

Abstract
We report a novel class of highly water-soluble decacationic methano[60]fullerene decaiodides capable of co-producing singlet oxygen (Type-II) and highly reactive hydroxyl radicals, formed from superoxide radicals in Type-I photosensitizing reactions, upon illumination at both UVA and white light wavelengths. The ·-production efficiency of 1-(I−)10 was confirmed by using an ·-reactive bis(2,4-dinitrobenzenesulfonyl)tetrafluorofluorescein probe and correlated to the photoinduced electron-transfer event going from iodide anions to leading to . Incorporation of a defined number (ten) of quaternary ammonium cationic charges per C60 in 1 was aimed to enhance its ability to target pathogenic Gram-positive and Gram-negative bacterial cells. We used the well-characterized malonato[60]fullerene diester mono-adduct C60[>M(t-Bu)2] as the starting fullerene derivative to provide a better synthetic route to via transesterification reaction under trifluoroacetic acid catalyzed conditions. These compounds may be used as effective photosensitizers and nano-PDT drugs for photoinactivation of pathogens.
Keywords: Decacationic C60 monoadducts; Photoinduced radicals; Transesterification; Di(N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)amido)malonate arm; Photosensitizer; Antimicrobial photodynamic therapy
1. Introduction
The fullerene cage moiety of C60 monoadducts retains most of the photophysical characteristics of pristine C60 with main absorption bands centered at UVA wavelength range. Photoexcitation of the fullerene cage promotes its electronic transition from the singlet excited state to a long-lived triplet excited state, via an intersystem crossing process. Subsequent intermolecular energy transfer to ground-state triplet molecular oxygen can occur yielding a reactive singlet oxygen (1O2) in Type-II reactions [1]. These processes are the basis of photodynamic therapy (PDT), a method of selectively killing either cancer cells or pathogenic microbial cells. Both singlet and triplet excited states of C60 and its derivatives are known to induce energy- and/or electron-transfer via either intramolecular or intermolecular processes [2] often involving either electron donors or electron-rich reductants, such as physiological NADH [3]. This process leads to the formation of anionic fullerenyl radical ( ·) intermediate and, subsequently, to more reactive radical-based reactive oxygen species (ROS), such as superoxide anion ( ·) and hydroxyl radical (HO·), detectable in water or buffer solution.
In the absence of electron-donating biosubstances, the primary PDT mechanism of most of fullerene-derived photosensitizers (FPSs), in general, follows triplet energy-transfer steps in Type-II reactions giving 1O2 as the main ROS which is toxic to pathogens. We are interested in developing a novel class of water-soluble fullerene adducts capable of producing highly reactive photoinduced radical species via Type-I photochemical reactions and have them used as effective photosensitizers (PSs) for photoinactivation of pathogens, such as multiantibiotic-resistant bacteria [4–8]. To increase the specificity for bacteria and other microbial cells, we need to consider the optimization of surface interactions between FPS and the bacteria cell wall for effective ROS attack endure in close contact with the cell membrane. The targeting approach chosen was based on static charge binding to negatively charged lipopolysaccharides, which are a part of the outer bilayer membrane characteristic of Gram-negative bacteria [9–12]. The exclusion of lipophilic structures and the anionic shielding provided by these lipopolysaccharide structures located outside the cytoplasmic membrane prohibit the penetration of some neutral and anionic PS resulting in marked Gram-negative resistance to photodynamic inactivation and lowered PDT efficiency [13]. Accordingly, the use of cationic PSs was proposed to facilitate the physical targeting to bacterial cells and was correlated with higher effectiveness in PDT-induced inactivation of Gram-negative bacteria [14,15]. PSs having a higher number of cationic charges on the PS molecule, such as polycationic porphyrins or phthalocyanines, were able to penetrate the outer barrier of Gram-negative bacteria and cause damage to the cell wall and the cytoplasmic membrane, considered to be the main causes of cell death [16]. These observations motivated our interest to develop polycationic nano-FPS with a well-defined number of cationic charges. However, the synthesis of polycationic [60]fullerenyl monoadducts having a high number of charges had previously remained challenging and has rarely been reported to date. In addition, it is unclear how photoinduced radicals could be produced by FPS in the absence of electron-rich chromophore addends.
Here we describe the synthesis and characterization of novel highly water-soluble decacationic [60]fullerenyl malonate mono-adducts, , capable of producing photoinduced superoxide radicals upon illumination. The compound 1-(I−)10 is in a form of methyl quaternary ammonium decaiodide salts bearing a well-defined high number of cationic charges, as shown in Scheme 1. They are able to function as effective nanophotosensitizers (nano-PS) in broad-spectrum, one-photon-excitation based photodynamic therapy (1γ-PDT)-mediated killing of pathogenic Gram-positive (e.g. Staphylococcus aureus) and Gram-negative (e.g. Escherichia coli) bacterial targets, examples of species that develop multiantibiotic-resistance [14,17].
Scheme 1.

The first synthetic steps of . Reagents and conditions: i. γ-butyrolactone (GBL), BF3·Et2O, triethylamine, CH2Cl2, 0 °C–r.t., 12 h; ii. malonyl chloride, pyridine, dichloromethane, 0 °C–r.t., 5 h; iii. CH3I, DMF, 45 °C, 3.0 d; iv. C60, CBr4, DBU, toluene–DMF, r.t., 10 h.
Accordingly, our choice of functional addends of FPS mono-adducts was designed to (1) tackle the importance of cell surface interactions between multicationic drug molecules and anionic peptide residues in the lipopolysaccharide of the cell wall, (2) increase the FPS solubility in physiologic media, (3) improve the FPS compatibility in tissue environment of bacterial infections, and (4) enhance the electron-transfer mechanism for photoinduced radical production. This hydrophilic structural modification of C60 to 1 should also enhance effective targeting selectivity, drug-delivery, and, thus, photodynamic inactivation of microorganism as compared with those of C60 itself [18,19]. For the synthetic preparation procedure of 1, we investigated two plausible routes for comparison of the overall reaction yield and complexity of the workup procedure. We also applied various spectroscopic techniques for the structural characterization of all intermediates and products. The new decacationic nano-PDT agent as the decaiodide salt 1-(I−)10 of was found to be capable of producing superoxide radicals upon illumination at the photoexcitation wavelength of the fullerene cage in both UVA and white light ranges.
2. Results and discussion
2.1. Structural design and synthesis
Organic chromophores exhibiting long-lived triplet excited states can be employed as photosensitizers for PDT. The biological efficacy of antimicrobial-PDT depends on several factors including the appropriate photophysical characteristics of the PS leading to the efficient production of ROS upon illumination in physiologic media, the ability to formulate the PS for administration, the ability of the PS to target bacteria following administration, and the availability of an appropriate light source for the treatment. [60] Fullerene monoadducts have, in general, been proven to be highly photostable molecules that make them suitable for multiple-PDT treatments using a single-dose administration. Desirable PDT properties of C60 monoadducts include a nearly quantitative efficiency of intersystem crossing process from the photoexcited singlet state of the C60 cage to the corresponding triplet excited state that subsequently undergo intermolecular energy transfer to triplet molecular oxygen, leading to the generation of reactive singlet oxygen (1O2) and subsequent highly reactive radical ROS [1,3]. This photoinduced mechanism of producing ROS can cause cell damage and be used for the effective killing of pathogenic bacteria and cancer cells. To facilitate the drug administration, appropriate chemical functionalization of highly hydrophobic C60 is necessary to enhance its solubility in water. This was made by synthetic incorporation of oligo(ethylene glycol) units [20–22], aminoacid moieties [23], or ionic functional groups [24–26] as addend attachments to the C60 cage. These chemical modifications can easily increase the hydrophilicity of resulting derivatives. The cage functionalization also transformed the character of derivatives to be more amphiphilic. This may drive the molecule to undergo different forms of solid or cluster aggregation in aqueous solution if the hydrophilic moiety of the addend is insufficiently large to overcome the high hydrophobicity of the fullerene cage. One approach to circumvent the aggregation problem is to increase the number of hydrophilic addend attachments on each C60 cage. However, the synthesis using multiple addends will also lead to formation of regioisomers. More crucially, the increase of the addend number may change significantly the molecular orbital configuration of the fullerene cage and, thus, its intersystem crossing efficiency of excited singlet to triplet states and reduce the related efficacy of the production of 1O2. To eliminate this concern, we restricted our approach to a very limited number of addends, i.e. to use only one fullerenyl olefinic bond, giving a monoadduct derivative, to preserve the low HOMO–LUMO energy gap level of the cage and optimize the 1γ-PDT efficiency. Therefore, to achieve the incorporation of a high number of cationic charges on a fullerenyl monoadduct structure, it would require the synthesis of a high number of cationic moieties on a single addend. Successful creation of such a molecule should provide sufficient hydrophilicity and high compatibility in water. The consideration led to our design of a well-defined water-soluble pentacationic N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)amine arm moiety , as a fully charged C3N6 (2) derivative, with the number of positive charge being fixed at five per arm and used as a key synthon for the preparation of nanophotosensitizers as in decacationic [60]full-erenyl malonate monoadducts 1, (Scheme 1). Furthermore, incorporation of decaiodide counter-anions to 1-(I−)10 may also allow the photoinduced oxidation of iodide (I−), as demonstrated via triplet excited states of the fullerene cage [3C60* or 3(C60>)*] in aqueous media, leading to the formation of full-erenyl anion radical intermediate [ · or (C60>)− ·] [27,28]. This anionic radical intermediate can serve as the precursor for the generation of superoxide radical.
Accordingly, we attached two quaternary alkylammonium multisalts to a malonate linker moiety forming a doubly-armed malonate ester precursor intermediate –malonate– , such as 5, as a plausible approach to assemble a well-defined decacationic addend. Synthesis of 5 was started with the ring-opening reaction of γ-butyrolactone using C3N6 (2) as the nucleophile in CH2Cl2 in the presence of Lewis acid BF3·Et2O at 25 °C overnight to afford 4-hydroxy-[N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)butanamide 3, C3N6C3–OH, in 60% yield. Subsequently, bis-[N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)amino] carbonylpropanoxy-malonate 4, M(C3N6C3)2, was synthesized in a yield of 78% by an esterification reaction between malonyl chloride and C3N6C3–OH in CH2Cl2 in the presence of pyridine at ambient temperature for a period of 5.0 h. One potential synthetic route to use the hydrophobic M(C3N6C3)2 intermediate in the reaction with C60 to give the corresponding neutral monoadduct was found to be problematic owing to the partial charge-transfer complex formation between deca(tertiary amine) moiety and the fullerene cage that resulted in uncharacterizable insoluble solids precipitated from the solution. Therefore, we proceeded to use an alternative intermediate by carrying out the quaternization reaction of 4 via the addition of an excess of iodomethane portionwise over a period of 3.0 d at 45 °C to afford bis[N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)amino]carbonylpropanoxymalonate deca(methyl quaternary ammonium iodide) salt 5, , in 92% yield. The use of multiple short hydrophobic n-propyl groups in 5 was intended to improve and compensate the large incompatibility of two pentacationic side-arms with the hydrophobic fullerene moiety in the subsequent reaction and allow the arm-wrapping around the surface of the C60 cage. These alkyl interactions along with in-chain charges should promote the solubility of mono-adducts in water. In the experiment, we discovered that large incompatibility and insolubility of 5 in toluene, as the preferred solvent for C60, incurred much challenge and difficulty for us to control homogeneity of the reaction for the exclusive monoadduct formation, the reaction efficiency and yield, and the product purification and characterization. Following many attempts to carry out the cyclopropanation reaction of C60 with 5 in the presence of CBr4 and 1.8-diazabicyclo[5.4.0]-undec-7-ene (DBU) under different conditions, we found that a reasonable yield of bis(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaazatricosan-23-yl)[60] fullerenyl malonate deca(methyl quaternary ammonium iodide) salt 1-(I−)10, , could be obtained in 65% yield (after recovery of unreacted C60) with a solvent polarity-balanced mixture of toluene–DMF.
2.2. Spectroscopic characterization
Functional characterization of 4 and 5 via FT-IR spectra (Supporting information) was accomplished by the detection of a clear strong malonyl ester carbonyl [–O–(C=O)–] vibrational absorption band centered at 1732 cm−1 accompanied with a strong malonyl amide carbonyl [–NH–C(=O)–] absorption band centered at 1660 cm−1, consistent with their structure. We were able to assign peaks at 2968, 2936, and 2877 cm−1 to the stretching absorption band of aliphatic –C–H. Anti-symmetric deformations of –CH3 groups and scissor vibrations of –CH2 groups appeared as medium intensity bands centered around 1456 cm−1, while symmetric deformations of CH3 groups exhibited the absorption around 1370 cm−1. The broad band at 1026 cm−1 was assigned to the absorption of a number of quaternary –C–N+–C– bonds. Meanwhile, attachment of to C60 introduced a clear new sharp band at 526 cm−1 corresponding to characteristic absorption of a half-cage moiety of C60 in structure of the monoadduct 1. This band can be easily differentiated from that of pristine C60 itself by the absence of three additional C60-associated characteristic bands at 1426, 1184, and 576 cm−1. In general, the 526 cm−1 band will disappear when the monoadduct is further reacted to form bisadducts or the derivative with more addends. Therefore, the detection this band provided evidence for our structural assignment of 1.
All proton peaks of compounds 2, 3, 4, 5, and 1-(I−)10 in 1H NMR spectra (Supporting information) were assigned according to corresponding proton symbols indicated on the compound structure shown in Scheme 1. Proton chemical shifts of the starting material 2 at δ 0.85 (Ha), 1.42 (Hb), 2.60–2.30 (Hc/Hc′), and 2.69 (Hd) were used as the reference for the characterization assignment of other compounds. Conversion of 2 to 3 with the change of methylene-amine protons, –CH2–NH2, to methyleneamide protons, –CH2– NH–(C=O)–, caused the down-fielded shift of Hd chemical shift from δ 2.69 to δ 3.27. It was accompanied with two new peaks at δ 3.66 and 1.83 corresponding to chemical shifts of hydroxylated methylene protons Hg, –CH2–OH, and methylene protons Hf, respectively. The peak of He [–NH–C(=O)–CH2–] was overlapped by a broad multiplet peak of Hc and Hc′ at δ 2.68–2.32. Upon esterification reaction of 3 with malonyl chloride, the chemical shift of Hg (–CH2–O–C=O of 4) shifted from δ 3.66 to δ 4.17 with the detection of a new singlet peak of malonate α-proton (Hα) at δ 3.35. All other proton peaks of 4 (Ha, Hb, Hc/Hc′, Hd, and Hf) remained in a similar range as those of 3. Methyl quaternization reaction of M(C3N6C3)2 to 5 resulted in a large down-fielded shift of all N-attached ethyleneamino –CH2– proton chemical shifts in more than 1.0 ppm from δ 2.20–2.79 to δ 3.09–4.50 (Hc, Hc′, and Hh) with the overlap of Hd, Hg, and Hα proton peaks as a broad band. Chemical shifts of all methyl (CH3–CH2–CH2–N+–) or methylene (CH3–CH2–CH2–N+–) proton peaks of 5 were also down-field shifted to δ 0.97 (Ha) and 1.63–1.96 (Hb), respectively. In the case of (1), a similar 1H NMR spectrum profile as that of 5 was observed owing to the fact of the reaction center being located at the malonate α-carbon that placed two arm moieties of 1 three atoms away from the C60 cage, giving a minimum electron-withdrawing effect.
We found that a better approach to the preparation of decacationic [60]fullerenyl malonate decaiodide salt 1-(I−)10 was to begin with a simple, well-defined fullerene monoadduct derivative, such as di(tert-butyl)fullerenyl malonates C60[>M(t-Bu)2] (6), capable of undergoing the chemical conversion reaction with derived intermediates, as shown in Scheme 2. Prior to the reactions, the structure of 6 was verified mainly by its 13C NMR spectrum (Fig. 1c) displaying a carbon peak at δ 162.31 corresponding to the chemical shift of ester carbonyl carbon and 16 peaks in different intensities (14 peaks each with 2C and two peaks each with 1C) in the region of δ 139–146 that were accounted for all 58 fullerenyl sp2 carbons. The data is consistent with a C60 monoadduct structure having a C2v-symmetrical structure, agreed well with that of 6. The structure also fitted well with FT-IR spectroscopic data (Supporting information) showing a clear strong malonyl ester carbonyl absorption band centered at 1741 cm−1 with a strong broad band at 1253 cm−1 corresponding to the asymmetric stretching vibrations of –C–C(=O)–O– moieties [i.e. mixed stretching vibrations of – C(=O)–O– and –C–C(=O)–], while the strong band centered at 1155 cm−1 was assigned for the absorption of C(C60)–C–C(=O)–deformations, rocking –CH3 vibrations, and –C–O–R (R = t-Bu) stretching vibrations. Furthermore, a clear sharp characteristic monoadduct absorption band of C60 was also detected at 526 cm−1 that excluded the other derivatives having a higher number of addends than one.
Scheme 2.

The second synthetic steps of and . Reagents and conditions: v. CF3COOH, 1,2-C2H4Cl2–DMF, reflux, 3.0 d; vi. (a) K2CO3 (aq.), (b) CH3I, CHCl3–DMF, 45 °C, 3.0 d; vii. CF3COONa, DMF.
Fig. 1.

13C NMR spectra of (a) 4 in CDCl3, (b) 1-(TFA−)10 in DMSO-d6–CDCl3–CS2, (c) 6 in CDCl3–CS2, and (d) 7 in DMF-d7–CDCl3–CS2.
A well-characterized structure of 6 should facilitate the product identification of subsequent reactions. Synthetically, in the next step reaction, we used a well-characterized penta(tertiary-amino)butanamide precursor arm, C3N6C3–OH (3), for the transesterification reaction with 6 under acidic conditions using trifluoroacetic acid as the catalytic reagent to afford the formation of decacationic bis(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaaza-tricosan-23-yl)[60]fullerenyl malonate (7) in 65% yield, in a form of deca(protonated quaternary ammonium trifluoroacetate) salt. The structure of 7 was subsequently verified by various spectroscopic techniques including 13C NMR spectrum (Fig. 1d) showing two peaks at δ 166.54 and 164.84 corresponding to the chemical shift of a carbonyl carbon of the amide [–HN–(C=O)–] and the ester [–O–(C=O)–] moieties, respectively, consistent with the structure of the arm. It also displayed a total of 30 peaks (one peak with 4C, 25 peaks each with 2C, and 4 peaks each with 1C) in different intensities in the region of δ 130–150 that were accounted for all 58 fullerenyl sp2 carbons. The spectral data indicated clearly a C2-symmetry across the cyclopropane-bridged [6,6]-bond of the cage moiety of 7 that belongs to a lower molecular symmetry than C2v-symmetry of 6. As the simple transesterification condition should not complicate the structural configuration of methano[60]fullerene cage moiety, slight decrease of the molecular symmetry may be indicative of strong interactions between fiexible hexapropyl-hexa(aminoethyl) butanamide arms and the C60 cage, as we proposed above. These interactions may induce a nonsymmetrical environment among cage carbons. Interestingly, we found that the use of C60-compatible deuterated solvents, such as CDCl3 and CS2, to enhance the detectability of fullerenyl carbon peak signals led to the full suppression of pentacationic arm moieties signals in the spectrum. Contrarily, the application of water-compatible deuterated solvents, such as D2O and DMF-d7, reduced significantly the signal intensity of fullerenyl carbon peaks, likely owing to clustering or aggregation of the cage.
Similar solvent-dependent phenomena were observed in 1H NMR spectrum of 7 (Fig. 2c with all assigned proton symbols indicated on the compound structure shown in Schemes 1 and 2), baring a certain degree of peak broadening. In the analysis, we applied all proton peak assignments of 2 (Fig. 2a) and 3 as the reference for correlation with those of 7 and 1. As shown in Fig. 2b, we assigned the peak at δ 3.27 to the chemical shift of methylene protons (Hd of 3) of –CH2–NH– and two peaks at δ 3.66 and 1.83 to methylene protons of –CH2–OH (Hg) and –(C=O)–CH2–CH2–CH2–OH (Hf), respectively. Transesterification reaction of 6 with 3 to 7 resulted in the introduction of protonated quaternary ammonium arm in the structure that caused a large down-fielded shift of chemical shifts (more than 1.0 ppm) of all N-attached ethyleneamino –CH2–N– protons from δ 2.32–2.68 to δ 3.40–4.10 as the chemical shifts of quaternary ammonium methylene protons Hc and Hc′ (–CH2–N+–), with the overlap of carboxylated methylene protons [–C(=O)–O–CH2–] (Hg) at δ 4.0– 4.25 in a broad band. The chemical shift of amidyl methylene protons (Hd) [–C(=O)–NH–CH2–] was found to be δ 3.11–3.20 appeared as multiplet peaks. Successful transesterification was also confirmed by infrared spectrum of 7 showing band profiles resembling those of the cationic precursor arm 5 as di(C3N6C3–O-yl)malonate trifluoroacetate salt, , with a strong band at 1621 cm−1 and a new band at 1714 cm−1 assigned to the absorption of amide carbonyl –NH–C(=O)– and malonyl ester carbonyl –O–(C=O)–, respectively. High retention in intensity of the sharp characteristic monoadduct absorption band at 526 cm−1 as that of 6 implied no interference of the reaction on the cage moiety and provided evidence of the transesterification product 7.
Fig. 2.

1H NMR spectra of (a) 2 in CDCl3, (b) 3 in CDCl3, (c) 7 in CDCl3–DMF-d7, (d) 1-(I−)10 in DMSO-d6, and (e) 1-(TFA−)10 in DMSO-d6.
Based on the characterized structure of 7, we carried out its subsequent conversion to the corresponding decacationic methyl quaternary ammonium decaiodide salt as in 92% yield by neutralization with K2CO3 and the subsequent treatment with CH3I. Methyl quaternization of neutral C60[>M(C3N6C3)2], having tertiary penta(ethylamino) arms, to 1-(I−)10 with decaiodide salts and the subsequent corresponding 1-(TFA−)10 with decatrifluoroacetate salts, upon ion-exchange of 1-(I−)10 with aqueous CF3COONa, showed retention of the most of IR absorption bands (Supporting information). It accompanied with a slight shift of –NH–C(=O)– absorption band to 1647 cm−1 and a new broad band at 1000–1200 cm−1 corresponding to the absorption of a number of quaternary –C–N+–C– bonds. These results provided a good agreement with the product structures of 1-(I−)10 and 1-(TFA−)10.
Conversion of the compound 7 to 1-(I−)10 resulted in a salt with multiple iodide anions. Unexpectedly, we were not able to detect any fullerenyl sp2 carbon signals in the 13C NMR spectrum of 1-(I−)10 under various deuterated solvent mixtures. However, these carbon signals re-appeared when all iodide anions were replaced by trifluoroacetate anions, leading to the corresponding salt 1-(TFA−)10. Accordingly, in the 13C NMR spectrum of 1-(TFA−)10 (Fig. 1b), we were able to assign two peaks at δ 168.33 and 165.30 to the chemical shift of an amide carbonyl carbon [–HN–(C=O)–] and the ester carbonyl carbon [–O–(C=O)–], respectively, which were in a similar range to those of 7. It also displayed a total of 30 peaks (one peak with 4C, 25 peaks each with 2C, and 4 peaks each with 1C) in the region of δ 136–148 that were accounted for a total of 58 fullerenyl sp2 carbons, indicating a similar C2-symmetry as that of the compound 7. These spectra provided the unambiguous support for structural assignments of 7 and 1-(TFA−)10. Furthermore, the observation of low 13C signal intensity in the spectrum of 1-(I−)10 led to our assumption on the possibility of reversible partial electron-transfer from the iodide (I−) donors to C60 cages forming a low degree of radicalized cage moieties that reduced significantly the resolution of 13C signals.
The mass spectroscopic data collection of [1-(I−)10 and 1-(TFA−)10] was proven to be difficult due to their polycationic nature and facile fragmentations occurring at the conjunction of the C60 cage and the decacationic malonate arm, giving mainly the highly detectable C60 ion mass at m/z 721, as displayed in MALDI-TOF (sinapic acid as the matrix) and electrospray ionization (ESI) mass spectra (Fig. 3). Fortunately, we were able to acquire several spectra in the high mass region showing the molecular ion mass at m/z 3220 (M+− I−) in low intensity (Fig. 3b) accompanied with several identifiable fragmentation ion mass peaks at m/z 1570, 1742, 1760, 2039, 2152, 2468, 2624, 2794, 2838, and 2852 (Fig. 3b and c). By variation of applied laser power intensity, spectra containing many ion mass peaks in significant signal intensity in the medium mass region were obtained, as shown in the inset of Fig. 3c. Structural elucidation of these ion fragments (the Supporting information) in the combined spectra of Fig. 3b and c allowed us to conclude (1) a monoadduct structure of 1-(I−)10 by the detection of an ion fragment mass of (100%) at m/z 871, (2) a facile process of nearly full dequaternization of 1-(I−)10 under MALDI-MS conditions giving mostly detectable monocationic mass fragments, and (3) the bond cleavage of side-chain alkyl group on the nitrogen atom was faster than that of the main chain moiety. Among them, a mass ion peak at m/z 1760 (ESI-MS) was elucidated by a mass loss of [10I + 6(CH2CH2CH3) + 4CH3] from the molecular mass ion (MH+), consistent with a mass of fully decationized 1 giving the evidence of a C60 malonate monoadduct with two 4-hydroxy-hexa(aminoethyl)butanamide main chains. It was followed by a mass ion peak at m/z 1570 assigned for the mass loss of [10I + 8(CH2CH2CH3) + 4HCH3 + HN(CH2CH2CH3)2] from the molecular mass ion (M+), consistent with further fragmentations from the peak of m/z 1760. Similarly, a mass ion peak at m/z 1682 in the MALDI-TOF mass spectrum also indicated a fragmented mass of fully decationized 1 by the mass loss of [10I + 8(CH2CH2 CH3) + 3CH3 + 3H2] from the molecular mass ion (M+). These data strongly support the molecular mass of 1-(I−)10 as a – malonate–C60 structure with many consistent (C3N6Cn)2–malonate–C60 (Cn: methyl or n-propyl group) fragments.
Fig. 3.

(a) MALDI-MS spectrum of C60[>M(t-Bu)2] (6) and (b) MALDI-MS and (c) ESI-MS spectra of .
2.3. ROS detection by fluorescent probes
Facile intersystem crossing efficiency from the methanofullerenyl singlet excited state 1(C60>)* to its triplet excited state 3(C60>)* was accounted for the efficient production of 1O2 in Type-II triplet energy-transfer processes. The possibility to achieve a Type-I photomechanism in the solution of will require photoinduced electron-transfer from iodide anions to the fullerene cage moiety in quasi-intramolecular processes to be carried out at either 1(C60>)* or 3(C60>)* excited states, leading to the formation of anionic methanofullerenyl radical (C60>)−· as a precursor for the generation of ·. Accordingly, we selected two reliable fluorescent (FL) probes for the detection of 1O2 and · separately in aqueous PBS solution of either 1-(I−)10 or 1-(TFA−)10 with high selectivity and specificity as a crucial measure for the correlation of its efficacy to 1γ-PDT-derived photokilling of pathogens in biological systems. For the detection of the former ROS, highly fluorescent tetrasodium α,α′-(anthracene-9,10-diyl)bis(methylmalonate) (ABMM, Fig. 4a) was used as the 1O2-reactive probe. The chemical trapping of 1O2 by highly fluorescent ABMM leads to the formation of non-fluorescent 9,10-endoperoxide product ABMM-O2. The conversion allows us to follow the loss of fluorescence emission intensity at 429 nm for its correlation to the proportional quantity of 1O2 produced due to the high kinetic rate constant of the trapping reaction in aqueous media [29], assuming the internal decay of 1O2 in the solvent system is identical among different probe experimental runs.
Fig. 4.
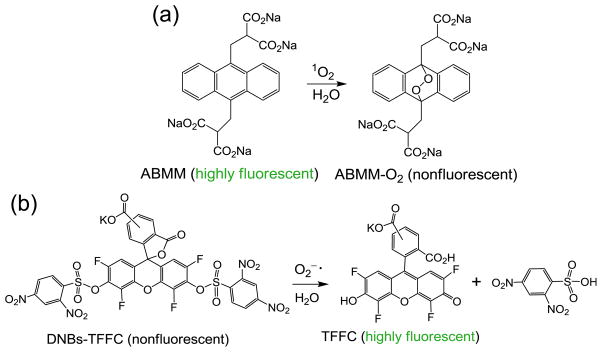
Chemical trapping reactions of (a) singlet oxygen and (b) superoxide radical using different fluorescent probes.
For monitoring the production of · by 1-(I−)10, superoxide radical-reactive fluorescent probes, potassium bis(2,4-dinitro-benzenesulfonyl)-2′,4′,5′,7′-tetrafluorofluorescein-10′ (or 11′)-carboxylate regioisomers (DNBs-TFFC, Fig. 4b) were used due to its high reaction selectivity with a good reported ·/1O2 sensitivity ratio of 46 [30]. A typical probe solution was prepared by diluting a stock solution of DNBs-TFFC in DMSO with phosphate buffered saline (PBS) solution (pH 7.4). Since DNBs-TFFC itself is photode-gradable, a dialysis film with the molecular weight cut-off (MWCO) of 100–500 Da was applied to separate the solution of 1-(I−)10 in H2O–DMSO (19:1) from the probe solution in H2O. The latter was kept in a cuvette with stirring during the fluorescent emission measurement. Only the solution of 1-(I−)10 in the membrane sack was subjected to the LED light exposure at the excitation wavelength of 365 nm or 400–700 nm (white light). Any superoxide radical produced was allowed to rapid diffuse in the probe solution and initiate the desulfonylation of DNBs-TFFC that led to the elimination of two dinitrobenzenesulfonyl moieties and yield the corresponding bisphenol intermediate after capturing two protons from the buffer medium. Rearrangement of bisphenol intermediate to the ring-opening of lactone afforded highly fluorescent potassium 2′,4′,5′,7′-tetrafluorofluorescein-10′-carboxylate regioisomers (TFFC), as shown in Fig. 4b, giving the fluorescence emission at 530 nm upon the excitation at 480 nm. As DNBs-TFFC is not a fluorescent compound, the detected emission photon counts can be correlated fully to the quantity of TFFC converted. By the assumption of constant kinetic rates of desulfonylation reaction among different experimental runs under similar conditions, measured total emission intensity can be used for the calculation of relative · generation.
In principle, continuous irradiation on the fullerene cage moiety of 1 in either UVA or white light should stimulate its photoexcitation from the ground to singlet excited state. Intersystem crossing from the singlet excited state of the cage moiety [1(C60>)*] of 1 to the corresponding long-lived triplet excited state [3(C60>)*] occurs in a time scale of 1.3 ns [1,31]. This duration is long enough to allow intermolecular triplet energy transfer from the 3(C60>)* moiety to O2 yielding a reactive 1O2. However, in the presence of electron-rich iodide anions, photoinduced electron-transfer from the iodide anion to the 3(C60>)* cage moiety of is also possible, as shown in Fig. 5, leading to the formation of anionic fullerenyl radical ( ·) intermediate. In the presence of molecular oxygen, subsequent electron-transfer from · intermediate to O2 should produce superoxide radical in a sequential multiple-step Type-I mechanism.
Fig. 5.
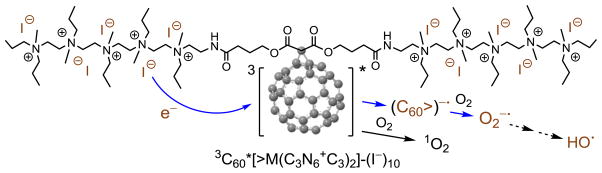
Photoinduced electron-transfer from the iodide anion to the triplet excited state of the cage moiety of as a mechanism to the generation of superoxide radical (blue pathway). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Indeed, we were able to detect rapid production of 1O2 by 1 upon illumination with white LED light. The FL-probe experiments were calibrated by the blank runs of different probe concentrations in the absence of 1 in the same illumination time scale to confirm slight photodegradation of the ABMM probe in H2O under the fluence rate and intensity of white LED light used, as shown in Fig. 6. In the presence of decaiodide salt (0.02 μmol or 8.33 μM based on the ABMM solution volume), a fast loss of fluorescence emission intensity of ABMM (λem 428 nm/λex 350 nm) was observed (Fig. 6a). The loss rate was roughly proportional to the cumulative fluence of light. As the calculated concentration of 1-(I−)10 increased to 20.8 μM (Fig. 6b), a roughly concentration-dependent loss rate was detected in 3.0–3.5-fold faster than the curve profile of Fig. 6a. In a short irradiation time scale of this curve in less than 25 s, the decay profile was almost linear in respect to the cumulative fluence indicating the approximately linear production of 1O2 by 1-(I−)10 with high photostability of FPS. As much of ABMM probe being consumed with the significant decrease of its concentration in the later stage of the irradiation, the FL emission loss rate was noticeably slower. Even though the lifetime of 1O2 in water was known to be as short as 2 μs, which is 10-fold–100-fold less than in organic phases [32], the use of ABMM as the 1O2 acceptor was proven to be effective for the determination of quantum efficiency in water [33] due to its extremely high water-solubility with four carboxylate salts per anthracene molecule in enabling the rapid trapping reaction. Therefore, ABMM can be a reliable FL-probe for the above concentration correlation of FPS, such as highly water-soluble 1.
Fig. 6.

Illumination time-dependent fluorescent intensity loss of ABMM in H2O in the presence of (a) (0.02 μmol or 8.33 μM), (b) (0.05 μmol or 20.8 μM), and (c) (0.05 μmol or 20.8 μM) by photoexcitation using white LED light (the collimated optical power output of >2000 mW in a diameter of 5.2 cm). The fluorometric traces were collected at λem 428 nm with λex 350 nm. The concentration value in parenthesis was calculated based on the aqueous solution volume of the ABMM probe.
Interestingly, a nearly identical curve profile (Fig. 6c) was obtained for 1-(TFA−)10 that revealed clearly low interference of decaiodide anions during the photoinduced production process of 1O2 by and negligible side-reactions of iodide with 1O2 in forming iodoperoxy anion I–O–O− intermediate and the protonated I–O–OH in H2O [34]. The former phenomenon can be understood by the much faster triplet energy-transfer efficiency between 3(C60>)* moiety and 3O2 than electron-transfer efficiency between 3(C60>)* moiety and I−. In addition, the quantity of I− is limited to ten per C60 cage, whereas 1O2 production by can be a continuous process during the illumination period due to the high photostability of 1. Accordingly, the electron-transfer process consisted of only a small fraction of overall photophysical events of 1-(I−)10. The latter conclusion was made by the fact that newly produced singlet oxygen should be in a close proximity to the fullerene cage and the counter iodide anion for capture by I−. A slightly faster decrease of FL intensity induced by 1-(I−)10 than 1-(TFA−)10 (Fig. 6b and c) at the early stage of illumination should exclude any loss of 1O2 in the membrane sack due to the presence of I− prior to the reaction with ABMM. Furthermore, clear detectable ·, stated below, also excluded the loss of I− as a nucleophile in this electrophilic side-reaction with 1O2.
Importantly, the subsequent superoxide radical-reactive fluorescent probe using DNBs-TFFC as the acceptor was found to be reactively sensitive enough for the clear detection of ·. Since the water-soluble probe DNBs-TFFC may suffer potential acid-induced hydrolysis and photohydrolysis, we undertook precaution in the experimental setup using the membrane barrier technique to separate it from the illuminated solution and minimize its light exposure. For the calibration of the former side-reaction, we used non-superoxide generating analogous as the reference sample to define the baseline of FL intensity under the same experimental conditions. As shown in Fig. 7a, in the presence of 1-(TFA−)10, a prolonged illumination period of LED (365 nm) light gave no increase of fluorescence intensity of TFFC in PBS media (pH 7.4) at λem 530 nm with λex 480 nm. A nearly constant FL intensity level of 1.1 × 105 counts/s can be regarded as a low degree of hydrolysis that debenzenesulfonylated DNBs-TFFC. Upon the addition of one equivalent of KI, no visible change of the FL intensity level was observed (Fig. 7b). Large increase of the KI addition to 50 equivalents began to induce partial electrolyte exchange that replaced a fraction of by I− to 1-(TFA−)10−x(I−)x with the FL intensity reaching roughly 2.0 × 105 counts/s (Fig. 7c). A higher FL intensity level of 5.0 × 105 counts/s (Fig. 7c) was achieved at the illumination time of 60 min only after the addition of a large excess amount of KI. The results implied (1) more than 100 equivalents (estimated based on the FL intensity counts) of KI were required to fully exchange ten counter-anions to produce 1-(I−)10 and (2) much slower electron-transfer efficiency from the external free iodide in PBS solution to the 3(C60>)* moiety as compared with that of the internal iodides functioning as the counter-anions to quaternary ammonium arms of 1.
Fig. 7.

Illumination time-dependent fluorescent intensity increase of fluorescein probe TFFC in PBS media (pH 7.4) in the presence of in a quantity of (a) 0.4 μmol, (b) 0.4 μmol with KI (0.4 μmol), (c) 0.4 μmol with KI (20 μmol), and (d) 0.4 μmol with KI (large excess) using a LED (365 nm) light source. The fluorometric traces were collected at λem 530 nm with λex 480 nm.
Under the same FL probe experimental conditions in the presence of at different concentrations, a systematic increase of FL emission intensity counts in roughly more than 4-fold was observed in line with the progressive increase of the FPS concentration from 8.33 to 167 μM under irradiation of 1-(I−)10 using a LED (365 nm) light, as shown in Fig. 8. The increase rate of accumulated incremental FL intensity from the baseline as that of Fig. 8a is approximately linear with the quantity (μmol) and the calculated concentration increase as depicted in the inset of Fig. 8. The lifetime of · seems to be long enough to allow its diffusion passing through the dialysis film membrane in the solution and react with the fluorescein probe. This linear correlation clearly indicated that the production of · was raised from photosensitization of 1-(I−)10 since the photoexcitation wavelength of 365 nm matched with the second absorption maximum range of the compound for specific excitation of the C60 > cage moiety to its singlet excited state . We found that photoexcitation of the C60 > cage by a white LED light (with collimated power output of >2000 mW over a beam diameter of 5.2 cm), having a broad emission spectrum covering the most of visible range with two extinction coefficient maxima centered at 451 and 530 nm, was able to induce the production of · in a sufficient quantity, as shown in Fig. 9, even though the optical absorption extinction coefficient of 1-(I−)10 is not particularly high over the full visible range (ε451 of 9.2 × 106 cm2/mol and ε530 of 4.9 × 106 cm2/mol) as compared with that at 365 nm (ε365 of 2.74 × 107 cm2/mol). Nevertheless, the accumulated incremental FL emission intensity of the fluorescein probe TFFC was, in general, about 3-fold less than that collected using a 365 nm light source with the same FPS concentration and illumination period. This can be interpreted as the requirement of a 3-fold longer of the irradiation time using a white light during the PDT treatment to match the total light dose. It is worthwhile to mention that the HOMO–LUMO energy level of the C60 > cage moiety of 1-(I−)10 is located in the range of 700–720 nm. Most of visible wavelengths are capable of initiating the photosensitization of 1-(I−)10, however, its higher absorption extinction coefficients in 400–550 nm should render this light wavelength range more 1γ-PDT effective than 550–700 nm.
Fig. 8.

Illumination time-dependent fluorescent intensity increase of fluorescein probe TFFC in PBS media (pH 7.4) in the presence of in a quantity of (a) blank, (b) 0.02 μmol (8.33 μM), (c) 0.05 μmol (20.8 μM), (d) 0.1 μmol (41.7 μM), (e) 0.15 μmol (62.5 μM), (f) 0.2 μmol (83.4 μM), (g) 0.3 μmol (125 μM), and (h) 0.4 μmol (167 μM) using a LED (365 nm) light source. The fluorometric traces were collected at λem 530 nm with λex 480 nm. The concentration value in parenthesis was calculated based on the aqueous solution volume (2.4 mL) of the DNBs-TFFC probe.
Fig. 9.

Illumination time-dependent fluorescent intensity increase of fluorescein probe TFFC in PBS media (pH 7.4) in the presence of in a quantity of (a) blank, (b) 0.02 μmol (8.33 μM), (c) 0.05 μmol (20.8 μM), (d) 0.1 μmol (41.7 μM), (e) 0.15 μmol (62.5 μM), (f) 0.2 μmol (83.4 μM), (g) 0.3 μmol (125 μM), and (h) 0.4 μmol (167 μM) using a white LED (λem 451 and 530 nm) light source. The fluorometric traces were collected at λem 530 nm with λex 480 nm. The concentration value in parenthesis was calculated based on the aqueous solution volume (2.4 mL) of the DNBs-TFFC probe.
It has been reported that 1O2 can give rise to several other types of ROS, including ·, H2O2, and HO·, in a sequential reaction with iodide anion (I−) [35]. The plausible mechanism of these conversions in the case of 1-(I−)10 will be the electron-transfer event from the counter-anion I− to newly produced 1O2 that led to the formation of ·. By this assumption, the yield of · may be proportional to the quantity of 1O2 produced since the concentration of I− is finite and decreasing upon consumption. Our previous observation of a higher 1O2 production yield of than in PBS solution under UVA irradiation giving a lower production yield of radical species revealed no direct correlation of 1O2 to · or HO· yields [18]. In fact, they were more likely in a competitive production process to each other in either the triplet energy-transfer (Type-II) or electron-transfer (Type-I) mechanism involving the electron-donating iodide anion (I−) and the electron-accepting fullerene cage. Apparently, the electron-accepting capability of cage moiety may induce electron-transfer from the iodide anion leading to the formation of prior to the further transfer of this electron to O2 that yields ·, consistent with the mechanism reported for solubilized C60-γ-cyclodextrin in water [27]. In a closed solution system, the reaction equation of 3(C60>)* and I− to yield (C60>)−· and I· was suggested to be reversible with the same rate constant for both forward and reverse pathway that gives no net reaction of iodide oxidation [28]. Similar arguments of the reverse dismutation of · by C60> giving backward products of (C60>)−· and O2 as several possible mechanisms in equilibration can also potentially complicate our elucidation of · production. Fortunately, the forward chemical trapping of · in current probe experiments effectively made its removal from the equilibration equation that allowed the forward reaction to move along in the reversible equation to produce more · product.
2.4. Antimicrobial effect of FPS-mediated PDT
Highly water-soluble decacationic was first applied in the PDT-killing of the Gram-positive S. aureus. In general, it is known that Gram-positive bacteria are more sensitive and susceptible to PDT destruction than Gram-negative bacteria. This allowed us to use a lower concentration (up to 10 μM) of 1-(I−)10 for S. aureus killing than that (up to 100 μM) needed for effective E. coli killing. Our previous PDT experiments have indicated a fast binding process of FPS to microbial cells [36]. Thus, a short incubation time of 30 min was applied for both species. As a result shown in Fig. 10, no decacationic fullerene derivative 1-(I−)10 gave any detectable toxicity to either bacterial species in the dark in a concentration up to 10 μM. A pronounced PDT effect was observed after illumination on S. aureus cells incubated with 1-(I−)10 using a broad-band white light source with total fluence intensity of 100 J/cm2, as shown in Fig. 10a, with high cytotoxicity apparent even at low fullerene concentration of 1.0 μM, giving 3 logs killing. As the administered dose increased to a concentration of 3.0 μM, a 5-log killing effectiveness was observed. Interestingly, 1-(I−)10 was able to eradicate the cells (>6 logs of killing) at concentrations higher than 3.0 μM.
Fig. 10.
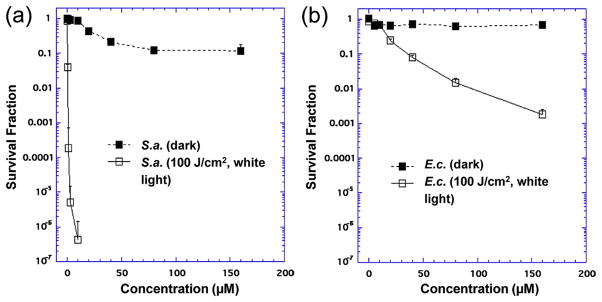
PDT killing of (a) Gram-positive bacteria, S. aureus and (b) Gram-negative bacteria, E. coli. Bacteria [10(8) cells/mL] were incubated with stated concentration of for 30 min followed by delivery or not of 100 J/cm2 of white light.
In the PDT experiments against E. coli shown in Fig. 10b, the need to use 8-times higher concentrations of 1-(I−)10 drug than needed for S. aureus led to the observation of some dark toxicity (1–2 logs) at the higher concentrations of 60 and 80 μM. White light delivery killed 1 log at 40 μM, 2 logs at 60 μM and 3 logs at 80 μM. These two curves (light and dark) were significantly different (p < 0.01). The killing curve of S. aureus was significantly different from the killing curve of E. coli (p < 0.01).
The efficacy of 1-(I−)10 as an antimicrobial photosensitizer compares favorably with many compounds that are well-accepted in the literature. For instance methylene blue, which is one of the few antimicrobial PS to have gained regulatory approval for clinical use in dentistry [37] requires 20 μM for S. aureus and 200 μM for E. coli [38], while toluidine blue needs 10 μM for Gram-positives and 35 μM for Gram-negatives [39].
3. Conclusion
We found that novel highly water-soluble decacationic methano [60]fullerene decaiodide is capable of co-producing singlet oxygen (Type-II) and superoxide, leading to highly reactive hydroxyl radicals via Type-I photochemical reactions, upon illumination at both UVA and white light wavelengths. We also confirmed the ·-production efficiency of 1-(I−)10 by using a water-soluble, ·-reactive bis(2,4-dinitrobenzenesulfonyl)tetrafluorofluorescein carboxylate (DNBs-TFFC) probe. Its generation was proposed to be correlated to the photoinduced electron-transfer event going from iodide anions to leading to the formation of as the precursor intermediate. The design of the compound 1 having well-defined ten methyl quaternary ammonium cationic charges per C60 was aimed to enhance its ability to target pathogenic Gram-positive and Gram-negative bacterial cells, especially, by binding to negatively charged lipopolysaccharides of the latter cells. This highly hydrophilic monoaddend attachment on C60 minimized the size of the hydrophobic fullerene–fullerene cluster formation and gave largely improved solubility in water to facilitate their utility as nano-PDT drugs. The synthesis was accomplished by the use of key synthons as either two-armed decacationic di(N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)amido)malonate or the corresponding one-armed pentacationic analogous, having a total of 12 attached propyl groups, instead of methyl or ethyl groups, in the former case. It was intended to provide a well-balanced hydrophobicity–hydrophilicity character of the decacationic malonate intermediates and to provide possible wrapping around the C60 cage moiety. We found that the use of well-characterized malonato[60]fullerene diester monoadduct C60[>M(t-Bu)2] as the starting fullerene derivative provided a better synthetic route for the preparation of 1-(I−)10 and 1-(TFA−)10 via transesterification reaction under trifluoroacetic acid catalyzed conditions. The reaction condition avoided the direct attack of tertiary-aminated butanamide precursor arm (C3N6C3–OH) on the C60 cage. This resulted in a clean decacationic protonated di[penta(ethylamino)]malonate derivative product of C60, , prior to its further conversion to 1-(I−)10. The compound 1-(I−)10 exhibiting unique photophysical properties to co-produce both photoinduced ·, subsequently to HO·, and 1O2 may be used as an effective photosensitizer and nano-PDT drug for photokilling of drug-resistant Gram-positive and Gram-negative bacteria.
4. Experimental section
4.1. Materials
Reagents of γ-butyrolactone (GBL), BF3·Et2O, triethylamine, pyridine, iodomethane, 1,8-diazabicyclo[5,4,0]-undec-7-ene (DBU), carbon tetrabromide (CBr4), trifluoroacetic acid, and potassium carbonate were purchased from Aldrich Chemicals and used without further purification. The reagent of malonyl chloride was purchased from TCI America. The chemical di(tert-butyl) malonate was purchased from Tokyo Chemical Industry Co., Ltd. A C60 sample with a purity of 99.0% was purchased from either Term USA, Inc. or Nano-C, Inc. Sodium sulfate was employed as a drying agent. Solvents were routinely distilled prior to the use. The fluorescent probe precursor, α,α′-(anthracene-9,10-diyl)bis(methylmalonic acid) (ABMA), for detecting singlet oxygen was synthesized by a modified literature procedure [40]. The other fluorescent probe precursor, bis(2,4-dinitrobenzenesulfonyl)-2′,4′,5′,7′-tetrafluoro-fluorescein-10′-carboxylic acid (DNBs-TFFA), for detecting super-oxide radical was also synthesized by a modified procedure [30].
4.2. Spectroscopic measurements
Infrared spectra were recorded as KBr pellets on a Thermo Nicolet Avatar 370 FT-IR spectrometer. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance Spectrospin–500 spectrometer. UV–vis spectra were recorded on a Perkin Elmer Lambda 750 UV–vis–NIR Spectrometer. MALDI-mass spectra were recorded on a WATERS Micromass MALDI-TOF mass spectrometer. Elemental analysis was taken by Galbraith Laboratories, Inc., Knoxville, TN.
4.3. Synthesis of 4-hydroxy-[N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)-butanamide] C3N6C3–OH (3)
To a solution of BF3·Et2O (1.98 g, 13.7 mmol) in anhydrous dichloromethane was added triethylamine (1.60 g, 15.8 mmol, predistilled) and stirred at 0 °C for a period of 30 min. In a separate flask, N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)amine (1.87 g, 3.9 mmol) and γ-butyrolactone (0.40 g, 4.6 mmol) were dissolved in dichloromethane (10 mL) and added to the above solution. Temperature of the mixture was slowly increased to 25 °C and kept for overnight. At the end of the reaction, it was quenched by the addition of cold aqueous sodium carbonate (10%). The organic layer was then washed sequentially with aq. sodium carbonate (10%) and water and subsequently dried over sodium sulfate. After removal of the solvent, the crude product was purified by column chromatography [neutral aluminum oxide using CHCl3–CH3OH (9.5:0.5, v/v) as the eluant] to give a viscous light yellow liquid of 4-hydroxy-[N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)butanamide] C3N6C3–OH (3) in 60% yield (1.32 g). Spectroscopic data: FT-IR (KBr) νmax 3304 (s, –OH), 2957 (vs, –C–H stretching), 2933 (vs, –C–H stretching), 2872 (vs), 2807 (vs), 1650 (vs, amide carbonyl –C= O), 1555 (s), 1540 (s), 1520 (m), 1505 (w), 1459 (s), 1381 (m), 1335 (w), 1295 (w), 1185 [m, –C–C(=O)–NH– and –C(C=O)–NH–C– stretchings], 1160 (m), 1114 (m), 1077 (s, –C–O– stretching), 954 (w), 923 (w), 866 (w), 755 (w), 749 (w), 622 (w), and 588 (w) cm−1;1H NMR (500 MHz, CDCl3, ppm) δ 3.66 (t, J = 5.65 Hz, 2H, methylene protons of –CH2–OH), 3.27 (t, J = 5.40 Hz, 2H, methylene protons of –CH2–NH–(C=O)−), 2.68–2.32 (br, 32H, amine N-attached –CH2– protons of ethyleneamino moiety), 1.83 (m, 2H, methylene protons of –CO–CH2–CH2–CH2–OH), 1.44 (m, 12H, CH3–CH2– CH2–N–), and 0.86 (m, 18H, CH3–CH2–CH2–N–). 13C NMR (500 MHz, CDCl3, ppm) δ 173.62 (amide carbonyl –NH–C=O–), 62.14, 57.45, 57.37, 57.00, 56.94, 56.93, 56.89, 56.84, 56.82, 56.77, 52.93, 52.63, 52.48, 52.43, 52.34, 51.74, 37.70, 34.19, 28.56, 20.44, 20.36, 20.34, 20.30, 20.20, 19.89, 12.12 (4C), and 12.05 (2C).
4.4. Synthesis of bis[N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl) amino]carbonyl-propanoxymalonate M(C3N6C3)2 (4)
To a solution of malonyl chloride (0.12 g, 0.85 mmol) in anhydrous dichloromethane (30 mL) at 0 °C was added a solution of C3N6C3–OH (3, 1.0 g, 1.75 mmol) and pyridine (0.14 g, 1.77 mmol) in anhydrous dichloromethane (10 mL). The reaction was kept for 5.0 h with the temperature slowly increased to 25 °C. The solution was washed by aq. sodium carbonate solution (10%) and water, and dried over sodium sulfate sequentially. The mixture was purified by alumina gel column using ethyl acetate–hexane (4:1, v/v) as the eluant to afford the product of bis[N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)amino]carbonylpropanoxymalonate, M(C3N6C3)2 (4) in a yield of 78% (0.80 g). Spectroscopic data: FT-IR (KBr) νmax 2957 (vs, –C–H stretching), 2933 (vs, –C–H stretching), 2872 (vs), 2807 (vs), 2729 (m), 1737 (s, ester carbonyl –C=O), 1658 (vs, amide carbonyl –C=O), 1545 (s), 1463 (s), 1378 (m), 1334 (m), 1292 (m), 1270 (m), 1118 (m), 1157 (m), 1114 (m), 1075 (s, –C–O– stretching), 1024 (m), 950 (w), 886 (w), 800 (w), 778 (w), and 747 (w) cm−1; 1H NMR (500 MHz, CDCl3, ppm) δ 4.17 [m, 4H, methylene protons of –CH2–O–(C=O)], 3.35 (s, 2H, α-proton), 3.27 [m, 4H, methylene protons of –CH2–NH–(C=O)], 2.20–2.79 (m, br, 64H, amine N-attached –CH2– protons of ethyleneamino moiety), 1.85 (m, 4H, methylene protons of –(C=O)–CH2–CH2–CH2–O–), 1.45 (m, 24H, CH3–CH2–CH2–N–), and 0.87 (m, 36H, CH3–CH2–CH2–N–). 13C NMR (500 MHz, CDCl3, ppm) δ 173.41 (2C, –NH–C=O–), 166.68 (2C, –O–C=O–), 64.52 (2C), 57.18 (2C), 57.10 (2C), 56.76 (2C), 56.71 (4C), 56.69 (4C), 56.60 (2C), 56.47 (2C), 52.76 (2C), 52.64 (2C), 52.34 (4C), 52.22 (2C), 52.13, 51.58, 41.74, 37.37 (2C), 33.58 (2C), 28.43 (2C), 20.24, 20.18, 20.13 (4C), 20.06 (4C), 19.93, 19.73, 11.85 (8C), and 11.76 (4C).
4.5. Synthesis of bis[N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl) amino]carbonyl-propanoxymalonate deca(quaternary methyl ammonium iodide) salt (5)
A solution of M(C3N6C3)2 (4, 0.60 g, 0.50 mmol) in anhydrous DMF was added iodomethane (12.0 mL, excess, in portions) and stirred at 45 °C for a period of 3.0 d. At the end of quaternization, the product bis[N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)amino]-carbonylpropanoxymalonate deca(quaternary methyl ammonium iodide) salt 5, , was obtained in 92% yield (1.20 g) after removing the remaining iodomethane on rotary evaporator, followed by precipitation using diethyl ether as the solvent. Spectroscopic data: FT-IR (KBr) νmax 3440 (vs, water peak), 2968 (vs), 2936 (vs), 2877 (s), 1732 (s), 1660 (vs), 1591 (s), 1538 (m), 1456 (vs), 1370 (m), 1315 (w), 1227 (m), 1174 (w), 1096 (m), 1053 (s), 1026 (s), 1006 (s), 947 (m), 878 (w), 823 (m), 759 (s), 659 (m), and 583 (m) cm−1; 1H NMR (500 MHz, DMSO-d6, ppm) δ 3.88–4.50 (m, br, 36H), 3.09–3.67 (m, br, 34H), 2.23 (m, 4H), 1.63–1.96 (m, br, 24H), and 0.97 (m, 36H); Anal. Calcd for C82.7H218.1N14O15I9.7 as C67H140N12O6·(CH3I)9.7·7H2O·2DMF (based on 97% quaternization on average): C, 34.74; H, 6.95; N, 6.86; I, 43.06; O, 8.39%. Found: C, 34.35; H, 6.34; N, 7.39; I, 43.69; O, 8.23% (by difference).
4.6. Synthesis of bis(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaazatricosan-23-yl)[60]fullerenyl malonate deca(quaternary ammonium iodide) salt, [1-(I−)10]
Finely divided [60]fullerene (0.50 g, 0.69 mmol, excess) was taken into a round bottom flask and added anhydrous toluene (500 mL) under nitrogen. The solution was stirred at ambient temperature for 12 h to ensure complete dissolution of C60. To the resulting purple-colored solution was added CBr4 (0.20 g, 0.59 mmol) followed by a solution of (5, 1.2 g, 0.46 mmol) in anhydrous DMF (100 mL). The solution mixture was stirred for an additional 30 min and slowly added 1.8-diazabicyclo[5.4.0]-undec-7-ene (DBU, 0.20 g, 1.3 mmol) over a period of 15 min. The color of solution slowly turned into brown in a reaction period of 10 h. The solution was then concentrated on rotary evaporator to roughly 30 mL. Upon the addition of methanol to this concentrated solution, the crude product was precipitated as brown solids which were collected via centrifugation. Unreacted C60 in the crude solids was removed by repeatedly washing with toluene until the washing solution became colorless. The remaining product of bis(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaazatricosan-23-yl)[60]fullerenyl malonate deca(quaternary ammonium iodide) salt 1-(I−)10, , was obtained as a brown solid in 65.0% yield (1.0 g). Spectroscopic data: MALDI-TOF-MS (sinapic acid as the matrix, rel. intensity) m/z 703 (50%), 721 (C60H+, 100%), 759 (20%), 928 (40%), 946 (20%), 1595, 1648, 1682 [a group of 4 peaks, 8%, M+ − 10I − 8(CH2CH2CH3) − 3CH3 − 3H2, consistent with a mass of fully decationized 1],1839,1989, 2039, 2152 (10%), 2359 (10%), 2468, 2626, 2794, 2838, 2852, and 3220 (M+− I−); ESI-MS (rel. intensity) m/z 630, 720 (C60), 871 [ , 100%], 872, 885, 942, 949, 984, 985, 1043 (30%), 1045, 1067, 1114, 1208, 1210, 1282, 1570 [a group of 6 peaks, 8%, M+− 10I − 8(CH2CH2CH3) − 4HCH3 − HN(CH2CH2CH3)2, consistent with further fragmentations from the peak of m/z 1760], 1587, 1591, 1608, 1623, 1625, 1741, 1742 (3%), and 1760 [a group of 7 peaks, 2%, MH+− 10I − 6(CH2CH2CH3) − 4CH3, consistent with a mass of fully decationized 1 giving the evidence of a C60 malonate monoadduct with two 4-hydroxy-hexa(aminoethyl)butanamide main chains]; FT-IR (KBr) νmax 3427 (vs, water peak), 2964 (s, –C–H stretching), 2928 (s, –C–H stretching), 2874 (m), 2850 (m), 1752 (w, malonyl ester –C=O), 1711 (w), 1650 (vs, amide carbonyl –C=O), 1457 (vs), 1373 (m), 1320 (w), 1182 (m), 1102 (w), 1080 (m), 1032 (s), 947 (m), 756 (w), 727 (m), 708 (w), 693 (w), 578 (m), and 526 (vs, a charac teristic band of C60 monoadduct) cm−1; UV–vis (DMF, cutoff at 268 nm, 1.0 × 10−5 M) λmax 323 nm (shoulder peak, ε = 3.58 × 107 cm2/mol); 1H NMR (500 MHz, DMSO-d6, ppm) δ 3.82– 4.43 [m, br, 36H, carboxylated methylene protons –C(=O)–O–CH2– and methylated quaternary ammonium ethylene protons –N+–CH2–], 3.10–3.69 [m, br, 32H, methylated quaternary ammonium ethylene protons –N+–CH2– and –C(=O)–NH–CH2–], 2.23 [m, 4H, –(C=O)–CH2–CH2–CH2–O–], 1.58–1.90 (m, 24H, CH3–CH2– CH2–N–), and 0.97 (m, 36H, CH3–CH2–CH2–N–). Elemental analysis of 1-(I−)10 giving C, 54.08; H, 4.53; N, 4.24; I, 29.14% indicated a measured I−/Nq (quaternary amine) ratio of 0.92 as the same degree of quaternization on average.
4.7. Synthesis of di(tert-butyl)[60]fullerenyl malonate, C60[>M(t-Bu)2] (6)
Finely divided [60]fullerene (2.0 g, 2.8 mmol) was taken into a round bottom flask and added anhydrous toluene (1.4 L) under nitrogen. The solution was stirred for 12 h at ambient temperature to ensure complete dissolution of C60. To the resulting purple-colored solution was added carbon tetrabromide (0.56 g, 1.68 mmol) followed by a solution of di-tert-butyl malonate (0.3 g, 1.38 mmol) in anhydrous toluene (100 mL). The solution was stirred for an additional 30 min and slowly added 1.8-diazabicyclo[5.4.0]-undec-7-ene (DBU, 0.52 g, 3.4 mmol) over a period of 15 min. The color of solution slowly turned into brown in a reaction period of 8.0 h. The solution was then concentrated on rotary evaporator to roughly 50 mL. Upon the addition of methanol to this concentrated solution, the crude product was precipitated as brown solids which were collected via centrifugation. Purification of di(tert-butyl)[60] fullerenyl malonate, C60[>M(t-Bu)2] (6) was made by column chromatography (silica gel using toluene–hexane (1:1, v/v) as the eluant) to afford brown solids in a yield of 62.0% (0.8 g). Spectroscopic data: MALDI-TOF-MS (sinapic acid as the matrix, rel. intensity) m/z 721 (C60H+, 70%), 734 ( , 100%), 747 ( , 37%), 763 (10%), 780 (90%), 794 (21%), 805 [C60C(CO)(CO2H)+, 24%], 824 (20%), 919 (12%), 921 (11%), 935 (MH+, 5%), and 937 (7%); FT-IR (KBr) νmax 3429 (vs, water peak), 2976 (m, –C–H stretching), 2924 (m, –C–H stretching), 2849 (m), 1741 (vs, malonyl ester –C=O), 1633 (m), 1454 (s, antisymmetric deformations of –CH3), 1427 (m), 1392 (m), 1367 (s, symmetric deformations of CH3), 1273 (vs), 1253 [vs, asymmetric stretching of –C–C(=O)–O–], 1155 [vs, –C–O-t-Bu stretching and C(C60)–C–C(=O)– deformations], 1113 (m), 1059 (w),1031 (w), 846 (m), 805 (w), 733 (m), 703 (m), 578 (m), 552 (m), and 526 (vs, a characteristic band of C60 monoadduct) cm−1; UV–vis (CHCl3, cutoff at 245 nm, 1.0 × 10−5 M) λmax 257 (ε = 1.18 × 108) and 323 (shoulder band, ε = 3.43 × 107 cm2/mol) nm; 1H NMR (500 MHz, CDCl3, ppm) δ 1.70 (s, 18H); 13C NMR (500 MHz, CDCl3–CS2, ppm) δ 162.31 (2C, –O–(C=O)–), 145.82 (4C), 145.39 (4C), 145.29 (4C), 145.21 (4C), 144.85 (4C), 144.75 (4C), 144.59 (4C), 143.96 (4C), 143.20, 143.18, 143.08 (4C), 143.06 (4C), 142.33 (4C), 141.99 (4C), 140.98 (4C), 139.03 (4C), 84.45 (2C), 72.29 (2C), 54.19, and 28.17 (6C), consistent with the structure of 6 in a C2v molecular symmetry.
4.8. Synthesis of bis(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaaza-tricosan-23-yl)[60]fullerenyl malonate deca(protonated quaternary ammonium trifluoroacetate) salt, (7)
To a solution of C60[>M(t-Bu)2] (6, 0.15 g, 0.16 mmol) in 1,2-dichloroethane was added C3N6C3–OH (3, 0.23 g, 0.40 mmol) and trifluoroacetic acid (1.0 g, 8.8 mmol). The solution was heated under reflux for a period of 3.0 d until no C60[>M(t-Bu)2] was detected on an analytical TLC plate (SiO2 with toluene as the eluant). Upon completion of the reaction, the mixture was washed by ice-cold aqueous potassium carbonate and water briefly, dried over sodium sulfate, and followed by solvent evaporation to give brown solids. An excess of C3N6C3–OH was removed by several ethyl acetate washings. The product was then quaternized by the addition of trifluoroacetic acid to afford bis(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaaza-tricosan-23-yl)[60]fullerenyl malonate deca(protonated quaternary ammonium trifluoroacetate) salt, (7) in a yield of 65% (0.32 g). Spectroscopic data: FT-IR (KBr) νmax 3436 (vs), 2957 (s, –C–H stretching), 2927 (s, –C–H stretching), 2868 (m), 2843 (m), 2809 (m), 1714 [m, malonyl ester carbonyl –O–(C=O)–], 1621 (vs, amide carbonyl –C=O), 1523 (w), 1456 (s, antisymmetric deformations of –CH3), 1428 (m), 1361 (m, symmetric deformations of CH3), 1322 (w), 1184 (m), 1152 [m, C(C60)–C–C(=O)– deformations and –C–O–C– stretching], 1046 (m), 740 (s), 706 (s), 573 (m), and 526 (vs, a characteristic band of C60 monoadduct) cm−1; UV–vis (DMF, cutoff at 268 nm, 1.0 × 10−5 M) λmax 323 nm (shoulder peak, ε = 4.10 × 107 cm2/mol); 1H NMR (500 MHz, CDCl3–CS2–DMF-d7, ppm) δ 3.32–4.25 [m, br, 60H, carboxylated methylene protons –C(=O)–O–CH2– and protonated quaternary ammonium ethylene protons –N+–CH2–], 3.20–3.11 [m, br, 8H, including –C(=O)–NH–CH2–], 2.34 (m, 4H, –CO–CH2–CH2–CH2–O–), 1.68 (m, 24H, CH3–CH2–CH2–N–), and 0.88 (m, 36H, CH3–CH2–CH2–N–); 13C NMR (500 MHz, CDCl3–CS2–DMF-d7, ppm) δ 166.54 (2C, amide carbonyl –NH–C= O–), 164.84 (2C, ester carbonyl –O–C=O–), 148.02 (2C), 145.82 (2C), 144.85 (2C), 144.39 (2C), 144.31 (2C), 144.17 (2C), 144.13 (2C), 144.08, 143.77, 143.69 (2C), 143.67 (2C), 143.58 (2C), 143.49 (2C), 143.31 (2C), 142.99 (2C), 142.91, 142.76 (2C), 142.28 (2C), 142.15 (4C), 141.99 (2C), 141.96 (2C), 141.85 (2C), 141.58 (2C), 141.26 (2C), 141.16 (2C), 140.10, 139.83 (2C), 139.51 (2C), 136.77 (2C), and 135.49 (2C) for the fullerenyl sp2 carbons, indicating a C2-symmetry of the fullerene cage. Signals of aliphatic carbons of the polycationic arm moiety were low due to high incompatibility of this moiety under the current solvent mixture used.
4.9. Synthesis of decacationic bis(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaaza-tricosan-23-yl)[60]fullerenyl malonate deca(methyl quaternary ammonium iodide) salt, [1-(I−)10]
A suspension solution of 7 (0.20 g, 0.065 mmol) in chloroform (20 mL) was added aqueous potassium carbonate (10 mL) and subsequently ultrasonicated for a period of 1.0 h. The organic layer was collected and dried over sodium sulfate. To this brown solution was added anhydrous DMF (20 mL) and iodomethane (12 mL, excess, addition in portions over the full reaction period). The reaction mixture was kept at 45 °C for 3.0 d. At the end of reaction, excessive iodomethane and solvent were removed via evaporation to give the product in a yield of 92% (0.20 g). Spectroscopic data: the same as those shown in 4.6.
4.10. Synthesis of decacationic bis(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaaza-tricosan-23-yl)[60]fullerenyl malonate deca(methyl quaternary ammonium trifluoroacetate) salt,
Ion-exchange reaction of the compound (30 mg, 0.0090 mmol) was carried out in DMF solution by the addition of an excess quantity of aqueous CF3COONa. The mixture was heated at 50 °C and sonicated for a period of 1.0 h. The product was precipitated from the solution upon the addition of methanol. The solids were washed by methanol and diethyl ether. The resulting solids were subjected to a repeated ion-exchange procedure and followed by drying under vacuum to afford the corresponding deca(methyl quaternary ammonium tri-fluoroacetate) salt in a nearly quantitative yield. Spectroscopic data: FT-IR (KBr) νmax 3448 (vs, water peak), 2970 (s, –C–H stretching), 2933 (s, –C–H stretching), 2877 (m), 2846 (m), 1728 (s, malonyl ester carbonyl – C=O), 1647 (vs, amide carbonyl –C=O), 1457 (vs), 1393 (w), 1368 (m), 1251 (w), 1213 (m), 1186 (m), 1154 (vs), 1111 (s), 1090 (s), 1019 (m), 944 (m), 752 (m), 613 (m), 576 (m), and 526 (vs, a characteristic band of C60 monoadduct) cm−1; 1H NMR (500 MHz, CDCl3–CS2– DMSO-d6, ppm) δ 4.09–4.39 [m, br, 36H, carboxylated methylene protons –C(=O)–O–CH2– and methylated quaternary ammonium ethylene protons –N+–CH2–], 2.86–3.50 [m, br, 32H, methylated quaternary ammonium ethylene protons –N+–CH2– and –C(= O)–NH–CH2–], 2.12 (m, 4H, –CO–CH2–CH2–CH2–O–), 1.45–1.95 (m, 24H, CH3–CH2–CH2–N–), and 0.74–1.11 (m, 36H, CH3–CH2– CH2–N–); 13C NMR (500 MHz, CDCl3–CS2–DMSO-d6, ppm) δ 168.33 (2C, –NH–C=O–), 165.30 (2C, –O–C=O–),147.12 (2C), 144.40 (2C), 143.88, 143.84 (2C), 143.72 (2C), 143.70, 143.64 (2C), 143.31 (2C), 143.26 (2C), 143.17 (2C), 143.06 (2C), 142.68, 142.38 (2C), 142.25 (2C), 141.92, 141.63 (4C), 141.51 (2C), 141.48 (2C), 140.73 (2C), 139.55 (2C), 139.41 (2C), 139.19 (2C), 138.80 (2C), 138.56 (2C), 138.26 (2C), 137.79 (2C), 137.36 (2C), 136.71 (2C), 136.70 (2C), and 136.61 (2C) for the fullerenyl sp2 carbons, indicating a C2-symmetry of the fullerene cage. Signals of aliphatic carbons of the polycationic arm moiety were low due to high incompatibility of this moiety under the current solvent mixture used.
4.11. Preparation of potassium bis(2,4-dinitrobenzenesulfonyl)-2′,4′,5′,7′-tetrafluorofluorescein-10′ (or 11′)-carboxylate (DNBs-TFFC)
The fluorescent DNBs-TFFC probe was prepared by treating the corresponding acid-phase of fluorescein probe DNBs-TFFA with saturated K2CO3 in DMF, prepared at 80 °C. In a typical procedure, DNBs-TFFA (50 mg, 0.05 mmol) was dissolved in DMF (1.0 mL) and added saturated K2CO3 in DMF (1.0 mL), followed by ultrasonication for a period of 5.0 min. It was dried by removal of DMF to solids. The product of DNBs-TFFC was obtained by quick extraction of solids with cold DMF to yield pale yellow solids in 77% (39 mg) after solvent removal. Spectroscopic data: FT-IR (KBr) νmax 3446 (s), 3108 (m), 3065 (w), 3040 (w), 2960 (m), 2928 (m), 2877 (w), 2849 (w), 1782 (vs), 1628 (m), 1609 (m), 1558 (vs), 1543 (vs), 1490 (vs), 1475 (vs), 1414 (s), 1385 (m), 1350 (s), 1298 (m), 1204 (s), 1158 (m), 1088 (s), 1048 (m), 1012 (s), 894 (w), 859 (w), 835 (w), 820 (w), 802 (w), 752 (m), 737 (m) cm−1. UV–vis (DMF, cutoff at 268 nm, 1.0 × 10−5 M) λmax 285 (ε = 1.98 × 107), 507 (shoulder band, ε = 2.0 × 106), and 544 (ε = 7.06 × 106 cm2/mol) nm; 1H NMR (500 MHz, DMSO-d6, ppm) δ 9.12 (s, 4H), 8.69 (d, 4H), 8.53 (d, 4H), 8.40 (s,1H), 8.27 (s,1H), 8.26 (s,1H), 8.14 (s, 1H), 8.09 (s, 1H), 7.80 (d, 1H), 7.40 (m, 2H), and 7.21 (d, 2H); 13C NMR (500 MHz, DMSO-d6, ppm) δ 176.77, 176.58, 167.96, 166.99, 152.78 (4C), 152.43 (2C), 152.15, 151.92 (2C), 150.44 (2C), 148.91 (4C), 148.41 (2C), 145.84 (2C), 145.69 (2C), 143.79 (2C), 138.73 (2C), 138.60,137.24 (2C),137.16 (2C), 134.59 (2C), 134.40 (2C), 132.96, 132.71, 132.04 (4C), 131.87, 131.66, 129.41 (2C), 128.89 (2C), 127.69, 127.59, 127.53, 127.45, 127.40, 127.14, 122.28 (2C), 122.19 (2C), 120.11 (2C), 120.05 (2C), 79.58, and 79.38, consistent with a mixture of two carboxylate regioisomers (10′-carboxylate and 11′-carboxylate) in a 1:1 ratio.
4.12. Preparation of tetrasodium α,α′-(anthracene-9,10-diyl) bis(methylmalonate) (ABMM)
The fluorescent ABMM probe was prepared by treating α,α′-(anthracene-9,10-diyl)bis(methylmalonic acid) (ABMA, 100 mg, 0.24 mmol), suspended in H2O (5.0 mL), with sodium hydroxide (0.05 g), followed by ultrasonication for a period of 5.0 min to give a clear solution. The tetrasodium salt product was precipitated upon the addition of ethanol. The solids were washed three times with ethanol and dried in vacuo to yield ABMM in 81% (97 mg). Spectroscopic data: FT-IR (KBr) νmax 3446 (vs, water peak), 2951 (w), 2923 (w), 2892 (w), 2844 (w), 1592 (vs), 1423 (m), 1339 (m), 1317 (s), 892 (m), 815 (w), 757 (m), 695 (w), 628 (w), 594 (w), 517 (w) cm−1. UV–vis (H2O,1.0 × 10−5 M) λmax 327 (shoulder band, ε = 9.62 × 105), 344 (ε = 2.54 × 106), 361 (ε = 5.65 × 106), 380 (ε = 9.38 × 106) and 402 (ε = 9.12 × 106 cm2/mol) nm; 1H NMR (500 MHz, D2O, ppm) δ 8.46 (d, 4H), 7.62 (dd, 4H), 4.16 (d, 4H), and 3.56 (t, 2H); 13C NMR (500 MHz, D2O, ppm) δ 179.00 (4C),132.37 (2C),129.40 (4C),125.53 (2C),125.41 (2C), 125.36 (2C), 125.30 (2C), 59.77 (2C), and 28.29 (2C).
4.13. ROS measurements using superoxide radical ( ·)-sensitive fluorescent probes
Fluorometric traces were collected using a PTI QuantaMaster™ 40 Fluorescence Spectrofluorometer. Two light sources were used, including an ultrahigh power white-light LED lamp (Prizmatix, operated at the emission peak maxima centered at 451 and 530 nm with the collimated optical power output of >2000 mW in a diameter of 5.2 cm) and an OmniCure LX400 UV LED spot curing lamp (operated at the emission peak maximum centered at 365 ± 5 nm with the peak irradiation intensity of 3000 mW/cm2 using a focus lens of 6 mm in diameter and the max power output of 320 mW), for the generation of emission spectra at white-light and UVA range, respectively.
Superoxide radical ( ·)-reactive fluorescent probes, potassium bis(2,4-dinitrobenzenesulfonyl)-2′,4′,5′,7′-tetrafluorofluorescein-10′ (or 11′)-carboxylate regioisomers (DNBs-TFFC), were used for the experiment. A typical probe solution was prepared by diluting a stock solution of DNBs-TFFC in DMSO (5.0 mM) by 300 times with phosphate buffered saline (PBS) solution (pH 7.4). A dialysis film with the molecular weight cut-off (MWCO) of 100–500 Da was used to separate the solution of [1, 1.0 mM in H2O– DMSO (19:1)] from the probe solution in H2O kept in a cuvette with stirring during the fluorescent measurement. Only the solution of 1 in the membrane sack was subjected to the LED light exposure at either the excitation wavelength of 365 nm or 400–700 nm (white light). The quantity of · generated was counted in association with its reaction with DNBs-TFFC that resulted in the product of potassium 2′,4′,5′,7′-tetrafluorofluorescein-10′ (or 11′)-carboxylate regioisomers (TFFC) with fluorescence emission at 530 nm upon excitation at 480 nm. The detected fluorescence intensity increase was then correlated to the relative quantity of · produced.
4.14. ROS measurements using singlet oxygen (1O2)-sensitive fluorescent probe
Highly water-soluble tetrasodium α,α′-(anthracene-9,10-diyl) bis(methylmalonate) (ABMM) was used as a fluorescent probe for singlet oxygen trapping. The quantity of 1O2 generated was monitored and counted by the relative fluorescence intensity decrease of ABMM at 428 nm under excitation wavelengths of 350–400 nm. A typical probe solution was prepared by diluting aqueous master solution of ABMM (5.0 mM in deionized water, 4 μL) with 600-folds in volume of PBS solution (pH 7.4, 2.4 mL) in a cuvette (10 × 10 × 45 mm). The solution was added by a pre-defined volume of in H2O–DMSO (1.0 mM), followed by illumination periodically using ultrahigh power white-light LED lamp (Prizmatix, operated at the emission peak maxima centered at 451 and 530 nm with the collimated optical power output of >2000 mW in a diameter of 5.2 cm). Progressive fluorescent spectra were taken on the PTI QuantaMaster™ 40 Fluorescence Spectrofluorometer.
4.15. Bacterial culture
S. aureus 8325-4 and E. coli K12 (both wild type) were obtained from ATCC. Planktonic bacterial cells were cultured in brain-heart infusion (BHI) broth with aeration at 37 °C to stationary phase overnight and refreshed to mid-log growth phase for 2.0 h the next day. Cell numbers were estimated by measuring the optical density (OD) at 600 nm [OD of 0.5 = 10(8) cells/mL].
4.16. Light source applied in in vitro experiments
We used a white (400–700 nm) broad-band light source (Lumacare, Newport Beach, CA) set to deliver a spot of 5.0 cm diameter at an irradiance of 100 mW/cm2. The power was measured with a power meter (Model DMM 199 with 201 Standard head, Coherent, Santa Clara, CA).
4.17. In vitro PDT experiments
Mid-log phase cells were collected through centrifugation (1000 g) for 5 min then suspended in PBS. A cell suspension consisting of 10(8) cells/mL for bacteria was incubated with for 30 min at room temperature in the dark. 500 μL aliquots of cell suspension were transferred to a 48-well plate followed by the addition of fullerene derivative 1-(I−)10 (up to 10 μM for S. aureus and up to 80 μM for E. coli) from a DMA stock solution prepared at 2.0 mM. The highest concentration (for 80 μM fullerene) of DMA used was 4% and this did not cause any toxicity to the cells. Cells were illuminated at room temperature for 16.6 min to deliver 100 J/cm2. At the completion of the illumination period, 100 μL aliquots were removed from illuminated and non-illuminated wells (cells incubated with fullerene but kept in 48-well plates covered with aluminum foil at room temperature for the duration of the illumination were used as control) and serially diluted 10-fold in PBS to give dilutions of 10−1–10−6 times the original concentrations and 10 μL aliquots of each of the dilutions were streaked horizontally on square BHI plates. Plates were streaked in triplicate and incubated for 24 h at 37 °C in the dark to allow colony formation. Controls groups included cells that were not treated with 1-(I−)10 or light, and cells treated with light but not with 1-(I−)10. Survival fractions (SF) were routinely expressed as ratios of CFU of microbial cells treated with light and 1-(I−)10 or treated with 1-(I−)10 in the dark, to CFU of microbes treated with neither.
4.18. Statistics
Values are means of three separate experiments, and bars presented in the graphs are standard errors of the means (SEM). Differences between means were statistically analyzed by one way ANOVA in Microsoft Excel, and p < 0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank the financial support of National Institute of Health (NIH) under the grant number R01 CA137108. We thank Jin Xu of UML for the collection of ESI-MS spectroscopic data.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.2013.01.052.
Contributor Information
Michael R. Hamblin, Email: hamblin@helix.mgh.harvard.edu.
Long Y. Chiang, Email: long_chiang@uml.edu.
References
- 1.Guldi DM, Prato M. Acc Chem Res. 2000;33:695–703. doi: 10.1021/ar990144m. [DOI] [PubMed] [Google Scholar]
- 2.Fujitsuka M, Ito O. In: Encyclopedia of Nanoscience and Nanotechnology. Nalwa HS, editor. Vol. 8. American Scientific Pub; 2004. pp. 593–615. [Google Scholar]
- 3.Yamakoshi Y, Umezawa N, Ryu A, Arakane K, Miyata N, Goda Y, Masumizu T, Nagano T. J Am Chem Soc. 2003;125:12803–12809. doi: 10.1021/ja0355574. [DOI] [PubMed] [Google Scholar]
- 4.Dolmans DE, Fukumura D, Jain RK. Nat Rev Cancer. 2003;3:380–387. doi: 10.1038/nrc1071. [DOI] [PubMed] [Google Scholar]
- 5.Hamblin MR, Hasan T. Photochem Photobiol Sci. 2004;3:436–450. doi: 10.1039/b311900a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Riordan K, Akilov OE, Hasan T. Photodiagnosis Photodynamic Ther. 2005;2:247–262. doi: 10.1016/S1572-1000(05)00099-2. [DOI] [PubMed] [Google Scholar]
- 7.Silva JN, Filipe P, Maziere P, Maziere JC, Freitas JP, Cine de Castro JP, Santus R. Bio-Med Mater Eng. 2006;16:S147–S154. [PubMed] [Google Scholar]
- 8.Sharma SK, Chiang LY, Hamblin MR. Nanomedicine. 2011;6:1813–1825. doi: 10.2217/nnm.11.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Navarre WW, Schneewind O. Microbiol Mol Biol Rev. 1999;63:174–229. doi: 10.1128/mmbr.63.1.174-229.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nikaido H. Microbiol Mol Biol Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scott JR, Barnett TC. Annu Rev Microbiol. 2006;60:397–423. doi: 10.1146/annurev.micro.60.080805.142256. [DOI] [PubMed] [Google Scholar]
- 12.Kell AJ, Stewart G, Ryan S, Peytavi R, Boissinot M, Huletsky A, Bergeron MG, Simard B. ACS Nano. 2008;2:1777–1788. doi: 10.1021/nn700183g. [DOI] [PubMed] [Google Scholar]
- 13.Malik Z, Ladan H, Nitzan Y. J Photochem Photobiol B. 1992;14:262–266. doi: 10.1016/1011-1344(92)85104-3. [DOI] [PubMed] [Google Scholar]
- 14.Hamblin MR, O’Donnell DA, Murthy N, Rajagopalan K, Michaud N, Sherwood ME, Hasan T. J Antimicrob Chemother. 2002;49:941–951. doi: 10.1093/jac/dkf053. [DOI] [PubMed] [Google Scholar]
- 15.Alves E, Costa L, Carvalho CMB, Tome JPC, Faustino MAF, Neves MGPMS, Tome AC, Cavaleiro JAS, Cunha A, Almeida A. BMC Microbiol. 2009;9:70–83. doi: 10.1186/1471-2180-9-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jori G, Coppellotti O. Anti-Infect Agents Med Chem. 2007;6:119–131. [Google Scholar]
- 17.Hamblin MR, O’Donnell DA, Murthy N, Contag CH, Hasan T. Photochem Photobiol. 2002;75:51–57. doi: 10.1562/0031-8655(2002)075<0051:rcowib>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 18.Wang M, Huang L, Sharma SK, Jeon S, Thota S, Sperandio FF, Nayka S, Chang J, Hamblin MR, Chiang LY. J Med Chem. 2012;55:4274–4285. doi: 10.1021/jm3000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thota S, Wang M, Jeon S, Maragani S, Hamblin MR, Chiang LY. Molecules. 2012;17:5225–5243. doi: 10.3390/molecules17055225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Da Ros T, Prato M, Novello F, Maggini M, Banfi E. J Org Chem. 1996;61:9070–9072. doi: 10.1021/jo961522t. [DOI] [PubMed] [Google Scholar]
- 21.Rio Y, Nicoud JF, Rehspringer JL, Nierengarten JF. Tetrahedron Lett. 2000;41:10207–10210. [Google Scholar]
- 22.Zhiguo Z, Schuster DI, Wilson SR. J Org Chem. 2003;68:7612–7617. doi: 10.1021/jo034542l. [DOI] [PubMed] [Google Scholar]
- 23.Bianco A, Da Ros T, Prato M, Toniolo C. J Pept Sci. 2001;7:208–219. doi: 10.1002/psc.313. [DOI] [PubMed] [Google Scholar]
- 24.Cusan C, Da Ros T, Spalluto G, Foley S, Janot JM, Seta P, Larroque C, Tomasini MC, Antonelli T, Ferraro L, Prato M. Eur J Org Chem. 2002;17:2928–2934. [Google Scholar]
- 25.Braun M, Atalick S, Guldi DM, Lanig H, Brettreich M, Burghardt S, Hatzimarinaki M, Ravanelli E, Prato M, van Eldik R, Hirsch AA. Chem Eur J. 2003;9:3867–3875. doi: 10.1002/chem.200204680. [DOI] [PubMed] [Google Scholar]
- 26.Carano M, Ceroni P, Paolucci F, Roffia S, Da Ros T, Prato M, Sluch MI, Pearson C, Petty MC, Bryce MR. J Mater Chem. 2000;10:269–273. [Google Scholar]
- 27.Guldi DM, Huie RH, Neta P, Hungerbuhler H, Asmus KD. Chem Phys Lett. 1994;223:511–516. [Google Scholar]
- 28.Texier I, Berberan-Santos MN, Fedorov A, Brettreich M, Schonberger H, Hirsch A, Leach S, Bensasson RV. J Phys Chem A. 2001;105:10278–10285. [Google Scholar]
- 29.Setsukinai KI, Urano Y, Kakinuma K, Majima HJ, Nagano T. J Biol Chem. 2003;278:3170–3175. doi: 10.1074/jbc.M209264200. [DOI] [PubMed] [Google Scholar]
- 30.Maeda H, Yamamoto K, Nomura Y, Kohno I, Hafsi L, Ueda N, Yoshida S, Fukuda M, Fukuyasu Y, Yamauchi Y, Itoh N. J Am Chem Soc. 2005;127:68–69. doi: 10.1021/ja047018k. [DOI] [PubMed] [Google Scholar]
- 31.Padmawar PA, Rogers JO, He GS, Chiang LY, Canteenwala T, Tan LS, Zheng Q, Lu C, Slagle JE, Danilov E, McLean DG, Fleitz PA, Prasad PN. Chem Mater. 2006;18:4065–4074. [Google Scholar]
- 32.Frimer AA. Singlet O2, Physical–Chemical Aspects. I. CRC Press; Boca Raton, FL: 1985. [Google Scholar]
- 33.Kuznetsova NA, Gretsova NS, Yuzhakova OA, Negrimovskii VM, Kaliya OL, Luk’yanets EA. Russ J Gen Chem. 2001;71:36–41. [Google Scholar]
- 34.Braathen G, Chou PT, Frei H. J Phys Chem. 1988;92:6610–6615. [Google Scholar]
- 35.Gupta AK, Rohatgi-Mukherjee KK. Photochem Photobiol. 1978;27:539–543. [Google Scholar]
- 36.Huang L, Dai T, Hamblin MR. Methods Mol Biol. 2010;635:155–173. doi: 10.1007/978-1-60761-697-9_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kharkwal GB, Sharma SK, Huang YY, Dai T, Hamblin MR. Lasers Surg Med. 2011;43:755–767. doi: 10.1002/lsm.21080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang L, St Denis TG, Xuan Y, Tanaka M, Huang YY, Zadlo A, Sama T, Hamblin MR. Free Radic Biol Med. 2012;53:2062–2071. doi: 10.1016/j.freeradbiomed.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Demidova TN, Hamblin MR. Antimicrob Agents Chemother. 2005;49:2329–2335. doi: 10.1128/AAC.49.6.2329-2335.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martinez GR, Garcia F, Catalani LH, Cadet J, Oliveira MCB, Ronsein GE, Miyamoto S, Medeiros MHG, Di Mascio P. Tetrahedron. 2006;62:10762–10770. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.