

Abstract
We conducted controlled crosses in populations of the long-lived clonal shrub, Vaccinium angustifolium (lowbush blueberry) to estimate inbreeding depression and mutation parameters associated with somatic deleterious mutation. Inbreeding depression level was high, with many plants failing to set fruit after self-pollination. We also compared fruit set from autogamous pollinations (pollen collected from within the same inflorescence) with fruit set from geitonogamous pollinations (pollen collected from the same plant but from inflorescences separated by several meters of branch growth). The difference between geitonogamous versus autogamous fitness within single plants is referred to as ‘autogamy depression' (AD). AD can be caused by somatic deleterious mutation. AD was significantly different from zero for fruit set. We developed a maximum-likelihood procedure to estimate somatic mutation parameters from AD, and applied it to geitonogamous and autogamous fruit set data from this experiment. We infer that, on average, approximately three sublethal, partially dominant somatic mutations exist within the crowns of the plants studied. We conclude that somatic mutation in this woody plant results in an overall genomic deleterious mutation rate that exceeds the rate measured to date for annual plants. Some implications of this result for evolutionary biology and agriculture are discussed.
Keywords: somatic mutation, deleterious mutation, inbreeding depression, long-lived plants
Introduction
Plants violate the Weismann's doctrine of the separation of germ line and soma (Weismann, 1893). Although in theory, this means that mutations can be passed from the soma to gametes, it is unclear how often mutations occur in somatic tissue during plant development, and whether and by how much intra-organismal selection filters out cells bearing such mutations before they are passed onto gametes (Otto and Orive, 1995). Somatic mutation and somatic selection are postulated to have important roles in plant evolution through their influence on total mutational load and plant defense (Whitham and Slobodchikoff, 1981; Antolin and Strobeck, 1985; Klekowski, 1988; Gill et al., 1995; Otto and Hastings, 1998). Despite frequent discussion of somatic mutation and selection in the plant evolutionary literature, published measurements of the magnitude of these processes are virtually nonexistent. This lack of information stems in part from the difficulty inherent in studying mutation and selection within the soma. It is also attributable to the fact that genomic mutation is an infrequent process, resulting in individual mutations whose effects are typically small and difficult to detect in reasonably sized experiments. Such obstacles are familiar to those who have attempted to estimate mutation rates from phenotypic data in mutation accumulation experiments (Drake et al., 1998).
In addition to the difficulties noted above, questions remain about the number of somatic cell divisions that arise between formation of the zygote and establishment of the plant germ line, an issue that is difficult to resolve without detailed knowledge of how plant meristems divide to produce additional stem cells and somatic tissue (Klekowski et al., 1985; Pineda-Krch and Lehtila, 2002). In some plant species, it has been estimated that there are fewer than a hundred somatic cell divisions, separating zygote formation from gamete formation, whereas, in others, there may be many thousands of such divisions. The actual number is difficult to determine with accuracy (Otto and Walbot, 1990; Cloutier et al., 2003; Scofield, 2006). In the case of long-lived plants, it has been argued that because of the large numbers of somatic cell divisions separating zygote from gamete formation, significant numbers of somatic mutations in theory can accumulate within the cell lineages that eventually differentiate into gametogenic tissue, and that this, in turn, should lead to a higher overall per-generation rate of mutation than observed in annual plants (Klekowski, 1988). For instance, Klekowski and Godfrey (1989) estimated that rates of chlorophyll deficiency-causing mutation per generation are 25 times higher in mangrove trees than in the annual plants barley and buckwheat.
The focus of this paper is on the rate of deleterious somatic mutation. Deleterious mutations may influence patterns of evolution and are also of concern in conservation biology (Kondrashov, 1988; Lynch and Gabriel, 1990; Schoen et al., 1998). For instance, deleterious somatic mutation may have a central role in mating system evolution. As a group, trees and shrubs are known to exhibit a higher rate of outcrossing than annual plants (Barrett et al., 1996). Scofield and Schultz (2006) suggested that this correlation of mating system and life history reflects evolved responses of the mating system to inbreeding depression levels. Specifically, they proposed that higher per-generation deleterious mutation rates arising as a consequence of somatic mutation accumulation in long-lived plants should lead to higher equilibrium levels of inbreeding depression compared with those in annual plants, and that this has favored the evolution of reproductive traits that enforce outcrossing (for example, self-incompatibility, dioecy). Data from the literature on the relationship between inbreeding depression levels and selfing rates support this idea. The hypothesis is further supported by models of mating system modifier selection in annual and perennial plants, that have shown that high rates of mitotic mutation promote the evolution of outcrossing in perennials (Morgan, 2001).
Rates of somatic mutation have been estimated by direct assay of marker locus genotypes in long-lived plants (for example, Cloutier et al., 2003; O'Connell and Ritland, 2004), but the fitness consequences of marker mutations are difficult to determine. In contrast, a more direct approach involves assaying the phenotypic effects of somatic mutation by measuring fitness of progeny (or gametophytes) produced by different portions of the plant (Schultz and Scofield, 2009). For instance, in a detailed study of the consequences of aging in the clonally spreading tree species Populus tremuloides, Ally et al. (2010) found that pollen grain viability decreased with plant age, an effect that they attributed to somatic mutation accumulation, though this hypothesis was not directly tested.
Schultz and Scofield (2009) formalized methods for estimating the contribution of somatic mutation accumulation to the overall per-generation rate of deleterious mutation. One method, the so-called ‘autogamy depression (AD) test', compares the fitness of progeny derived by within-flower self-fertilization to that derived by between-flower self-fertilization (that is, progeny fitness following autogamy versus geitonogamy). AD can be defined as 1−(wa/wg), where wa is the fitness of progeny derived via autogamy and wg is the fitness of progeny derived via geitonogamy (in parallel with the definition of inbreeding depression), or alternatively, as D=wg−wa, the definition we use in the present study. AD is a direct outcome of the somatic mutational process, given that certain conditions hold. First, in diploid organisms it is expected that three-quarters of progeny produced via autogamous pollination will inherit a de novo somatic mutation one-half the time as heterozygotes and one-quarter the time as homozygotes (Schultz and Scofield, 2009), whereas, progeny produced via geitonogamy will only bear such mutations in a heterozygous state. Second, if the inherited somatic mutations are not expressed in the tissues of the crown that differentiate into meiocytes, then somatic selection will be ineffective at removing mutations from the cell lineages that give rise to gametes. Third, if the somatic mutations are deleterious and recessive (or partially recessive), and expressed in the progeny, then AD should become manifest.
Although appropriate experimental designs to assess rates of somatic mutation in plants have now been described (Schultz and Scofield, 2009), there have been no studies published to date that employ these methods to estimate somatic mutation parameters. In this study, we present measurements of inbreeding depression obtained from the clonal woody plant, Vaccinium. angustifolium (lowbush blueberry). The pollinations and fitness assays conducted were first used to assess overall levels of early-acting inbreeding depression. In a separate experiment, we applied pollination treatments required to obtain measurements of AD. We derive a maximum-likelihood approach for estimating somatic mutation parameters from AD data, and use the data obtained from experiments with V. angustifolium to estimate somatic mutation parameters.
Materials and methods
Target species and study site
V. angustifolium is a clonal woody plant that spreads through a system of underground rhizomes. Separate genotypes within the same population often differ in leaf morphology as well as leaf and flower color, and clones can often be distinguished from one another in the field. The work reported here was conducted in commercial blueberry fields in the region around Neguac, New Brunswick, Canada. V. angustifolium occurs as an understory plant in the forests of the region, and commercial blueberry fields are established by clearing the forest and encouraging growth of the blueberry plants. The blueberry clones in these fields were established from naturally occurring plants over 100 years ago, and many clones are large, covering more than 20 m2 in surface area.
Hokanson and Hancock (1993) studied inheritance in V. angustifolium. On the basis of several crosses, they were unable to distinguish with certainty between tetrasomic and disomic modes of inheritance. Accordingly, we base our conclusions on the possibility that either mode of inheritance could apply to this species.
Estimation of early-acting inbreeding depression
Pollinations were carried out in the summer of 2011 to determine the level of early-acting inbreeding depression in lowbush blueberry plants. Twenty-four blueberry fields were selected. Twelve blueberry clones per field were selected at 5 m intervals along 60 m transects. Inflorescences used for pollination were identified and labeled when flowers were in bud. Pollinations were conducted with inflorescences that were bagged prior to anthesis and kept in bags until flowers senesced. In inflorescences selected for autogamous pollination, pollen was collected into sterile tubes and then applied to stigmas with a fine paintbrush, cleaned with 70% ethanol between pollinations. Outcrossed pollinations were conducted using a similar procedure, but with pollen from clones located at least 5 m away. Flower counts and pollinations were conducted a minimum of three times per inflorescence during the flowering season.
It has been previously shown that the closely related blueberry species, V. corymbosum (highbush blueberry), exhibits early-acting inbreeding depression, manifested as a high rate of fruit abortion following selfing. We therefore used percent fruit set per inflorescence as the fitness measure for estimating inbreeding depression. Percent fruit set was calculated by counting the number of flowers pollinated per inflorescence and comparing that value to the number of mature berries later in the season. Inbreeding depression (∂) was calculated using the formula ∂=1−(Ws/Wo) (CharlesworthCharlesworth and Charlesworth, 1987), where Ws represents percent fruit set in self-pollinated inflorescences and Wo represents percent fruit in outcross-pollinated inflorescences.
Clone identification for the AD test
In 2012, separate (putative) clones from one field were selected for the pollination treatments. These clones ranged in size from 9 to 30 m2 in surface area. For each clone, 20 paired autogamous and geitonogamous pollinations (Figure 1) were conducted. The physical limits of each putative clone were initially determined from observations on homogeneity of growth form (upright or spreading branches), leaf pigmentation (green or red-purple, anthocyanin-rich) and timing of leaf development (early in season or late), together with the presence of bare ground or other species of plants between blueberry stems. Each putative clone was then genotyped at two highly polymorphic microsatellite loci by sampling leaf tissue from four widely spaced edges of the clone. Leaf samples subtended the infloresences used in the controlled pollinations.
Figure 1.

Experimental design. Shown is a diagram of a single clone. An oval represents a pair of inflorescences located close-by near the tips of a branch. One inflorescence was pollinated with pollen from that same inflorescence pair (=autogamy treatment), whereas the other inflorescence was pollinated with pollen from one of the inflorescences located on the opposite side of the clone (=geitonogamy treatment). This set of autogamous and geitonogamous pollination treatments was replicated 20 times within each clone. Also shown are the positions where Uc ‘crown mutations' are assumed to occur with respect to the position of the last common meristematic cell shared by the cell lineages that produced the flowers used in the geitonogamy treatment.
Genomic DNA was extracted separately from each leaf sample using a QIAcube robot and spin column kits (DNAeasy Plant Mini prep) with the appropriate preloaded standard plant DNA extraction protocol (Qiagen, Valencia, CA, USA). The two variable microsatellite loci used (CA 344F and CA 794F) are described in Boches et al. (2005) and primer sequences also are listed in that publication. PCR reactions were carried out in a final volume of 10 μl. The reaction mixture consisted of 2 μl of DNA solution, 5 U μl−1 of Taq DNA polymerase, 1 μl of 10 × PCR buffer, 10 mM of dNTP mix, 2.5 mM MgCl2, 2 μM M13 Forward primer, 2 μM M13 5′-IRDye-labeled forward primer (at 700 nm). The cycling program consisted of 300 s of initial denaturation at 94 °C and 25 cycles of 95 °C for 40 s, 61 °C for 40 s and 72 °C for 40 s. This was followed by 15 cycles of 94 °C for 40 s, 53 °C for 40 s and 72 °C for 40 s, with a final single cycle of 71 °C for 600 s. Microsatellite genotypes were determined using the LI-COR NEN Model 4300 DNA analyzer and the SAGA software package (LI-COR Biosciences, Lincoln, NE, USA).
Allelic richness at the microsatellite loci was high, and each of the 20 putative clones selected possessed a multilocus genotype that was unique to the sample. The initial assignment of ramets, as belonging to a specific clone based on morphology and plant spacing alone, was verified in 19 out of the 20 cases by genotypes obtained at the marker loci—that is, all four of the leaf samples collected from each of the 20 putative clones exhibited the same unique and diagnostic multilocus marker genotypes. In the case of the one remaining clone, one out of the four leaf samples was shown to exhibit a multilocus genotype that differed from that of the other three leaf samples. Although this could be due to a somatic microsatellite mutation, we opted to be conservative and not consider data from pollinations conducted with inflorescences in that portion of the clone.
Pollinations for the AD test
Our implementation of the AD test involved pollinations made with inflorescences pairs, either close together on the same branch or widely separated, but within the same clone. The inflorescences used for geitonogamous pollinations were typically separated by several meters of branch growth, and thus could acquire non-shared somatic mutations (Figure 1). For the purpose of the AD test, all within-branch inflorescence pair pollinations were treated as ‘autogamous', whereas all between-branch inflorescence pair pollinations were treated as ‘geitonogamous'. Although this implementation of the AD test does not adhere to the strict botanical definition of autogamy and geitonogamy, it does, nevertheless, lead to contrast between treatments in the expected incidence of homozygosity of somatic mutations, and was the only practical way in which to proceed. We refer below to such a pair of pollinations as an ‘autogamous–geitonogamous' crossing pair.
As in the case of the inbreeding depression measurements, inflorescences were identified, labeled and bagged prior to and during the flowering period. Upon flowering, inflorescences selected for the autogamous pollination treatment were pollinated with pollen collected from a within-branch inflorescence pair, placed in sterile tubes and then applied to stigmas with a fine paintbrush. For geitonogamous pollinations, pollen was collected in sterile tubes from a distantly positioned inflorescence (an inflorescences on an opposite side of the clone, as shown in Figure 1) and then applied to stigmas.
Fitness measures for the AD test
As in the inbreeding depression study, percent fruit set per inflorescence was used as the fitness measure, and so the data obtained pertain only to mutations that influence early survival. To obtain this information, flower counts and pollinations were conducted every third or fourth day until senescence of all flowers occurred (early June). Bags were then removed. To minimize error in estimating percent fruit set due to fruit loss by bird foraging, fruit set counts were obtained when the fruits were green, but fully developed in size (early August).
A potential problem with the AD test when fruit set is the fitness component is that fruits may fail to set for reasons not related to inbreeding depression. For example, problems with animal predation or the transfer of pollen could result in failed fruit set. This could result in negative AD. We lost about half of our pollinated infloresences due to damage from animal predation, and discarded <10% of undamaged inflorescences due to problems we encountered in transferring pollen between flowers.
Maximum-likelihood estimation of mutation parameters
Somatic deleterious mutations influence both the mean and the variance of fitness of progeny produced via autogamy and geitonogamy. Together the fitness difference (the measured AD) along with the (co-) variances in autogamous and geitonogamous fitnesses can provide the information required to estimate mutation parameters, paralleling the classic Bateman–Mukai approach to estimating mutation parameters (Mukai, 1964; Schultz and Scofield, 2009). Alternatively, a maximum-likelihood-based approach may be applied to estimate mutation parameters (Keightley, 1994), particularly when mutation rates are large and approximations based on observed change in fitness and fitness variance breaks down—conditions that hold for the data collected in this study.
We derived the likelihood function following the method of Keightley (1994). For simplicity, we assume diploid inheritance. If inheritance in V. angustifolium is actually tetrasomic rather than disomic, violation of this assumption would lead to an underestimate of the somatic mutation rate, as the probability of autozygosity under tetrasomic inheritance is lower (Haldane, 1930). This is acceptable because our aim is to provide a conservative estimate of the somatic mutation rate.
Let Dk denote the measured fitness difference between progeny produced in the kth autogamous–geitonogamous crossing pair. We assume that there are Ub deleterious ‘background' mutations present in a heterozygous state in the zygoyte that gave rise to the plants where autogamously and geitonogamously produced progeny were obtained. In addition, we assume that there are Uc deleterious ‘crown' mutations have occurred in the independent branches of this plant that bear the flowers used in the geitonogamous crosses, and that arose since these branches last shared a common meristematic cell in the crown (Figure 1). The rate of crown mutation is assumed to be Poisson distributed with mean λ=Uc per branch. We note that it is also possible to formulate the model below using λ=Uc per unit length of branch growth (rather than per branch), but we have opted not to do so because we lacked precise information on the branch lengths. All mutations are assumed to have a constant additive effect on progeny fitness denoted by s, and to be expressed in the progeny with dominance coefficient h. Moreover, we assume that these newly arising mutations do not experience somatic selection. Thus, our estimates apply to the class of mutations with fitness effects that are not expressed during branch growth. Accordingly, we have:
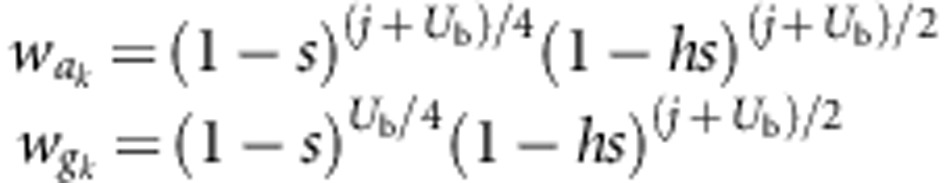 |
where 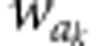 and
and 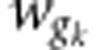 denote autogamous and
geitonogamous fitnesses from the kth crossing pair, respectively, and
j is a Poisson deviate with parameter
λ=Uc. We assume that environmental effects
influencing autogamous and geitonogamous fitness (and their differences) are normally
distributed with variance Ve. Thus, the likelihood associated with
the autogamous fitness and AD measured from the kth
autogamous–geitonogamous pair is:
denote autogamous and
geitonogamous fitnesses from the kth crossing pair, respectively, and
j is a Poisson deviate with parameter
λ=Uc. We assume that environmental effects
influencing autogamous and geitonogamous fitness (and their differences) are normally
distributed with variance Ve. Thus, the likelihood associated with
the autogamous fitness and AD measured from the kth
autogamous–geitonogamous pair is:
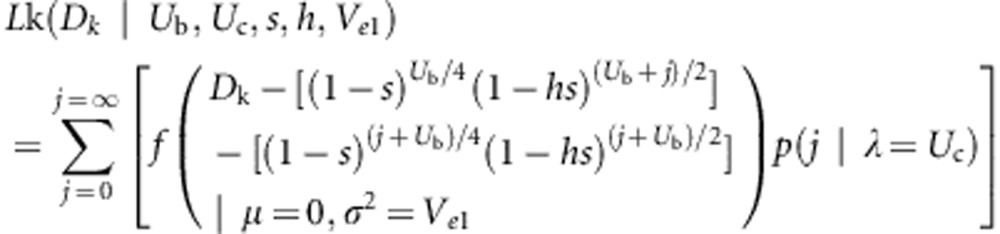 |
where f is the Gaussian probability density function, and p(j) is the Poisson probability that the branch in question has accumulated j new (heterozygous) somatic mutations. Two other likelihoods were also used in estimating the mutation parameters. These are:
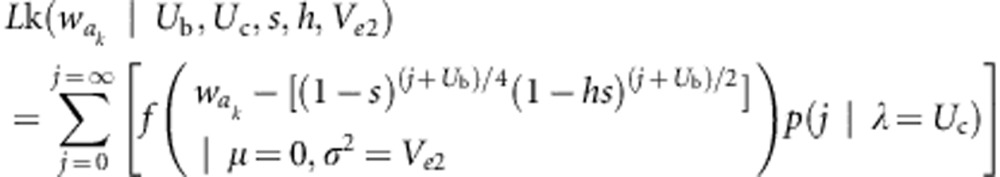 |
and
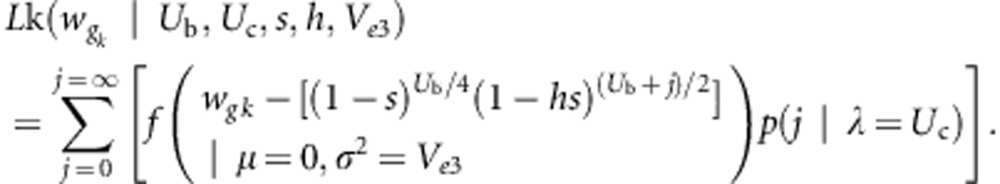 |
Finally, we have the log likelihood of the intersection of each of the three quantities above:
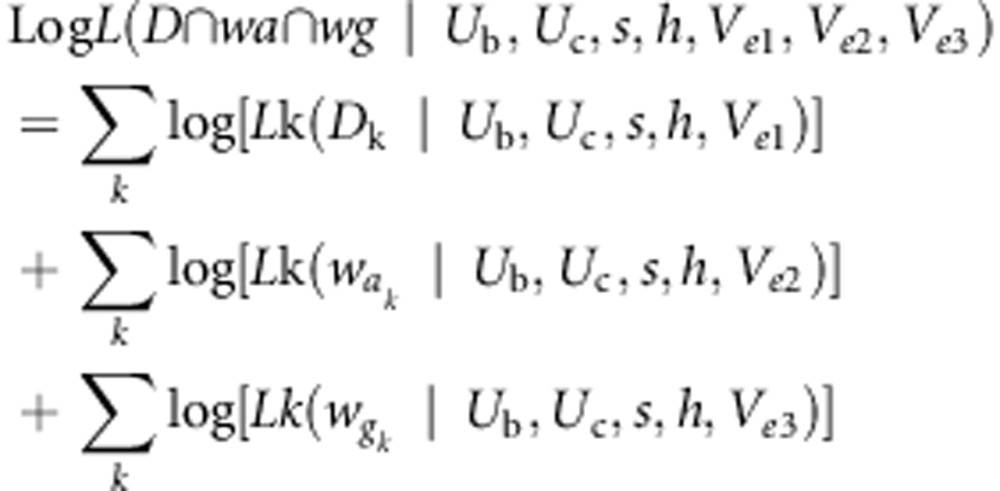 |
A four-step procedure was used to find mutation parameters that maximized the likelihood of the data. First, a random grid search of the parameter space was performed, in which the Nelder–Mead downhill simplex method was applied to 15 000 random starting points satisfying 0<h<0.5, 0<s<1, 0<Uc or Ub<30, and 0<Ve's<0.5 (Nelder and Mead, 1965; Haag-Liautard). The resulting likelihood values were used to find the maximum-likelihood estimates of the environmental variance parameters Ve's to construct the overall loess-interpolated likelihood surface and to construct the profile likelihoods. Profile likelihoods are the projection of the multidimensional likelihood surface into the plane relating a particular mutational parameter to the log maximum likelihood of the surface (Nocedal and Wright, 1999). Profile likelihoods were constructed for Uc, Ub, Uc s, h, hs, and s. Second, the Nelder–Mead method was used to find the maximum-likelihood estimates of Uc, Ub, s and h while holding the environmental variance components fixed at their maximum-likelihood values obtained from the grid search. The standard errors of these estimates were obtained using the likelihood ratio method (Weir, 1996). Third, these results were checked and confirmed by two additional methods, the quasi-Newton or variable metric algorithm developed by Broyden (1970), Fletcher (1970), Fletcher and Reeves (1964), Goldfarb (1970), Shanno and Kettler (1970) and Shanno (1970) (the Broyden-Fletcher-Goldfarb-Shanno algorithm (BFGS)) and an intelligent grid search method (Heim, 2007; R Core Team, 2012), both of which gave results to within less than 1% of the values found by the Nelder–Mead method. Fourth, the Nelder–Mead, BFGS and the intelligent grid search algorithms were each used separately to find the maximum-likelihood estimate of the product of Uc and s, assuming fixed values of h and Ub that differed from their maximum-likelihood estimates to assess the sensitivity to the estimate of Uc s to variation in these parameter values. These methods again gave results that differed from each other by less than one percent. All numerical optimization procedures were executed in R using the functions optim(), mle() and cleversearch() (Heim, 2007; R Core Team, 2012).
In theory, a more refined maximum-likelihood estimation procedure to estimate mutation parameters, one that entails a model with variable as opposed to constant mutation effects, could be obtained by extending estimation along the lines described by Keightley (1994), in which mutational effects are assumed to be gamma distributed (with shape parameter-β, scale parameter-α). But in view of the relatively modest-sized data set and the lower statistical power available for estimating additional parameters, we have opted not to do so here.
Results
Inbreeding and AD
The majority of the selfed plants showed low fruit set (mean=0.13±0.16) compared with the outcrossing treatment (mean=0.53±0.24). A Wilcoxon-signed rank test showed that the difference was highly significant (P<0.0001). Consequently, inbreeding depression level is high in the populations studied with many values near 1 (Figure 2). Assuming diploid inheritance and based on the approach by Morton et al. (1956), the estimate of the number of lethal equivalents per zygote (2B) is 2B=−4 ln(Ws/Wo)=2.41. Assuming tetrasomic inheritance, in the absence of double reduction, autozygosity is approached at approximately one-third the rate compared with diploidy (Haldane, 1930). This leads to an estimate of numbers of lethal equivalents per zygote (based on tetrasomy) of 2B=−12 ln(Ws/Wo)=7.22.
Figure 2.

Distribution of inbreeding depression (delta) estimated from self and outcross fruit set in 288 plants across 24 field populations of V. angustifolium.
AD was found to be approximately normally distributed with a mean value of 0.155 (95% confidence intervals=0.106–0.204), and significantly different from 0 by a one-sample t-test (t=6.218, df=185, P<0.0025) (Figure 3).
Figure 3.

Distribution of AD (D=Wg−Wa) estimated from self- and outcross fruit set in 186 autogamous–geitonogamous cross pairs from 20 plants.
Mutation parameter estimates
The maximum-likelihood profile surface of Uc against s showed a prominent likelihood ridge corresponding to the product Ucs∼2–3, suggesting strong support for 2–3 lethal equivalent mutations affecting fruit set within the crown of our study plants (Figure 4a). This surface also showed very pronounced peaks for the individual mutation parameters, yielding strong maximum-likelihood estimates of Uc=3.1 (s.e.=0.1) crown mutations, homozygous effect s=0.86 (s.e.=0.01) and dominance h=0.43 (s.e.=0.02) within the fixed-effects mutational model (Figures 4a and c). The number of background mutations affecting fruit set was estimated to be near zero, with maximum-likelihood estimate by the Nelder–Mead method yielding Ub=0.005 (s.e.=0.02). The sensitivity of the product Ucs to variation in Ub and h is moderate, with a minimum value of ∼2 for partially dominant canopy mutations, regardless of the number of background mutations up to 3 (Figure 4d).
Figure 4.

Maximum-likelihood profiles. (a) Loess-interpolated maximum-likelihood surface of Uc and s from Nelder–Mead-assisted, random grid search. (b) Likelihood profiles for number of canopy mutations Uc, number of background mutations Ub, and the product of Uc and s, the homozygous loss in fitness. (c) Likelihood profiles of the homozygous loss in fitness s, the dominance coefficient h, and hs, the heterozygous loss in fitness. (d) The maximum-likelihood estimate of the product of Uc and s for the indicated values of h and Ub.
Discussion
Inbreeding depression
Inbreeding depression estimates in the populations of V. angustifolium studied here are high, as reported by Bell et al. (2010) for populations of this species located in the northeastern United States. Prior studies with this species suggest that this result is not due to a system of self-pollen rejection (self-incompatibility). For instance, to distinguish self-incompatibility from early-acting inbreeding depression in V. angustifolium, Hokanson and Hancock (2000) emasculated flowers and then compared fruit set after several pollination treatments, including: (1) application of self-pollen to stigmas on day 0, (2) application of cross-pollen to stigmas on day 0, (3) application of cross-pollen to stigmas on day 3, (4) application of self-pollen to stigmas on day 0 followed by cross-pollen on day 1 and (5) application of self-pollen to stigmas on day 0 followed by cross-pollen on day 3. The latter two treatments produced statistically significantly greater seed per fruit than either the selfed treatment alone or the self followed by cross-pollen on day 3, suggesting that low self-fertility is not due to self-incompatibility.
AD and somatic mutation parameter estimates
The somatic mutation parameter estimation conducted here suggests that several mutations arise during growth of the crown. These are mutations of large effect (s⩾0.8) and partially dominant (h⩾0.3). It is notable that this large value of Uc results in a total genomic deleterious mutation rate that exceeds that measured for most annual plants by up to an order of magnitude (Baer et al., 2007). The actual discrepancy could be greater than this, as we examined only the effect of early-acting mutations, whereas studies of deleterious mutation in annuals have generally examined fitness effects that accrue over the entire lifespan of the plant. The higher estimated rate of somatic mutation accords with suggestions that woody plants do indeed undergo higher overall mutation rate per generation than annuals (Klekowski, 1988), and suggests that cell lineage selection may not be efficient at removing the class of mutations examined in this study.
Sources of bias in the estimation of somatic mutation parameters
The somatic mutation parameter estimates obtained here may potentially be biased downward. First, the estimates are based on autogamous–geitongamous cross pairs obtained from 20 different plants. We have pooled the results from these 20 plants, thereby assuming uniform values of Ub and Uc for each plant; one-way analysis of variance treating clone as a random factor shows that there is some variation in D among clones (F19166=1.89, P<0.02). Second, with respect to the estimation of Uc, the sizes of the plants sampled differ (there is about a twofold range of plant surface area), so that even if the rate of somatic mutation per cell is constant, the number of mutations accumulating along the branches could vary, with larger plants accumulating more mutations in the crown. Because our estimation of mutation parameters assumes that selection does not influence the estimation of the rate of somatic mutation (or alternatively, that if somatic selection does act, it does so equally for each plant), selection combined with such size variability could introduce error. That is, if somatic selection occurs and the opportunity for selection increases as the branch grows, mutations in flowers subtended by longer branches may experience more selection than those subtended by shorter ones. This would tend to bias down the estimate of Uc, though such an effect could be offset by the greater opportunity for somatic mutation to occur in longer branches. Third, the selective effects and dominance coefficients associated with individual mutations likely vary for each mutation. Variation in the selective effects would lead to downward bias in the estimate of mutation rate, with the degree of bias depending on the shape of the distribution of mutational effects (Simmons and Crow, 1977; Keightley, 1994). Fourth, the methods may underestimate the true rate of somatic mutation, as we have assumed diploidy. As noted above, there is some evidence to suggest that V. angustifolium exhibits tetrasomic inheritance, which if present, is expected to give rise to a lower probability of homozygosity of mutations upon selfing, the actual amount depending on the degree of double reduction (Haldane, 1930). If inheritance is tetrasomic, our estimates of mutation rates would be biased downward. Unfortunately, the exact degree of potential bias cannot be calculated, as there is no method for determining the frequency of double reduction of the deleterious mutations.
Other autogamy depression studies
Although there exist no other detailed studies of AD, such as the one we have conducted here, a survey of literature revealed a number of studies in which both autogamy and geitonogamy were conducted in the same plants. These studies show that D>0 is a common finding (Table 1). Although these studies do not provide explicit information on within-plant distances (number of somatic cell divisions) separating flowers used in the geitonogamous crosses, it is reasonable to expect that it would tend to be larger in woody plants, where there is greater latitude for using widely separated flowers. In this regard, it is noteworthy that D for woody plants tends to be larger than D for herbs (t=1.74, df=20, P<0.10), as expected if somatic mutation is a general feature of plants, though it is important to note that this pattern could also be an artifact of investigators unknowingly using different plants (that is, different ramets) when conducting geitonogamous crosses.
Table 1. Autogamy depression values from published studies.
| Plant species | Growth form | Autogamy depression | Reference a |
|---|---|---|---|
| Abronia ammophila | Herb | 0 | Saunders and Sipes (2006) |
| Anthyllis vulneraria | Herb | 0.687 | Navarro (1999) |
| Astragalus filipes | Herb | 0.57 | Watrous and Cane (2011) |
| Canarina canariensis | Herb | 0.50 | Rodriguez-Rodriguez and Valido 2011 |
| Epipactis palustris | Herb | 0 | Talalaj and Brzosko (2008) |
| Oxalis corniculata | Herb | 0 | Abid (2010) |
| Petrocoptis viscosa | Herb | 0 | Navarro and Guitian (2002) |
| Serpia vomeracea | Herb | 0 | Pellegrino et al. (2005) |
| Silene acutifolia | Herb | 0.12 | Buide and Guitan (2002) |
| Iris pumila | Herb (clonal) | 0.46 | Tarasjev (2005) |
| Banksia spinulosa | Shrub | 0 | Vaughton (1988) |
| Bulnesia retama | Shrub | 0.77 | Debrandi et al. (2002) |
| Cneorum triococcon | Shrub | 0.80 | Traveset (1995) |
| Erica multiflora | Shrub | 0.87 | Santandreu and Lloret (1999) |
| Helianthemum squamatum | Shrub | 0.5 | Aragon and Escudero (2008) |
| Hormathophylla spinosa | Shrub | 1 | Gomez et al. (1996) |
| Ligaria cuneifolia | Shrub | 0.49 | Rivera et al. (1996) |
| Lonicera etusca | Shrub | 1 | Guitan et al. (1993) |
| Withania somnifera | Shrub | 0 | Mir et al. (2012) |
| Acacia senegal | Shrub (tree) | 0 | Tandon and Shivanna (2001) |
| Erythrina crista-galli | Shrub (tree) | 0 | Galetto et al. (2000) |
| Pterocarpus santalinus | Shrub (tree) | 0.532 | Rao et al. (2001) |
See Supplementary Information for literature cited in this table.
Conclusions and future work
Our results provide whole-genome estimates of the rate of somatic deleterious mutations. There have also been recent studies suggesting that somatic mutation accumulation can reduce fitness through its effect on pollen viability (Ally et al., 2010). We have used phenotypic methods to estimate the deleterious mutation rate. An alternative approach would be to use DNA sequencing to assay the number of mutations directly. We note that molecular and phenotypic estimates of mutation rate are both informative, but they provide fundamentally different kinds of information. Molecular estimates inform us about processes that have occurred at the molecular level, whereas phenotypic studies tell us the fitness effect of mutations. Unfortunately, there is no reliable way to convert estimates of point mutation and insertion–deletion events detected with sequencing into estimates of fitness effects.
Our results are limited to one fitness component, an early-acting one at that, where somatic selection may have limited power to remove mutations. It will be important to extend this type of estimation to other fitness components, such as growth rate, flowering and so on. In these cases, there may be more opportunity for somatic selection to reduce the realized number of mutations passed onto gametes.
Finally, we note that our study was conducted in a commercially valuable clonal species rather than an annual species. There has been much discussion of the merits of relying more upon perennial plants for agricultural production, as the use of such plants is expected to reduce soil erosion and reliance on high-input chemical fertilizers and herbicides, while promoting above- and below-ground soil biodiversity and ecological processes (Glover et al., 2010). If, however, somatic mutation is a general feature of plant growth and occurs at a high rate, a switch to perennial or clonal crop plants could have the effect of reducing seed and/or fruit yields (McKey et al., 2010). Indeed, in light of the results obtained here, it is reasonable to ask, for the blueberry plantations investigated in this study, whether the practice of allowing unlimited vegetative expansion of clones may contribute to limiting yield, through its effect on augmenting somatic mutation and consequent reduction in fruit set due to early-acting inbreeding depression. Smaller clone size could perhaps lead to lower inbreeding depression. Another possible strategy to reduce the impact of inbreeding depression in clonal or tree crops is to increase pollen dispersal rates by deploying pollinator species that take longer flights on average between foraging bouts so that more of the pollen transfer occurs between genets.
Data archiving
Data deposited in the Dryad repository: doi:10.5061/dryad.t8q7t.
Acknowledgments
We thank Dr Douglas Scofield for assistance during the planning stages of this work, Dr Doyle McKey for comments on the manuscript and Dr Sier-Ching Chantha for assistance with microsatellite genotyping. The following persons provided help and logistics with the field and lab work: Dr Linley Jesson, Melissa Fulton, Kathryn Leyens, Duncan McGillivray, Irene McKechnie, Natasha Salter and Bonnie Sim. We especially thank Mr Bernard Savoie for allowing us to work in his blueberry fields and also for arranging access to fields owned by growers in Neguac, New Brunswick. KB was supported by an educational grant from the Mississauga First Nation Education Department. This work was supported by an NSERC Discovery Grant and an by an NSERC Strategic Network grant to the Canadian Pollination Initiative (CANPOLIN). This is CANPOLIN paper number 86.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Heredity website (http://www.nature.com/hdy)
Supplementary Material
References
- Ally D, Ritland K, Otto SP. Aging in a long-lived clonal tree. PLoS Biol. 2010;8:e1000454. doi: 10.1371/journal.pbio.1000454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antolin MF, Strobeck C. The population genetics of somatic mutation in plants. Am Nat. 1985;126:52–62. [Google Scholar]
- Baer CF, Miyamoto MM, Denver DR. Mutation rate variation in multicellular eukaryotes: causes and consequences. Nat Rev Genet. 2007;8:619–631. doi: 10.1038/nrg2158. [DOI] [PubMed] [Google Scholar]
- Barrett SCH, Harder LD, Worley A. The comparative biology of pollination and mating in flowering plants. Phil Trans R Soc Ser B. 1996;351:1271–1280. [Google Scholar]
- Bell DJ, Rowland LJ, Stommel J. Yield variation among clones of lowbush blueberry as a function of genetic similarity and self-compatibility. J Amer Soc Hort Sci. 2010;135:259–270. [Google Scholar]
- Boches PS, Bassil NV. Microsatellite markers for Vaccinium from EST and genomic libraries. Mol Ecol Res. 2005;5:657–660. [Google Scholar]
- Broyden CG. The convergence of a class of double-rank minimization algorithms 1. General considerations. IMA J Appl Math. 1970;6:76–90. [Google Scholar]
- Charlesworth D, Charlesworth B. Inbreeding depression and its evolutionary consequences. Ann Rev Ecol Syst. 1987;18:237–268. [Google Scholar]
- Cloutier D, Rioux D, Beaulieu J, Schoen DJ. Somatic stability of microsatellite loci in Eastern white pine, Pinus strobus L. Heredity. 2003;90:247–252. doi: 10.1038/sj.hdy.6800221. [DOI] [PubMed] [Google Scholar]
- Drake JW, Charlesworth B, Charlesworth D, Crow JF. Rates of spontaneous mutation. Genetics. 1998;148:1667–1686. doi: 10.1093/genetics/148.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher R. A new approach to variable metric algorithms. Comp J. 1970;13:317–322. [Google Scholar]
- Fletcher R, Reeves C. Function minimization by conjugate gradients. Comp J. 1964;7:149–154. [Google Scholar]
- Gill DE, Chao L, Perkins SL, Wolf JB. Genetic mosaicism in plants and clonal animals. Ann Rev Ecol Syst. 1995;26:423–444. [Google Scholar]
- Glover JD, Culman SW, DuPont ST, Broussard W, Young L, Mangan ME, et al. Harvested perennial grasslands provide ecological benchmarks for agricultural sustainability. Agric Ecosyst Environ. 2010;137:3–12. [Google Scholar]
- Goldfarb D. A family of variable-metric methods derived by variational means. Math Comp. 1970;24:23–26. [Google Scholar]
- Haag-Liautard C, Coffey N, Houle D, Lynch M, Charlesworth B, Keightley PD. Direct estimation of the mitochondrial DNA mutation rate in Drosophila melanogaster. PLoS Biol. 2008;6:1706–1714. doi: 10.1371/journal.pbio.0060204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane JBS. Theoretical genetics of autopolyploids. J Genet. 1930;22:359–372. [Google Scholar]
- Heim S.2007. svcm: 2d and 3sd Space-Varying Coefficient Models in R. R package version 0.1.2 http://CRAN.R-project.org/package=svcm .
- Hokanson K, Hancock J. The common lowbush blueberry, Vaccinium angustifolium, may be an autotetraploid. Can J Plant Sci. 1993;73:889–891. [Google Scholar]
- Hokanson K, Hancock J. Early-acting inbreeding depression in three species of Vaccinium (Ericaceae) Sex Plant Reprod. 2000;13:145–150. [Google Scholar]
- Keightley PD. The distribution of mutation effects on viability in Drosophila melanogaster. Genetics. 1994;138:1315–1322. doi: 10.1093/genetics/138.4.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klekowski EJ., Jr . Mutation, Developmental Selection, and Plant Evolution. Columbia University Press: New York; 1988. [Google Scholar]
- Klekowski EJ, Jr, Godfrey PJ. Aging and mutation in plants. Nature. 1989;340:389–390. [Google Scholar]
- Klekowski EJ, Jr, Kazarinova-Fukshansky NK, Mohr H. Shoot apical meristems and mutation: Selective loss of advantageous cell genotypes. Am J Bot. 1985;71:28–34. [Google Scholar]
- Kondrashov AS. Deleterious mutation and the evolution of sexual reproduction. Nature. 1988;336:435–440. doi: 10.1038/336435a0. [DOI] [PubMed] [Google Scholar]
- Lynch M, Gabriel W. Mutation load and the survival of small populations. Evolution. 1990;44:1725–1737. doi: 10.1111/j.1558-5646.1990.tb05244.x. [DOI] [PubMed] [Google Scholar]
- McKey D, Elias M, Pujol B, Duputie A. The evolutionary ecology of clonally propagated domesticated plants. New Phytol. 2010;186:318–332. doi: 10.1111/j.1469-8137.2010.03210.x. [DOI] [PubMed] [Google Scholar]
- Morgan MT. Consequences of life history for inbreeding depression and mating system evolution in plants. Proc Biol Sci. 2001;268:1817–1824. doi: 10.1098/rspb.2001.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton NE, Crow JF, Muller HJ. An estimate of the mutational damage in man from data of consanguineous marriages. Proc Natl Acad Sci USA. 1956;42:855–863. doi: 10.1073/pnas.42.11.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai T. The genetic structure of natural populations of Drosophila melanogaster. I. Spontaneous mutation rate of polygenes controlling viability. Genetics. 1964;72:333–355. doi: 10.1093/genetics/50.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelder JA, Mead R. A simplex method for function minimization. Comp J. 1965;7:308–313. [Google Scholar]
- Nocedal J, Wright SJ.1999Numerical Optimization2nd edn.Springer: New York, USA [Google Scholar]
- O'Connell LM, Ritland K. Somatic mutations at microsatellite loci in western Redcedar (Thuja plicata: Cupressaceae) J Hered. 2004;95:172–176. doi: 10.1093/jhered/esh024. [DOI] [PubMed] [Google Scholar]
- Otto SP, Orive ME. Evolutionary consequences of mutation and selection within an individual. Genetics. 1995;141:1173–1187. doi: 10.1093/genetics/141.3.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto SP, Hastings IM. Mutation and selection within the individual. Genetica. 1998;102-103:507–524. [PubMed] [Google Scholar]
- Otto SP, Walbot V. DNA methylation in eukaryotes: kinetics of demethylation and de novo methylation during the life cycle. Genetics. 1990;124:429–437. doi: 10.1093/genetics/124.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda-Krch M, Lehtila K. Cell lineage dynamics in stratified shoot apical meristems. J Theor Biol. 2002;219:495–505. doi: 10.1006/jtbi.2002.3139. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2012R: A Language and Environment For Statistical Computing. R Foundation for Statistical Computing ISBN: Vienna, Austria; ISBN. 3-900051-07-0 http://www.R-project.org/ . [Google Scholar]
- Schoen DJ, Bataillon TM, David JL. Deleterious mutation accumulation and the regeneration of genetic resources. Proc Natl Acad Sci USA. 1998;95:394. doi: 10.1073/pnas.95.1.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz ST, Scofield DG. Mutation accumulation in real branches: fitness assays for genomic deleterious mutation rate and effect in large-statured plants. Am Nat. 2009;174:163–175. doi: 10.1086/600100. [DOI] [PubMed] [Google Scholar]
- Scofield DG, Schultz ST. Mitosis, stature, and evolution of plant mating systems: low-Φ and high-Φ plants. Proc Biol Sci. 2006;273:275–282. doi: 10.1098/rspb.2005.3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scofield DG. Medial pith cells per meter in twigs as a proxy for mitotic growth rate (Φ/m) in the apical meristem. Am J Bot. 2006;93:1740–1747. doi: 10.3732/ajb.93.12.1740. [DOI] [PubMed] [Google Scholar]
- Shanno DF. Conditioning of quasi-Newton methods for function minimization. Math Comp. 1970;24:647–656. [Google Scholar]
- Shanno DF, Kettler PC. Optimal conditioning of Quasi-Newton methods. Math Comp. 1970;24:657–664. [Google Scholar]
- Simmons MJ, Crow JF. Mutations affecting fitness in Drosophila populations. Ann Rev Genet. 1977;11:49–78. doi: 10.1146/annurev.ge.11.120177.000405. [DOI] [PubMed] [Google Scholar]
- Weir BS. Genetic Data Analysis II: Methods for Discrete Population Genetic Data. Sinauer Associates: Sunderland, MA, USA; 1996. [Google Scholar]
- Weismann A. The germ-plasm: a theory of heredity. Charles Scribner's and Sons: New York; 1893. [Google Scholar]
- Whitham TA, Slobodchikoff CN. Evolution by individuals, plant/herbivoreinteractions, and mosaics of variability: the adaptive significance of somatic mutations in plants. Oecologia. 1981;49:287–292. doi: 10.1007/BF00347587. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.