degraded and GFP is left largely undegraded. Therefore, the production of free GFP from GFP-Atg8 can be used as a monitor for autophagy and Cvt. As this assay is a reporter for both autophagy and Cvt these experiments were performed in an atg11Δ atg17Δ to prevent any effects from Cvt. In agreement with the Pho8Δ60 assay, the monomeric Atg17 constructs are unable to initiate autophagy (Figure 5E). The effects of the observed phenotypes were not due to decreased stability of Atg17, as total expression of the Atg17-GFP constructs was similar (Figure 5F).
Membrane curvature sensing and complex formation by the Atg1 EAT domain
Based on its crescent shape, we anticipated that Atg17-Atg31-Atg29 might act as a lipid vesicle curvature sensor. To test this hypothesis we performed liposome sedimentation assays with the Atg17-Atg31-Atg29 complex against a range of Folch liposome sizes including multilamellar vesicles (MLVs), large unilamellar vesicles (LUVs) and small unilamellar vesicles (SUVs) prepared by sonication, which typically range in size from 20 to 30 nm. The Atg17-Atg31-Atg29 complex did not to bind liposomes of any size (Figure 6A). Previously, it has been reported that the C-terminal domain of ULK1, the mammalian homologue of Atg1, is involved in membrane targeting in vivo (Chan et al., 2009) and we therefore tested whether the lipid binding activity of the Atg1 complex was contained within the Atg1 C-terminal domain. As described below, this domain is capable of tethering vesicles. Given its previously demonstrated membrane targeting properties and the newly described tethering activity, and we have dubbed it the “Early Autophagy Targeting/Tethering” (EAT) domain.
Figure 6.
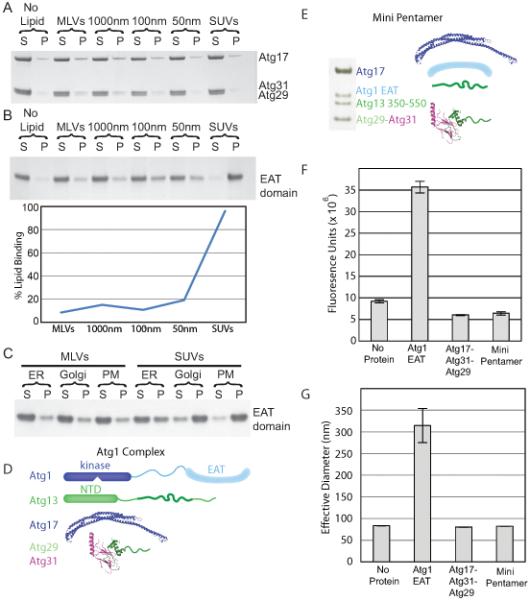
Regulated high curvature vesicle tethering by the EAT domain
(A) Liposome sedimentation assays for the full-length Atg17-Atg31-Atg29 complex with Folch liposomes of varying diameters. Atg17-Atg31-Atg29 does not bind to Folch liposomes of any size.
(B) Liposome sedimentation assay for the EAT domain with Folch liposomes of varying diameters. Atg1 shows a strong preference for small sonicated Folch liposomes.
(C) Liposome sedimentation assay for the EAT domain with liposomes mimicking the endoplasmic reticulum (ER), golgi apparatus and plasma membrane (PM) of S. cerevisiae.
(D) SDS-PAGE of the minimal Atg1 complex. The bands for Atg1, 13, 17, 29 and 31 are labeled. Atg29 and Atg31 run at identical locations on SDS-PAGE.
(E) Schematic of the full-length Atg1 complex, for comparison to the mini-pentamer expressed in (D).
(F) Sonicated liposomes containing biotin were mixed with fluorescently labeled sonicated liposomes and buffer, the EAT domain, full-length Atg17-Atg31-Atg29 or the mini-pentamer. The EAT domain potently tethers vesicles, but Atg17-Atg31-Atg29 does not. Atg17-Atg31-Atg29 inhibits tethering by the EAT domain. Biotin liposomes were captured by streptavidin resin and the amount of fluorescent lipid tethered to the biotin liposomes was quantified.
(G) The effective diameter of SUVs obtained from light scattering with no protein, the EAT domain, full-length Atg17-Atg31-Atg29 or the mini-pentamer. The increase in the effective liposome diameter induced by the EAT domain shows that it tethers liposomes. The lack of increase above baseline shows that the other complexes tested do not tether liposomes, consistent with results in (F).
Error bars in (F) and (G) represent the standard deviation of triplicate experiments.
The Atg1 EAT domain was expressed and purified as a stable domain, which was found by light scattering to be a dimer (Table S2). Liposome sedimentation assays show that the EAT domain binds weakly to MLVs and LUVs, but much more tightly to SUVs (Figure 6B). The EAT domain shows a striking preference for binding to small highly curved vesicles. As Folch liposomes are representative of mammalian lipid compositions we also tested three lipid mixtures designed to mimic the plasma membrane (PM), endoplasmic reticulum (ER), and Golgi apparatus from S. cerevisiae (Klemm et al., 2009; Zinser et al., 1991). The EAT domain shows the same preference for binding to SUVs over larger vesicles in the S. cerevisiae-based liposomes as was observed for Folch liposomes (Figure 6C).
The EAT domain also contains the binding site for Atg13 (Yeh et al., 2011) suggesting that this domain is crucial for both formation of the full Atg1 complex and targeting of the complex to membranes. To determine how Atg1 and Atg13 assemble with Atg17-Atg31-Atg29, we reconstituted a minimal Atg1-Atg13-Atg17-Atg31-Atg29 complex containing the EAT domain and 200 amino acid residues from the predicted unstructured central portion of Atg13. This central region of Atg13 binds to both Atg1 and the full length Atg17-Atg31-Atg29 complex (Figure 6D and E). This construct, which we refer to as the mini Atg1 complex pentamer, or mini-pentamer for short, was expressed in E. coli. All five components could be co-purified using the single His6 tag on the C-terminus of Atg31. The Atg1 mini-pentamer elutes from SEC using a Superose 6 column at 47 ml, indicating that the mini-pentamer is soluble and not aggregated.
To test whether the Atg1 mini-pentamer or the EAT domain were able to tether liposomes, we performed two tethering assays. In the first assay protein was mixed with two distinct sonicated liposomes. The first liposome contained Biotin-PEG2000-DSPE, and the second liposome contained the fluorescent marker DID. The protein-liposome mixture was passed over strepdavidin resin, which was thoroughly washed prior to quantifying the amount of DID liposomes remaining on the resin by fluorescence (Figure 6F). The EAT domain strongly increased the fluorescence signal in the tethering assay demonstrating that the EAT domain can pull down fluorescent liposomes by streptavidin resin, thus demonstrating that the EAT domain is able to link liposomes. In the second assay protein was mixed with sonicated liposomes and the effective diameter of the liposomes was determined by light scattering (Figure 6G) as has been recently used to analyze liposome tethering (Lo et al., 2011). Light scattering revealed that the effective diameter of sonicated liposomes alone is 85 nm. As sonicated liposomes are a mixture of small (20-30 nm) vesicles and larger (>50 nm) vesicles, the effective diameter represents the entire pool of liposomes. The EAT domain increased the effective diameter of SUVs to 320 nm, demonstrating that the EAT domain is tethering vesicles. Both assays demonstrate that the EAT domain alone is able to tether liposomes, while neither the Atg17-Atg31-Atg29 complex or the mini-pentamer are capable of tethering liposomes. This data also suggests a means for regulating and enhancing affinity for highly curved Atg9 vesicles, as discussed below.
DISCUSSION
The molecular mechanism of phagophore initiation is one of the central mysteries in the autophagy field. Here we provide a structural framework for understanding how the Atg1 complex scaffolds the earliest steps in phagophore biogenesis. The highest resolution insights pertain to the Atg17-Atg31-Atg29 complex, which is the earliest-acting portion of the larger Atg1 complex. The Atg17-Atg31-Atg29 complex forms an extended 343Å double crescent shaped dimer. The formation of this dimer is essential for both PAS localization and autophagy initiation. We identified the EAT domain as a dimeric membrane curvature sensor capable of binding to and tethering synthetic ~20-30 nm liposomes.
On the basis of the structure and interactions described above, it was possible to construct provisional models for generic lipid vesicle tethering by the EAT domain, autoinhibition of tethering by the crystallized conformation of Atg17-Atg31-Atg29, and for Atg9-positive vesicle tethering by an activated and fully assembled Atg1 complex. The structure of the EAT domain is not known, but has been schematized on the basis of its dimerization and the predicted helical structure of its 270 residues. A likely mode of lipid vesicle tethering by EAT domain dimers is shown in Fig. 7A. We refer to this as generic vesicle tethering because a variety of simple lipid mixtures bind to EAT domain, and Atg9 is not required for binding or tethering by EAT domain. Fig. 7A is intended as a model for the mechanism of in vitro vesicle tethering by EAT domain, and is not intended to suggest that the isolated EAT domain is capable of nucleating autophagy in cells. Indeed, while EAT domain can target membranes autonomously in cells, its overexpression inhibits autophagy (Chan et al., 2009).
Figure 7.
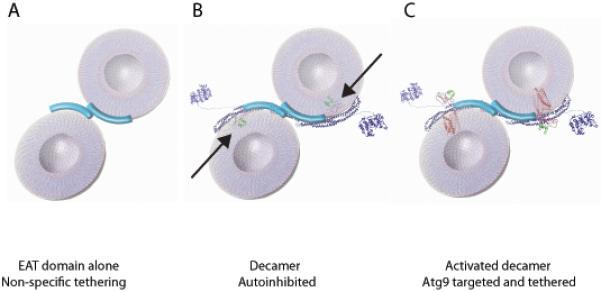
Model for vesicle tethering at the PAS by the Atg1 complex.
(A) Two 20 nm vesicles are shown being coordinated by the EAT domain dimer. (B) The mini-pentamer is unable to bind lipid vesicles in this model due to steric hinderance of Atg29-Atg31 highlighted by arrows. Atg17-Atg31-Atg29 is shown as a ribbon structural model. The kinase domain of Atg1 (KD) represented by the coordinates of a homologous protein kinase catalytic domain of known structure (PDB 4DC2). Unstructured regions of Atg1 are represented as lines. Atg13 bridges Atg1 and Atg17, but is omitted from the graphic for simplicity.
(C) In this hypothetical model, the minipentamer is predicted to be able to coordinate two vesicles containing Atg9 following a change in position of the Atg29-Atg31 subcomplex to unblock the Atg17 crescent. The structure of rhodopsin (PDB 1U19) is used as a stand-in for the structure of Atg9 in order to represent the likely dimensions of the transmembrane domain of Atg9, although Atg9 has no sequence homology to rhodopsin.
Incorporation of the EAT domain into the mini-pentamer completely reversed its ability to tether generic vesicles. Understanding the mechanism of inhibition will ultimately require crystallization of the Atg1-Atg13-Atg17-Atg31-Atg29 complex. Fig. 7B shows a reasonable provisional model for an inhibitory mechanism. In this model, it is postulated that the putative membrane-binding surfaces of the EAT domain and Atg17 dimers are aligned to bind to the same vesicles. The consequence of this postulate is that the Atg29-Atg31 subcomplex (black arrows in Fig. 7B) sterically overlaps with the membrane, which prevents binding in this conformation.
Cell homeostasis requires that autophagy be tightly regulated, and therefore, autophagy is nucleated in yeast from a single specialized pool of as few as three Atg9 positive vesicles (Yamamoto et al., 2012). Thus, stringent regulatory mechanisms must be in place to prevent ectopic autophagy initiation at inappropriate cellular locations. One function of Atg29-Atg31 might be to gate vesicle access to the Atg17 crescent in response to the presence of Atg9 or some other regulatory signal. The structure of an activated Atg1 complex is shown in Fig. 7C. The key inputs to this model are the observations of mobility in the Atg29-Atg31 subcomplex, suggesting is capable of being displaced, and the known interaction between Atg17 and Atg9 (Sekito et al., 2009). Reconstitution of Atg9 positive vesicles has been a major technical challenge in the field. Resolution of this obstacle, and clarification of the mechanism by which autoinhibition of Atg17-Atg31-Atg29 is reversed, will be essential to test the predictions made by this model.
The EAT domain is conserved in metazoan orthologs, where, as in yeast, it is the locus for incorporation into the larger Atg1 complex (Chan et al., 2009; Hara et al., 2008; Hosokawa et al., 2009; Jung et al., 2009). Moreover, the EAT domain is important for membrane targeting of the human and C. elegans Atg1 ortholog, ULK1 (Chan et al., 2009). This region is predicted to be almost entirely helical, but does not correspond to any known fold, including that of Atg17. It will be important to investigate the structural basis for the exceptional ability of EAT domain to bind so tightly and selectively to highly curved liposomes.
Phagophore biogenesis in mammalian cells differs from yeast in that there is not a PAS in mammalian cells. On the other hand, phagophore nucleation does depend on the fusion of small precursor vesicles (Moreau et al., 2011), as in yeast. Mammalian cells have conserved orthologs of Atg1 and Atg13 in the ULK 1/2 kinases and mAtg13. In particular, the key EAT domain of the ULKs is highly conserved with respect to Atg1 and is known to play a role in membrane targeting in mammals. The functional counterpart of Atg17 is FIP200 (Ganley et al., 2009; Hara et al., 2008; Hosokawa et al., 2009; Jung et al., 2009), a protein that has been shown to act as both an oncogenic factor and a tumor suppressive factor (Choi et al., 2012; Liu et al., 2010; Wei et al., 2011). The current consensus is that the ULK1 complex in metazoa is the earliest-acting factor in autophagosome biogenesis, in a close analogy with the yeast Atg1 complex (Itakura and Mizushima, 2010). FIP200 is similar to Atg17 in that it is a large predicted all-helical protein. The functional analogy and similar secondary structure and size characteristics of Atg17 and FIP200 leads us to predict that FIP200 will be a curved scaffold like Atg17. Experimental structure determination will be necessary to confirm if this is true. Atg29 and Atg31 have no known counterparts in metazoa and likely play a more specialized role in the regulation of autophagy in yeast. Thus, the general outlines of the architecture of the Atg1 complex and its role as an early-acting membrane curvature scaffold in autophagy are likely to be conserved in yeast and mammals, but with considerable difference in detail.
One of the important elaborations in mammalian cells is the presence of additional curvature sensors that are either not present or not required for autophagy in yeast. The mammalian BAR protein endophilin B1/Bif-1 (Takahashi et al., 2007), which binds to the PI 3-kinase subunit, UVRAG has been reported to regulate autophagy. Similarly, the metazoan ATG14L/BARKOR subunits of the mammalian class III PI 3-kinase complex contain a C-terminal curvature-sensing ALPS motif also known as a “BATS domain” (Fan et al., 2011) that is not present in the shorter sequence of yeast Atg14. This might foster PI(3)P synthesis at the rim of the growing phagophore downstream of ULK1 (Matsunaga et al., 2010). The model outlined above suggests that the initial scaffolding of membrane curvature in mammalian cells occurs through a conserved mechanism, and that the specialized adaptations just noted are more likely to function in downstream steps.
The inner and outer membranes of the phagophore differ in their protein content. Atg8 is present on both membranes, while nearly all of the other autophagic proteins appear to be confined to the outer membrane. The molecular mechanism for segregating most Atg proteins to the outer membrane has been mysterious. The observation that Atg17 is an asymmetric membrane scaffold suggests a hypothesis for the mechanism of protein segregation. It is currently thought that the Atg1 complex remains present on the phagophore until late in its maturation. The curvature of the Atg17 fold and the curvature-dependent binding of the EAT domain leads us to predict that the Atg1 complex would be localized to the rim of the growing phagophore. By binding to the rim with unique directionality, the Atg1 complex would be ideally poised to serve as a marker and divider between the inner and outer membrane. This model explains why Atg17 evolved as a unique asymmetric curvature scaffold, which fulfills a function that could not have been carried out by symmetric BAR domain homodimers.
In summary, the structural and biochemical insights into Atg1 complex assembly, the discovery of a curved scaffold structure within Atg17, the finding that Atg1 itself is a potent curvature sensor, and finally the discovery of the Atg17-regulated tethering activity of EAT domain, provide a structural framework for the first steps in the nucleation and growth of the phagophore. We were able to probe the function of one of the key features of the scaffold, the Atg17 dimer interface. We found definitively that Atg17 dimerization is absolutely required for PAS formation and autophagic function. This is consistent with a model in which Atg1-Atg13-Atg17-Atg31-Atg29 dimers tether multiple Atg9 vesicles to one another at the PAS, positioning them for fusion into the early phagophore. Other predictions of the model remain to be tested by in vitro reconstitution with Atg9-positive vesicles.
EXPERIMENTAL PROCEDURES
See supplemental methods for details.
Crystallographic structure determination
Atg17-Atg31-Atg29 crystallized in space group P21 with two copies in the asymmetric unit and a solvent content of 0.75. Se SAD datasets were collected at the Se edge (determined each time by a fluorescence scan) at the APS SER-CAT 22-ID and 22-BM beamlines. The data were initially processed by HKL2000 (HKL Research). The dffraction pattern for these crystals was visibly anisotropic, so ellipsoidal truncation and anisotropic scaling were applied to the data using the diffraction anisotropy server (http://services.mbi.ucla.edu/anisoscale/) (Strong et al., 2006). Data were subjected to ellipsoidal truncation with resolution limits of 3.05 Å along a* and c* and 3.3 Å along b*.
Initial phases were determined to 4.5 Å resolution by single anomalous dispersion (SAD) from a selenomethionyl crystal using AutoSol (Terwilliger et al., 2009), which was able to locate all 22 ordered Met sites. Prior to building the initial model the phases were extended to higher resolution by a combination of phase extension and two fold noncrystallographic symmetry averaging using the program DM-multi (Cowtan and Main, 1998) with a higher resolution native dataset. The model was built manually using iterative rounds of manual building with COOT (Emsley et al., 2010) followed by refinement with Refmac (Murshudov et al., 1999). The final model containing two additional methionine residues in Atg31 was solved by a combination of SAD and molecular replacement using AutoSol (Terwilliger et al., 2009). The refinement was carried out in Refmac and the final model was refined to Rwork/Rfree = 30.6/33.4 %.
Supplementary Material
HIGHLIGHTS.
Crystal structure of the yeast Atg17-Atg31-Atg29 autophagy initiation complex
Atg17 is crescent shaped and functions as a dimer of two crescents
C-terminal EAT domain of Atg1 binds and tethers highly curved vesicles
Atg1 EAT domain binds Atg17 via Atg13, leading to a model for the PAS
ACKNOWLEDGMENTS
We thank D. Klionsky and W. Prinz for strains, constructs, and technical advice, X. Zuo for assistance with SAXS, X.-F. Ren and R. Ghirlando for light scattering assistance, B. Canagarajah for assistance with Fig. 7, and T. R. Serio for a vector. Crystallographic data were collected at the Southeast Regional Collaborative Access Team 22-ID and 22-BM beamlines at the Advanced Photon Source, Argonne National Laboratory. SAXS data were collected at NCI beamline 12ID-B. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38 and was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. This research was supported by the Intramural Program of the NIH, NIDDK (J.H.H.), a Damon Runyon Cancer Research Fellowship (R. E. S.), and Ruth Kirschtein NRSA fellowship GM099319 (M. J. R.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Crystallographic coordinates have been deposited in the RCSB data bank with accession code 4HPQ.
REFERENCES
- Chan EY, Longatti A, McKnight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. MolCell Biol. 2009;29:157–171. doi: 10.1128/MCB.01082-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong H, Nair U, Geng JF, Klionsky DJ. The Atg1 kinase complex is involved in the regulation of protein recruitment to initiate sequestering vesicle formation for nonspecific autophagy in Saccharomyces cerevisiae. Molecular Biology of the Cell. 2008;19:668–681. doi: 10.1091/mbc.E07-08-0826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JD, Ryu M, Ae Park M, Jeong G, Lee JS. FIP200 inhibits beta-catenin-mediated transcription by promoting APC-independent beta-catenin ubiquitination. Oncogene. 2012 doi: 10.1038/onc.2012.262. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Cowtan K, Main P. Miscellaneous algorithms for density modification. Acta Crystallogr Sect D-Biol Crystallogr. 1998;54:487–493. doi: 10.1107/s0907444997011980. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr Sect D-Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Nassiri A, Zhong Q. Autophagosome targeting and membrane curvature sensing by Barkor/Atg14(L) Proc Natl Acad Sci U S A. 2011;108:7769–7774. doi: 10.1073/pnas.1016472108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost A, Unger VM, De Camilli P. The BAR Domain Superfamily: Membrane-Molding Macromolecules. Cell. 2009;137:191–196. doi: 10.1016/j.cell.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley IG, Lam DH, Wang J, Ding X, Chen S, Jiang X. ULK1-ATG13-FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009 doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Takamura A, Kishi C, Iemura SI, Natsume T, Guan JL, Mizushima N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181:497–510. doi: 10.1083/jcb.200712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura SI, Natsume T, Takehana K, Yamada N, et al. Nutrient-dependent mTORC1 Association with the ULK1-Atg13-FIP200 Complex Required for Autophagy. Molecular Biology of the Cell. 2009;20:1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6:764–776. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 Complexes Mediate mTOR Signaling to the Autophagy Machinery. Molecular Biology of the Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Kamada Y, Baba M, Takikawa H, Sasaki M, Ohsumi Y. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Molecular Biology of the Cell. 2005;16:2544–2553. doi: 10.1091/mbc.E04-08-0669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Kawarnata T, Suzuki K, Ohsumi Y. Cis1/Atg31 is required for autophagosome formation in Saccharomyces cereviside. Biochem Biophys Res Commun. 2007;356:405–410. doi: 10.1016/j.bbrc.2007.02.150. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Noda NN, Fujioka Y, Suzuki K, Inagaki F, Ohsumi Y. Characterization of the Atg17-Atg29-Atg31 complex specifically required for starvation-induced autophagy in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 2009;389:612–615. doi: 10.1016/j.bbrc.2009.09.034. [DOI] [PubMed] [Google Scholar]
- Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol. 2000;150:1507–1513. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata T, Kamada Y, Kabeya Y, Sekito T, Ohsumi Y. Organization of the pre-autophagosomal structure responsible for autophagosome formation. Molecular Biology of the Cell. 2008;19:2039–2050. doi: 10.1091/mbc.E07-10-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata T, Kamada Y, Suzuki K, Kuboshima N, Akimatsu H, Ota S, Ohsumi M, Ohsumi Y. Characterization of a novel autophagy-specific gene, ATG29. Biochem Biophys Res Commun. 2005;338:1884–1889. doi: 10.1016/j.bbrc.2005.10.163. [DOI] [PubMed] [Google Scholar]
- Klemm RW, Eising CS, Surma MA, Kaiser HJ, Gerl MJ, Sampaio JL, de Robillard Q, Ferguson C, Proszynski TJ, Shevchenko A, et al. Segregation of Sphingolipids and Sterols During Formation of Secretory Vesicles at the trans-Golgi Network. J Cell Biol. 2009;185:601–612. doi: 10.1083/jcb.200901145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ. Monitoring autophagy in yeast: The Pho8Δ60 assay. Methods in Molecular Biology. 2007;390:363–371. doi: 10.1007/978-1-59745-466-7_24. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy. 2007;3:181–206. doi: 10.4161/auto.3678. [DOI] [PubMed] [Google Scholar]
- Koch MHJ, Vachette P, Svergun DI. Small-angle scattering: a view on the properties, structures and structural changes of biological macromolecules in solution. Q Rev Biophys. 2003;36:147–227. doi: 10.1017/s0033583503003871. [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Liu F, Lee JY, Wei HJ, Tanabe O, Engel JD, Morrison SJ, Guan JL. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood. 2010;116:4806–4814. doi: 10.1182/blood-2010-06-288589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo SY, Brett CL, Plemel RL, Vignali M, Fields S, Gonen T, Merz AJ. Intrinsic Tethering Activity of Endosomal Rab Proteins. Nat Struct Mol Biol. 2011;19:40–47. doi: 10.1038/nsmb.2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari M, Griffith J, Rieter E, Krishnappa L, Klionsky DJ, Reggiori F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol. 2010;190:1005–1022. doi: 10.1083/jcb.200912089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, Noda T, Yoshimori T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol. 2010;190:511–521. doi: 10.1083/jcb.200911141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132–139. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Ohsumi Y. The Role of Atg Proteins in Autophagosome Formation. Annual Review of Cell and Developmental Biology. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- Moreau K, Ravikumar B, Renna M, Puri C, Rubinsztein DC. Autophagosome Precursor Maturation Requires Homotypic Fusion. Cell. 2011;146:303–317. doi: 10.1016/j.cell.2011.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr Sect D-Biol Crystallogr. 1999;55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- Nair U, Jotwani A, Geng JF, Gammoh N, Richerson D, Yen WL, Griffith J, Nag S, Wang K, Moss T, et al. SNARE Proteins Are Required for Macroautophagy. Cell. 2011;146:290–302. doi: 10.1016/j.cell.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nature Reviews Molecular Cell Biology. 2009;10:458–467. doi: 10.1038/nrm2708. [DOI] [PubMed] [Google Scholar]
- Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJG, Evans PR, McMahon HT. BAR domains as sensors of membrane curvature: The amphiphysin BAR structure. Science. 2004;303:495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Shpilka T, Elazar Z. Mechanisms of Autophagosome Biogenesis. Curr Biol. 2012;22:R29–R34. doi: 10.1016/j.cub.2011.11.034. [DOI] [PubMed] [Google Scholar]
- Sekito T, Kawamata T, Ichikawa R, Suzuki K, Ohsumi Y. Atg17 recruits Atg9 to organize the pre-autophogsomal structure. Genes Cells. 2009;14:525–538. doi: 10.1111/j.1365-2443.2009.01299.x. [DOI] [PubMed] [Google Scholar]
- Strong M, Sawaya MR, Wang S, Phillips M, Cascio D, Eisenberg D. Towards the structural genomics of complexes: Crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2006;103:8060–8065. doi: 10.1073/pnas.0602606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Kubota Y, Sekito T, Ohsumi Y. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells. 2007;12:209–218. doi: 10.1111/j.1365-2443.2007.01050.x. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Ohsumi Y. Current knowledge of the pre-autophagosomal structure (PAS) FEBS Lett. 2010;584:1280–1286. doi: 10.1016/j.febslet.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mul JJ, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. NatCell Biol. 2007;9:1142–1151. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Adams PD, Read RJ, McCoy AJ, Moriarty NW, Grosse-Kunstleve RW, Afonine PV, Zwart PH, Hung L-W. Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr Sect D-Biol Crystallogr. 2009;65:582–601. doi: 10.1107/S0907444909012098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei HJ, Wei S, Gan BY, Peng X, Zou WP, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011;25:1510–1527. doi: 10.1101/gad.2051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo-Kakuta C, Ichikawa R, Kinjo M, Ohsumi Y. Atg9 Vesicles are an Important Membrane Source During Early Steps of Autophagosome Formation. J Cell Biol. 2012;198:219–233. doi: 10.1083/jcb.201202061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh YY, Shah KH, Herman PK. An Atg13 Protein-mediated Self-association of the Atg1 Protein Kinase Is Important for the Induction of Autophagy. J Biol Chem. 2011;286:28931–28939. doi: 10.1074/jbc.M111.250324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinser E, Sperka-Gottlieb CD, Fasch EV, Kohlwein SD, Paltauf F, Daum G. Phospholipid Synthesis and Lipid Composition of Subcellular Membranes in the Unicellular Eukaryote Saccharomyces cerevisiae. J Bacteriol. 1991;173:2026–2034. doi: 10.1128/jb.173.6.2026-2034.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.