

Abstract
Understanding the regulation of airway epithelial barrier function is a new frontier in asthma and respiratory viral infections. Despite recent progress, little is known about how respiratory syncytial virus (RSV) acts at mucosal sites, and very little is known about its ability to influence airway epithelial barrier function. Here, we studied the effect of RSV infection on the airway epithelial barrier using model epithelia. 16HBE14o- bronchial epithelial cells were grown on Transwell inserts and infected with RSV strain A2. We analyzed (i) epithelial apical junction complex (AJC) function, measuring transepithelial electrical resistance (TEER) and permeability to fluorescein isothiocyanate (FITC)-conjugated dextran, and (ii) AJC structure using immunofluorescent staining. Cells were pretreated or not with protein kinase D (PKD) inhibitors. UV-irradiated RSV served as a negative control. RSV infection led to a significant reduction in TEER and increase in permeability. Additionally it caused disruption of the AJC and remodeling of the apical actin cytoskeleton. Pretreatment with two structurally unrelated PKD inhibitors markedly attenuated RSV-induced effects. RSV induced phosphorylation of the actin binding protein cortactin in a PKD-dependent manner. UV-inactivated RSV had no effect on AJC function or structure. Our results suggest that RSV-induced airway epithelial barrier disruption involves PKD-dependent actin cytoskeletal remodeling, possibly dependent on cortactin activation. Defining the mechanisms by which RSV disrupts epithelial structure and function should enhance our understanding of the association between respiratory viral infections, airway inflammation, and allergen sensitization. Impaired barrier function may open a potential new therapeutic target for RSV-mediated lung diseases.
INTRODUCTION
Respiratory syncytial virus (RSV) is the most common respiratory pathogen in infants and young children (1) and an important cause of death in childhood (2). RSV has been identified as a source of morbidity and mortality in elderly and high-risk adults (3). RSV infects airway epithelial cells and is thought to cause tissue pathology by inducing the expression of proinflammatory mediators, leading to airway inflammation and, ultimately, an antiviral immune response (4). RSV also induces the expression of antiapoptotic genes and promotes epithelial cell survival, which is probably a strategy to ensure viral replication in infected cells (5).
Emerging evidence points to a role for airway barrier dysfunction during respiratory viral infections (6), as well as in stable asthmatics (7). The airway barrier is made up of the surface mucus layer, as well as apical junction complexes (AJC) that regulate paracellular permeability (8). Previously we demonstrated that polyinosinic-polycytidylic acid [poly(I-C)], a synthetic double-stranded RNA and viral mimetic, induces potent breakdown of the airway epithelial AJC in a protein kinase D (PKD)-dependent manner (9). PKD, formerly known as PKCμ, is a serine/threonine protein kinase family consisting of three isoforms (PKD1 to -3) (10). The PKD family is involved in a number of important cell functions, including survival, migration, differentiation, proliferation, and membrane trafficking (11). Interestingly, PKD was recently shown to be an upstream regulator of cortactin, an actin binding protein involved in actin polymerization and regulation of junctional structures in other cell types (12, 13). Although activation of epithelial PKC plays a role in the early stages of RSV infection (14, 15), we have limited understanding of the expression and function of PKD in epithelial cells in the context of naturally occurring viral infections. Furthermore, whether cortactin-dependent actin polymerization is involved in AJC disassembly in the airway is not known.
In the current study we sought to address these gaps in our knowledge by studying the effect of RSV infection on airway epithelial AJC structure and function. We tested the hypothesis that RSV mediates AJC disassembly and remodeling of the perijunctional F-actin cytoskeleton in a PKD-dependent manner. We show that RSV induces potent breakdown of AJC structure and function in the absence of cell death, and we propose a model in which RSV replication leads to sustained PKD activation, phosphorylation of cortactin, actin remodeling, and AJC disassembly. These findings provide new knowledge about RSV effects on the airway barrier and identify new pharmacologic targets to explore in the treatment of RSV-induced lung infections.
MATERIALS AND METHODS
Antibodies.
The following primary monoclonal antibodies (MAbs) and polyclonal antibodies (PAbs) were used to detect junctional and signaling proteins by immunofluorescent labeling and immunoblotting: anti-occludin, anti-zonula occludens protein 1 (ZO-1), and anti-E-cadherin MAbs (Invitrogen, Camarillo, CA); anti-β-catenin MAb (BD Bioscience); anti-Toll-like receptor 3 (TLR3) (Abcam, Cambridge, MA); phospho-PKD/PKC (Ser744/748) and PKD/PKCμ and cleaved caspase 3 PAb (Cell Signaling, Danvers, Mass); phospho-cortactin PAb (anti-cortactin [pS405] phosphospecific PAb) (Protea Bioscience, San Jose, CA); and anti-cortactin (p80/85) clone 4F11 MAb (Millipore, Billerica, MA). Fluorescently labeled phalloidin 594 and 488 (Invitrogen, Camarillo, CA) were used to visualize actin filaments. Anti-rabbit and anti-mouse secondary antibodies conjugated to Alexa-488 or Alexa-568 were obtained from Invitrogen (Camarillo, CA). Mouse and rabbit horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from GE Healthcare (Fairfield, CT).
Chemicals and reagents.
The PKC inhibitors Gö6976 and Gö6983 were obtained from EMD Biosciences (Billerica, MA). The PKC inhibitor GF109203X was obtained from Calbiochem. The PKD inhibitor CID755673 was a kind gift from Elizabeth R. Sharlow (University of Virginia; it is also commercially available from Tocris [number 3327]). Fluorescein-conjugated 3-kDa dextran was obtained from Invitrogen (Camarillo, CA). Palivizumab (Synagis) was obtained from MedImmune.
Airway epithelial cell culture.
16HBE14o- human bronchial epithelial cells (a gift from Dieter C. Gruenert, University of California—San Francisco) were cultured in collagen-coated transwells as previously described (9). The cells were cultured on 0.4-μm- or 5.0-μm-pore-size filters (Transwell; Costar, Cambridge, MA) for immunofluorescent staining and permeability studies, respectively. For biochemical experiments, the cells were cultured in 24-well plastic plates.
Respiratory syncytial virus.
Polarized human airway epithelial cells were infected apically with RSV derived from RSV strain A2 (a kind gift from Edward E. Walsh, University of Rochester, Rochester, NY). In some experiments, we used rgRSV (RSV derived from RSV A2 expressing the green fluorescent protein gene), a kind gift from Mark Peeples (Nationwide Children's Hospital Research Institute, Columbus, OH) and Peter Collins (National Institutes of Health, Bethesda, MD), as described previously (16, 17). UV-inactivated RSV (UV-RSV) was used as a negative control. UV inactivation was performed by exposure of RSV to UVB irradiation for 20 min. Inactivation of RSV replication was confirmed by a plaque-forming assay.
TEER.
Transepithelial electrical resistance (TEER) was measured with an EVOMX volt-ohm meter (World Precision Instruments, Sarasota, FL) as previously described (9). Data are presented as percent changes compared to time zero. Experiments were performed when TEER reached >500 Ω × cm2.
Paracellular flux of fluorescent markers.
Permeability assays were performed by measuring the flux of apically added fluorescein isothiocyanate (FITC)-conjugated 3-kDa dextran (Invitrogen, Camarillo, CA) from the apical to the basal chamber across epithelial monolayers (9). The results were expressed as the fold changes from baseline.
Cell cytotoxicity and apoptosis assays.
All cultures were monitored daily for cytopathic effect by light microscopy. Following the indicated time points, supernatant was collected for cytotoxicity assay by measuring the release of the cytosolic enzyme lactate dehydrogenase (LDH) extracellularly (LDH detection kit; Clontech, CA) (9).
To analyze cell death by apoptosis, protein was extracted and Western blotting was performed with a cleaved caspase 3 PAb (Cell Signaling, Danvers, MA).
Immunofluorescence staining of junctional proteins.
As in previous studies (9), after the indicated treatments, cell monolayers were fixed and analyzed by immunofluorescence microscopy. The fixed cells were incubated with specific primary antibodies directed against ZO-1, occludin, E-cadherin, and β-catenin, followed by incubation with Alexa Fluor-labeled secondary antibodies and mounting with Prolong Gold antifade mounting medium (Invitrogen, Camarillo, CA). Immunofluorescently labeled cell monolayers were examined with a scanning confocal microscope. Single-plane confocal images were taken with an Olympus FV1000 laser scanning confocal microscope at the University of Rochester Medical Center Confocal and Conventional Microscopy Core. A 100× U Plan S Apo 1.4-numerical-aperture (NA) (oil) objective was used, and images were taken at 512 by 512 resolution with a Kalman setting of 14. All images were optimized so that the fluorescence intensity remained in the linear range. Images were processed with Adobe Photoshop software.
Analysis of TJ proteins by immunoblotting.
After the indicated treatments, cell monolayers grown in cell culture plates were washed with cold phosphate-buffered saline (PBS) and lysed in RIPA lysis buffer (Sigma). The protein concentration was quantified by bicinchoninic acid protein assay (Pierce, Cheshire, United Kingdom), followed by resolution on SDS-PAGE and transfer to polyvinylidene difluoride (PVDF) membranes (Bio-Rad). The membranes were incubated overnight at 4°C with the indicated primary antibodies and then for 1 h at room temperature with horseradish peroxidase-conjugated secondary antibodies. The blots were exposed to enhanced chemiluminescence (ECL) (RPN 2106; GE Healthcare, Fairfield, CT) and subjected to autoradiography. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Abcam, Cambridge, MA) was used as a lane-loading control. Densitometric quantification was performed using NIH Image J software.
Extraction of RNA and quantitative real-time PCR analysis.
Total RNA was extracted from epithelial cells using an E.Z.N.A total RNA kit (Omega Bio-Tek, Norcross, GA), and cDNA was synthesized using an iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA) and amplified by real-time PCR with a iQ5 multicolor Real-Time PCR Detection System (Bio-Rad, Hercules, CA) using iQ SYBR green Supermix (Bio-Rad, Hercules, CA) and primers targeting the 87-bp sequence of the RSV strain A2 genome, which encodes the viral fusion (F) protein (5′ CACCCTGTTGGAAAC 3′ and 5′ CTCTGTCAGTTCTTG 3′; Sigma Aldrich).
Statistical analysis.
Data were analyzed using Prism software (GraphPad, San Diego, CA) and Microsoft Excel. The data are representative of three or more experiments and are means ± standard errors of the mean (SEM). The data were evaluated statistically with analysis of variance (ANOVA) and the Student t test, with Bonferroni correction for multiple comparisons. Significance was considered to be a P value of less than 0.05.
RESULTS
RSV infection decreased resistance and increased the permeability of polarized airway epithelial cells.
RSV is known to infect human and mouse airway epithelial cells, leading to cytokine/chemokine production and airway inflammation (4). We previously showed that poly(I-C), a mimic of viral infection, induced paracellular permeability of airway epithelial cells (9). Here, we hypothesized that RSV can disrupt AJC. To test this possibility, we used polarized 16HBE140- human bronchial epithelial cells, which maintain tight junctions (TJ) and adherens junctions (AJ) similarly to primary human bronchial epithelial cells (6, 9). We began by growing 16HBE cells on semipermeable inserts (see Materials and Methods) and infected them apically with RSV strain A2 (courtesy of Edward Walsh, University of Rochester, Rochester, NY) at multiplicities of infection (MOIs) ranging from 0.05 to 1.0. RSV induced a marked decrease in TEER, consistent with barrier disruption (Fig. 1A). The effect of RSV was both dose and time dependent, resulting in sustained decreases in TEER for up to 72 h (Fig. 1A). RSV infection also led to significant increases in the paracellular flux of fluorescein-conjugated 3-kDa dextran (Fig. 1B), indicating increased permeability to macromolecules. In subsequent studies, we used RSV at an MOI of 0.5.
Fig 1.
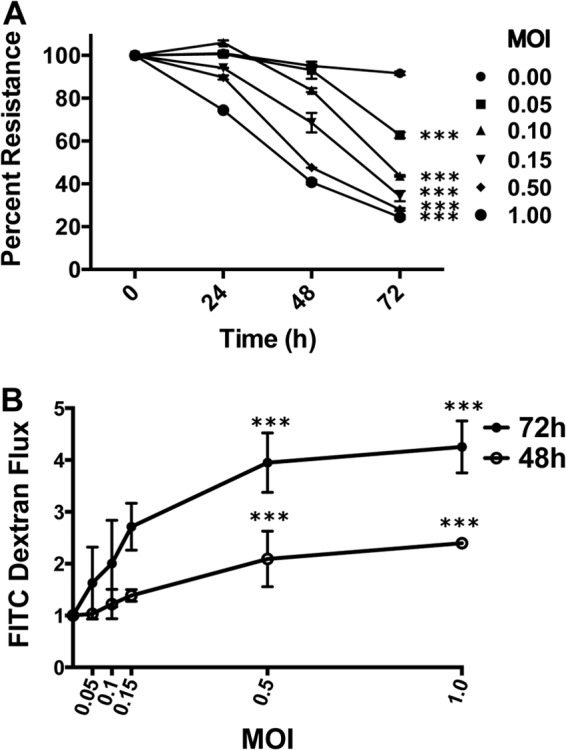
RSV infection disrupts the barrier function of airway epithelial cells. Confluent epithelial cell monolayers were infected with RSV at MOIs of 0.05 to 1.0 for 72 h. (A) TEER was measured with a volt-ohm meter at different time points after infection. RSV causes a dose- and time-dependent decrease in TEER when data are expressed relative to TEER measured at time zero (values of approximately 500 Ω × cm2, which was set as 100%). (B) Permeability assays were performed with 3-kDa fluorescein-conjugated dextran at 48 and 72 h post-RSV infection. The data show that RSV causes a time- and dose-dependent increase in permeability. The data are representative of 2 or 3 independent experiments. Differences between control and RSV-infected cells were analyzed by ANOVA. ***, P < 0.001. The error bars indicate SEM.
RSV infection did not induce cell cytotoxicity or apoptosis.
To investigate whether RSV infection induced cytotoxicity or apoptosis in epithelial cells, we infected cells with RSV at an MOI of 0.5 and collected cell supernatants at different time points for cytotoxicity assays (see Materials and Methods). As a positive control, we treated the cells with 2% Triton, whereas cell culture medium was used as a negative control. Importantly, similar to other reports (5, 18, 19), we found no evidence of RSV-induced cell cytotoxicity (Fig. 2A). In an independent experiment, confluent polarized epithelial cells were infected with RSV (MOI, 0.5), and whole-cell lysates were analyzed by Western blotting for cleaved caspase 3 as a marker of apoptosis. We found no evidence of apoptosis in RSV-infected epithelia (Fig. 2B).
Fig 2.

RSV infection does not induce cell cytotoxicity or apoptosis. (A) 16HBE140- cell monolayers were infected with RSV at an MOI of 0.5 or with control medium for 0 to 48 h. (A) Results showing cytotoxicity indexes of infected cells compared with untreated cells. Medium alone was used as a negative control, and cells lysed with Triton served as a positive control. The error bars indicate SEM. (B) Confluent polarized epithelial cells were infected with RSV at an MOI of 0.5 or with control medium. The cell lysates were immunoblotted and probed with antibodies against cleaved caspase 3. Cell lysate from cytochrome c-treated and untreated Jurkat cells (Cell Signaling) were used as positive (+con) and negative (−con) controls. A representative blot from 3 independent experiments is shown.
RSV infection induces AJC disassembly in airway epithelial cells, which requires live replicating virus.
We next examined the effect of RSV or UV-inactivated RSV on AJC structure and function. Polarized16HBE140- cell monolayers were infected apically with live RSV (MOI, 0.5) or equivalent amounts of UV-irradiated RSV, and the AJC structure was determined using immunofluorescence labeling and confocal microscopy for different TJ (occludin and ZO-1) or AJ (E-cadherin and β-catenin) proteins. In comparison with control uninfected cells, which showed a normal “chicken wire” appearance of AJ and TJ structure, RSV-infected cells exhibited marked disruption of the AJC, with evidence of gaps and breaks in junctional strands (Fig. 3A, arrowheads). In spite of the breakdown of the AJC, epithelial nuclei remained intact (data not shown).
Fig 3.

RSV-induced AJC disassembly requires live replicating virus. Polarized 16HBE140- monolayers were infected with RSV at an MOI of 0.5, control medium, or an equal amount of UV-irradiated RSV for 48 h. (A) Apical junction complexes were visualized by immunofluorescent staining and confocal microscopy. The arrows indicate normal chicken wire appearance of membrane AJC. The arrowheads indicate gaps in AJC integrity and ZO-1 localized to intracellular vacuoles. (B) TEER was measured using a volt-ohm meter at the indicated time points. UV-inactivated RSV did not cause epithelial junction disassembly or changes in TEER. The images are representative of at least 3 independent experiments. Differences between control, UV-irradiated RSV-, and RSV-infected cells were analyzed by ANOVA. ***, P < 0.001. The error bars indicate SEM. (C) RSV infection did not change AJC protein expression. Total cell lysates of control and RSV-infected epithelial cells were analyzed by Western blotting with antibodies directed against different TJ and AJ components. Immunoblots representative of different time points postinfection are shown. (D) Densitometric quantification of at least 3 independent experiments.
Interestingly, UV-irradiated RSV did not disrupt apical junction structure, and in contrast to nonirradiated intact virus, UV-RSV also did not induce junctional dysfunction, as determined by measuring TEER (Fig. 3B) and paracellular flux of fluorescein-conjugated 3-kDa dextran (data not shown).
RSV infection induced PKD phosphorylation.
In other cell types, apical junction complex dysfunction can occur via suppression of expression of junctional components or by expression-independent mechanisms. Western blotting of whole-cell lysates revealed that total expression of key junctional components was largely unaltered following RSV infection (Fig. 3C and D). This contrasted with marked disruption of AJC formation at the apical membrane as detected using confocal microscopy (Fig. 3A). Expression-independent AJC disruption often involves junctional internalization driven by actin cytoskeletal remodeling (20). When we analyzed actin structure in RSV-infected epithelial cells, we found marked redistribution of cortical actin fibers (Fig. 4A).
Fig 4.

RSV infection induces redistribution of cortical actin fibers and PKD phosphorylation. Confluent polarized epithelial cells were infected with RSV at an MOI of 0.5 or with medium control at the indicated time points. (A) The actin cytoskeleton was visualized by fluorescently labeled phalloidin and confocal microscopy. (B) Cell lysates were analyzed by Western blotting with antibodies against phospho-PKD (Ph-PKD)/PKC (Ser744/748), total PKD, and GAPDH. RSV infection induced PKD phosphorylation beginning at 3 h, with another peak at 24 h. The expression of total PKD also slightly increased at later time points (≥24 h). The images are representative of 3 independent experiments. (C) Densitometric quantification of at least 3 independent experiments for Ph-PKD and PKD Western blotting using RSV as shown in panel B. (D) Polarized epithelial cells were infected with UV-irradiated RSV. Like live virus, UV-irradiated virus led to increased expression of phospho-PKD at early time points. However, the second peak was not observed in UV-inactivated RSV, confirming that the late effect requires live replicating virus. (E) Densitometric quantification of at least 3 independent experiments using UV-irradiated virus as shown in panel D. The error bars indicate SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
PKD is now known to play an important role in cell motility and cytoskeletal rearrangements (11, 12), but very little is known about this signaling molecule in the airway. We examined activation of PKD in RSV-infected epithelial cells by monitoring PKD phosphorylation (Ser744/748) by Western blotting. Interestingly, RSV infection resulted in biphasic PKD phosphorylation with both early (2- to 3-h) and late (48-h) kinetics (Fig. 4B and C). Only the early wave of PKD phosphorylation occurred using UV-inactivated RSV (Fig. 4D and E), suggesting that this early phosphorylation depended on viral entry whereas the later phase required viral replication (see below).
RSV-induced epithelial permeability is mediated by PKD.
Previous studies have shown the role of PKC signaling in epithelial cell tight-junction integrity (20), and activation of different PKC family members plays a role in the early stages of RSV infection (14, 15). In order to determine whether these protein kinases contributed to RSV-induced AJC dysfunction, we used a panel of inhibitors targeting both classical and atypical PKC family members (Gö6983 and GF109203X), as well as PKD (Gö6976). Interestingly, only Gö6976 (Fig. 5A), and not other inhibitors (data not shown), attenuated the effect of RSV infection on epithelial permeability and AJC structure. To confirm the role of PKD signaling in RSV-induced disrupted barrier structure, we next used a novel benzoxoloazepinolone PKD inhibitor (CID755673, provided by E. Sharlow, University of Virginia [UVA]), which is selective, cell permeable, and structurally unrelated to Gö6976 (21, 22). This compound also strikingly prevented RSV-induced barrier disruption, confirming a role for PKD in RSV-induced AJC disassembly (Fig. 5A) and increased permeability (Fig. 5B). Similarly RSV, but not UV-RSV, upregulated expression of the key viral sensor TLR3 (Fig. 6A and B). This indicates that live replicating virus is required to induce epithelial junction dysfunction. Interestingly the PKD antagonist also blocked the upregulation of TLR3 (Fig. 6A).
Fig 5.

RSV-induced epithelial junction disassembly is mediated by PKD. (A) Confluent 16HBE140- cell monolayers were infected with RSV at an MOI of 0.5 for 48 h in the presence or absence of two structurally unrelated PKD inhibitors (Gö6976 [10 μM; Calbiochem] and CID755673 [10 μM; Tocris]). TJ and AJ were visualized by immunofluorescent staining and confocal microscopy. Note the intact AJ and TJ staining in control noninfected cells (arrows), which was disrupted in RSV-infected cells (arrowheads). AJ and TJ surface expression were restored in RSV-infected cells pretreated with two structurally unrelated PKD inhibitors. (B) Cells were infected with RSV at an MOI of 0.5 for 48 h in the presence or absence of PKD inhibitors, and a permeability assay with 3-kDa fluorescein-conjugated dextran was performed at 48 h posttreatment. Both inhibitors significantly attenuated the barrier-disrupting effects of RSV. The images are representative of at least 3 independent experiments. The data are presented as means plus SEM of 3 experiments. ***, P < 0.001.
Fig 6.
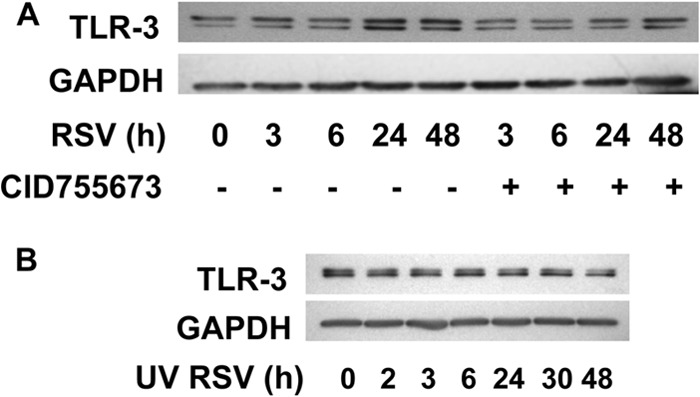
RSV induces upregulation of TLR3, which requires infection with live replicating virus and is inhibited by PKD inhibitor. (A) Confluent polarized epithelial cells were infected with RSV at an MOI of 0.5 or with medium control at the indicated time points. Cell lysates were immunoblotted and probed with antibodies against TLR3. (B) UV irradiation of RSV did not change virus-induced TLR expression. Polarized epithelial cells were infected with equal amounts of UV-irradiated RSV, and the protein was subjected to Western blot analysis. No change in TLR3 expression was noted with UV irradiation, which again indicates the requirement for live replicating virus in this process. The images are representative of at least 3 independent experiments.
RSV-induced phosphorylation of cortactin is dependent on PKD signaling.
To further dissect the mechanism of RSV-induced AJC disruption, we sought to examine the molecular events involved in AJC disruption induced by RSV and dependent on PKD. PKD was recently shown to be an upstream regulator of cortactin, an actin binding protein involved in actin polymerization (12, 13). We next assessed the expression of total cortactin and phospho-cortactin (Ser405) at different time points following RSV infection. Similar to the case of PKD, RSV infection resulted in a biphasic pattern of cortactin phosphorylation (Fig. 7A and B). Importantly, both phases of cortactin phosphorylation were dependent upon PKD, but only the early phase occurred using UV-RSV (Fig. 7C and D). Confocal microscopy revealed increased colocalization of actin and cortactin in response to RSV infection (Fig. 7E, yellow staining). Taken together, these data support a model in which RSV infection and replication induce sustained PKD phosphorylation, leading to cortactin-dependent remodeling of the actin cytoskeleton, destabilizing membrane apical junction complexes.
Fig 7.

RSV infection induced phosphorylation of cortactin, which was inhibited by a PKD inhibitor. Confluent polarized epithelial cells were infected with RSV at an MOI of 0.5 or with medium control at the indicated time points. Cell lysates were analyzed by Western blotting with antibodies directed against phospho-cortactin (pS405), total cortactin (p80/85; clone 4F11), and GAPDH. (A) RSV infection caused cortactin phosphorylation, which was blocked by PKD inhibition. (B) Densitometric quantification of at least 3 independent experiments as shown in panel A. The error bars indicate SEM. **, P < 0.01. (C) UV inactivation of RSV changed the pattern of cortactin phosphorylation. Polarized epithelial cells were infected with UV-irradiated RSV. Like live virus, irradiated virus led to expression of phospho-cortactin at early time points. However, the second peak was not observed in UV-inactivated RSV, confirming that the late effect requires live replicating virus. (D) Densitometric quantification of at least 3 independent experiments as shown in panel C. *, P < 0.05; ***, P < 0.001. (E) Confocal images of epithelial cells show marked redistribution of cortical actin fibers and redistribution of cortactin in RSV-infected cells into intracellular vesicles. The bottom row shows increased colocalization of actin and cortactin in response to RSV infection (yellow staining). The data are representative of 3 independent experiments.
PKD inhibitor did not inhibit early RSV infection or RSV replication.
One possible explanation for the observed effects of PKD inhibitors is that they prevent RSV uptake or replication. To test this, epithelial cells were infected with RSV at a multiplicity of infection of 0.05 to 0.5 in the presence or absence of PKD inhibition. Palivizumab, a known monoclonal antibody against RSV fusion protein, was used as a positive control for suppression of viral infection. Importantly, we observed no significant differences in RSV infectivity in the presence of PKD inhibition (Fig. 8A and B). In addition, PKD inhibition did not affect viral replication, as similar viral loads were recovered from RSV-infected cells in the presence or absence of the inhibitor (Fig. 8C).
Fig 8.

PKD inhibitor did not inhibit early RSV infection or viral replication in epithelial cells. Polarized epithelial cells were preincubated with PKD inhibitor for 2 h, followed by infection with rgRSV at an MOI of 0.05 to 0.5, and rinsed twice with PBS after 2 h, followed by incubation at 37°C for 16 h. Palivizumab, a monoclonal antibody against RSV fusion (F) protein, was used as a positive control, which was also added 2 h before RSV infection. Sixteen hours post-RSV infection, live cells were subjected to fluorescence microscopy. (A and B) GFP+ cells are readily apparent after rgRSV infection, which was completely inhibited using pavilizumab. In contrast, the PKD inhibitor did not affect the number of GFP+ cells. A representative example (A, lower right) and averages of 3 experiments (B) are shown. (C) In parallel, inhibition of PKD did not affect RSV replication, as determined in cells infected with RSV A2 and by RT-PCR with primers that amplify the RSV A2 F protein. The images are representative of 3 independent experiments. The data are representative of three experiments (n = 3 per experiment). The error bars indicate SEM.
DISCUSSION
Apical junctional complexes are an integral part of the airway epithelial barrier. Although epithelial barrier dysfunction is increasingly associated with disease states, including asthma (7), the molecular mechanisms involved are not well understood. Tight junctions and adherens junctions are dynamic structures that are responsive to diverse environmental stimuli (23). Here, we show that RSV, a common respiratory virus associated with substantial morbidity, induces epithelial barrier dysfunction in a PKD-dependent manner. RSV infection causes barrier dysfunction without altering the expression of key junctional components and instead appears to induce disassembly of epithelial junction complexes by destabilizing the cortical actin cytoskeleton in a cortactin-dependent manner. Our results highlight a novel molecular pathway of airway barrier dysfunction that likely contributes to RSV-induced airway inflammation.
Respiratory viruses, including rhinovirus (RV) and RSV, have been previously shown to cause airway epithelial barrier dysfunction via different mechanisms. Hershenson and colleagues have shown that RV causes epithelial junction dysfunction in a NOX1-dependent manner (6, 24), whereas Singh et al. showed that RSV infection reduced transepithelial electrical resistance across airway epithelial monolayers (25). Our results clearly show that RSV infection leads to marked disassembly of AJC, resulting in increased paracellular permeability (Fig. 1A and B). Inducible junction dysfunction may promote viral replication by allowing paracellular escape of basolateral viral particles (26). We speculate that another consequence of virus-induced epithelial permeability might be increased “outside-in” translocation of inhaled particles that are deposited in the airway. In the case of inhaled aeroallergens, increased paracellular translocation facilitates uptake by intraepithelial dendritic cells and may help explain the association between respiratory viral infection and allergen sensitization in asthma.
The observation that RSV also paradoxically promotes epithelial cell survival (5, 18, 19, 27) raises the intriguing possibility that a “leaky airway” may persist after acute infection. At present, there are no assays of outside-in airway epithelial barrier function in human subjects in routine clinical use, but development of such assays may prove useful in the future to determine the clinical significance of the mechanisms we report here.
Originally known as PKCμ, this molecule was renamed PKD because it has a different structure and substrate specificity than other PKC family members (10, 28). In other cell types, PKD regulates cell shape and motility, in part by controlling actin dynamics, but it has not been well studied in the lung (12, 29–32). A recent study uncovered a key role for PKD in a mouse model of hypersensitivity pneumonitis (33), and the Caenorhabiditis elegans PKD homolog DKF-2 was found to regulate intestinal innate immunity (34). Thus, PKD may play a broader role in regulating airway mucosal immune responses, and further study of the expression and function of this signaling molecule in asthma and other airway diseases seems warranted. The recent development of selective benzoxoloazepinolone PKD inhibitor (Fig. 5 and 7) should accelerate research in this area (22, 35–37). The compound is a potent and cell-permeable inhibitor of PKD. Although any chemical inhibitor might have potential off-target effects, we used two structurally unrelated antagonists that prevented RSV-induced junctional disassembly, as well as PKD phosphorylation, thus fulfilling two key criteria of inhibitor specificity (38).
We found that RSV, similar to the synthetic double-stranded RNA poly(I-C) (9), induced phosphorylation of PKD on Ser744/748, a key step in PKD activation (39). Both RSV infection and exposure disrupted apical junction complexes in a PKD-dependent manner, and both also led to PKD-dependent cortactin phosphorylation (Fig. 7 and data not shown). Thus, even though they engage different pattern recognition receptors, RSV and poly(I-C) converge on PKD and cortactin to cause epithelial barrier dysfunction.
Our kinetic analysis uncovered temporally distinct patterns of activation of signaling molecules in response to RSV and UV-inactivated RSV, which allowed us to construct the following model. UV-RSV induced only the early phase of PKD and cortactin phosphorylation (Fig. 4D and 7C) without affecting AJC structure or function (Fig. 3A and B). In contrast, nonirradiated RSV induced both early and sustained PKD activation and cortactin phosphorylation (Fig. 4B and 7A). This suggests that live replicating virus is required to cause sustained PKD activation and sufficient cortactin-dependent actin remodeling to cause AJC disassembly. This model is consistent with earlier work by Monick et al. showing that RSV induced biphasic PKC and MAPK activation in A549 cells (14). Since RSV (and not UV-RSV) also resulted in upregulation of TLR3 (Fig. 6A and B), which we previously found was responsible in part for poly(I-C)-induced AJC disassembly (9), we speculate that sustained viral replication generates a positive-feedback loop amplifying signaling modules that lead to barrier dysfunction.
RSV was discovered more than 50 years ago and continues to be a chief cause of hospitalization worldwide, not only in infants, but also in older and high-risk adults (3). RSV infection in childhood has been linked to chronic inflammation and asthma (40–42), and although we have a very detailed understanding of RSV immunopathogenesis (43–47), current treatment strategies for RSV infection are largely supportive. Palivizumab, which is approved for RSV prophylaxis in high-risk infants, has only moderately decreased hospital admission due to RSV infection (48, 49). Here, we report a new pathway of RSV-induced junctional complex disassembly that likely contributes to airway epithelial barrier dysfunction and inflammation in vivo. Therapeutic strategies that interfere with virus-induced barrier dysfunction and restore epithelial barrier integrity may hold promise in the future.
ACKNOWLEDGMENTS
We thank Dieter C. Gruenert (University of California) for providing 16HBE14o- and Elizabeth R. Sharlow (University of Virginia) for providing protein kinase D inhibitor (CID755673). We gratefully acknowledge Edward E. Walsh for providing RSVA2 and for his helpful suggestions during the course of these studies. We gratefully acknowledge Mark Peeples (Nationwide Children's Hospital Research Institute, OH) and Peter Collins (NIH) for providing the rgRSV244. We thank Linda Callahan and Paivi Jordan (URMC Confocal Imaging Core) for their helpful assistance.
This work was supported by NIH T32 HD057821 (F.R.), NIH K12HD068373 (F.R.), NIH R01 HL071933 (S.N.G.), NIH P30 ES001247 (S.N.G.), NIH DK084953 (A.I.I.), DK083968 (A.I.I.), and NIH F32 HL110718 (T.J.C.).
All of the authors approved the final submitted version of the manuscript.
Footnotes
Published ahead of print 7 August 2013
REFERENCES
- 1.Hall CB, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, Auinger P, Griffin MR, Poehling KA, Erdman D, Grijalva CG, Zhu Y, Szilagyi P. 2009. The burden of respiratory syncytial virus infection in young children. N. Engl. J. Med. 360:588–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nair H, Nokes DJ, Gessner BD, Dherani M, Madhi SA, Singleton RJ, O'Brien KL, Roca A, Wright PF, Bruce N, Chandran A, Theodoratou E, Sutanto A, Sedyaningsih ER, Ngama M, Munywoki PK, Kartasasmita C, Simoes EA, Rudan I, Weber MW, Campbell H. 2010. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet 375:1545–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. 2005. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 352:1749–1759. [DOI] [PubMed] [Google Scholar]
- 4.Lotz MT, Peebles RS., Jr 2012. Mechanisms of respiratory syncytial virus modulation of airway immune responses. Curr. Allergy Asthma Rep. 12:380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Domachowske JB, Bonville CA, Mortelliti AJ, Colella CB, Kim U, Rosenberg HF. 2000. Respiratory syncytial virus infection induces expression of the anti-apoptosis gene IEX-1L in human respiratory epithelial cells. J. Infect. Dis. 181:824–830. [DOI] [PubMed] [Google Scholar]
- 6.Sajjan U, Wang Q, Zhao Y, Gruenert DC, Hershenson MB. 2008. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am. J. Respir. Crit. Care Med. 178:1271–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiao C, Puddicombe SM, Field S, Haywood J, Broughton-Head V, Puxeddu I, Haitchi HM, Vernon-Wilson E, Sammut D, Bedke N, Cremin C, Sones J, Djukanovic R, Howarth PH, Collins JE, Holgate ST, Monk P, Davies DE. 2011. Defective epithelial barrier function in asthma. J. Allergy Clin. Immunol. 128:549–556. [DOI] [PubMed] [Google Scholar]
- 8.Niessen CM. 2007. Tight junctions/adherens junctions: basic structure and function. J. Investig. Dermatol. 127:2525–2532. [DOI] [PubMed] [Google Scholar]
- 9.Rezaee F, Meednu N, Emo JA, Saatian B, Chapman TJ, Naydenov NG, De Benedetto A, Beck LA, Ivanov AI, Georas SN. 2011. Polyinosinic:polycytidylic acid induces protein kinase D-dependent disassembly of apical junctions and barrier dysfunction in airway epithelial cells. J. Allergy Clin. Immunol. 128:1216–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valverde AM, Sinnett-Smith J, Van Lint J, Rozengurt E. 1994. Molecular cloning and characterization of protein kinase D: a target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc. Natl. Acad. Sci. U. S. A. 91:8572–8576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eiseler T, Schmid MA, Topbas F, Pfizenmaier K, Hausser A. 2007. PKD is recruited to sites of actin remodelling at the leading edge and negatively regulates cell migration. FEBS Lett. 581:4279–4287. [DOI] [PubMed] [Google Scholar]
- 12.Eiseler T, Hausser A, De Kimpe L, Van Lint J, Pfizenmaier K. 2010. Protein kinase D controls actin polymerization and cell motility through phosphorylation of cortactin. J. Biol. Chem. 285:18672–18683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bougneres L, Girardin SE, Weed SA, Karginov AV, Olivo-Marin JC, Parsons JT, Sansonetti PJ, Van Nhieu GT. 2004. Cortactin and Crk cooperate to trigger actin polymerization during Shigella invasion of epithelial cells. J. Cell Biol. 166:225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monick M, Staber J, Thomas K, Hunninghake G. 2001. Respiratory syncytial virus infection results in activation of multiple protein kinase C isoforms leading to activation of mitogen-activated protein kinase. J. Immunol. 166:2681–2687. [DOI] [PubMed] [Google Scholar]
- 15.San-Juan-Vergara H, Peeples ME, Lockey RF, Mohapatra SS. 2004. Protein kinase C-alpha activity is required for respiratory syncytial virus fusion to human bronchial epithelial cells. J. Virol. 78:13717–13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hallak LK, Spillmann D, Collins PL, Peeples ME. 2000. Glycosaminoglycan sulfation requirements for respiratory syncytial virus infection. J. Virol. 74:10508–10513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rezaee F, Gibson LF, Piktel D, Othumpangat S, Piedimonte G. 2011. Respiratory syncytial virus infection in human bone marrow stromal cells. Am. J. Respir. Cell Mol. Biol. 45:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Groskreutz DJ, Monick MM, Yarovinsky TO, Powers LS, Quelle DE, Varga SM, Look DC, Hunninghake GW. 2007. Respiratory syncytial virus decreases p53 protein to prolong survival of airway epithelial cells. J. Immunol. 179:2741–2747. [DOI] [PubMed] [Google Scholar]
- 19.Bitko V, Shulyayeva O, Mazumder B, Musiyenko A, Ramaswamy M, Look DC, Barik S. 2007. Nonstructural proteins of respiratory syncytial virus suppress premature apoptosis by an NF-kappaB-dependent, interferon-independent mechanism and facilitate virus growth. J. Virol. 81:1786–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ivanov AI. 2008. Actin motors that drive formation and disassembly of epithelial apical junctions. Front. Biosci. 13:6662–6681. [DOI] [PubMed] [Google Scholar]
- 21.George A, Pushkaran S, Li L, An X, Zheng Y, Mohandas N, Joiner CH, Kalfa TA. 2010. Altered phosphorylation of cytoskeleton proteins in sickle red blood cells: the role of protein kinase C, Rac GTPases, and reactive oxygen species. Blood Cells Mol. Dis. 45:41–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharlow ER, Giridhar KV, LaValle CR, Chen J, Leimgruber S, Barrett R, Bravo-Altamirano K, Wipf P, Lazo JS, Wang QJ. 2008. Potent and selective disruption of protein kinase D functionality by a benzoxoloazepinolone. J. Biol. Chem. 283:33516–33526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turner JR. 2009. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 9:799–809. [DOI] [PubMed] [Google Scholar]
- 24.Comstock AT, Ganesan S, Chattoraj A, Faris AN, Margolis BL, Hershenson MB, Sajjan US. 2011. Rhinovirus-induced barrier dysfunction in polarized airway epithelial cells is mediated by NADPH oxidase 1. J. Virol. 85:6795–6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh D, McCann KL, Imani F. 2007. MAPK and heat shock protein 27 activation are associated with respiratory syncytial virus induction of human bronchial epithelial monolayer disruption. Am. J. Physiol. Lung Cell. Mol. Physiol. 293:L436–L445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walters RW, Freimuth P, Moninger TO, Ganske I, Zabner J, Welsh MJ. 2002. Adenovirus fiber disrupts CAR-mediated intercellular adhesion allowing virus escape. Cell 110:789–799. [DOI] [PubMed] [Google Scholar]
- 27.Zhang L, Peeples ME, Boucher RC, Collins PL, Pickles RJ. 2002. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J. Virol. 76:5654–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishikawa K, Toker A, Johannes FJ, Songyang Z, Cantley LC. 1997. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J. Biol. Chem. 272:952–960. [DOI] [PubMed] [Google Scholar]
- 29.Sinnett-Smith J, Rozengurt N, Kui R, Huang C, Rozengurt E. 2011. Protein kinase D1 mediates stimulation of DNA synthesis and proliferation in intestinal epithelial IEC-18 cells and in mouse intestinal crypts. J. Biol. Chem. 286:511–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sinnett-Smith J, Jacamo R, Kui R, Wang YM, Young SH, Rey O, Waldron RT, Rozengurt E. 2009. Protein kinase D mediates mitogenic signaling by Gq-coupled receptors through protein kinase C-independent regulation of activation loop Ser744 and Ser748 phosphorylation. J. Biol. Chem. 284:13434–13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ziegler S, Eiseler T, Scholz RP, Beck A, Link G, Hausser A. 2011. A novel protein kinase D phosphorylation site in the tumor suppressor Rab interactor 1 is critical for coordination of cell migration. Mol. Biol. Cell 22:570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matthews SA, Navarro MN, Sinclair LV, Emslie E, Feijoo-Carnero C, Cantrell DA. 2010. Unique functions for protein kinase D1 and protein kinase D2 in mammalian cells. Biochem. J. 432:153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim YI, Park JE, Brand DD, Fitzpatrick EA, Yi AK. 2010. Protein kinase D1 is essential for the proinflammatory response induced by hypersensitivity pneumonitis-causing thermophilic actinomycetes Saccharopolyspora rectivirgula. J. Immunol. 184:3145–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren M, Feng H, Fu Y, Land M, Rubin CS. 2009. Protein kinase D is an essential regulator of C. elegans innate immunity. Immunity 30:521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bravo-Altamirano K, George KM, Frantz MC, Lavalle CR, Tandon M, Leimgruber S, Sharlow ER, Lazo JS, Wang QJ, Wipf P. 2011. Synthesis and structure-activity relationships of benzothienothiazepinone inhibitors of protein kinase D. ACS Med. Chem. Lett. 2:154–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.George KM, Frantz MC, Bravo-Altamirano K, Lavalle CR, Tandon M, Leimgruber S, Sharlow ER, Lazo JS, Wang QJ, Wipf P. 2011. Design, synthesis, and biological evaluation of PKD inhibitors. Pharmaceutics 3:186–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharlow ER, Mustata Wilson G, Close D, Leimgruber S, Tandon M, Reed RB, Shun TY, Wang QJ, Wipf P, Lazo JS. 2011. Discovery of diverse small molecule chemotypes with cell-based PKD1 inhibitory activity. PLoS One 6:e25134. 10.1371/journal.pone.0025134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cohen P. 2010. Guidelines for the effective use of chemical inhibitors of protein function to understand their roles in cell regulation. Biochem. J. 425:53–54. [DOI] [PubMed] [Google Scholar]
- 39.Iglesias T, Waldron RT, Rozengurt E. 1998. Identification of in vivo phosphorylation sites required for protein kinase D activation. J. Biol. Chem. 273:27662–27667. [DOI] [PubMed] [Google Scholar]
- 40.Sigurs N, Bjarnason R, Sigurbergsson F, Kjellman B. 2000. Respiratory syncytial virus bronchiolitis in infancy is an important risk factor for asthma and allergy at age 7. Am. J. Respir. Crit. Care Med. 161:1501–1507. [DOI] [PubMed] [Google Scholar]
- 41.Stein RT, Sherrill D, Morgan WJ, Holberg CJ, Halonen M, Taussig LM, Wright AL, Martinez FD. 1999. Respiratory syncytial virus in early life and risk of wheeze and allergy by age 13 years. Lancet 354:541–545. [DOI] [PubMed] [Google Scholar]
- 42.Peebles RS., Jr 2004. Viral infections, atopy, and asthma: is there a causal relationship? J. Allergy Clin. Immunol. 113:S15–S18. [DOI] [PubMed] [Google Scholar]
- 43.Becker S, Reed W, Henderson FW, Noah TL. 1997. RSV infection of human airway epithelial cells causes production of the beta-chemokine RANTES. Am. J. Physiol. 272:L512–L520. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y, Luxon BA, Casola A, Garofalo RP, Jamaluddin M, Brasier AR. 2001. Expression of respiratory syncytial virus-induced chemokine gene networks in lower airway epithelial cells revealed by cDNA microarrays. J. Virol. 75:9044–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Piedimonte G, Hegele RG, Auais A. 2004. Persistent airway inflammation after resolution of respiratory syncytial virus infection in rats. Pediatr. Res. 55:657–665. [DOI] [PubMed] [Google Scholar]
- 46.King KA, Hu C, Rodriguez MM, Romaguera R, Jiang X, Piedimonte G. 2001. Exaggerated neurogenic inflammation and substance P receptor upregulation in RSV-infected weanling rats. Am. J. Respir. Cell Mol. Biol. 24:101–107. [DOI] [PubMed] [Google Scholar]
- 47.Piedimonte G. 2003. Contribution of neuroimmune mechanisms to airway inflammation and remodeling during and after respiratory syncytial virus infection. Pediatr. Infect. Dis. J. 22:S66–S75. [DOI] [PubMed] [Google Scholar]
- 48.Bauer G, Bossi L, Santoalla M, Rodriguez S, Farina D, Speranza AM. 2009. Impact of a respiratory disease prevention program in high-risk preterm infants: a prospective, multicentric study. Arch. Argentin. Pediatr. 107:111–118. (In Spanish). [DOI] [PubMed] [Google Scholar]
- 49.IMpact-RSV Study Group. 1998. Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. Pediatrics 102:531–537. [PubMed] [Google Scholar]