

Abstract
The 44-amino-acid E5 protein of bovine papillomavirus is a dimeric transmembrane protein that exists in a stable complex with the platelet-derived growth factor (PDGF) β receptor, causing receptor activation and cell transformation. The transmembrane domain of the PDGF β receptor is required for complex formation, but it is not known if the two proteins contact one another directly. Here, we studied a PDGF β receptor mutant containing a leucine-to-isoleucine substitution in its transmembrane domain, which prevents complex formation with the wild-type E5 protein in mouse BaF3 cells and inhibits receptor activation by the E5 protein. We selected E5 mutants containing either a small deletion or multiple substitution mutations that restored binding to the mutant PDGF β receptor, resulting in receptor activation and growth factor independence. These E5 mutants displayed lower activity with PDGF β receptor mutants containing other transmembrane substitutions in the vicinity of the original mutation, and one of them cooperated with a receptor mutant containing a distal mutation in the juxtamembrane domain. These results provide strong genetic evidence that the transmembrane domains of the E5 protein and the PDGF β receptor contact one another directly. They also demonstrate that different mutations in the E5 protein allow it to tolerate the same mutation in the PDGF β receptor transmembrane domain and that a mutation in the E5 protein can allow it to tolerate different mutations in the PDGF β receptor. Thus, the rules governing direct interactions between transmembrane helices are complex and not restricted to local interactions.
INTRODUCTION
Proteins that span cellular membranes are thought to comprise up to 30% of the proteome (1). The membrane-spanning segments of most of these transmembrane proteins adopt an α-helical conformation and can undergo highly specific, lateral interactions to form oligomeric protein complexes or properly folded multipass transmembrane proteins (2, 3). These helical interactions are often essential for biological activity, but the structural basis of the vast majority of transmembrane interactions is not understood because of difficulties in obtaining high-resolution structures of transmembrane helical bundles. Mutational analysis showed that homodimeric helix-helix interactions can be determined by highly specific interactions between amino acid side chains (4). Formation of heteromeric transmembrane complexes can be mediated by interactions between hydrophilic amino acid side chains (5–7), but the structural basis for the specificity of heteromeric transmembrane interactions has not been studied in detail. In our laboratory, we have developed genetic methods to analyze heteromeric transmembrane interactions in mammalian cells and showed that artificial transmembrane proteins can undergo specific functional interactions with native transmembrane proteins (8–12).
The 44-amino-acid E5 oncoprotein of bovine papillomavirus type 1 (BPV) is essentially an isolated transmembrane domain that forms a homodimer and transforms mouse cells to tumorigenicity (13, 14). In transformed cells, the E5 protein is present in a stable complex with the cellular platelet-derived growth factor (PDGF) β receptor, a receptor tyrosine kinase whose extracellular domain normally binds the soluble protein ligand PDGF (15, 16). The interaction between the E5 protein and the PDGF β receptor causes dimerization and trans-autophosphorylation of the receptor, resulting in sustained receptor activation and cell transformation or growth factor independence (17–21). The E5 protein–PDGF β receptor interaction is highly specific, and previous studies of mutant and chimeric receptors in cells showed that the receptor transmembrane domain is required for complex formation with the E5 protein (17, 22–26).
Although the E5 protein and the PDGF β receptor are present in a physical complex in transformed cells, it is not known if the transmembrane domains of these two proteins interact directly or if this interaction is mediated by an as-yet-unidentified cellular protein. A direct interaction is suggested by mutational analysis, which identified four specific amino acids as being important for complex formation and transforming activity: glutamine 17 and aspartic acid 33 of the E5 protein, and juxtamembrane lysine 499 and transmembrane threonine 513 of the PDGF β receptor (27–30). The PDGF β receptor is a type I transmembrane protein (i.e., with the N terminus outside the cell or in the lumen of intracellular organelles), whereas the E5 protein is thought to have the opposite, type II orientation (31). Therefore, if the transmembrane domains of these two proteins interact directly, they would adopt an antiparallel arrangement, which would put E5 Asp33 in proximity to PDGF β receptor Lys499 in the extracellular/luminal juxtamembrane region, where they might form a salt bridge, and E5 Gln17 near Thr513 in the transmembrane segment of the PDGF β receptor, where they might form a hydrogen bond (Fig. 1) (28, 32, 33). Computational modeling supports these assignments (34, 35). However, swapping of the charged amino acids in the juxtamembrane region of the E5 protein and the PDGF β receptor does not allow activity, perhaps because the mutant side chains are not properly oriented or because the two charged amino acids do not in fact interact directly.
Fig 1.

Sequences of the wild-type and mutant E5 proteins and the transmembrane domain of the PDGF β receptor. The sequence of the transmembrane domain of the wild-type and mutant murine PDGF β receptors is from amino acids 495 to 527. The sequence labeled E5 is the “wild-type” E5 protein containing a proline-to-alanine mutation at position 2 to optimize the Kozak translation initiation sequence. The LRM mutants are E5 mutants isolated from the limited random mutagenesis library. Mutations in the E5 protein and PDGF β receptor are shown in red. Deletions are represented by hyphens. Reflecting the proposed antiparallel orientation of the E5 protein and the PDGF β receptor, the N terminus of the PDGF β receptor segment is at the right, and the N terminus of the E5 protein is at the left. Putative interactions between specific amino acids in the E5 protein and the PDGF β receptor are boxed in green.
Here, we employed genetic selection in mouse cells to isolate E5 mutants that physically and functionally interact with a PDGF β receptor containing a transmembrane mutation that inhibits binding to the wild-type E5 protein. These E5 mutants displayed the highest activity with the defective receptor mutant which they were selected against and with the wild-type PDGF β receptor and displayed lower activity with receptors containing other nearby substitutions that inhibit the interaction with the wild-type E5 protein. In addition, one of these E5 mutants cooperated with a PDGF β receptor mutant containing a substitution 13 positions away from the original receptor mutation. These experiments provide strong genetic evidence that the transmembrane domains of the E5 protein and the PDGF β receptor interact directly. In addition, our results demonstrate that the rules governing direct interactions between transmembrane helices are complex.
MATERIALS AND METHODS
Retrovirus production.
Human 293T cells were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum (FBS), 10 mM HEPES, penicillin-streptomycin, and gentamicin (DMEM-10). Cells were cotransfected with retroviral DNA and the pCL-Eco and VSV-g retroviral packaging plasmids, as described previously (36). After 48 h, pseudotyped retrovirus was collected, filtered through a 0.45-μm filter, and stored at −80°C. Virus used for library infections was concentrated approximately 20-fold by using ultracentrifugal concentrators (Millipore).
Generation of stable cell lines expressing the wild-type or mutant PDGF β receptor.
BaF3 cells, an interleukin-3 (IL-3)-dependent murine cell line, were grown in suspension in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 50 μM β-mercaptoethanol, 5% WEHI-3B conditioned supernatant as a source of IL-3, and antibiotics (RPMI/IL-3 medium). PDGF β receptor mutants and the PR-βαβ chimera were constructed by replacing a 28-codon segment of the full-length murine PDGF β receptor clone with an annealed oligonucleotide containing the desired mutation(s). A total of 5 × 105 BaF3 cells were infected with LXSN retrovirus expressing the murine PDGF β receptor, chimeric PR-βαβ, or one of the mutant PDGF β receptors: L512I, L512V, T513L, T513I, or K499D. After 24 h, cells were selected with 1 mg/ml G418 (Gibco) for 10 days. The stable lines were grown for approximately 1 week in RPMI medium lacking IL-3 but containing 40 ng/ml PDGF-BB to ensure receptor activity. Cells were then maintained in RPMI/IL-3 medium containing 0.5 mg/ml G418.
Library construction.
A PCR-based approach was used to construct a library expressing a large collection of BPV E5 proteins, each with a limited number of random substitutions in the transmembrane domain (12). A degenerate oligonucleotide was designed that contained 12 fixed 5′ nucleotides, including an AvrII recognition site immediately upstream of a mutagenized stretch of 54 nucleotides (corresponding to E5 codons 12 to 30), followed by 26 nucleotides encoding amino acids 31 to 39 of E5. At each position in the mutagenized segment, the wild-type E5 nucleotide was incorporated approximately 85% of the time, and each of the three incorrect ones was incorporated 5% of the time. In addition, position 33 of the E5 protein was mutagenized to generate proteins with a variety of hydrophilic amino acids at this position by using an equimolar mixture of all 4 nucleotides at codon position 1, an equimolar mixture of A and G at position 2, and an equimolar mixture of C and G at position 3. A nondegenerate oligonucleotide encoding the antisense sequence of the E5 C terminus and containing a BamHI site at its 5′ end was annealed to the degenerate oligonucleotide. After extension to form double-stranded DNA, short 5′ and 3′ primers that annealed to the fixed ends of the oligonucleotides were used for PCR amplification. The amplification products were then digested with AvrII and BamHI, purified on a Qiagen PCR purification column, and ligated into pT2H-F13 (8), a hygromycin-resistant retroviral vector containing the 5′ end of the E5 gene, to reconstitute the intact E5 gene with a mutagenized central segment. Ligation reaction mixtures were purified and used to transform Escherichia coli DH10β cells (Invitrogen, Carlsbad, CA). After ∼90,000 ampicillin-resistant colonies were pooled, the plasmid DNA was purified and used for retrovirus production. This yielded a library (E5-LRM [limited random mutagenesis]) averaging 4 amino acid changes per transmembrane domain, as confirmed by sequencing of randomly chosen clones from the library. The sequences of oligonucleotides used in this study and details of construction are available from the authors upon request.
Selection of compensatory E5 mutants.
Four pools of 5 × 105 BaF3 cells expressing the PDGF β receptor L512I mutant were mock infected or infected with 500 μl 20×-concentrated E5-LRM retrovirus and incubated at 37°C in RPMI/IL-3 medium lacking antibiotics for 24 h. Hygromycin B sulfate (American Bioanalytical) was then added to a final concentration of 1 mg/ml. Two to three days later, the cells were pelleted and resuspended with RPMI/IL-3 medium containing 1 mg/ml G418 and hygromycin B. The cells were incubated until the mock-infected cells died (5 to 7 days). The infected cells were then pelleted, washed twice in phosphate-buffered saline (PBS), and resuspended in RPMI medium lacking IL-3. After 5 days, genomic DNA was extracted (DNeasy kit; Qiagen) from the surviving cells, and library sequences were amplified by PCR using primers for the invariant portion of the vector. These sequences were then cloned back into the retroviral vector as a mixture, and a secondary library was packaged into retrovirus particles and used to conduct a second round of selection, as described above. This cycle of library construction and screening was repeated once more, and individual clones recovered from the DNA of IL-3-independent cultures were sequenced.
Clonal infections and interleukin-3 independence assay.
A total of 5 × 105 BaF3 cells expressing the wild-type or mutant PDGF β receptors were mock infected or infected with 1 ml of unconcentrated retrovirus in a 25-cm2 flask as described above and maintained in RPMI/IL-3 medium and the appropriate drugs until the mock-infected cells died. A total of 200,000 cells from each infection were collected, washed, and resuspended in 10 ml RPMI medium lacking IL-3 and supplemented with 6% FBS, penicillin, streptomycin, and 0.25 mg/ml hygromycin. Live cells were counted on a hemocytometer after growth factor removal. Multiple independent experiments were performed with independently derived cell lines, with cell numbers recorded for any one experiment on day 5, 6, or 7 after growth factor removal, on the day before the cells expressing BPV E5 and the PDGF β receptor became overgrown and started to die. The results shown in the graphs are the aggregated data for all experiments. In some cases, the results for a particular E5 protein-PDGF receptor combination are shown in more than one graph to group related experiments together for clarity of presentation. Statistical significance was assessed by unpaired nonparametric Mann-Whitney tests.
Construction of specific mutations in the E5 protein.
To construct E5-WLQ, E5-LEK, and deletion mutations in the E5 gene, long oligonucleotides containing the desired sequence changes were phosphorylated, annealed, and ligated into the appropriate pT2H-F13-based retroviral vector to reconstitute the mutant E5 gene. E5-SIS and the component mutations in E5-LEK were constructed by using QuikChange site-directed mutagenesis (Aligent, Santa Clara, CA). Sense and antisense oligonucleotides encoding the desired mutations were mixed with the DNA template, Pfu Turbo polymerase, and PCR nucleotide mix (Roche) and subjected to 18 cycles of PCR, followed by DpnI digestion. Three microliters of each mutagenesis reaction mixture was used to transform E. coli DH10β cells (Invitrogen). Plasmid DNA was extracted from individual ampicillin-resistant colonies (Nucleobond kit; Clontech) and sequenced.
Immunoprecipitation and immunoblotting.
BaF3 cells were grown to a high density (∼106 cells/ml) in RPMI/IL-3 medium and then pelleted and washed twice in ice-cold PBS supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF), 250 μM pervanadate, and 5 μg/ml aprotinin, and leupeptin. The cells were then lysed in radioimmunoprecipitation assay-morpholinepropanesulfonic acid (RIPA-MOPS) buffer (20 mM MOPS, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1% deoxycholate, and 0.1% SDS) containing protease and phosphatase inhibitors, and extracted protein was quantitated by using a bicinchoninic acid (BCA) assay (Pierce). For phosphotyrosine (PY) and PDGF receptor blotting, 0.75 mg protein was immunoprecipitated with 7 μl anti-PR rabbit serum raised against the carboxyl-terminal 13 amino acids of the murine PDGF β receptor. For E5 immunoprecipitation, 10 μl of anti-E5 rabbit serum raised against the C-terminal 14 amino acids of the wild-type E5 protein was added to 1 mg of protein. Protein-antibody mixtures were rotated overnight at 4°C, and complexes were collected with protein A-Sepharose, washed, and resuspended in 2× Laemmli sample buffer. Proteins were denatured and separated on a 7.5% (for PDGF receptor and phosphotyrosine blotting) or 20% (for E5 blotting) SDS–polyacrylamide gel and transferred onto a polyvinylidene difluoride (PVDF) (E5) or nitrocellulose (PDGF β receptor or PY) membrane for blotting, as described previously (37). No SDS was included for transfer of the E5 protein. Membranes were incubated with primary antibodies overnight, washed, incubated with a horseradish peroxidase-conjugated secondary antibody for 1 h, washed again, and detected by using West Pico chemiluminescent reagents (Pierce), as previously described (37).
RESULTS
Identification of a defective PDGF β receptor transmembrane mutant.
Some mutations in the transmembrane domain of the PDGF β receptor prevent binding and activation by the wild-type E5 protein (23, 30). We reasoned that it would be possible to isolate E5 mutants that specifically bound and activated such a receptor mutant if the E5 protein normally interacts directly with the PDGF β receptor transmembrane domain. To identify such mutants, we used murine BaF3 cells, which are strictly dependent on interleukin-3 (IL-3) for growth and do not express endogenous PDGF β receptor. The productive interaction between the E5 protein and an ectopically expressed PDGF β receptor confers IL-3 independence in these cells (17). This activity forms the basis of a genetic selection for compensating mutations in the E5 protein and the PDGF β receptor.
To identify a murine PDGF β receptor mutant that did not respond to the E5 protein, we tested a series of mutant receptors with single hydrophobic amino acid substitutions in the transmembrane domain at or near position 513 (Fig. 1, top sequences). These PDGF β receptor mutants were expressed in BaF3 cells and tested for their ability to confer IL-3 independence in response to the wild-type E5 protein or v-sis, a viral homologue of PDGF, which binds to the extracellular domain of the receptor (Fig. 2). As expected, parental BaF3 cells lacking the PDGF β receptor failed to respond to E5 or v-sis, and BaF3 cells expressing the wild-type murine PDGF β receptor (BaF3-PR cells) displayed growth factor independence in response to either the E5 protein or v-sis. None of the mutant PDGF β receptors conferred growth factor independence in BaF3 cells infected with the empty vector, confirming that they were not constitutively active, and all of them conferred growth factor independence in response to v-sis, demonstrating that they were functional, ligand-dependent PDGF receptors. Notably, the receptor transmembrane mutants failed to confer growth factor independence in response to the E5 protein. The mutant receptor containing a leucine-to-isoleucine substitution at position 512 (PR-L512I) was reproducibly the most defective in its response to the E5 protein, so we focused on this mutant in further experiments.
Fig 2.
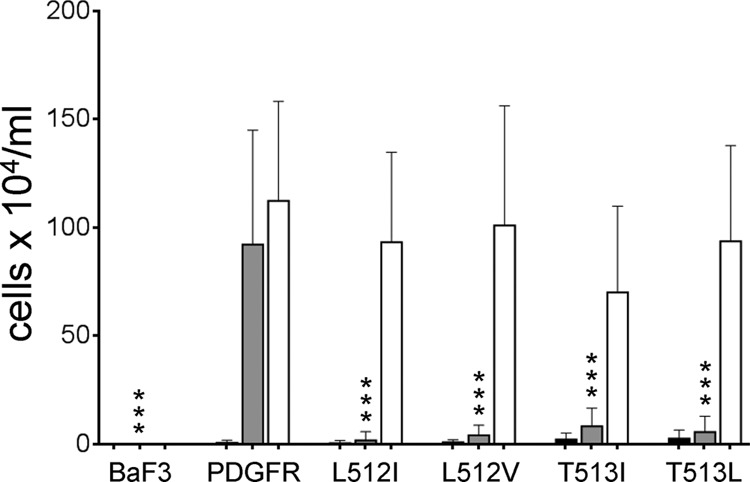
Identification of PDGF β receptor mutants unable to respond to E5 protein. Parental BaF3 cells or cells expressing the wild-type PDGF β receptor (PDGFR) or the indicated receptor mutants were infected with the T2H-F13 retrovirus vector expressing no insert (black bars), the wild-type E5 protein (gray bars), or the PDGF homologue, v-sis (white bars). Viable cells were counted after the cultures were incubated for up to 7 days in the absence of growth factors. The averages of data from numerous independent replicate experiments with standard deviations are shown. Statistical significance of the results of the E5 tests compared to the PDGFβR/E5 combination is indicated by three asterisks (P ≤ 0.002).
To determine whether the E5 protein activated PR-L512I, we immunoprecipitated the PDGF β receptor from extracts of cells expressing the wild-type PDGF β receptor or this mutant and conducted phosphotyrosine blotting. The E5 protein caused tyrosine phosphorylation of the slowly migrating mature form of the wild-type PDGF β receptor and the more rapidly migrating, intracellular precursor form, but it did not induce tyrosine phosphorylation of PR-L512I (Fig. 3, top, compare lanes 2 and 6), even though the E5 protein was abundantly expressed in both cell lines (Fig. 3, bottom). To determine whether the L512I mutation prevented the physical interaction with the E5 protein, we conducted coimmunoprecipitation experiments, in which we immunoprecipitated detergent extracts with anti-E5 antiserum and performed immunoblotting with anti-PDGF receptor antiserum. An association between the wild-type E5 protein and the precursor form of the wild-type PDGF β receptor was readily detectable, reflecting the primary localization of the E5 protein to intracellular membranes of the endoplasmic reticulum and Golgi apparatus (31). Strikingly, the E5 protein was markedly impaired in its ability to form a complex with PR-L512I (Fig. 3, second panel from the top, compare lanes 2 and 6). All cell lines expressed similar levels of PDGF β receptor. These results demonstrated that the L512I mutation in the PDGF β receptor transmembrane domain inhibited productive interaction between the E5 protein and the receptor.
Fig 3.
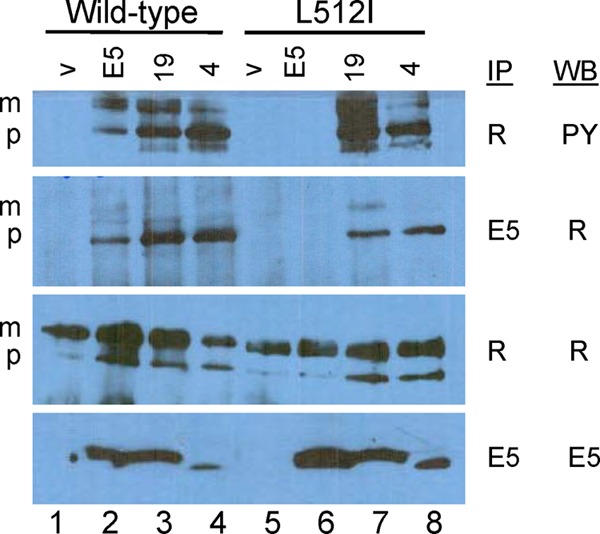
Binding and activation of the PDGF β receptor. Detergent extracts were prepared from BaF3 cells expressing the wild-type PDGF β receptor or PR-L512I and coexpressing either the empty vector (v), the wild-type E5 protein (E5), E5.LRM19 (19), or E5.LRM4 (4). Samples were immunoprecipitated (IP) with antibody recognizing the PDGF β receptor (R) or the E5 protein (E5) and then immunoblotted (Western blot [WB]) with antibody recognizing phosphotyrosine (PY), the PDGF β receptor, or the E5 protein. m and p indicate the mature and precursor forms of the PDGF β receptor, respectively.
Isolation of E5 mutants that interact with PR-L512I.
To isolate compensatory E5 mutants, we constructed a library of mutants that contained a limited number of random substitution mutations throughout the transmembrane domain of the E5 protein and screened this library for clones that conferred growth factor independence in BaF3-PR-L512I cells. To construct this library, we used a “doped” oligonucleotide in which the segment encoding positions 12 to 30 of the E5 protein was synthesized with a mixture of nucleotides consisting of approximately 85% of the wild-type nucleotide at each position and 5% of each of the three incorrect nucleotides. In addition, position 33 was mutagenized so that approximately 87% of the clones encoded a hydrophilic amino acid at this position. A second oligonucleotide was annealed to the fixed 3′ end of the doped oligonucleotide and extended to generate double-stranded DNA, which was amplified with flanking primers and cloned into the pT2H-F13 retrovirus expression vector (8). Individual sequenced clones encoded E5 proteins that contained on average four amino acid substitution mutations in the transmembrane domain (data not shown). DNA was isolated from approximately 90,000 pooled bacterial colonies and cotransfected into 293T cells with retroviral packaging plasmids to generate the E5-LRM (limited random mutagenesis) retroviral expression library.
We used serial selection for growth factor independence in BaF3-L512I cells infected with the E5-LRM library to isolate compensatory E5 mutants (Fig. 4). Four pools of BaF3-L512I cells growing in medium containing IL-3 were infected with the E5-LRM library and selected for hygromycin resistance carried on the retroviral vector. After mock-infected cells died, hygromycin-resistant cells were incubated in medium lacking IL-3. After 8 days in the absence of IL-3, most cultures infected with the library of E5 mutants contained actively growing cells, whereas cells infected with the empty vector failed to proliferate. Retroviral inserts were recovered by PCR from genomic DNA isolated from each pool of proliferating cells and cloned as mixtures into the retrovirus vector to generate secondary libraries, which were packaged and used to infect naive BaF3-L512I cells. After a second round of selection for growth factor independence, retroviral inserts were again recovered, cloned as mixtures, and reintroduced into BaF3-L512I cells, which were selected for IL-3 independence. After this third round of selection, we identified several full-length E5 clones containing up to seven missense mutations (Fig. 1, bottom sequences). In addition, alanine 14 and alanine 15 were deleted in two clones. One of these deletion clones, E5.LRM9, also contained a leucine-to-valine substitution at position 19 in the E5 sequence (L19V), and the other, E5.LRM19, contained three different missense mutations (F23S, L29I, and D33S).
Fig 4.
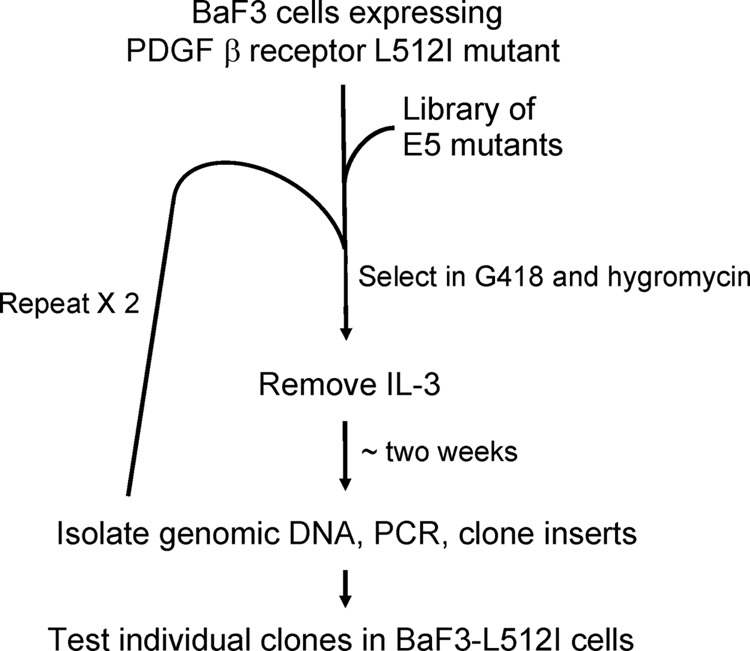
Schematic diagram of the genetic screen to isolate compensatory mutants of the E5 protein. See the text for details.
The recovered clones were tested individually for activity in BaF3 cells expressing wild-type or mutant PDGF β receptors, including a chimeric receptor containing the transmembrane domain of the PDGF α receptor (PR-βαβ), which is not recognized by the wild-type E5 protein (37). As expected, the wild-type E5 protein was inactive in parental BaF3 cells lacking the PDGF β receptor, BaF3-L512I cells, and BaF3-βαβ cells but conferred growth factor independence in BaF3 cells expressing the wild-type PDGF β receptor (Fig. 5A and data not shown). None of the clones recovered from the library were active in parental BaF3 cells or in BaF3-βαβ cells. Strikingly, three clones, E5.LRM4, E5.LRM9, and E5.LRM19, conferred robust growth factor independence in cooperation with either the wild-type PDGR β receptor or PR-L512I (Fig. 5A and B). As noted above, E5.LRM9 and E5.LRM19 share the same 2-amino-acid deletion. E5.LRM4 contained six missense mutations. E5.LRM3, which differs from clone E5.LRM4 only at position 33, was active in cells expressing the wild-type PDGF β receptor and displayed moderate activity in cells expressing PR-L512I. The other clones did not support growth factor independence in any of the cell lines (Fig. 5A and data not shown). Thus, we had selected several E5 mutants that, unlike the wild-type E5 protein, cooperated with PR-L512I. The ability of these E5 mutants to cooperate with PR-L512I and their inability to cooperate with the chimeric βαβ receptor, which contains a foreign transmembrane domain, indicated that these clones required the transmembrane domain of the PDGF β receptor for activity. Because of these results, we focused our remaining studies on two of the most active clones, E5.LRM4 and E5.LRM19.
Fig 5.
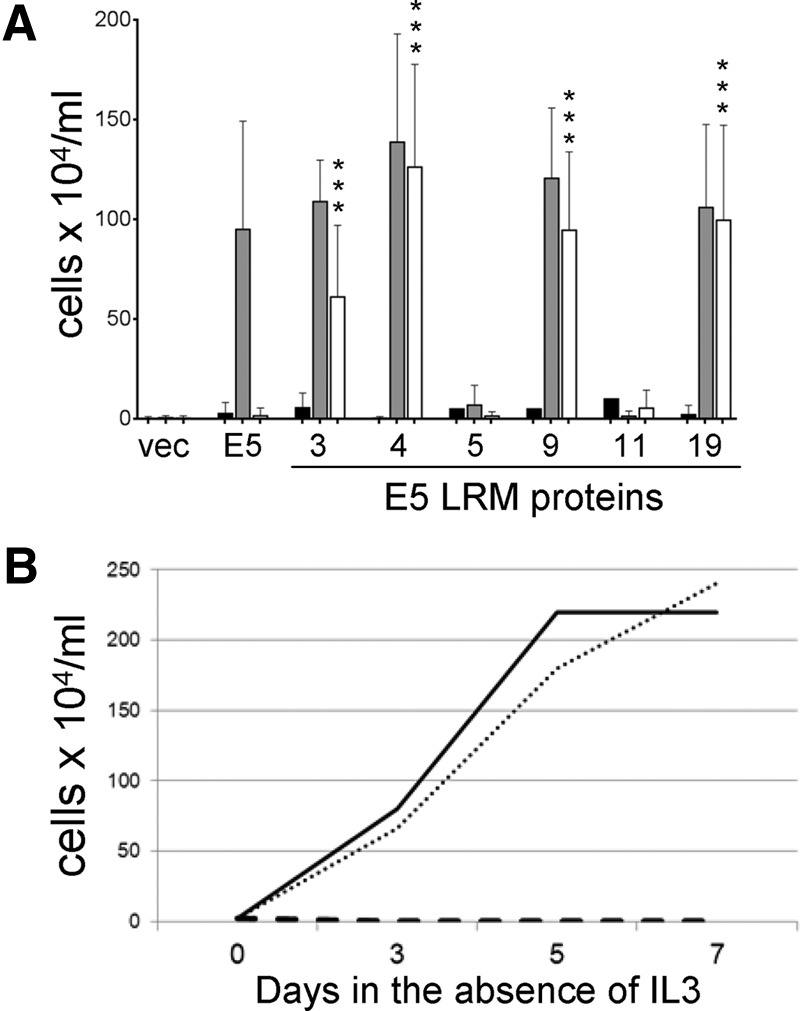
Identification of E5 mutants that cooperate with PR-L512I. (A) BaF3 cells expressing the βαβ chimeric receptor (black bars), the wild-type PDGF β receptor (gray bars), or PR-L512I (white bars) were infected with retroviruses expressing no insert (vec), the wild-type E5 protein, or the indicated clone recovered from the E5-LRM library. Viable cells were counted and recorded as described in the legend of Fig. 2. Statistical significance of the results of the PR-L512I tests compared to the E5/PR-L512I combination is indicated by three asterisks (P ≤ 0.002). (B) BaF3 cells expressing PR-L512I were infected with retroviruses expressing the wild-type E5 protein (dashed line), E5.LRM4 (solid line), and E5.LRM19 (dotted line). After selection for hygromycin resistance, viable cells were counted at various times after removal of growth factors.
Biochemical analysis of BaF3 cells expressing E5 mutants.
We analyzed cells expressing the E5 mutants to determine whether the mutants physically interacted with PR-L512I and caused biochemical activation of this mutant receptor. First, the PDGF β receptor was immunoprecipitated from cell extracts and immunoblotted with an antiphosphotyrosine antibody (Fig. 3, top). As noted above, the wild-type E5 protein caused tyrosine phosphorylation of the mature and precursor forms of the wild-type PDGF β receptor but not PR-L512I. In striking contrast, E5.LRM4 and E5.LRM19, both of which cooperated with PR-L512I in conferring IL-3 independence, caused tyrosine phosphorylation of the mature form and primarily the precursor form of the mutant PDGF β receptor as well as the wild-type receptor (Fig. 3, lanes 7 and 8). Similar levels of wild-type and mutant PDGF β receptors were expressed in all of the cell lines (Fig. 3, third panel from the top). Thus, according to both physiological and biochemical criteria, E5.LRM4 and E5.LRM19 activated PR-L512I.
We used coimmunoprecipitation to assess complex formation with the PDGF β receptor. Detergent extracts of cells expressing various combinations of wild-type and mutant proteins were immunoprecipitated with anti-E5 antibody (which recognizes an epitope retained in E5.LRM4 and E5.LRM19) and then immunoblotted with PDGF receptor antibody to detect receptor physically associated with the E5 protein (Fig. 3, second panel from the top). As noted above, the precursor form and a small amount of the mature form of the wild-type PDGF receptor were coimmunoprecipitated from cells expressing the wild-type E5 protein but not from cells expressing the empty vector, indicating that the E5 protein and the wild-type PDGF β receptor were present in a physical complex. E5.LRM4 and E5.LRM19 also interacted with the precursor form of the wild-type PDGF β receptor, consistent with the ability of these mutants to cooperate with the wild-type receptor. Strikingly, although the wild-type E5 protein did not associate with PR-L512I, both E5 mutants formed a complex with the precursor form of PR-L512I (Fig. 3, compare lanes 7 and 8 to lane 6). These differences were not due to a lower expression level of the wild-type E5 protein in PR-L512I cells (Fig. 3, bottom). Thus, the mutations in E5.LRM4 and E5.LRM19 allowed these E5 mutants to interact with a PDGF β receptor mutant that was not recognized by the wild-type E5 protein. These results showed that the two E5 mutants physically associated with PR-L512I and induced its tyrosine phosphorylation, thereby accounting for their ability to cooperate with this mutant in the growth factor independence assay.
Specificity for other PDGF β receptor mutants.
To determine the specificity of E5.LRM4 and E5.LRM19, we assayed their activity against the βαβ chimeric receptor and a panel of defective PDGF β receptor point mutants. These receptor mutants carried a different amino acid substitution at position 512 (PR-L512V) or a substitution at a neighboring residue (PR-T513L and PR-T513I), which, as shown in Fig. 2, prevented them from responding to the wild-type E5 protein. PR-βαβ was not constitutively active, nor did it respond to the E5 protein or either of its mutants (Fig. 6), and all of the receptors cooperated with v-sis to confer growth factor independence.
Fig 6.
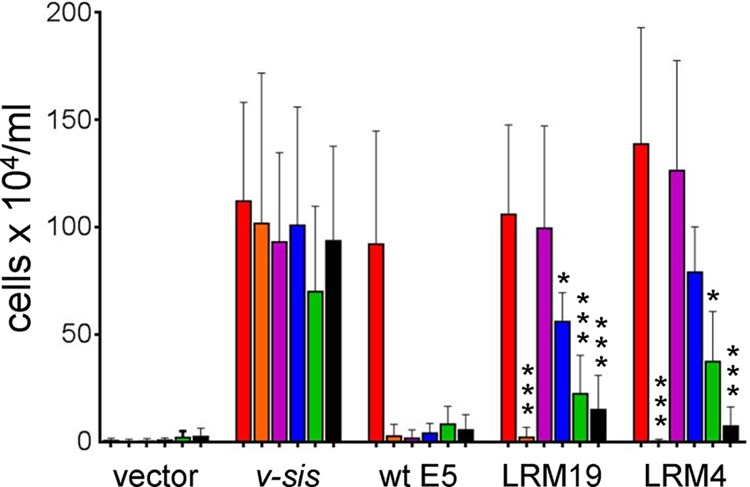
Specificity of E5 mutants LRM4 and LRM19. BaF3 cells expressing the wild-type PDGF β receptor (red bars), the βαβ chimera (orange bars), PR-L512I (purple bars), PR-L512V (blue bars), PR-T513I (green bars), or PR-T513L (black bars) were infected with retroviruses expressing no insert (vector), v-sis, the wild-type (wt) E5 protein, E5.LRM19, or E5.LRM4, as indicated. Viable cells were counted and recorded as described in the legend of Fig. 2. Statistical significance of the LRM19 and LRM4 tests compared to the cognate LRM/PR-L512I combination is indicated by one asterisk (P ≤ 0.05) or three asterisks (P ≤ 0.002).
As well as cooperating with the wild-type PDGF β receptor and PR-L512I, E5.LRM4 and E5.LRM19 showed a moderate ability to confer growth factor independence in cells expressing PR-L512V (Fig. 6). Both E5 mutants displayed low activity with receptors carrying a mutation at position 513, suggesting that, like the wild-type E5 protein, they still required threonine 513 in the transmembrane domain of the PDGF β receptor. Thus, both of the selected E5 mutants were most active with the wild-type PDGF β receptor and PR-L512I, which was used in the genetic screen.
Mapping of the mutations responsible for recognition of PR-L512I.
Because both of the active E5 mutants contained multiple mutations, we determined which mutations were responsible for their ability to cooperate with PR-L512I. E5.LRM4 contained three substitutions clustered in the N-terminal segment of the transmembrane domain and three in the C-terminal segment. Each set of three mutations was introduced separately into the E5 protein (Fig. 7), and the resulting mutants were tested for activity. E5-LEK carrying the N-terminal mutations from E5.LRM4 (V13L, A14E, and Q17K) cooperated with either the wild-type PDGF β receptor or PR-L512I, demonstrating that the N-terminal substitutions were sufficient for activity with the mutant receptor (Fig. 8A). None of the three substitutions in E5-LEK individually conferred activity with PR-L512I, and the mutant containing the glutamic acid and lysine substitutions was partially active (Fig. 8B), demonstrating that the combination of all three mutations was required for full activity. In contrast, E5-WLQ containing only the three C-terminal substitutions showed low activity with the wild-type PDGF β receptor and was inactive with PR-L512I (Fig. 8A). Thus, the C-terminal mutations in E5.LRM4 were not responsible for recognition of the PR-L512I receptor. In addition, these mutations also severely inhibited the ability of the E5 protein to functionally interact with the wild-type PDGF β receptor, unless they were paired with the N-terminal substitutions in E5.LRM4 (Fig. 8A, compare gray bars for the E5, LRM4, and E5-WLQ samples).
Fig 7.

Sequences of mutations used in mapping experiments. The top line shows the sequence of the wild-type E5 protein. The next three lines show E5.LRM4 and related mutants. The middle set of three lines shows E5.LRM19 and related mutants. The final set of three lines shows additional E5 deletion mutants. Mutations are shown in red, and deletions are represented by hyphens.
Fig 8.
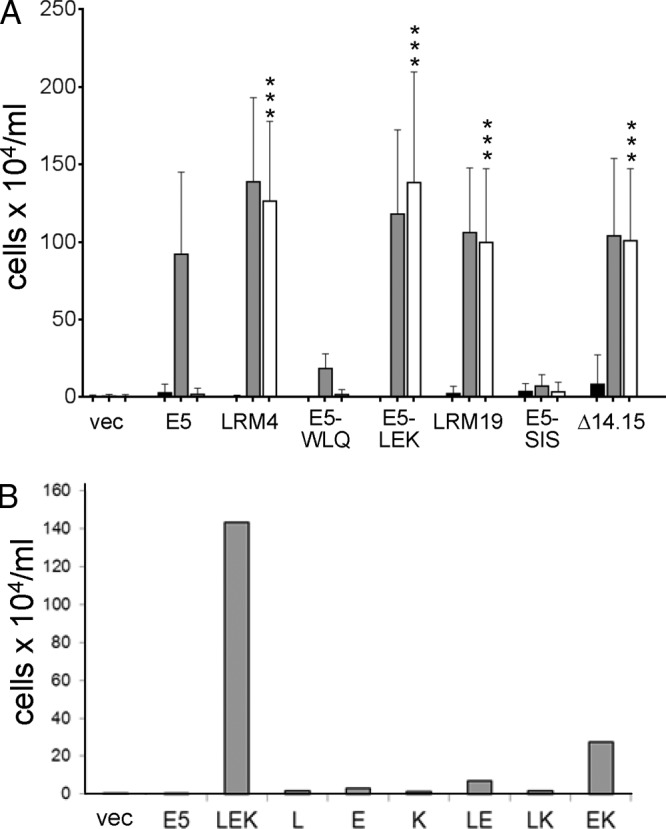
Mapping of mutations that confer activity with PR-L512I. (A) BaF3 cells expressing the βαβ chimera (black bars), the wild-type PDGF β receptor (gray bars), or PR-L512I (white bars) were infected with retroviruses expressing no insert (vec), the wild-type E5 protein, or the indicated E5 mutant. Viable cells were counted and recorded as described in the legend of Fig. 2. Statistical significance of the results of the PR-L512I tests compared to the E5/PR-L512I combination is indicated by three asterisks (P ≤ 0.002). (B) BaF3 cells expressing PR-L512I were infected with retroviruses expressing the indicated E5 mutant and analyzed as described above for panel A. The graph shows the averaged results of multiple independent experiments.
Because E5.LRM9 and E5.LRM19 shared the deletion of alanine 14 and alanine 15, it seemed likely that this deletion was responsible for the activity with PR-L512I. Therefore, we deleted these alanines from the wild-type E5 protein to generate E5Δ14.15 and assayed its activity. As shown in Fig. 8A, E5Δ14.15 conferred growth factor independence in both BaF3-PR and BaF3-L512I cells, demonstrating that deletion of alanines 14 and 15 was sufficient to allow the E5 protein to cooperate with PR-L512I. In contrast, an E5 mutant (E5-SIS) containing only the three C-terminal missense mutations from E5.LRM19 (F23S, L29I, and D33S) cooperated with neither PR-L512I nor the wild-type PDGF β receptor. Thus, the three missense mutations in E5.LRM19 prevented the E5 protein from activating the wild-type PDGF β receptor unless they were paired with the deletion of amino acids 14 and 15 (Fig. 8A, compare gray bars for the E5, LRM19, and E5-SIS samples).
The inability of E5-WLQ to cooperate with the wild-type PDGF β receptor, while the parental mutant E5.LRM4 was active, demonstrated that the three N-terminal substitutions in E5.LRM4 overcame the deleterious effects of the C-terminal mutations. Similarly, E5-SIS was severely impaired in its ability to functionally interact with the wild-type PDGF β receptor, demonstrating that the deletion of alanines 14 and 15 overcame the deleterious effects of the C-terminal missense mutations in E5-SIS. Therefore, certain mutations in the N-terminal region of the transmembrane domain of the E5 protein not only allowed it to recognize the mutant PDGF β receptor but also overcame the effects of loss-of-function mutations in the C-terminal portion of the transmembrane domain of the E5 protein.
Transmembrane deletions are not tolerated throughout the E5 protein.
We next determined whether the specific deletion of alanines 14 and 15 was responsible for the ability of E5Δ14.15 to cooperate with PR-L512I or if deleting any two consecutive, identical residues in the E5 protein conferred activity. We constructed 2-amino-acid deletions at several positions in the E5 protein: the two leucines at positions 7 and 8, upstream of the predicted transmembrane segment (designated E5Δ7.8); two leucines in the stretch of five leucines from positions 18 to 22 (designated E5Δ18.19); and two leucines in the stretch of three leucines from positions 24 to 26 (designed E5Δ24.25) (Fig. 7). We first tested the activity of these deletion mutants in cells expressing the wild-type PDGF β receptor (Fig. 9). Like E5Δ14.15, E5Δ7.8 conferred growth factor independence in BaF3 cells expressing the wild-type PDGF β receptor, but E5Δ18.19 and E5Δ24.25 displayed markedly lower activities. These results demonstrated that the transmembrane segment of the E5 protein between glutamine 17 and aspartic acid 33 cannot tolerate 2-amino-acid deletions, whereas deletions upstream of position 17 were compatible with strong biological activity.
Fig 9.
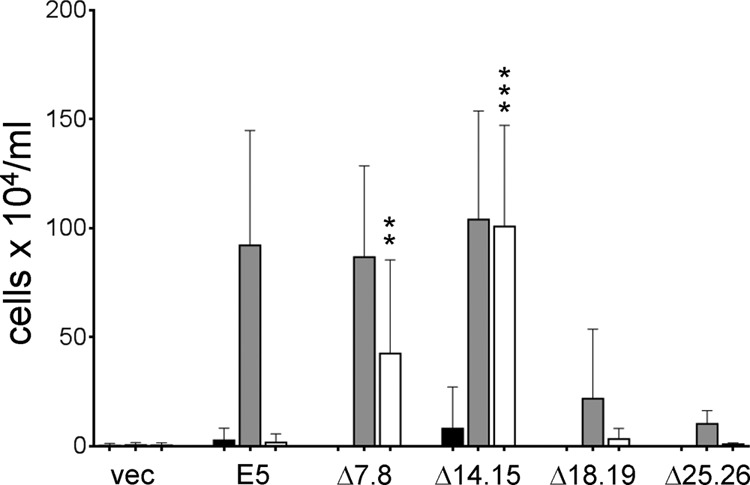
Activity of E5 deletion mutants. BaF3 cells expressing the βαβ chimera (black bars), the wild-type PDGF β receptor (gray bars), or PR-L512I (white bars) were infected with retroviruses expressing no insert (vec), the wild-type E5 protein, or the indicated E5 deletion mutant. Viable cells were counted and recorded as described in the legend of Fig. 2. Statistical significance of the results of the PR-L512I tests compared to the E5/PR-L512I combination is indicated by two asterisks (P ≤ 0.01) or three asterisks (P ≤ 0.002).
The deletion mutants were also tested in BaF3-PRL512I cells. In contrast to E5Δ14.15, which was highly active in conferring growth factor independence, E5Δ7.8 showed intermediate activity with PR-L512I, and the other deletion mutants were inactive (Fig. 9). These results demonstrated that the specific deletion of alanines 14 and 15, and not simply the shorter length of E5Δ14.15, is responsible for its robust activity with PR-L512I.
Effect of E5 mutants with a distal PDGF β receptor mutant.
To test whether the active E5 mutants could cooperate with a PDGF β receptor mutant containing a mutation distal to L512I, they were coexpressed with PR-K499D, which contains a substitution in the juxtamembrane region that prevents the action of the wild-type E5 protein (30). As shown in Fig. 10, E5.LRM4, unlike the wild-type E5 protein, induced growth factor independence in cooperation with PR-K499D. In contrast, E5.LRM19 displayed little activity with this mutant receptor. Thus, the specificities of E5.LRM4 and E5.LRM19 are different. To determine whether the N-terminal or C-terminal missense mutations in E5.LRM4 were responsible for its activity with PR-K499D, we tested the ability of E5-LEK and E5-WLQ to cooperate with this receptor mutant. As shown in Fig. 10, E5-LEK conferred growth factor independence in cooperation with PR-K499D, but E5-WLQ was devoid of activity. These results demonstrated that the N-terminal mutations in E5-LEK are responsible for its activity with PR-K499D, as they are for activity with PR-L512I.
Fig 10.
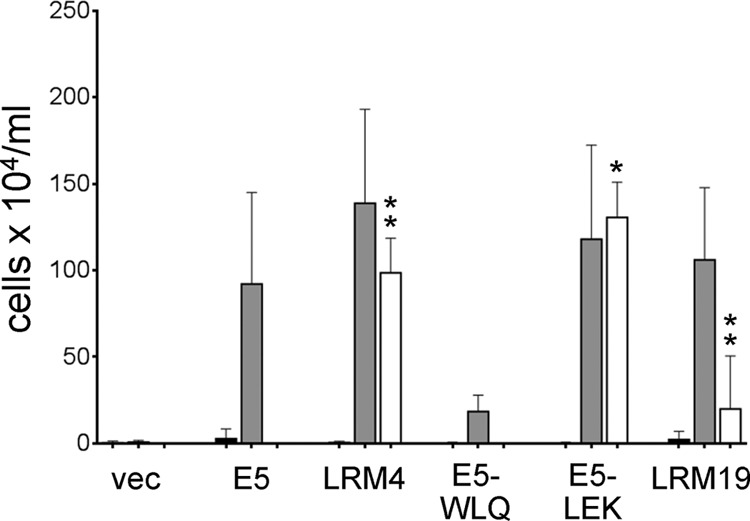
E5.LRM4 cooperates with a juxtamembrane PDGF β receptor mutant. BaF3 cells expressing the βαβ chimera (black bars), the wild-type PDGF β receptor (gray bars), or PR-K499D (white bars) were infected with retroviruses expressing no insert (vec), the wild-type E5 protein, or the indicated E5 mutant. Viable cells were counted and recorded as described in the legend of Fig. 2. Statistical significance of the results of the PR-K499D tests compared to the E5/PR-K499D combination is indicated by one asterisk (P ≤ 0.05) or two asterisks (P ≤ 0.01).
DISCUSSION
Specific interactions between transmembrane helices play an important role in the assembly of transmembrane protein complexes and in the proper folding of multipass transmembrane proteins. Because transmembrane proteins comprise a large fraction of the proteome, such interactions are essential for normal cellular function. However, studies of the molecular basis of specific recognition between membrane-spanning protein segments have been hindered by the difficulties in obtaining high-resolution structures of such segments and in conducting biochemical and biophysical experiments with intact transmembrane proteins or hydrophobic peptides. Despite these limitations, considerable progress has been made in studying the sequence basis for homodimerization of transmembrane helices, but the study of heteromeric interactions has lagged (2, 3, 38).
The small size of the BPV E5 protein and its ability to form a specific complex with the PDGF β receptor and activate it in trans provide a powerful genetic system to explore transmembrane domain interactions within mammalian cell membranes, because the sequences of the two interacting domains can be manipulated independently and the biological consequences of the interaction can be readily scored. To investigate the factors that control specific recognition between transmembrane domains, we selected E5 mutants, which can tolerate PDGF β receptor mutations that inhibit the interaction with the wild-type E5 protein.
We and others previously reported that some mutations in the transmembrane and juxtamembrane regions of the PDGF β receptor interfered with its recognition by the E5 protein (23, 24, 30). Here, we describe several additional, conservative mutations in the transmembrane domain of the receptor that inhibit E5-mediated receptor activation. These mutations substitute a hydrophobic amino acid for another hydrophobic amino acid (L512I and L512V) or one β-branched amino acid for another β-branched amino acid (T513I). The dramatic phenotypes caused by these relatively subtle, single-amino-acid substitutions emphasize the highly specific nature of the interaction between the E5 protein and the transmembrane domain of the PDGF β receptor. All of these mutant receptors responded to v-sis, which binds to the extracellular domain of the receptor, demonstrating that the natural ligand can tolerate mutations in the transmembrane domain of the receptor that interfere with the E5 protein.
The E5 mutants isolated here were selected for their ability to cooperate with a mutant receptor, PR-L512I, which does not respond to the wild-type E5 protein. The E5 mutations also restored binding to the mutant receptor, confirming that binding is required for activation of the mutant receptor and biological activity. These E5 mutants showed lower activity with mutants containing different substitutions at the same or a nearby position in the PDGF β receptor transmembrane domain, and one of them was active with a juxtamembrane receptor mutant. Thus, the compensatory E5 mutants were not simply hyperactive or promiscuous in their ability to recognize a broad range of receptor mutants but rather were specific for the transmembrane domain of the wild-type PDGF β receptor and the receptor mutant used in the original screen (and, in one case, a mutant with a distal mutation). These genetic results establish an essential feature of the interaction between the E5 protein and the PDGF β receptor. If a third protein mediated complex formation between the E5 protein and the PDGF β receptor, the L512I receptor mutation might inhibit complex formation by interfering with the ability of the PDGF β receptor to associate with this putative intermediary protein. However, this model cannot readily explain the ability of mutations in the E5 protein to specifically restore binding to the mutant receptor. Therefore, our results provide strong genetic evidence that the transmembrane domains of the E5 protein and the PDGF β receptor contact one another directly.
Our results also provide insight into rules that govern specific interactions between transmembrane domains. We previously isolated small, artificial proteins with totally randomized transmembrane domains that activated the PDGF β receptor, and we showed that one of them had more strict sequence specificity than the E5 protein itself (11), whereas one with an entirely different transmembrane sequence appeared to be less specific (10). However, because the transmembrane sequences of these proteins were unrelated to the sequence of the wild-type E5 protein, it was not possible to ascribe this altered specificity to discrete sequence changes in the viral protein. Therefore, we undertook the analysis described here, in which specificity was changed by a limited number of mutations in the E5 protein.
In the best-understood situations, helix-helix interactions in the hydrophobic core of a membrane are dominated by the fit of the helix surfaces with each other and by various types of interhelical polar interactions, including hydrogen bonds (e.g., see references 4, 38, and 39). In addition, since a beta-branched side chain (such as isoleucine and valine) has fewer rotational degrees of freedom than an unbranched one (such as leucine), differences in side chain branching can also introduce entropy terms. Thus, changes in side chain volume or orientation that alter the fit between helices, the addition or removal of potential hydrogen bonding groups, or alterations in beta branching can affect the total free energy of association. Most compensatory mutations between two helices are presumably best interpreted as acting locally, but it is formally possible that a loss of interaction energy at one location along the helix-helix interface could be compensated by a gain at another location, since the sum of all interactions governs the strength of association. Consistent with these considerations, the E5 mutations responsible for recognizing PR-L512I were clustered between positions 13 and 17 in both independent mutants, suggesting that the mutations exert a local effect, which permitted recognition of PR-L512I, possibly by allowing this segment of the E5 protein to interact directly with the mutant PDGF β receptor transmembrane domain in the vicinity of position 512. For example, Glu14 and Lys17 in E5-LEK may form a hydrophilic environment that favors the interaction with the polar threonine 513 in the PDGF β receptor, thereby minimizing the disruptive effect of the L512I mutation. This model would also explain why E5-LEK works poorly with PR-T513I and PR-T513L, which lack the threonine, and why E5-EK displays partial activity (Fig. 8B). However, the ability of E5-LEK to cooperate not only with PR-L512I but also with PR-K499D demonstrates that local interactions are not solely responsible for specific transmembrane recognition. It seems implausible that the middle of this E5 mutant is able to interact directly with the aspartic acid at position 499 in PR-K499D as well as with the PDGF β receptor transmembrane domain near position 512. Rather, we hypothesize that that the mutations in E5-LEK cause a conformational change in the E5 protein or alter the energetics of the interaction so that a salt bridge between Asp33 of E5 and Lys499 of the PDGF β receptor is no longer required for a productive interaction.
The model that substitutions in the E5 transmembrane domain can cause global structural alterations in the E5 protein that affect recognition of the PDGF β receptor is consistent with our finding that the glutamine-to-arginine difference at position 33 in E5.LRM4 and E5.LRM3 affected the ability of these otherwise identical variants to cooperate with PR-L512I (Fig. 5A). Similarly, our previous analysis of small proteins with randomized transmembrane domains showed that a single-amino-acid difference in a small artificial transmembrane protein could affect its interaction with PDGF β receptor mutants containing different mutations along the length of the receptor transmembrane domain (11).
Our results also indicate that the sequence basis for transmembrane domain recognition in this system is complex. There are at least two different combinations of mutations that allow the E5 protein to interact with the same mutant target, demonstrating that there is not a unique structural solution demanded by the starting transmembrane mutation in the PDGF β receptor. In one case, deletion of two adjacent small amino acids at a specific site conferred activity, whereas in the other, the combination of three different substitution mutations was required for full activity. Thus, specificity is not determined by a one-to-one correspondence between individual amino acids on the interacting helices, but rather, in the two mutants studied, several different mutations in the small protein are required to accommodate a single substitution in the target. Interestingly, although these two E5 mutants displayed similar patterns of activity with mutations at positions 512 and 513, only one of them cooperated with the distal K499D mutation, suggesting that the structural and/or energetic consequences of the two sets of E5 mutations are quite different. The availability of compensatory pairs of E5 and PDGF β receptor mutants should facilitate the development and evaluation of improved structural models of this interaction.
In addition, we found that mutations that allowed the E5 protein to recognize a mutant PDGF β receptor also allowed it to tolerate inhibitory C-terminal mutations in the E5 transmembrane domain itself. This unexpected finding provided further evidence that mutations in one portion of the E5 transmembrane domain can influence distal sequences within the same transmembrane domain. The structural basis for this behavior is not clear but again suggests that the rules governing transmembrane interactions are not restricted to local effects. For example, if the N-terminal mutations in the E5 mutant strengthen the interaction with the PDGF β receptor in the vicinity of position 512, this might compensate for C-terminal E5 mutations that otherwise weaken the interaction between these two proteins. Isolation and analysis of additional compensatory E5 mutants will reveal if the ability to recognize a mutant PDGF β receptor is consistently associated with the ability to tolerate distal mutations in the E5 protein itself.
Our analysis of E5 deletion mutants suggests that the correct spacing of Gln17 and Asp33 is critical for PDGF β receptor activation, perhaps to ensure the correct distance or orientation of these two residues to allow them to simultaneously engage particular sites on the PDGF β receptor (34, 35). A 2-amino-acid deletion between Gln17 and Asp33 would shorten the distance between these two residues by 3 Å and change their relative orientation by ∼200°, presumably disrupting their interaction with Thr513 and/or Lys499 in the PDGF β receptor transmembrane region. Notably, the glutamine and the aspartic (or glutamic) acid and the spacing between them are absolutely conserved in the E5 proteins encoded by all of the fibropapillomaviruses from different animal species, and the lysine and threonine in the PDGF β receptor are absolutely conserved in mammals, suggesting that these interactions have been conserved for millions of years of evolution since the divergence of the animal host species for these viruses. We also note that E5Δ7.8 displays moderate activity with PR-L512I. The deletions in E5Δ7.8 and E5Δ14.15 are 7 residues apart, two full turns of the helix. This would place the E5 segment upstream of position 7 and downstream of position 15 in the same relative orientation in the two mutants, which might contribute to their activity with PR-L512I.
Taken together, our results demonstrate that the rules that govern direct specific interactions between transmembrane helices are complex and that further analysis is required to understand the molecular basis of specific transmembrane domain recognition and activity. The genetic approach described here provides a route to explore these rules and could be extended to additional PDGF β receptor mutants and to other pairs of interacting transmembrane proteins, perhaps combined with high-throughput sequencing to identify a larger number of compensating mutations.
ACKNOWLEDGMENTS
We thank Dawn Mattoon for essential reagents; Lisa Petti, Francisco Barrera Olivares, and Donald Engelman for helpful discussions; and Jan Zulkeski for assistance in preparing the manuscript.
This work was supported by a grant from the NIH to D.D. (grant CA037157).
Footnotes
Published ahead of print 7 August 2013
REFERENCES
- 1.Lehnert U, Xia Y, Royce TE, Goh CS, Liu Y, Senes A, Yu H, Zhang ZL, Engelman DM, Gerstein M. 2004. Computational analysis of membrane proteins: genomic occurrence, structure prediction and helix interactions. Q. Rev. Biophys. 37:121–146. [DOI] [PubMed] [Google Scholar]
- 2.MacKenzie KR. 2006. Folding and stability of α-helical integral membrane proteins. Chem. Rev. 106:1931–1977. [DOI] [PubMed] [Google Scholar]
- 3.Schneider D, Finger C, Prodohl A, Volkmer T. 2007. From interactions of single transmembrane helices to folding of alpha-helical membrane proteins: analyzing transmembrane helix-helix interactions in bacteria. Curr. Protein Pept. Sci. 8:45–61. [DOI] [PubMed] [Google Scholar]
- 4.Lemmon MA, Flanagan JM, Treutlein HR, Zhang J, Engelman DM. 1992. Sequence specificity in the dimerization of transmembrane α-helices. Biochemistry 31:12719–12725. [DOI] [PubMed] [Google Scholar]
- 5.Call ME, Pyrdol J, Wiedmann M, Wucherpfennig KW. 2002. The organizing principle in the formation of the T cell receptor-CD3 complex. Cell 111:967–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cosson P, Lankford SP, Bonifacino JS, Klausner RD. 1991. Membrane protein association by potential intramembrane charge pairs. Nature 351:414–416. [DOI] [PubMed] [Google Scholar]
- 7.Manolios N, Bonifacino JS, Klausner RD. 1990. Transmembrane helical interactions and the assembly of the T cell receptor complex. Science 249:274–277. [DOI] [PubMed] [Google Scholar]
- 8.Cammett TJ, Jun SJ, Cohen EB, Barrera FN, Engelman DM, DiMaio D. 2010. Construction and genetic selection of small transmembrane proteins that activate the human erythropoietin receptor. Proc. Natl. Acad. Sci. U. S. A. 107:3447–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freeman-Cook L, Dixon AM, Frank JB, Xia Y, Ely L, Gerstein M, Engelman DM, DiMaio D. 2004. Selection and characterization of small random transmembrane proteins that bind and activate the platelet-derived growth factor β receptor. J. Mol. Biol. 338:907–920. [DOI] [PubMed] [Google Scholar]
- 10.Freeman-Cook LL, Edwards APB, Dixon AM, Yates KE, Ely L, Engelman DM, DiMaio D. 2005. Specific locations of hydrophilic amino acids in constructed transmembrane ligands of the platelet-derived growth factor β receptor. J. Mol. Biol. 345:907–921. [DOI] [PubMed] [Google Scholar]
- 11.Ptacek JB, Edwards APB, Freeman-Cook LL, DiMaio D. 2007. Packing contacts can mediate highly specific interactions between artificial transmembrane proteins and the PDGF β receptor. Proc. Natl. Acad. Sci. U. S. A. 104:11945–11950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scheideman EH, Marlatt SA, Xie Y, Hu Y, Sutton RE, DiMaio D. 2012. Transmembrane protein aptamers that inhibit CCR5 expression and HIV coreceptor function. J. Virol. 86:10281–10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schlegel R, Wade-Glass M, Rabson MS, Yang Y-C. 1986. The E5 transforming gene of bovine papillomavirus encodes a small hydrophobic protein. Science 233:464–467. [DOI] [PubMed] [Google Scholar]
- 14.Talbert-Slagle K, DiMaio D. 2009. The bovine papillomavirus E5 protein and the PDGF β receptor: it takes two to tango. Virology 384:345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldstein DJ, Andresson T, Sparkowski JJ, Schlegel R. 1992. The BPV-1 E5 protein, the 16 kDa membrane pore-forming protein and the PDGF receptor exist in a complex that is dependent on hydrophobic transmembrane interactions. EMBO J. 11:4851–4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petti L, DiMaio D. 1992. Stable association between the bovine papillomavirus E5 transforming protein and activated platelet-derived growth factor receptor in transformed mouse cells. Proc. Natl. Acad. Sci. U. S. A. 89:6736–6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drummond-Barbosa DA, Vaillancourt RR, Kazlauskas A, DiMaio D. 1995. Ligand-independent activation of the platelet-derived growth factor beta receptor: requirements for bovine papillomavirus E5-induced mitogenic signaling. Mol. Cell. Biol. 15:2570–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldstein DJ, Kulke R, DiMaio D, Schlegel R. 1992. A glutamine residue in the membrane-associating domain of the bovine papillomavirus type 1 E5 oncoprotein mediates its binding to a transmembrane component of the vacuolar H(+)-ATPase. J. Virol. 66:405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lai CC, Henningson C, DiMaio D. 1998. Bovine papillomavirus E5 protein induces oligomerization and trans-phosphorylation of the platelet-derived growth factor β receptor. Proc. Natl. Acad. Sci. U. S. A. 95:15241–15246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nilson LA, DiMaio D. 1993. Platelet-derived growth factor receptor can mediate tumorigenic transformation by the bovine papillomavirus E5 protein. Mol. Cell. Biol. 13:4137–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petti L, Nilson LA, DiMaio D. 1991. Activation of the platelet-derived growth factor receptor by the bovine papillomavirus E5 transforming protein. EMBO J. 10:845–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goldstein DJ, Li W, Wang L-M, Heidaran MA, Aaronson SA, Shinn R, Schlegel R, Pierce JH. 1994. The bovine papillomavirus type 1 E5 transforming protein specifically binds and activates the beta-type receptor for platelet-derived growth factor but not other tyrosine kinase-containing receptors to induce cellular transformation. J. Virol. 68:4432–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nappi VM, Petti LM. 2002. Multiple transmembrane amino acid requirements suggest a highly specific interaction between the bovine papillomavirus E5 oncoprotein and the platelet-derived growth factor beta receptor. J. Virol. 76:7976–7986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nappi VM, Schaefer JA, Petti LM. 2002. Molecular examination of the transmembrane requirements of the platelet-derived growth factor beta receptor for a productive interaction with the bovine papillomavirus E5 oncoprotein. J. Biol. Chem. 277:47149–47159. [DOI] [PubMed] [Google Scholar]
- 25.Petti L, DiMaio D. 1994. Specific interaction between the bovine papillomavirus E5 transforming protein and the beta receptor for platelet-derived growth factor in stably transformed and acutely transfected cells. J. Virol. 68:3582–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Staebler A, Pierce JH, Brazinski S, Heidaran MA, Li W, Schlegel R, Goldstein DJ. 1995. Mutational analysis of the beta-type platelet-derived growth factor receptor defines the site of interaction with the bovine papillomavirus type 1 E5 transforming protein. J. Virol. 69:6507–6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horwitz BH, Burkhardt AL, Schlegel R, DiMaio D. 1988. 44-amino-acid E5 transforming protein of bovine papillomavirus requires a hydrophobic core and specific carboxyl-terminal amino acids. Mol. Cell. Biol. 8:4071–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meyer AN, Xu Y-F, Webster MK, Smith AS, Donoghue DJ. 1994. Cellular transformation by a transmembrane peptide: structural requirements for the bovine papillomavirus E5 oncoprotein. Proc. Natl. Acad. Sci. U. S. A. 91:4634–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nilson LA, Gottlieb RL, Polack GW, DiMaio D. 1995. Mutational analysis of the interaction between the bovine papillomavirus E5 transforming protein and the endogenous beta receptor for platelet-derived growth factor in mouse C127 cells. J. Virol. 69:5869–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petti LM, Reddy V, Smith SO, DiMaio D. 1997. Identification of amino acids in the transmembrane and juxtamembrane domains of the platelet-derived growth factor receptor required for productive interaction with the bovine papillomavirus E5 protein. J. Virol. 71:7318–7327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burkhardt A, Willingham M, Gay C, Jeang K-T, Schlegel R. 1989. The E5 oncoprotein of bovine papillomavirus is oriented asymmetrically in Golgi and plasma membranes. Virology 170:334–339. [DOI] [PubMed] [Google Scholar]
- 32.Klein O, Kegler-Ebo D, Su J, Smith S, DiMaio D. 1999. The bovine papillomavirus E5 protein requires a juxtamembrane negative charge for activation of the platelet-derived growth factor beta receptor and transformation of C127 cells. J. Virol. 73:3264–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klein O, Polack GW, Surti T, Kegler-Ebo D, Smith SO, DiMaio D. 1998. Role of glutamine 17 of the bovine papillomavirus E5 protein in platelet-derived growth factor beta receptor activation and cell transformation. J. Virol. 72:8921–8932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.King G, Oates J, Patel D, van den Berg HA, Dixon AM. 2011. Towards a structural understanding of the smallest known oncoprotein: investigation of the bovine papillomavirus E5 protein using solution-state NMR. Biochim. Biophys. Acta 1808:1493–1501. [DOI] [PubMed] [Google Scholar]
- 35.Surti T, Klein O, Aschheim K, DiMaio D, Smith SO. 1998. Structural models of the bovine papillomavirus E5 protein. Proteins 33:601–612. [PubMed] [Google Scholar]
- 36.Naviaux RK, Costanzi E, Haas M, Verma IM. 1996. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol. 70:5701–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Talbert-Slagle K, Marlatt S, Barrera FN, Khurana E, Oates J, Gerstein M, Engelman DM, Dixon AM, DiMaio D. 2009. Artificial transmembrane oncoproteins smaller than the bovine papillomavirus E5 protein redefine the sequence requirements for activation of the platelet-derived growth factor beta receptor. J. Virol. 83:9773–9785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacKenzie KR, Fleming KG. 2008. Association energetics of membrane spanning alpha-helices. Curr. Opin. Struct. Biol. 18:412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.MacKenzie KR, Engelman DM. 1998. Structure-based prediction of the stability of transmembrane helix-helix interactions: the sequence dependence of glycophorin A dimerization. Proc. Natl. Acad. Sci. U. S. A. 95:3583–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]