

Abstract
Interferon (IFN)-mediated innate immune defense is a potent antiviral mechanism. Viruses evade innate immunity and limit secretion of beta interferon (IFN-β) to replicate and survive in the host. The largest tegument protein of herpes simplex virus 1 (HSV-1), UL36, contains a novel deubiquitinase (DUB) motif embedded in its N terminus, denoted UL36 ubiquitin-specific protease (UL36USP). In the present study, we demonstrate that HSV-1 UL36USP inhibits Sendai virus (SeV)-induced interferon regulatory factor 3 (IRF3) dimerization, promoter activation, and transcription of IFN-β. The DUB activity of UL36USP is essential to block IFN-β production. UL36USP also inhibited IFN-β promoter activity induced by overexpression of the N terminus of RIG-I (RIG-IN) and MAVS, but not TBK-1, IκB kinase ε (IKKε), and IRF3/5D. UL36USP was subsequently shown to deubiquitinate TRAF3 and prevent the recruitment of the downstream adaptor TBK1. The recombinant HSV-1 lacking UL36USP DUB activity was generated. Cells infected with the mutant virus produced more IFN-β than wild-type (WT) HSV-1-infected cells. These findings demonstrate HSV-1 UL36USP removes polyubiquitin chains on TRAF3 and counteracts the IFN-β pathway.
INTRODUCTION
Innate immunity is the first line of host defense against pathogen invasion. The type I interferon (IFN-I) system plays a crucial role for vertebrates in controlling viral infections. Pattern recognition receptors (PRRs) of the host cells mediate the innate recognition of viruses and initiate a series of signaling cascades, activating the transcription factors NF-κB and interferon regulatory factors 3 and 7 (IRF3 and IRF7). The activated NF-κB and IRF3/7 collaborate to trigger the expression of IFN-I, which upregulates a diverse set of interferon-stimulated genes (ISGs) and protects host cells against the invading virus (1–4).
Ubiquitination is a widely used posttranslational protein modification that regulates many physiological processes, including immune responses (5, 6). Ubiquitination has a crucial role in regulating the RIG-I signaling pathway. It is reported that ubiquitin (Ub) ligase tripartite motif-containing protein 25 (TRIM25) and RNF135 catalyze K63-linked polyubiquitination of RIG-I, and this enhances the binding of RIG-I to MAVS (IPS-1/Cardiff/VISA) (7, 8). Downstream of RIG-I, K63-linked polyubiquitination of TRAF3 recruits the kinases TBK1 and IκB kinase ε (IKKε), leading to IRF3 phosphorylation and subsequent IFN-I production (9–13). TRIM56 stimulates K63-linked polyubiquitination of STING, helping to recruit TBK1 (14, 15). TBK1 and IKKε also undergo polyubiquitination, which has been suggested to promote IRF3 activation (16, 17). Furthermore, TRIM23 is involved in polyubiquitination of NEMO, enhancing beta interferon (IFN-β) production (18).
Herpes simplex virus 1 (HSV-1) is the archetypal member of the Alphaherpesvirinae subfamily, with a large, linear double-stranded DNA (dsDNA) virus genome of about 152 kb. HSV-1 is an extremely successful human pathogen and has evolved multiple immune evasion strategies that allow it to exist for the lifetime of its host. For example, HSV-1 ICP0 targets IRF3 and blocks IFN production (19–22). Previous studies from our lab have demonstrated that varicella-zoster virus (VZV) immediate early protein open reading frame 61 (ORF61), the homologue of HSV-1 ICP0, antagonizes the IFN-β pathway by degradation of activated IRF3 (23), and HSV-1 US11 serves as a novel antagonist of the IFN-β pathway via direct binding to RIG-I and MDA-5 (24). HSV-1 ICP34.5 binds and sequesters TBK-1 to inhibit IFN production (25, 26). The virion host shutoff (vhs) protein of HSV-2 suppresses IFN and ISG induction by degrading cellular mRNA (27, 28). ICP27 was also suggested to inhibit IFN production, and HSV-1 lacking functional ICP27 induces higher levels of IFN-α and IFN-β in macrophages than wild-type (WT) virus does (29). HSV-1 US3 is suggested to play an important role in immune evasion during HSV-1 infection, and US3 null HSV-1 resulted in strong activation of IRF3 and IFN-I responses (30).
The largest tegument protein of HSV-1, VP1/2, the product of the UL36 gene, is essential for HSV-1 replication and is conserved across the Herpesviridae family. VP1/2, a large multifunctional protein, plays crucial roles in HSV-1 entry, capsid transport, and virion assembly, formation of mature virions, microtubule transport of capsids, neuroinvasion, pathogenesis, etc. (31–41). Kattenhorn et al. have identified an approximately 500-amino-acid peptide that exhibits unique deubiquitinase (DUB) activity (denoted as UL36USP, for UL36 ubiquitin-specific protease), which is embedded within the N-terminal region of HSV-1 VP1/2 (42). UL36USP is detectable as early as 12 h postinfection and only after cleavage of UL36USP from full-length UL36. HSV-1 UL36USP is highly specific for ubiquitin and cleaves K48- and K63-linked polyubiquitin chains but not ubiquitin-like proteins, such as SUMO 1, Nedd8, or ISG15 (38, 42–44). A purified UL36USP expressed in Escherichia coli has also been shown to specifically bind to ubiquitin and cleave ubiquitin-based substrates. UL36USP contains the core catalytic residues, including C65 (in HSV), that are required for its deubiquitinase activity (42).
Homologues of HSV-1 UL36USP have been confirmed in several other members of herpesviruses, including pseudorabies virus (PRV) (45), Marek's disease virus (46), human cytomegalovirus (HCMV) (44, 47), murine cytomegalovirus (MCMV) (48), simian cytomegalovirus (47), Epstein-Barr virus (EBV) (38), Kaposi's sarcoma-associated herpesvirus (KSHV), (49), and mouse herpesvirus strain 68 (MHV68) (50). Although the precise role of UL36USP remains unclear, its strict conservation through all Herpesviridae subfamilies suggests an important role during viral infection.
Ubiquitination has a crucial role in regulating the innate immune response. Thus, it is not surprising that viruses have evolved strategies to manipulate the Ub ligation process to thwart host defense mechanisms (51, 52). It was recently reported that EBV-encoded BPLF1 deubiquitinates TRAF6 to inhibit NF-κB signaling during lytic infection, leading to promotion of viral lytic DNA replication (53). However, the mechanism by which HSV-1 UL36USP is involved in immune evasion is still poorly understood. In this study, we demonstrate that ectopic expression of UL36USP significantly downregulates Sendai virus (SeV)-activated IFN-β promoter activity and that the deubiquitinase activity of UL36USP is indispensable for the inhibitory activity. Additionally, UL36USP is demonstrated to cleave both the K63- and K48-linked polyubiquitin chains of TRAF3 and abrogate TRAF3 mediation of IFN-β production. Finally, a recombinant HSV-1 lacking deubiquitinase activity, denoted C40A HSV-1, was generated, and infection by mutant HSV-1 resulted in higher production of IFN-β. These findings reveal a novel mechanism for HSV-1 to evade the host's antiviral immunity.
MATERIALS AND METHODS
Cells, viruses, and antibodies.
HEK293T cells, Vero cells, and HeLa cells were grown in Dulbecco's modified Eagle medium (DMEM) (Gibco-BRL) supplemented with 10% fetal bovine serum (FBS) and 100 U/ml of penicillin and streptomycin. The WT HSV-1 F strain and its derivative UL36USP mutant HSV-1 strain were propagated in Vero cells and titrated as described previously (54). Sendai virus (SeV) was propagated and titrated as previously described (24). The protease inhibitor cocktail mixture was purchased from CST (Boston, MA). Mouse anti-Myc, anti-Flag, and antihemagglutinin (anti-HA) monoclonal antibodies (MAbs) were purchased from ABmart (Shanghai, China). Mouse monoclonal IgG1 and IgG2b isotype control antibodies were purchased from eBioscience, Inc. (San Diego, CA). Rabbit anti-TRAF3 polyclonal antibody (pAb), rabbit anti-Ub pAb, and mouse anti-β-actin MAb were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit antibody against IRF3-S396 was previously described (23).
Plasmid construction.
All enzymes used for cloning procedures, except for T4 DNA ligase (New England BioLabs, MA), were purchased from TaKaRa (Dalian, China). To construct UL36USP-Flag, the N-terminal 500-amino-acid (aa) region of UL36 was amplified from plasmid UL36-EYFP (expressing enhanced yellow fluorescent protein [EYFP]) as previously described (55) and cloned into the HindIII and EcoRI sites of the pCMV-Flag vector. Commercial reporter plasmids include NF-κB-Luc (expressing luciferase (Luc) (Stratagene, La Jolla, CA) and pRL-TK plasmid (expressing thymidine kinase [TK]) (Promega). Gift plasmids include the following: (PRDIII-I)4-Luc (56), pcDNA3.1-FlagTBK1 and pcDNA3.1/Zeo-MAVS (57), pcDNA3.1-FlagIKKε (58), pEF-Flag-RIG-IN (59), IRF3/5D (60), pCAGGS-NS1 (61), and IFN-β promoter reporter plasmid p125-luc (62).
RNA isolation, semiquantitative RT-PCR, and quantification of gel image.
Total RNA was extracted from HEK293T cells with TRIzol (Invitrogen, CA) according to the manufacturer's manual. Samples were digested with DNase I and subjected to reverse transcription (RT) (63). The cDNA was used as a template for semiquantitative PCR to investigate the expression patterns of human IFN-β, ISG54, and ISG56. The details of the protocols have been described previously (23).
Transfection and dual luciferase reporter (DLR) assay.
HEK293T cells were plated on 24-well dishes (Corning, NY) in DMEM (Gibco-BRL, MD) with 10% FBS at a density of 1 × 105 cells per well overnight before transfection, as previously described (23). Cells were then cotransfected with 1 μg expression plasmid, 500 ng reporter plasmid, such as those expressing IFN-β-Luc, NF-κB-Luc, or (PRDIII-I)4-Luc, and 50 ng of pRL-TK Renilla luciferase reporter plasmid to normalize transfection efficiency, as indicated by standard calcium phosphate precipitation (64, 65). At 24 h posttransfection, cells were infected with SeV (100 hemagglutination units [HAU]/ml) for 16 h, and then luciferase assays were performed as previously described (23) with a luciferase assay kit (Promega, Madison, WI). Poly(I · C) and poly(dA-dT) were purchased from InvivoGen.
Co-IP and WB analysis.
Coimmunoprecipitation (co-IP) assays were performed as previously described (54). Briefly, HEK293T cells (∼5 ×106) were cotransfected with 10 μg of the indicated expression plasmids. Transfected cells were harvested at 24 h posttransfection and lysed on ice with 500 μl of lysis buffer. The lysates were incubated with the indicated antibodies and 30 μl of a 1:1 slurry of protein A/G PLUS-agarose (Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C. The beads were washed four times with 1 ml of lysis buffer containing 500 mM NaCl, and Western blot (WB) analysis was performed to detect the interaction of proteins. The co-IP assays were repeated twice; a typical blot is shown.
WB analysis was performed as previously described (54). Briefly, the protein samples were subjected to 10% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) or nitrocellulose membranes, followed by blocking with 5% nonfat milk in Tris-buffered saline-Tween (TBST) and probed with the indicated primary antibodies at 37°C for 2 h. After being washed with TBST, the membrane was incubated with alkaline phosphatase (AP)-conjugated goat anti-rabbit IgG or goat anti-mouse IgG. Protein bands specific to the antibody were developed by 5-bromo-4-chloro-3-indolylphosphate (BCIP)-nitroblue tetrazolium (NBT) and terminated by distilled water.
Native PAGE.
Native PAGE was carried out using ReadyGels (7.5%; Bio-Rad). The gel was prerun with 25 mM Tris and 192 mM glycine (pH 8.4), with 1% deoxycholate (DOC) in the cathode chamber for 30 min at 40 mA. Samples in native sample buffer (10 μg protein, 62.5 mM Tris-Cl [pH 6.8], 15% glycerol, 1% DOC) were size fractionated by electrophoresis for 60 min at 25 mA and transferred to nitrocellulose membranes for WB analysis as previously described (66).
Recombinant virus construction.
Two-step Red-mediated recombination was applied to construct UL36(C40A) HSV-1 (67). The Kanr cassette was amplified by PCR with a pair of primers containing 40-bp-homology flanking sequence of mutant sites. Then the PCR product was transformed into E. coli GS1783 competent cells carrying the pHSV-1 bacterial artificial chromosome (BAC) via electroporation. PCR assays were used to identify the positive clones. l-Arabinose was used to induce the second Red recombination to delete the Kanr cassette (see Fig. 5). To analyze the integrity of the BAC clones, 15 μl of BAC DNA was digested with HindIII or BamHI, and the restriction pattern of BAC DNA was compared to that of WT BAC (data not shown). The recombinant viruses were validated by PCR and sequencing using primers upstream or downstream of the UL36 mutant sites (data not shown). Then viruses were harvested, and the growth kinetics of the recombinant viruses were characterized by both traditional plaque assay and luciferase activity assay in Vero cells at a multiplicity of infection (MOI) of 0.1 or 1. The luciferase activity assay was performed with a luciferase assay kit (Promega, Madison, WI).
Fig 5.

The UL36USP C40A mutation reduces the replication of recombinant virus in HEK293T cells. (A) Schematic diagram of construction of the C40A HSV-1 BAC. Cys40 of UL36 was substituted for by Ala in the HSV-1 genome. Confluent Vero cells were infected with the indicated viruses at MOI of 0.1 (B) and 1.0 (C). Growth curves were generated by luciferase activity assays (B) and traditional plaque assays (C). (D, E, and F) HEK293T cells were infected with WT HSV-1 or C40A HSV-1 at an MOI of 0.1. Growth curves were generated by luciferase activity assays. (D) The expression of ICP0, UL42, and UL46 was detected by Western blotting. (E) The transcription levels of ISG54 and ISG56 were detected by semiquantitative PCR (F).
ELISA for IFN-β.
An enzyme-linked immunosorbent assay (ELISA) to quantify secreted IFN-β was carried out with culture supernatants collected from infected cells as previously described (24). Briefly, cell culture medium was collected and centrifuged to remove cell debris. A human IFN-β ELISA kit (PBL InterferonSource, Piscataway, NJ) was used to detect the IFN-β according to the manufacturer's instructions. Four-week-old female C57BL/6 mice were purchased from the Experimental Animal Center, Wuhan Institute of Virology, Chinese Academy of Sciences. The mice were injected with 106 PFU of the indicated virus. After 24 h, the mice were sacrificed, and a Legend Max mouse IFN-β ELISA kit (BioLegend, San Diego, CA) was used to detect the IFN-β in serum. The animal study proposal was approved by the Institutional Animal Care and Use Committee (IACUC) of the Experimental Animal Center, Wuhan Institute of Virology, Chinese Academy of Sciences. The approved protocol no. is IACUC2013012.
RESULTS
UL36USP inhibits the SeV-mediated activation of IFN-β and interferon-stimulated response element promoter activity.
To investigate the function of UL36USP in the regulation of virus-mediated activation of IFN-β promoter, UL36USP was coexpressed in HEK293T cells in the presence of IFN-β reporter, and the pRL-TK Renilla luciferase reporter plasmid. DLR assays showed that Sendai virus (SeV) infection led to an ∼170-fold induction of the IFN-β-Luc reporter activity. Ectopic expression of UL36USP significantly suppressed SeV-mediated activation of the IFN-β promoter activity (Fig. 1A). The NS1 served as a positive control. Furthermore, UL36USP also inhibited the poly(I · C)-induced IFN-β promoter activity (Fig. 1B). It has been reported that the core catalytic residues, including C65 (in HSV-1), are important for the deubiquitinase activity of UL36USP, and the mutation of Cys65 to Ala abolishes its deubiquitinase activity (42). However, in the HSV-1 F strain, we found that residue Cys40 is required for the DUB activity of UL36USP. In Fig. 1C, ubiquitination level of total protein was significantly reduced by transfection of UL36USP but not the C40A mutant. In order to determine whether the DUB activity of UL36USP is required for the inhibition of IFN production, C40A mutants were generated. DLR assays showed that ectopic expression of C40A did not affect the SeV-mediated activation of the IFN-β promoter activity (Fig. 1D).
Fig 1.

HSV-1 UL36USP inhibits SeV-mediated IFN-β induction. UL36USP inhibits activation of IFN-β promoter, dimerization of IRF3, and transcription of ISGs. (A) HEK293T cells were cotransfected with IFN-β-Luc reporter plasmid, the pRL-TK control plasmid along with empty vector, and plasmids encoding UL36USP or influenza virus NS1 protein. Twenty-four hours after transfection, cells were infected with 100 HAU/ml SeV or mock infected, luciferase activity was measured 16 h postinfection, and fold activation was determined compared to that of the empty vector with mock infection. (B) UL36USP inhibited poly(I · C)-induced IFN-β promoter activity. IFN-β-Luc, pRL-TK, TLR3, UL36USP, and empty vector were transfected as indicated. After 24 h, 100 ng/ml poly(I · C) was transfected as indicated. Luciferase activity was measured as in panel A. (C) HEK293T cells were cotransfected with HA-Ub and control vector, UL36USP, or UL36USP with the C40A mutant. Western blotting was performed to examine the ubiquitination of total cell lysates. (D) DLR assays showed that the C40A mutant did not inhibit the SeV-mediated activation of the IFN-β promoter activity. (E) HEK293T cells were transfected with the UL36USP and C40A expression plasmid. Twenty-four hours posttransfection, cells were mock infected or infected with SeV for 16 h. Whole-cell extracts were subjected to native PAGE and probed with anti-IRF3 antibody to detect IRF3 dimerization. (F) Semiquantitative RT-PCR analysis was then performed to detect the mRNA levels of ISG54 and ISG56.
IRF3 is a key transcription factor in the IFN-β signaling pathway, and its dimerization is a hallmark of the early activation of the antiviral response. Native PAGE assays were performed to examine whether UL36USP inhibited IRF3 dimerization. As shown in Fig. 1E, IRF3 existed as a monomer in mock-treated cells, and SeV infection induced the dimerization of IRF3. However, the presence of UL36USP inhibited the SeV-mediated IRF3 dimer formation. Ectopic expression of C40A failed to inhibit dimerization of IRF3.
The IFN-I induces a diverse set of IFN-stimulated genes (ISGs) to mediate the innate antiviral response. Whether UL36USP inhibited SeV-induced transcription of ISG54 and ISG56 was detected by semiquantitative PCR. Expression of wild-type UL36USP significantly inhibited SeV-induced ISG54 and ISG56 mRNA expression, whereas the presence of the C40A mutant did not affect the expression of ISG54 and ISG56 (Fig. 1F). Taken together, these results indicate that UL36USP was sufficient to inhibit SeV-mediated activation of IFN-β activities and that the DUB activity of UL36USP was required for its inhibitory activity.
UL36 inhibits the IFN-β signaling pathway at the level between MAVS and TBK1.
The transcription of the IFN-β gene required transcription factor IRF3 and NF-κB binding to distinct regulatory domains in the IFN-β promoter. To investigate whether UL36USP inhibited the SeV-induced activation of IRF3 and NF-κB, reporter gene assays were performed in HEK293 cells using the luciferase reporter plasmid driven by the tandem IRF binding sites PRD(III-I) and the NF-κB regulatory element from the IFN-β promoter. The result showed that SeV infection induces strong IRF-responsive PRD(III-I) and NF-κB promoter activities, and transfection of UL36USP remarkably inhibited both SeV-induced PRD(III-I) (Fig. 2A) and NF-κB (Fig. 2B) reporter activities.
Fig 2.
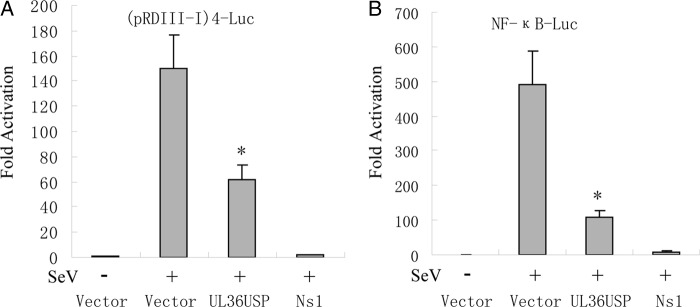
UL36USP inhibits SeV-mediated IRF3 and NF-κB promoter activation. HEK293T cells were cotransfected with either pRDIII-I-Luc (A) or NF-κB reporter plasmid (B) along with pRL-TK control plasmid and empty vector or plasmids encoding the indicated viral proteins. Twenty-four hours after transfection, cells were infected with 100 HAU/ml SeV or mock infected for 16 h, and luciferase activity was measured and fold activation was determined compared to those of the empty vector with mock infection.
To determine at what level in the pathway UL36USP blocked IFN-β production, UL36USP, and expression plasmids of RIG-I signaling pathway components, including the active CARD domain-containing form of RIG-I (RIG-IN), MAVS, IKKε kinase, TBK1 kinase, or the active form of IRF3 (IRF3/5D), were cotransfected into HEK293T cells. All expression constructs resulted in a 110- to 800-fold induction of the IFN-β-Luc reporter activity (Fig. 3A to E). IFN-β promoter activation driven by RIG-IN or MAVS was inhibited about 90% by UL36USP (Fig. 3A and B). Whereas UL36USP could not significantly suppress the IFN-β promoter activation driven by TBK1, IKKε, or IRF3/5D (Fig. 3C to E). These results suggested that UL36USP inhibited the IFN antiviral response at the level between MAVS and TBK1.
Fig 3.

UL36USP inhibits the IFN-β promoter activity between the level of MAVS and TBK1/IKKε. HEK293T cells were cotransfected with IFN-β-Luc reporter, PRL-TK and RIG-IN (A), IPS-1 (B), TBK1 (C), IKKε (D), or IRF3/5D (E) expression plasmids along with UL36USP expression plasmid. Luciferase activity was analyzed as described for Fig. 1A. The data represent means ± standard deviations for three replicates. Statistical analysis was performed using the t test. *, P < 0.05.
UL36USP deubiquitinates TRAF3 and inhibits recruitment of TBK1.
In the RLR-mediated signaling pathway, TRAF3 links upstream IFN signaling responses of IPS-1 (MAVS/Cardiff/VISA) to TBK1. The K63-linked polyubiquitination of TRAF3 is crucial for signaling by MAVS and recruitment of the kinases TBK1 and IKKε (9, 11). Thus, we investigated the possibility that UL36USP cleaved the polyubiquitin chain of TRAF3. In the co-IP experiment, TRAF3-Flag, Ub-HA, and UL36USP or the C40A expression plasmid were cotransfected into HEK293T cells. Following SeV infection, TRAF3 was immunoprecipitated by anti-Flag antibody, and the Western blot assay was performed to detect the ubiquitination of TRAF3. As shown in Fig. 4A, the presence of UL36USP reduced the SeV-induced ubiquitination of TRAF3, whereas the C40A mutant did not affect the ubiquitination level of TRAF3. Then co-IP assays were carried out to investigate whether UL36USP removed the K63- or K48-linked polyubiquitin chain of TRAF3. TRAF3-Flag was transfected with the linkage-specific ubiquitin HA-K63-Ub or HA-K48-Ub expression plasmid into HEK293T cells. The results showed that UL36USP could inhibit both K48- and K63-linked ubiquitination of TRAF3 (Fig. 4B).
Fig 4.

UL36USP deubiquitinates TRAF3. (A) HEK293T cells were transfected with HA-Ub, TRAF3-Flag along with empty vector, UL36USP-Myc, or C40A-Myc. Twenty-four hours posttransfection, cells were infected with SeV or mock infected. Flag-tagged TRAF3 was immunoprecipitated using an anti-FLAG antibody and subjected to immunoblot (IB) analysis using an anti-HA antibody to detect ubiquitination of TRAF3. (B) UL36 deubiquitinates both K48- and K63-linked polyubiquitin chains of TRAF3. TRAF3-Flag, UL36USP-Myc, and HA-Ub or the linkage-specific ubiquitin HA-K63-Ub or HA-K48-Ub expression plasmid was transfected as indicated. After SeV infection, co-IP assays were performed to detect the ubiquitination of TRAF3. (C) Co-IP experiments indicated that UL36USP inhibited the SeV-mediated recruitment of TBK1 by TRAF3.
We further investigated whether deubiquitinating activity of UL36USP impaired the interaction between TRAF3 and TBK1. As shown in Fig. 4C, more TBK1 was coimmunoprecipitated by TRAF3 after SeV stimulation than that shown in the lane without SeV infection. Cotransfection with UL36USP, but not the C40A mutant, inhibited the SeV-induced recruitment of TBK1 by TRAF3. Taken together, the results indicated that UL36USP deubiquitinated TRAF3 and abrogated the recruitment of TBK1.
UL36 C40A mutant virus induced a larger amount of IFN-β than WT virus.
To investigate the physiological functions of UL36USP in context with HSV-1 infection, the UL36 C40A point mutant, denoted C40A HSV-1, was generated using our HSV-1 BAC system as previously described (Fig. 5A) (68). Recombinant HSV-1 BAC contains a firefly luciferase cassette, and the luciferase activity could be easily quantified in vitro. Luciferase activity assays were used to determine the replication kinetics of C40A HSV-1 and WT HSV-1. When Vero cells were infected with the corresponding viruses at an MOI of 0.1, mutation of the key cysteine residue of UL36USP did not affect the viral replication (Fig. 5B). Traditional plaque assays were also performed at an MOI of 1.0 in Vero cells, and the results correlated with the luciferase activity (Fig. 5C). Because Vero cells are deficient in IFN production, we examined the replication of C40A HSV-1 in HEK293T cells to further study the function of UL36USP. Interestingly, the luciferase activity assay showed that the replication of C40A HSV-1 was remarkably impaired compared with that of WT HSV-1 (Fig. 5D). ICP0, UL42, and UL46 expression in C40A HSV-1-infected HEK293T cells was also reduced (Fig. 5E), confirming the reduced replication of C40A HSV-1. Semiquantitative PCR was performed to detect the transcription of ISG54 and ISG56 in C40A HSV-1-infected HEK293 cells. The results showed that a remarkably higher level of transcription of ISG54 and ISG56 was observed in C40A HSV-1-infected HEK293T cells at 12 and 24 h postinfection (Fig. 5F), implying that the replication of C40A HSV-1 was suppressed by the higher level of ISGs.
To detect the ubiquitination of TRAF3 in the context of viral infection, HEK293T cells were transfected with TRAF3-Flag and HA-Ub expression plasmid and subsequently infected with SeV, WT HSV-1, or C40A HSV-1 at an MOI of 5 for 16 h. Then co-IP experiments were performed to detect the ubiquitination of TRAF3. As expected, WT HSV-1 infection abrogated TRAF3 ubiquitination, whereas C40A HSV-1 infection did not (Fig. 6A). Co-IP assays were also performed using endogenous TRAF3 and Ub, and the results demonstrated that WT HSV-1 infection, but not C40A HSV-1 infection, abrogates the ubiquitination of TRAF3 (Fig. 6B).
Fig 6.

Cells and C57BL/6 mice infected with C40A HSV-1 produce more IFN-β than those infected with WT HSV-1. (A and B) WT HSV-1 infection, but not C40A HSV-1 infection, affected the ubiquination of TRAF3. (A) TRAF3-Flag and HA-Ub-expressing plasmids were cotransfected into HEK293T cells, and the cells were infected with SeV, WT HSV-1, or C40A HSV-1 at an MOI of 5 for 16 h. Co-IP experiments were performed to detect the ubiquitination of TRAF3. (B) HEK293T cells were infected as in panel A, and co-IP experiments were performed using TRAF3 and Ub pAb to detect the ubiquitination of endogenous TRAF3. (C) HEK293T cells, MEF cells, and HeLa cells were infected with the indicated viruses for 16 h, and semiquantitative PCR assays were performed to detect the mRNA level of IFN-β. (D) HEK293T cells in a 24-cell plate were infected with WT HSV-1 or C40A HSV-1 at an MOI of 5 for 20 h. Medium from the cells was analyzed by ELISA for IFN-β secretion. (E) C57BL/6 mice were infected with 106 PFU of the indicated virus. After 24 h, blood serum was collected and subjected to ELISA to detect IFN-β production. The data represent means ± standard deviations for three replicates. Statistical analysis was performed using the t test. *, P < 0.05.
Semiquantitative PCR was performed to measure the IFN-β mRNA expression in HEK293T cells, mouse embryonic fibroblast (MEF) cells, and HeLa cells infected with wild-type or C40A HSV-1. SeV induced a high level of IFN-β mRNA as a positive control. The WT HSV-1 infection induced only a trace amount of IFN-β mRNA. The C40A mutant viruses induced a significantly higher level of IFN-β mRNA than WT HSV-1 did in all three cell lines (Fig. 6C). Then ELISAs were also performed to measure the secretion of IFN-β when HEK293T cells were infected by those viruses at an MOI of 5 for 10 h. The results indicated that C40A HSV-1 induced a remarkably larger amount of IFN-β secretion than the wild type (Fig. 6D). In addition, C57BL/6 mice were infected with 106 PFU of the indicated virus for 24 h, and serum IFN-β was detected with an ELISA kit. WT HSV-1 only induced a little IFN-β production. C40A HSV-1 infection induced the production of approximately 12 ng/ml IFN-β, which was remarkable higher than the level in WT virus infection (Fig. 6E).
Taken together, these pieces of evidence demonstrated that HSV-1 UL36USP deubiquitinates TRAF3 and suppresses IFN-β production, contributing to immune evasion during HSV-1 infection.
DISCUSSION
Innate immunity is a conserved, rapid response mechanism against pathogen invasion. IFN-β plays a crucial role in mediating antiviral response through the induction of a diverse set of ISGs. The PRRs of host cells recognize a pathogen-associated molecular pattern, which usually is viral nucleic acid. PRRs subsequently initiate a series of signaling cascades and finally activate IRF3 and NF-κB, inducing the transcription of IFN-β (69, 70). Ubiquitination is a crucial regulatory mechanism in the innate antiviral response, and many viruses encode DUBs to counteract the innate immunity. A number of viruses encode DUBs to manipulate cellular processes, and several viral DUBs have been shown to play crucial roles in immune evasion. HSV-1 UL36USP and the homologues in other herpesviruses, including MCMV M48, HCMV UL48, EBV BPLF1, and KSHV and MHV68 ORF64 (44, 47–49, 53), could cleave both K48- and K63-linked polyubiquitin chains. It is recently reported that the N-terminal 325-aa region of EBV BPLF1 carries DUB activity, interacts with, and deubiquitinates TRAF6 to inhibit NF-κB signaling during lytic infection (53). KSHV ORF64 reduces the ubiquitination of RIG-I, counteracting RIG-I-mediated IFN signaling (71). The papain-like protease domain 2 (PLP2) of murine hepatitis virus A59 (MHV-A59) deubiquitinated TBK1 and reduced its kinase activity (72). Papain-like protease (PLpro) of foot-and-mouth disease virus (FMDV), a papain-like proteinase which acts as a viral DUB, was reported to decrease IRF3/7 expression and inhibit activation of NF-κB, suppressing dsRNA-induced IFN-I production (73). Hepatitis B virus (HBV) X protein cleaves Lys63-linked polyubiquitin chains of many proteins to negatively regulate IFN-I production (74). The cysteine protease domain of porcine reproductive and respiratory syndrome virus (PRRSV) nonstructural protein 2 possesses deubiquitinating and interferon antagonism functions (75). PLpro from the severe acute respiratory syndrome coronavirus (SARS-CoV) removes K48-linked polyubiquitin chains; PLpro mutations enhanced innate immune signaling (76, 77). Arterivirus papain-like protease 2 (PLP2) can also remove ubiquitin from cellular proteins and is also involved in innate immune evasion (78). All of these facts indicate that encoding of viral DUBs is one of the preferred mechanisms to subvert the host's innate immune response.
We found that ectopic expression of UL36USP was sufficient to downregulate SeV-activated IFN-β promoter activity. UL36USP inhibited the activities of RIG-IN and MAVS, but not TBK1, IKKε, and IRF3/5D-mediated IFN-β promoter activities, suggesting UL36 inhibits IFN-β production at a level between those of MAVS and TBK1. Subsequently, UL36USP was identified as deubiquitinating TRAF3 and inhibiting the recruitment of TBK1 by TRAF3. Also, a recombinant HSV-1 lacking DUB activity of UL36USP induced more IFN-β than WT HSV-1.
HSV-1 UL36USP is embedded within the N-terminal region of the VP1/2 HSV-1 large tegument protein, and the core catalytic residue, including C65 (in HSV), is required for its deubiquitinase activity (42). Whereas the Cys65 did not exist in VP1/2 of HSV-1 F strain, Cys40 was further identified to be required for the DUB activity. The C40A mutation in UL36USP abolished its inhibitory activity to IFN-β production. The C40A HSV-1, harboring the C40A mutation in UL36USP and lacking DUB activity, was generated. Luciferase activity assays and plaque assays showed that C40A HSV-1 replicated in Vero cells with little difference from WT HSV-1. This result is similar to that found with recombinant HSV-1 with a C65A mutation at the active site of HSV-1 UL36USP. The single-step growth curve assays of VP1/2 C65A mutant HSV-1 in rabbit skin cells (RSC) indicated that the C65A mutation of HSV-1 did not affect viral replication (43). Because the Vero cell is deficient in IFN production, the replication of C40A HSV-1 was also examined in HEK293T cells. Interestingly, the replication of C40A HSV-1 in HEK293T cells was reduced, compared to that in the WT HSV-1 strain. Western blot assays were performed to detect the expression of ICP0, UL42, and UL46 in HEK293T cells, and the same result was received. Thus, we speculated that C40A HSV-1 induced more IFN-β production, which inhibited the replication of the mutant virus. Subsequently, we examined the IFN-β production induced by C40A HSV-1. As expected, ELISA and semiquantitative PCR demonstrated that cells infected with C40A HSV-1 produced remarkably more IFN-β than cells infected with WT HSV-1.
UL36USP was shown to inhibit the production of IFN-β by removing polyubiquitin chain on TRAf3, and C40A HSV-1 infection failed to deubiquitinate TRAF3, inducing more IFN-β and ISGs expression than WT virus did. HSV-1 evolves multiple immune evasion strategies, and several HSV-1 proteins have been demonstrated to inhibit IFN-β production and contribute to immune evasion at different levels, including ICP0 (19–22), US11 (24), ICP34.5 (25, 26), vhs (27, 28), ICP27 (29), and US3 (30). Most of these viral proteins, including UL36, are components of the tegument and are released into the cytoplasm immediately after viral entry, contributing to the inhibition of the early antiviral response. It was reported that HSV-1 ICP0 dampened IFN production by sequestering IRF3 and CBP/p300 away from the normal binding sites on the promoter of host genes, and the ICP0 mutant HSV-2 led to an enhanced antiviral response (19–22). However, the role of ICP0 in immune evasion might be exaggerated in the context of HSV-1 infection, because the mutation of ICP0 abolished its transactivation function and decreased the expression of other early and late genes. Although the contributions of those proteins to immune evasion remain uncharacterized, we inferred that UL36USP and those HSV-1 proteins collaborated to evade the host antiviral response.
Mutation of the conserved cysteine residue of USPs in most herpesviruses impaired viral replication in vitro. HEK293 cells infected by BPLF1-deficient recombinant EBV exhibited poor viral DNA replication compared with the wild type. Knockdown of p65 in cells restored DNA replication of BPLF1-deficient viruses (53). MHV68 carrying an enzymatically inactive ORF64 protein was cleared faster than revertant viruses in an in vivo mouse infection model (50). HCMV harboring a mutation in the pUL48 USP domain (C24I or H162A) replicated more slowly and produced 10-fold-lower level of progeny virus (36, 44). PRV with a VP1/2 USP C26S or C26A mutation resulted in about a 20- to 30-fold or 10- to 20-fold reduction in virus replication, respectively (45, 79).
In recent years, herpesvirus tegument USPs have attracted much interest. Several USPs have been shown to play an important role in virus pathogenesis and have been suggested to be possible targets for antiviral therapy (48). For example, in the absence of USP activity, PRV is deficient for neuroinvasion properties in the mouse model system and exhibits delayed onset of pathogenic features after intranasal infection of mice (45). In this study, UL36USP was identified to deubiquitinate TRAF3 and suppress IFN-β production. These findings reveal a novel mechanism for HSV-1 to evade host antiviral immunity and will help to develop new drug targets for anti-HSV-1 therapy.
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (81371795, 81171584, 81101263, and 31300886), the Program for Changjiang Scholars and Innovative Research Team in Soochow University (PCSIRT and IRT1075), and the Jiangsu Provincial Innovative Research Team.
We thank Yi-Ling Lin for the gift of plasmid IRF3/5D, S. Ludwig for (PRDDIII-)4-Luc, and Takashi Fujita for pEF-Flag-RIG-IN, pMyc-MAVS, and IFN-β-Luc. We thank You Li for help with constructing HSV-1 mutant viruses. We thank Karen Mossman and Rongtuan Lin for help with this study. We greatly appreciate the comments from anonymous reviewers for improving our manuscript.
Footnotes
Published ahead of print 28 August 2013
REFERENCES
- 1.Kadowaki N, Antonenko S, Lau JY, Liu YJ. 2000. Natural interferon alpha/beta-producing cells link innate and adaptive immunity. J. Exp. Med. 192:219–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1–47 [DOI] [PubMed] [Google Scholar]
- 3.Sarkar SN, Sen GC. 2004. Novel functions of proteins encoded by viral stress-inducible genes. Pharmacol. Ther. 103:245–259 [DOI] [PubMed] [Google Scholar]
- 4.Takeuchi O, Akira S. 2009. Innate immunity to virus infection. Immunol. Rev. 227:75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhoj VG, Chen ZJ. 2009. Ubiquitylation in innate and adaptive immunity. Nature 458:430–437 [DOI] [PubMed] [Google Scholar]
- 6.Jiang X, Chen ZJ. 2012. The role of ubiquitylation in immune defence and pathogen evasion. Nat. Rev. Immunol. 12:35–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. 2007. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446:916–920 [DOI] [PubMed] [Google Scholar]
- 8.Oshiumi H, Matsumoto M, Hatakeyama S, Seya T. 2009. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J. Biol. Chem. 284:807–817 [DOI] [PubMed] [Google Scholar]
- 9.Hacker H, Redecke V, Blagoev B, Kratchmarova I, Hsu LC, Wang GG, Kamps MP, Raz E, Wagner H, Hacker G, Mann M, Karin M. 2006. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439:204–207 [DOI] [PubMed] [Google Scholar]
- 10.Kayagaki N, Phung Q, Chan S, Chaudhari R, Quan C, O'Rourke KM, Eby M, Pietras E, Cheng G, Bazan JF, Zhang Z, Arnott D, Dixit VM. 2007. DUBA: a deubiquitinase that regulates type I interferon production. Science 318:1628–1632 [DOI] [PubMed] [Google Scholar]
- 11.Oganesyan G, Saha SK, Guo B, He JQ, Shahangian A, Zarnegar B, Perry A, Cheng G. 2006. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 439:208–211 [DOI] [PubMed] [Google Scholar]
- 12.Saha SK, Pietras EM, He JQ, Kang JR, Liu SY, Oganesyan G, Shahangian A, Zarnegar B, Shiba TL, Wang Y, Cheng G. 2006. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J. 25:3257–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682 [DOI] [PubMed] [Google Scholar]
- 14.Barber GN. 2011. Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Curr. Opin. Immunol. 23:10–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsuchida T, Zou J, Saitoh T, Kumar H, Abe T, Matsuura Y, Kawai T, Akira S. 2010. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33:765–776 [DOI] [PubMed] [Google Scholar]
- 16.Friedman CS, O'Donnell MA, Legarda-Addison D, Ng A, Cardenas WB, Yount JS, Moran TM, Basler CF, Komuro A, Horvath CM, Xavier R, Ting AT. 2008. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 9:930–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang C, Chen T, Zhang J, Yang M, Li N, Xu X, Cao X. 2009. The E3 ubiquitin ligase Nrdp1 ‘preferentially' promotes TLR-mediated production of type I interferon. Nat. Immunol. 10:744–752 [DOI] [PubMed] [Google Scholar]
- 18.Arimoto K, Funami K, Saeki Y, Tanaka K, Okawa K, Takeuchi O, Akira S, Murakami Y, Shimotohno K. 2010. Polyubiquitin conjugation to NEMO by triparite motif protein 23 (TRIM23) is critical in antiviral defense. Proc. Natl. Acad. Sci. U. S. A. 107:15856–15861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin R, Noyce RS, Collins SE, Everett RD, Mossman KL. 2004. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J. Virol. 78:1675–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Melroe GT, DeLuca NA, Knipe DM. 2004. Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. J. Virol. 78:8411–8420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mossman K. 2005. Analysis of anti-interferon properties of the herpes simplex virus type I ICP0 protein. Methods Mol. Med. 116:195–205 [DOI] [PubMed] [Google Scholar]
- 22.Paladino P, Collins SE, Mossman KL. 2010. Cellular localization of the herpes simplex virus ICP0 protein dictates its ability to block IRF3-mediated innate immune responses. PLoS One 5:e10428. 10.1371/journal.pone.0010428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu H, Zheng C, Xing J, Wang S, Li S, Lin R, Mossman KL. 2011. Varicella-zoster virus immediate-early protein ORF61 abrogates the IRF3-mediated innate immune response through degradation of activated IRF3. J. Virol. 85:11079–11089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xing J, Wang S, Lin R, Mossman KL, Zheng C. 2012. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J. Virol. 86:3528–3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mossman KL, Smiley JR. 2002. Herpes simplex virus ICP0 and ICP34.5 counteract distinct interferon-induced barriers to virus replication. J. Virol. 76:1995–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verpooten D, Ma Y, Hou S, Yan Z, He B. 2009. Control of TANK-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1. J. Biol. Chem. 284:1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elgadi MM, Hayes CE, Smiley JR. 1999. The herpes simplex virus vhs protein induces endoribonucleolytic cleavage of target RNAs in cell extracts. J. Virol. 73:7153–7164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yao XD, Rosenthal KL. 2011. Herpes simplex virus type 2 virion host shutoff protein suppresses innate dsRNA antiviral pathways in human vaginal epithelial cells. J. Gen. Virol. 92:1981–1993 [DOI] [PubMed] [Google Scholar]
- 29.Melchjorsen J, Siren J, Julkunen I, Paludan SR, Matikainen S. 2006. Induction of cytokine expression by herpes simplex virus in human monocyte-derived macrophages and dendritic cells is dependent on virus replication and is counteracted by ICP27 targeting NF-kappaB and IRF-3. J. Gen. Virol. 87:1099–1108 [DOI] [PubMed] [Google Scholar]
- 30.Peri P, Mattila RK, Kantola H, Broberg E, Karttunen HS, Waris M, Vuorinen T, Hukkanen V. 2008. Herpes simplex virus type 1 Us3 gene deletion influences Toll-like receptor responses in cultured monocytic cells. Virol. J. 5:140. 10.1186/1743-422X-5-140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abaitua F, O'Hare P. 2008. Identification of a highly conserved, functional nuclear localization signal within the N-terminal region of herpes simplex virus type 1 VP1-2 tegument protein. J. Virol. 82:5234–5244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chou J, Roizman B. 1989. Characterization of DNA sequence-common and sequence-specific proteins binding to cis-acting sites for cleavage of the terminal a sequence of the herpes simplex virus 1 genome. J. Virol. 63:1059–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Desai PJ. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 74:11608–11618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fuchs W, Klupp BG, Granzow H, Mettenleiter TC. 2004. Essential function of the pseudorabies virus UL36 gene product is independent of its interaction with the UL37 protein. J. Virol. 78:11879–11889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JI, Luxton GW, Smith GA. 2006. Identification of an essential domain in the herpesvirus VP1/2 tegument protein: the carboxy terminus directs incorporation into capsid assemblons. J. Virol. 80:12086–12094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luxton GW, Lee JI, Haverlock-Moyns S, Schober JM, Smith GA. 2006. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 80:201–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roberts AP, Abaitua F, O'Hare P, McNab D, Rixon FJ, Pasdeloup D. 2009. Differing roles of inner tegument proteins pUL36 and pUL37 during entry of herpes simplex virus type 1. J. Virol. 83:105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schlieker C, Korbel GA, Kattenhorn LM, Ploegh HL. 2005. A deubiquitinating activity is conserved in the large tegument protein of the Herpesviridae. J. Virol. 79:15582–15585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shanda SK, Wilson DW. 2008. UL36p is required for efficient transport of membrane-associated herpes simplex virus type 1 along microtubules. J. Virol. 82:7388–7394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zaichick SV, Bohannon KP, Hughes A, Sollars PJ, Pickard GE, Smith GA. 2013. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe 13:193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou ZH, Chen DH, Jakana J, Rixon FJ, Chiu W. 1999. Visualization of tegument-capsid interactions and DNA in intact herpes simplex virus type 1 virions. J. Virol. 73:3210–3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kattenhorn LM, Korbel GA, Kessler BM, Spooner E, Ploegh HL. 2005. A deubiquitinating enzyme encoded by HSV-1 belongs to a family of cysteine proteases that is conserved across the family Herpesviridae. Mol. Cell 19:547–557 [DOI] [PubMed] [Google Scholar]
- 43.Bolstad M, Abaitua F, Crump CM, O'Hare P. 2011. Autocatalytic activity of the ubiquitin-specific protease domain of herpes simplex virus 1 VP1-2. J. Virol. 85:8738–8751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim ET, Oh SE, Lee YO, Gibson W, Ahn JH. 2009. Cleavage specificity of the UL48 deubiquitinating protease activity of human cytomegalovirus and the growth of an active-site mutant virus in cultured cells. J. Virol. 83:12046–12056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bottcher S, Maresch C, Granzow H, Klupp BG, Teifke JP, Mettenleiter TC. 2008. Mutagenesis of the active-site cysteine in the ubiquitin-specific protease contained in large tegument protein pUL36 of pseudorabies virus impairs viral replication in vitro and neuroinvasion in vivo. J. Virol. 82:6009–6016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jarosinski K, Kattenhorn L, Kaufer B, Ploegh H, Osterrieder N. 2007. A herpesvirus ubiquitin-specific protease is critical for efficient T cell lymphoma formation. Proc. Natl. Acad. Sci. U. S. A. 104:20025–20030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang J, Loveland AN, Kattenhorn LM, Ploegh HL, Gibson W. 2006. High-molecular-weight protein (pUL48) of human cytomegalovirus is a competent deubiquitinating protease: mutant viruses altered in its active-site cysteine or histidine are viable. J. Virol. 80:6003–6012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schlieker C, Weihofen WA, Frijns E, Kattenhorn LM, Gaudet R, Ploegh HL. 2007. Structure of a herpesvirus-encoded cysteine protease reveals a unique class of deubiquitinating enzymes. Mol. Cell 25:677–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gonzalez CM, Wang L, Damania B. 2009. Kaposi's sarcoma-associated herpesvirus encodes a viral deubiquitinase. J. Virol. 83:10224–10233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gredmark-Russ S, Isaacson MK, Kattenhorn L, Cheung EJ, Watson N, Ploegh HL. 2009. A gammaherpesvirus ubiquitin-specific protease is involved in the establishment of murine gammaherpesvirus 68 infection. J. Virol. 83:10644–10652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shackelford J, Pagano JS. 2005. Targeting of host-cell ubiquitin pathways by viruses. Essays Biochem. 41:139–156 [DOI] [PubMed] [Google Scholar]
- 52.Sulea T, Lindner HA, Menard R. 2006. Structural aspects of recently discovered viral deubiquitinating activities. Biol. Chem. 387:853–862 [DOI] [PubMed] [Google Scholar]
- 53.Saito S, Murata T, Kanda T, Isomura H, Narita Y, Sugimoto A, Kawashima D, Tsurumi T. 2013. Epstein-Barr virus deubiquitinase downregulates TRAF6-mediated NF-kappaB signaling during productive replication. J. Virol. 87:4060–4070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xing J, Wang S, Lin F, Pan W, Hu CD, Zheng C. 2011. Comprehensive characterization of interaction complexes of herpes simplex virus type 1 ICP22, UL3, UL4, and UL20.5. J. Virol. 85:1881–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xing J, Wang S, Li Y, Guo H, Zhao L, Pan W, Lin F, Zhu H, Wang L, Li M, Zheng C. 2011. Characterization of the subcellular localization of herpes simplex virus type 1 proteins in living cells. Med. Microbiol. Immunol. 200:61–68 [DOI] [PubMed] [Google Scholar]
- 56.Ehrhardt C, Kardinal C, Wurzer WJ, Wolff T, von Eichel-Streiber C, Pleschka S, Planz O, Ludwig S. 2004. Rac1 and PAK1 are upstream of IKK-epsilon and TBK-1 in the viral activation of interferon regulatory factor-3. FEBS Lett. 567:230–238 [DOI] [PubMed] [Google Scholar]
- 57.Paz S, Vilasco M, Arguello M, Sun Q, Lacoste J, Nguyen TL, Zhao T, Shestakova EA, Zaari S, Bibeau-Poirier A, Servant MJ, Lin R, Meurs EF, Hiscott J. 2009. Ubiquitin-regulated recruitment of IkappaB kinase epsilon to the MAVS interferon signaling adapter. Mol. Cell. Biol. 29:3401–3412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao T, Yang L, Sun Q, Arguello M, Ballard DW, Hiscott J, Lin R. 2007. The NEMO adaptor bridges the nuclear factor-kappaB and interferon regulatory factor signaling pathways. Nat. Immunol. 8:592–600 [DOI] [PubMed] [Google Scholar]
- 59.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5:730–737 [DOI] [PubMed] [Google Scholar]
- 60.Chang TH, Liao CL, Lin YL. 2006. Flavivirus induces interferon-beta gene expression through a pathway involving RIG-I-dependent IRF-3 and PI3K-dependent NF-kappaB activation. Microbes Infect. 8:157–171 [DOI] [PubMed] [Google Scholar]
- 61.Kochs G, Garcia-Sastre A, Martinez-Sobrido L. 2007. Multiple anti-interferon actions of the influenza A virus NS1 protein. J. Virol. 81:7011–7021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin R, Lacoste J, Nakhaei P, Sun Q, Yang L, Paz S, Wilkinson P, Julkunen I, Vitour D, Meurs E, Hiscott J. 2006. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKepsilon molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3-4A proteolytic cleavage. J. Virol. 80:6072–6083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang S, Liu N, Chen AJ, Zhao XF, Wang JX. 2009. TRBP homolog interacts with eukaryotic initiation factor 6 (eIF6) in Fenneropenaeus chinensis. J. Immunol. 182:5250–5258 [DOI] [PubMed] [Google Scholar]
- 64.Jordan M, Schallhorn A, Wurm FM. 1996. Transfecting mammalian cells: optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res. 24:596–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, Shu HB. 2008. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29:538–550 [DOI] [PubMed] [Google Scholar]
- 66.Iwamura T, Yoneyama M, Yamaguchi K, Suhara W, Mori W, Shiota K, Okabe Y, Namiki H, Fujita T. 2001. Induction of IRF-3/-7 kinase and NF-kappaB in response to double-stranded RNA and virus infection: common and unique pathways. Genes Cells 6:375–388 [DOI] [PubMed] [Google Scholar]
- 67.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step Red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197 [DOI] [PubMed] [Google Scholar]
- 68.Li Y, Wang S, Zhu H, Zheng C. 2011. Cloning of the herpes simplex virus type 1 genome as a novel luciferase-tagged infectious bacterial artificial chromosome. Arch. Virol. 156:2267–2272 [DOI] [PubMed] [Google Scholar]
- 69.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732–738 [DOI] [PubMed] [Google Scholar]
- 70.Takahasi K, Yoneyama M, Nishihori T, Hirai R, Kumeta H, Narita R, Gale M, Jr, Inagaki F, Fujita T. 2008. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol. Cell 29:428–440 [DOI] [PubMed] [Google Scholar]
- 71.Inn KS, Lee SH, Rathbun JY, Wong LY, Toth Z, Machida K, Ou JH, Jung JU. 2011. Inhibition of RIG-I-mediated signaling by Kaposi's sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol. 85:10899–10904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang G, Chen G, Zheng D, Cheng G, Tang H. 2011. PLP2 of mouse hepatitis virus A59 (MHV-A59) targets TBK1 to negatively regulate the cellular type I interferon signaling pathway. PLoS One 6:e17192. 10.1371/journal.pone.0017192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang D, Fang L, Li P, Sun L, Fan J, Zhang Q, Luo R, Liu X, Li K, Chen H, Chen Z, Xiao S. 2011. The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J. Virol. 85:3758–3766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jiang J, Tang H. 2010. Mechanism of inhibiting type I interferon induction by hepatitis B virus X protein. Protein Cell 1:1106–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sun Z, Chen Z, Lawson SR, Fang Y. 2010. The cysteine protease domain of porcine reproductive and respiratory syndrome virus nonstructural protein 2 possesses deubiquitinating and interferon antagonism functions. J. Virol. 84:7832–7846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barretto N, Jukneliene D, Ratia K, Chen Z, Mesecar AD, Baker SC. 2005. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 79:15189–15198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lindner HA, Fotouhi-Ardakani N, Lytvyn V, Lachance P, Sulea T, Menard R. 2005. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J. Virol. 79:15199–15208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van Kasteren PB, Bailey-Elkin BA, James TW, Ninaber DK, Beugeling C, Khajehpour M, Snijder EJ, Mark BL, Kikkert M. 2013. Deubiquitinase function of arterivirus papain-like protease 2 suppresses the innate immune response in infected host cells. Proc. Natl. Acad. Sci. U. S. A. 110:E838–E847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee JI, Sollars PJ, Baver SB, Pickard GE, Leelawong M, Smith GA. 2009. A herpesvirus encoded deubiquitinase is a novel neuroinvasive determinant. PLoS Pathog. 5:e1000387. 10.1371/journal.ppat.1000387 [DOI] [PMC free article] [PubMed] [Google Scholar]