

Abstract
Sequences and structures within the terminal genomic regions of plus-strand RNA viruses are targets for the binding of host proteins that modulate functions such as translation, RNA replication, and encapsidation. Using murine norovirus 1 (MNV-1), we describe the presence of long-range RNA-RNA interactions that were stabilized by cellular proteins. The proteins potentially responsible for the stabilization were selected based on their ability to bind the MNV-1 genome and/or having been reported to be involved in the stabilization of RNA-RNA interactions. Cell extracts were preincubated with antibodies against the selected proteins and used for coprecipitation reactions. Extracts treated with antibodies to poly(C) binding protein 2 (PCBP2) and heterogeneous nuclear ribonucleoprotein (hnRNP) A1 significantly reduced the 5′-3′ interaction. Both PCBP2 and hnRNP A1 recombinant proteins stabilized the 5′-3′ interactions and formed ribonucleoprotein complexes with the 5′ and 3′ ends of the MNV-1 genomic RNA. Mutations within the 3′ complementary sequences (CS) that disrupt the 5′-3′-end interactions resulted in a significant reduction of the viral titer, suggesting that the integrity of the 3′-end sequence and/or the lack of complementarity with the 5′ end is important for efficient virus replication. Small interfering RNA-mediated knockdown of PCBP2 or hnRNP A1 resulted in a reduction in virus yield, confirming a role for the observed interactions in efficient viral replication. PCBP2 and hnRNP A1 induced the circularization of MNV-1 RNA, as revealed by electron microscopy. This study provides evidence that PCBP2 and hnRNP A1 bind to the 5′ and 3′ ends of the MNV-1 viral RNA and contribute to RNA circularization, playing a role in the virus life cycle.
INTRODUCTION
Noroviruses (NoVs) are the causative agents of nonbacterial gastroenteritis in humans and are responsible for almost all viral gastroenteritis outbreaks worldwide (1–3). The genus Norovirus within the family Caliciviridae comprises nonenveloped icosahedral viruses with a single-stranded positive-polarity RNA genome. NoV genomic RNA typically contains three open reading frames (ORFs): ORF1 encodes a polyprotein precursor that is processed to give rise to 6 or 7 nonstructural proteins. ORF2 and ORF3 encode the major and minor capsid proteins VP1 and VP2, respectively. Both VP1 and VP2 are synthesized from a subgenomic RNA (4). In the case of murine norovirus 1 (MNV-1), a fourth potential ORF (ORF4) has been identified that is highly conserved between strains and encodes a protein (VF1) involved in the regulation of the innate immune response to NoV infection (5).
Attempts to grow human noroviruses (HuNoVs) in cell culture have been largely unsuccessful (6); therefore, the finer details of the NoV replication cycle remain unclear. However, the identification of the first MNV-1 strain and its routine laboratory propagation in the murine macrophage cell line Raw264.7 provide a cell culture system to investigate the molecular mechanisms of NoV translation and replication (6, 7). The NoV genome is flanked by a very short 5′ untranslated region (UTR) covalently linked to the viral VPg protein and by a polyadenylated 3′ UTR (8). VPg functions as a proteinaceous cap substitute and is able to bind to eukaryotic initiation factors to promote viral translation (9–12).
MNV-1 is an enteric pathogen that shares many molecular and biological properties with HuNoV (13). The 5′-end sequence of both the genomic and subgenomic RNAs is highly conserved among several members of the family Caliciviridae (14, 15). Moreover, their 5′- and 3′-end regions contain several conserved RNA secondary structures, with various sizes and positions, implicated in viral replication (16–18), as well as viral pathogenesis (14).
Translation and replication of the positive-strand RNA viruses take place in the cytoplasm of the infected cells. Several lines of evidence support the hypothesis that an interaction between the 5′ and 3′ ends of viral RNA genomes regulates the translation and RNA replication of many viruses (19–21). However, the mechanism by which the 5′ and 3′ ends associate and the fine details of how different conformations of the RNA participate in viral translation and replication remain unclear. Several variations in the way in which this interaction occurs have been observed, with each virus developing its own strategies to allow these 5′-3′-end contacts. Some of these interactions can occur via direct RNA-RNA contacts, as in the case of dengue virus genomic RNA, where complementary sequences (CS) present in the viral genome ends are necessary for RNA replication and viral viability (22, 23). Sequence complementarity is necessary but not sufficient in some cases to direct these 5′-3′-end interactions (24); therefore, cellular proteins act as facilitators to maintain the interactions. In the case of bovine viral diarrhea virus (BVDV) and hepatitis C virus (HCV), with RNA genomes that lack a 5′ cap structure and a 3′ poly(A) tail, 5′-3′ genomic-RNA contacts are mediated by the NFAR proteins (25, 26). More recently, the role of the cellular poly(rC) binding protein 2 (PCBP2) in the circularization of the HCV RNA genome has been described (27). Members of the family Picornaviridae contain genomic RNAs that are polyadenylated at their 3′ ends and do not have a cap structure at their 5′ ends. They contain highly structured 5′ UTRs able to bind to cellular and viral proteins that promote and regulate viral translation, as well as replication (24, 28). In the case of poliovirus, PCBP2 forms a complex on the 5′ UTR with the viral protease-polymerase precursor (3CD), which can then interact with the cellular poly(A) binding protein (PABP) bound to the 3′ poly(A) tail, promoting the synthesis of the negative-polarity RNA (29–32).
Recently, an interaction between the 5′ and 3′ ends of the Norwalk virus (NV) genomic RNA has been reported, where the RNA-RNA interactions are further stabilized by cellular proteins (33), although the identities of the cellular proteins involved in this process have yet to be determined. Here, we report the identification of similar long-range RNA-RNA interactions between the 5′ and 3′ ends of the MNV-1 genome. We also demonstrate that the cellular factors PCBP2 and heterogeneous nuclear ribonucleoprotein (hnRNP) A1 play key roles in this interaction that are required for efficient viral replication.
MATERIALS AND METHODS
Computer-generated secondary-structure analysis.
Computational analysis of the RNA secondary structures was performed using the Mfold2 software (34) with default settings through the Web interface at http://frontend.bioinfo.rpi.edu/applications/mfold/.
The following 50 full-length genomic sequences were downloaded from GenBank and used to evaluate the degree of conservation of nucleotides (nt) 64 to 69 (ACAAGA) and 7323 to 7328 (UUUUGU) among different MNV isolates: group 1, EU004662.1, EU004661.1, EU004660.1, EU004659.1, EU004658, EU004657.1, EU004656.1, EU004655.1, EU004654.1, EF014462.1, EU004662.1, AY228235.2 EU004675.1, EU004672.1, EU004681.1, and EU004682.1; group 2, FJ446720.1, HQ317203.1, AB435515.1, AB435514.1, EU004683.1, EU004677.1, and EU004674; group 3, JF320653.1; group 4, JF320651.1, JF320650.1, JF320648.1, JF320647.1, JF320645.1, JF320646.1, JF320644.1, JF320649.1, and EU004663.1; group 5, EU004673.1, EU004664.1, FJ446719.1, EU004671.1, EF531291.1, EU004680.1, EU004679.1, and EU004678.1; group 6, EU004668.1 and EU004670.1; group 7, EU004667.1, EU004666.1, and EU004669.1; group 8, EF531290.1 and DQ911368.1; group 9, EU004670.1 and EU004668.1 (Table 1).
Table 1.
Conserved bases in the complementary regions of 50 different MNV isolatesa

*, amino acid N was changed to S; **, isolates in which the predicted contacts involve alternative 5′ sequences. The groups were formed with isolates having the same nucleotides in the sequences shown.
Cell culture.
The murine leukemia macrophage-like cell line Raw264.7 (obtained from ATCC) and baby hamster kidney cells expressing T7 DNA polymerase (BSR-T7 cells) used during reverse-genetics recovery of mutant viruses (obtained from Klaus Conzelmann, Ludwig-Maximilians-University, Munich, Germany) (14, 35) were cultured as described previously. The murine microglial BV-2 cell line was cultured as described previously (36, 37).
Protein expression and purification.
For the expression of His-hnRNP A1, Escherichia coli DH5α cells were transformed with the pProEX Hta hnRNP A1 plasmid, generated in our laboratory. Cells from a single colony were grown in LB medium at 37°C at an optical density at 600 nm (OD600) of 0.6. Isopropyl-β-d-thiogalactopyranoside (IPTG) was added at a final concentration of 0.7 mM, and protein expression was induced for 3 h. Expression and purification of His-PCBP2 from the pET22B:PCBP2 plasmid (kindly provided by Bert Semler, University of California, Irvine, CA) was carried out as described previously (29). Expression and purification of His-tagged PABP from the expression vector (a kind gift from R. Andino, University of California, San Francisco, CA) was performed as described previously (31). Protein concentrations were determined using the Bradford assay (38). Proteins were stored at 4°C until they were required.
Preparation of Raw264.7 S10 cell extracts.
Raw264.7 cell extracts were prepared essentially as described previously (39).
In vitro transcription of regions of the MNV-1 genomic RNA.
Two RNA molecules corresponding to the first 146 nt from the MNV-1 5′ end and the complete 3′ UTR without the poly(A) tail of the MNV-1 genomic RNA were synthesized by in vitro transcription using T7 RNA polymerase from the PCR-amplified cDNAs containing the respective regions. The PCR was performed using the complete MNV-1 cDNA, generously donated by H. W. Virgin (Washington University School of Medicine, St. Louis, MO, USA), as a template. All sense primers used in the PCR contained the bacteriophage T7 promoter sequence (TAATACGACTCACTATAGG). Oligonucleotides T7 Fwd (TAATACGACTCACTATAGG) and 3′UTR-Mut Rev (AAAATGCATCTAACTACCACAAAGAAAAGAAAGCAGTAAGCAGAAATCATTTTCGTGGGGGGTTTCTCTTCCAACCCTATAGTGAGTCGTATTA) were used for the synthesis of the mutant 3′-UTR RNA. Oligonucleotides 5′-Comp Fwd (TAATACGACTCACTATAGGGTGAAATGAGGATGGCAACGCCATCTTCTGCGCCCTCTGTGCGCAACACAGAGAAACGCAAAAGTGGGGAGGCTTCGTCTAAAGCTAGTG) and 5′-Comp Rev (GTAATTAATTTCGTCTTCGCTCTCCGAAGAGAGGGGGCTAGGTGCTCCAAAGGAGACACTAGCTTTAGACGAAGCCTCCC) were used for the synthesis of the mutant 5′-end RNA. The resulting PCR products were purified with a QIAquick gel extraction G-50 kit (Qiagen) before they were used as templates for RNA synthesis. After transcription, the reaction mixture was treated with DNase (Epicentre Biotechnologies) at 37°C for 30 min to remove the DNA template in the presence of RNase inhibitors (Roche). Unincorporated nucleotides were removed by phenol extraction, followed by isopropanol precipitation. For the synthesis of radiolabeled and biotin-labeled RNA transcripts, [α-32P]UTP and biotin-16-UTP were included in the transcription reaction mixture. In some cases, the ferritin H-chain iron-responsive element (IRE-fer) (40) or an RNA of 60 nt that corresponded to the FL-LAP sequence of the C/EBPβ transcription factor RNA (41) was used as the nonrelated RNA, as indicated.
RNA coprecipitation assay.
The RNA coprecipitation assay was performed as described previously (26, 33). The amount of His-hnRNP A1 or His-PCBP2 recombinant protein used is indicated for each experiment. Quantification of the precipitated α-32P-labeled RNA under different conditions was obtained from band intensities in the scanned images using ImageJ software (http://rsb.info.nih.gov/ij) and expressed as arbitrary intensity units, with the positive-control samples designated 100 units. In the assays performed in the presence of antibodies, 0.2 μg of each antibody was preincubated with the S10 extracts for 15 min at room temperature prior to the addition of the RNA.
EMSA.
Electrophoretic mobility shift assays (EMSAs) were performed as described previously (14).
RNA in vitro transcription and capping for the recovery of infectious MNV-1.
The pT7:MNV-13′Rz infectious clone containing the MNV-1 sequence was linearized with the NheI restriction enzyme and used for in vitro transcription as previously described (42). RNA was capped using the ScriptCap system (Epicentre) according to the manufacturer's instructions (42).
For RNA transfection, BSRT7 cells, previously reported to produce the highest virus yields (43), were seeded in a 6-well plate at 2.5 × 105 cells/well and incubated for 24 h in antibiotic-free Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS). The cells were transfected with the capped RNA, using Lipofectamine 2000 following the manufacturer's instructions (Invitrogen) (42). For virus recovery, cells were incubated for 24 h at 37°C prior to freezing at −80°C. Virus titration was performed by 50% tissue culture infective dose (TCID50) in Raw264.7 cells. Viral protein expression was analyzed by harvesting cells in RIPA buffer and performing Western blot analysis of a normalized sample.
Coimmunoprecipitation of viral RNA during infection.
The interaction of hnRNP A1 and PCBP2 with MNV-1 RNA was studied during infection using RNA coimmunoprecipitation. Briefly, Raw264.7 cells were infected with MNV-1 at a multiplicity of infection (MOI) of 5. Cell lysates were prepared 12 h postinfection (p.i.) using lysis buffer (10 mM Tris, pH 7.0, 150 mM NaCl, 1% Triton, 1 mM dithiothreitol [DTT], and protease inhibitors) and used in subsequent immunoprecipitations. Immunoprecipitations were carried out using a rabbit polyclonal antibody to hnRNP A1 (Santa Cruz Biotechnology, Santa Cruz, CA), a monoclonal antibody to PCBP2 (Santa Cruz Biotechnology, Santa Cruz, CA), and a rabbit polyclonal antibody to the viral RNA polymerase NS7 (or control purified rabbit IgG using protein A/G-Sepharose beads) (Santa-Cruz Biotechnology, Santa Cruz, CA). Viral RNA coimmunoprecipitated in the immune complexes was isolated using Trizol reagent (Invitrogen) and subsequently amplified by reverse transcription (RT)-PCR. Reverse transcription was performed using the Titan one-tube RT-PCR system (Roche) with primers 3D-FWD (CCCCCCATGCTTCCCCGCCC) and 3D-REV (TCACTCATCCTCATTCACAAAG) to amplify the complete NS7 region of the MNV-1 genome (44).
Immunofluorescence assays.
Raw264.7 cells were grown on glass coverslips and infected with MNV-1 at an MOI of 5 for 16 h. After this time, the coverslips were treated with cytoskeleton buffer (CB) (10 mM morpholineethanesulfonic acid [MES] [Sigma M-8250], 150 mM NaCl, 5 mM EGTA, 5 mM MgCl2, and 5 mM glucose) for 5 min and permeabilized in 4% paraformaldehyde solution containing 0.2% Triton X-100 for an additional 5 min at room temperature. The coverslips were washed three times for 5 min with phosphate-buffered saline (PBS), and the cells were fixed with 4% paraformaldehyde for 20 min at room temperature. The coverslips were washed three times for 5 min each time with PBS, blocked with 5% gelatin from porcine skin (Sigma) in PBS for 40 min at room temperature, washed three times for 5 min each time with PBS, and incubated with the primary antibody (anti-hnRNP A1 or anti-PCBP2) in PBS at 1% at 4°C overnight. Then, the coverslips were washed three times for 5 min each time with cold PBS and incubated with the corresponding secondary antibody (Alexa Fluor 488 [green]; Invitrogen) for 1 h at room temperature, washed three times with PBS, and incubated with the anti-MNV-1 NS7 antibody diluted 1:100 in 5% gelatin-PBS solution at 37°C for 1 h. The coverslips were washed three times with PBS, incubated with Alexa Fluor 594 (red) (Invitrogen) for 1 h at room temperature, washed three times with PBS, and incubated with 1 μg/μl of 4′6′-diamidino-2-phenylindole (DAPI) for 2 min. The coverslips were washed six times with PBS and three times with distilled water. Finally the coverslips were removed and treated with Vecta-Shield (Vector Laboratories A.C.). Samples were examined with a Zeiss LSM-700 confocal microscope.
siRNA-mediated knockdown of hnRNP A1 and PCBP2. For siRNA-mediated knockdown of hnRNP A1 and PCBP2 expression, actively growing BV-2 cells were transfected with siRNAs (hnRNP-E2:sc-38271, hnRNP-A1:sc-35576, and control nonspecific siRNA [GCGCGCUUUGUAGGAUUCG]) according to the protocol recommended by the manufacturer using the Neon Transfection System (Invitrogen). Cells (6 × 106) were electroporated with 100 pmol of siRNA duplexes at 1,700 V, 10 ms, and 3 pulses. Twelve hours posttransfection, the cells were infected with MNV-1 (MOI = 5 TCID50/cell), and samples were harvested for RNA isolation, virus titer determination, and Western blotting at 18 h p.i. The virus titer was determined by TCID50, and the viral RNA copy number was quantified using RT-quantitative PCR (qPCR) as described previously (14, 45). The results were plotted using Sigma Plot, and two-way analysis of variance (ANOVA) was performed as a method of statistical analysis. The P value obtained was <0.001 for qPCR experiments. For the TCID50 experiments, the P value was <0.01. The viability of untreated and small interfering RNA (siRNA)-treated cells was also determined at 48 h using Cell Titer 96 (Promega), following the manufacturer's instructions, and found to be unaffected.
Preparation of the RNA template for EM.
For visualization of RNA by electron microscopy (EM), an RNA template was designed, which consisted of the 5′ end (nt 1 to 146) and 3′ UTR of the MNV-1 genome flanking a 930-bp region of double-stranded RNA (dsRNA) to avoid an RNA secondary structure. The 930-bp fragment from the luciferase-coding sequence was amplified from the plasmid pRS424LucIresDen2CapDeleted by PCR (kindly donated by Rosa del Angel) using primers MNV-1-LUC-5′-FWD (GCGAAGACGAAATTAATTACGCTTCCAAGGTGTACGACCC) and MNV-1-LUC 3′-REV (AAAATGCATCTAACTACCACAAAGAAAAGAAAGCAGTAAGCAGAAATCATTTTCACAAAAGGTTTCTCTTCCAACTTACTGCTCGTTCTTCAGCA), which allow the addition of nt 127 to 146 and the complete 3′ UTR from the MNV-1 genome in the 5′ end and downstream of the luciferase gene, respectively. The complete 146-nt sequence from the 5′ end was amplified using the primer pair MNV-15′TR-FWD (TAATACGACTCACTATAGGGTGAAATGAGGATGGCAACGCCATCTTCTGCGCCCTCTGTGCGCAACACAGAGAAACGCAAAAACAAGAAGGCTTCGTCTAAAGCTAGTG), which contains the sequence of the T7 polymerase promoter (underlined) upstream of the MNV-1 5′-end sequence, and MNV-15′TR-REV (GTAATTAATTTCGTCTTCGCTCTCCGAAGAGAGGGGGCTAGGTGCTC CAAAGGAGACACTAGCTTTAGACGAAGCC). Both amplified cDNAs were subsequently fused by PCR to produce a complete amplicon of 1,173 bp containing the luciferase gene preceded by a T7 polymerase promoter fused to the MNV 5′ end and followed by the complete 3′-UTR sequence of MNV-1. The amplicon to produce antisense luciferase RNA was generated using a similar strategy from the plasmid pRS424LucIresDen2CapDeleted. PCR was performed using the primer pair LUC-NEG-FWD (ATGGCTTCCAAGGTGTACGA) and LUC-NEG-REV (TAATACGACTCACTATAGGGTTACTGCTCGTTCTTCAGCA) containing aT7 polymerase promoter sequence (underlined).
The complete 1,173 bp and the luciferase amplicons were in vitro transcribed as described above, and both the complete RNA and the RNA that consists of the negative sequence of the luciferase gene were treated with DNase I to remove the DNA templates. Partially double-stranded RNA molecules were prepared by annealing equal amounts of positive- and negative-strand RNA in annealing buffer (10 mM HEPES, pH 7.4, 100 mM KCl, 0.2 mM EDTA) at 95°C and slowly cooling them to 4°C. The integrity of the RNA was confirmed by gel electrophoresis prior to use.
Electron microscopy.
Protein-RNA complexes were formed as previously described with some modifications (27). One hundred nanograms of bovine serum albumin (BSA) or a mixture of 100 ng each of hnRNP A1 and PCBP2 was incubated with 1.5 μg of RNA in 15 μl of RNA-protein binding buffer (20 mM HEPES and 50 mM KCl) for 20 min at room temperature and stabilized by cross-linking with 1% glutaraldehyde at 4°C overnight. Fifty-microliter droplets, consisting of 10 ng/μl RNA, 10 mM Tris-HCl, pH 8, 1 mM EDTA, 50% formamide, and 100 ng/μl cytochrome c to maintain the RNA conformation, were incubated for 2 min at 4°C (27).
RNA diffused to a hypophase surface and was picked up on Formvar-coated grids (300 mesh). The grids were stained with 10 nM uranyl acetate in 90% ethanol for 20 s and dried with isopentane prior to rotary shadowing with platinum (35 A of platinum-palladium [Pt/Pl] at 8°C). The RNA conformation was observed with a transmission electron microscope (JEM 1400; JEOL Company). Micrographs were taken at ×100,000 magnification.
RESULTS
Identification of RNA-RNA contacts.
We have previously described the occurrence of 5′-3′ RNA interactions within the NV genome that initially occur by direct RNA-RNA interactions but are further stabilized by cellular proteins (33). To determine the relevance of these 5′-3′-end contacts in NoV replication, the presence of potential interactions in the MNV-1 genome was examined, as MNV-1 remains the only NoV that can undergo a full infectious life cycle in cell culture. In silico analysis was carried out using the Mfold-2 program (34) on RNA sequences consisting of the first 146 nt of the 5′ end and the complete 3′ UTR of the MNV-1 genome (Fig. 1A). The limit of the 5′ RNA sequence was chosen because it contains the majority of the most conserved predicted stem-loop elements in the 5′ end (18). A spacer sequence consisting of 45 N nucleotides between the 5′ and 3′ RNA sequences was used to facilitate RNA folding (33). A folding prediction with a ΔG of −70.6 was obtained, where all stem-loop structures predicted previously and confirmed by biochemical mapping were conserved (18, 45). In addition, a base-pairing region formed from a sequence of 6 nt (64 to 68) of the 5′ end (ACAAGA) and nt 7323 to 7328 within the 3′ UTR (UUUUGU) was predicted (Fig. 1B). To evaluate the degree of conservation of the 5′ ACAAGA and the 3′ UUUUGU nucleotide sequences between MNV-1 isolates, 49 sequences were analyzed (Table 1). The majority of the nucleotide variations in the 5′ CS represent synonymous changes, with the exception of 2 isolates, indicating the importance of the amino acid conservation in this coding region. The 3′ UUUGU sequence is also well conserved, although a C insertion in the middle of the region is present in half of the MNV-1 isolates analyzed (Table 1). Despite these changes, all the genomes retain the ability to form the predicted 5′-3′-end contacts, although in 1 isolate, the contacts involve alternative 5′ sequences (Fig. 1C).
Fig 1.


Computer analysis of the 5′-3′-end contacts of the MNV-1 genomic RNA wt region. (A) Genome schematic of the secondary structures formed within the first 146 nt and the last 75 nt (3′ UTR) of the MNV-1 genomic RNA. The positions of the CS and the AUG initiation codon are highlighted. (B) Predicted secondary structure formed between the first 146 nt and the last 75 nt (3′ UTR) of the MNV-1 genomic RNA. An expanded drawing shows CS formed in the predicted structure. The secondary structures were predicted using Mfold2 software (http://mfold.rna.albany.edu) with a ΔG value of −70.6 kcal/mol. The Ns represent the genome sequence between the 5′ and 3′ ends. (C) Predicted secondary structures formed between the first 146 nt and the last 75 nt (3′ UTR) of 9 different representative isolates and/or strains of MNV-1, corresponding to groups 1 to 9 shown in Table 1 and in Materials and Methods. The numbers in parentheses are the number of isolates in each group.
Cellular proteins mediate the interaction between the 5′ and 3′ ends of the MNV-1 genomic RNA.
To further determine if the interactions between the 5′ and 3′ ends of the MNV-1 genomic RNA predicted by the in silico analysis also occurred in vitro, coprecipitation assays were performed in which biotin-labeled 5′ MNV-1 RNA was used to precipitate α-32P-labeled 3′-UTR RNA. Under the conditions previously described (26, 33), a modest interaction between the 5′ and 3′ ends of the MNV-1 genomic RNA was observed (Fig. 2B, lane 2), which was increased significantly in the presence of cellular proteins from MNV-1 permissive Raw264.7 cells (Fig. 2B, lane 5). In addition, the observed RNA-RNA interactions in the presence of cellular proteins were specific, since a nonrelated protein, such as BSA (Fig. 2B, lane 3), or an irrelevant biotin-labeled RNA did not allow complex formation (Fig. 2B, lane 4) (41).
Fig 2.

The interaction between the 5′ and 3′ ends of the MNV-1 genomic RNA requires cellular proteins and CS complementarity, and antibodies to hnRNP A1 and PCBP2 affect these interactions. (A) Schematic representation of the 5′-3′ CS. Mutations introduced in the 5′ end (solid circles) and the compensatory mutations in the 3′ end (open circles) are highlighted. (B) Coprecipitation assay of the α-32P-labeled wt 3′ UTR (lanes 1 to 5) and the biotin-labeled wt 5′ end (lanes 2, 3, and 5) of the MNV-1 genomic RNA or an irrelevant related biotin-labeled RNA (lane 4) in the absence (lanes 1 and 2) or presence (lane 3) of 10 μg BSA and 10 μg Raw264.7 S10 (lanes 4 and 5). ext, extract; *, α-32P-labeled; b, biotin-labeled. The error bars indicate standard deviations from three independent experiments. (C) Coprecipitation assay of the α-32P-labeled wt 3′-UTR (lanes 1 to 3) or the α-32P-labeled mutant (mut) 3′-UTR (lanes 4 to 8) RNAs and the biotin-labeled wt 5′-end (lanes 2, 3, 5, and 6) or the mut 5′-end (5′ comp) RNAs (lanes 7 and 8) in the absence (lanes 1, 2, 4, 5, and 7) or presence (lanes 3, 6, and 8) of 10 μg Raw264.7 S10 extracts. (D) Complex formation in the presence of specific antibodies. Coprecipitation assay of the α-32P-labeled wt 3′ UTR and the biotin-labeled wt 5′ end of the MNV-1 genomic RNA with Raw264.7 cell extracts (all lanes), alone (lane 1 from left) or incubated in the presence of anti-La (lane 2), anti-hnRNP A1 (lane 3), anti-PCBP2 (lane 4), anti-NCL (lane 5), and anti-PTB (lane 6) antibodies prior to addition to the coprecipitation reaction mixtures. Precipitated α-32P-labeled RNA levels were quantified using ImageJ software and expressed as arbitrary intensity units. The error bars represent the standard deviations from three independent experiments. (E) RNA integrity in the presence of antibodies. The α-32P-labeled 3′-UTR RNA was interacted alone (far left lane) or in the presence of 0.2 μg of anti-La (La), anti-hnRNP A1 (A1), anti-PCBP2 (PCBP2), anti-NCL (NCL), anti-PTB (PTB), and anti-PABP (PABP) antibodies or 0.2 μg of micrococcal nuclease (far right lane).
To analyze if complementarity of the RNA was required for the observed coprecipitation, similar RNA coprecipitation studies were performed using a 3′-UTR RNA containing six mutations predicted to disrupt the interaction with the 5′ end (Fig. 2A and C). Changes introduced in the 3′ UTR were chosen to conserve the stem-loop structures in the region. Replacement of the 6 bases in the α-32P-labeled 3′ UTR RNA (3′ mutant), as shown in Fig. 2A, reduced the level of coprecipitation with wild-type (wt) biotin-labeled 5′ end RNA (Fig. 2C, lane 6), indicating that the sequence or the complementarity of the region is important for interaction with the 3′ UTR in the presence of cellular proteins. However, when compensatory mutations were included in the 5′-end RNA (5′ comp), restoring complementarity with the 3′-mutated region (ΔG, −79.6, compared to the ΔG value of −70.6 of the wt sequences), the ability to coprecipitate the 3′ UTR was restored (Fig. 2C, lane 8), indicating that complementarity and not the specific sequence is required for efficient 5′-3′ RNA interactions to occur in the presence of cellular proteins.
Identification of cellular proteins that interact with the 5′ end and the 3′ UTR of the MNV-1 genomic RNA.
We hypothesized that the primary interaction between the 5′ and the 3′ ends of the MNV-1 genome require RNA-RNA contacts in the CS but the participation of cellular proteins is required to stabilize these interactions. As our study was focused on the identification of factors that may contribute to the stabilization of the 5′-3′ RNA-RNA interactions, we selected proteins for further study on the basis of whether the proteins were known to bind the MNV-1 genome (references 14, 17, and 45 and unpublished data). Based on these criteria and the availability of reagents, we initially selected La, hnRNP A1, PCBP2, nucleolin, and PTB for further analysis. To determine if any of the selected targets might be involved directly in the stabilization of the 5′-3′ RNA-RNA interactions, cell extracts were preincubated with antibodies specific to the various proteins prior to the RNA coprecipitation assay (Fig. 2D). We hypothesized that antisera to proteins involved in this interaction would reduce the ability of the biotin-labeled 5′-end RNA to coprecipitate the α-32P-labeled 3′ UTR RNA, on the assumption that the antisera block the RNA binding or the protein-binding interaction domains required for stabilization. The reactivity of each antibody used was previously tested by Western blotting (data not shown). Protein extracts preincubated with anti-La and anti-PTB antibodies were able to precipitate labeled RNA with an efficiency similar to that of the protein extract alone (Fig. 2D, lanes 2, 6, and 1, respectively). However, preincubation with the anti-hnRNP A1 or anti-PCBP2 antibody decreased complex formation. This reduction in complex formation was observed in at least 3 independent experiments and was not due to the presence of RNAs, as the α-32P-labeled 3′-UTR RNA integrity was not affected by the presence of each antibody alone (Fig. 2E).
Role of recombinant proteins PCBP2 and hnRNP A1 in the stabilization of the 5′-3′-end contacts from the MNV-1 genomic RNA.
Given that anti-hnRNP A1 and anti-PCBP2 were able to reduce complex formation and that both proteins bind to the MNV-1 genomic RNA and have been implicated in the stabilization of viral RNA-RNA interactions (27, 49), we next examined if these proteins could promote the interaction of the 5′ and the 3′ ends of the MNV-1 genomic RNA. Therefore, a coprecipitation assay was performed using biotin-labeled 5′-end and α-32P-labeled 3′-UTR RNA probes in the presence of both PCBP2-His and His-hnRNP A1 recombinant proteins (Fig. 3A). The 3′-UTR probe contains the last 77 nt from the MNV-1 genomic RNA without the poly(A) tail, since no differences in the coprecipitation complexes formed with or without the poly(A) tail from NV (33) or MNV-1 (data not shown) were observed. Low levels of RNA coprecipitation between the 5′ and 3′ ends of the MNV-1 genomic RNA were observed in the presence of the individual recombinant protein His-hnRNP A1, PABP, or PCBP2-His (used as a control) (Fig. 3A, lanes 2, 3, and 4, respectively), as well as with both His-hnRNP A1 and PABP or PCBP2-His and PABP (Fig. 3A, lanes 6 and 8, respectively). However, when the interaction was performed in the presence of the three recombinant proteins or with both hnRNP A1 and PCBP2, increased complex formation was observed (Fig. 3A, lanes 5 and 7, respectively), indicating that hnRNP A1 and PCBP2 contributed to the MNV-1 5′- and 3′-end contacts. The efficiency of RNA coprecipitation observed using the Raw264.7 cell extracts was greater than that observed with the recombinant proteins PCBP2-His and His-hnRNP A1, suggesting that other proteins present in the cell extract may contribute to the stabilization of the RNA-RNA interactions (Fig. 3A, lane 1). The differences in the intensities of the complexes formed with the three recombinant proteins or with His-hnRNP A1 and PCBP2-His (Fig. 3A, compare lanes 6 and 8) could be related to a negative effect of PABP in the reaction mixture, due to its association with one or both recombinant proteins or to the RNAs (48).
Fig 3.

hnRNP A1 and PCBP2 contribute to CS binding, and mutations that disrupt the CS affect the formation of the coprecipitated complex. Coprecipitation assay between α-32P-labeled wt 3′ UTR and the biotin-labeled wt 5′ end (A), α-32P-labeled mut 3′ UTR and the biotin-labeled wt 5′ end (B), and α-32P-labeled mut 3′ UTR and the biotin-labeled comp 5′ end (C) were carried out in the presence of cell extracts (lanes 2), His-hnRNP A1 (lanes 2), PABP (lanes 3), PCBP2-His (lanes 4), all recombinant proteins (lanes 5), His-hnRNP A1 and PABP (lanes 6), His-hnRNP A1 and PCBP2-His (lanes 7), and PABP and PCBP2-His (lanes 8). Precipitated α-32P-labeled RNA levels were quantified using ImageJ software and are expressed as percent arbitrary intensity units. The error bars represent the standard deviations from three independent experiments.
To determine if the mutations included in the 3′-UTR RNA that disrupt the 5′-3′-end contacts affect complex formation, similar coprecipitation assays were performed in the presence of the α-32P-labeled 3′ UTR mutant RNA. Notably, the labeled wt and mutant 3′ UTRs had similar specific activities. Low levels of complex formation were observed in the assays with His-hnRNP A1, PCBP2-His, or PABP alone or with both His-hnRNP A1 and PABP or PCBP2-His and PABP, as well as with the three recombinant proteins (Fig. 3B). Finally, to determine if the mutations included in both the 5′-end and the 3′-UTR RNAs, which compensate for the 5′-3′-end contacts, restore the ability for complex formation, similar coprecipitation assays were performed in the presence of the α-32P-labeled 3′-UTR mutant and biotin-labeled 5′-end RNAs. No complex formation was observed in the assays with His-hnRNP A1, PCBP2-His, or PABP alone or with both His-hnRNP A1 and PABP or PCBP2-His and PABP. However, when the assays were performed with both His-hnRNP A1 and PCBP2-His, complex formation was observed (Fig. 3C). Taken together, these results indicate that both CS in the 5′ and 3′ end RNAs and the presence of the His-hnRNP A1 and PCBP2-His recombinant proteins are required for the in vitro stabilization of the 5′-3′-end contacts of the MNV-1 genomic RNA.
Mutations in the complementary sequences affect hnRNP A1 and/or PCBP2 binding and virus replication.
Since both PCBP2-His and His-hnRNP A1 recombinant proteins were able to stabilize the 5′-3′-end interactions of the MNV-1 genomic RNA in vitro, the abilities of both proteins to interact directly with the 5′ and 3′ ends of the MNV-1 genomic RNA were further analyzed. PCBP2 has been previously identified as a component of an RNP formed on the 5′ and the 3′ ends of the NV (14, 17) and the MNV-1 genome (14, 39), and hnRNP A1 was recently reported to bind to the MNV-1 3′ UTR (45). Therefore, the ability of the recombinant His-hnRNP A1 to bind to the 5′ or the 3′ end of the MNV-1 genomic RNA was confirmed by EMSA (Fig. 4A to D). His-hnRNP A1 was able to interact with both the 5′ and the 3′ wild-type ends of the MNV-1 genomic RNA in a dose-dependent manner, forming stable RNP complexes (Fig. 4A and B, lanes 1 to 4). In addition, the ability of the His-PCBP2 protein to bind in a dose-dependent manner to both the 5′ and the 3′ ends of the MNV-1 genome was also established (Fig. 4C and D, lanes 1 to 4). While both hnRNP A1-His and His-PCBP2 were able to bind to the corresponding RNAs that contain the mutations GGGGUG and CCCCAC in the 5′ and 3′ CS, respectively (Fig. 4A to D, lanes 5 to 8), a reduction in complex formation was observed with all the 5′ and 3′ mutant RNAs.
Fig 4.

Mutations in the CS affect hnRNP A1 and/or PCBP2 binding to the 5′ and 3′ ends of the MNV-1 genomic RNA and virus replication. (A to D) EMSA showing the recombinant hnRNP A1-His (A and B) and His-PCBP2 (C and D) interaction with the [α-32P]UTP-labeled wt 5′ end (A and C, lanes 1 to 4) and [α-32P]UTP-labeled mut 5′ end (A and C, lanes 5 to 8) and with the [α-32P] UTP-labeled wt 3′ UTR (B and D, lanes 1 to 4) and [α-32P]UTP-labeled mut 3′ UTR (B and D, lanes 5 to 8). (E and F) EMSA showing the specific interaction of the recombinant hnRNP A1-His with the MNV-1 [α-32P]UTP-labeled 5′ end (E, lanes 2 to 4) and [α-32P]UTP-labeled 3′ UTR (E, lanes 6 to 8), alone (lanes 2) or in the presence of 30× homologous (lanes 3 and 7) or heterologous (lanes 4 and 8) unlabeled RNAs. The EMSAs were performed using 2 μg of protein. Lanes 1 and 5, free probes. Unrelated [α-32P]UTP-labeled RNA (see Materials and Methods) was interacted alone (F, lanes 1 and 7) or with increasing amounts of hnRNP A1-His (F, lanes 2 to 5) or His-PCBP2 (F, lanes 8 to 11) or 3 μg BSA (lanes 6). The positions of the free probe (P) and various RNA-protein complexes (C) are highlighted.
The specificity of the interaction of His-hnRNP A1 with the MNV-1 5′-end and 3′-UTR RNAs was confirmed by both the inability of heterologous competitor RNA to compete for RNP complex formation and the ability of nonlabeled homologous RNA to compete (Fig. 4E). Moreover, none of the proteins were able to bind to an irrelevant stem-loop-labeled RNA of 60 nt that corresponds to the FL-LAP sequence of the C/EBPβ transcription factor RNA (Fig. 4F) (41).
Taken together, the results shown above suggest that the viral genome contains CS in the 5′ and 3′ ends of the MNV-1 genomic RNA that are stabilized by cellular proteins. To determine if these interactions were important during authentic viral RNA replication, two mutant cDNA constructs were produced (Fig. 5A and B), containing changes in the 3′-UTR sequence that would disrupt the 5′-3′ interactions (Table 1). The CS UUUUGU present in the 3′ UTR was changed to CCCCAC (mutant M1) and to AAAAAA (mutant M2), both predicted to disrupt complementarity with the 5′ region. Virus recovery, expressed as TCID50/ml 24 h posttransfection, was used as a measure of a single cycle of virus replication, as the cells used (a derivative of baby hamster kidney cells, BSRT7), although permissive for virus replication, lack a suitable receptor to allow multicycle replication. Therefore, variations in the virus titer in this assay are reflective of a difference in the viral translation/replication kinetics only. Viral titers from the mutant viruses were significantly lower (>100-fold) than those of the wild-type virus (P < 0.05) (Fig. 5B). Detection of the viral NS7 protein by Western blotting carried out on lysates of transfected BSRT7 cells was also performed as an indicator of the transfection efficiency, confirming that equal levels of primary cap-dependent translation occurred (Fig. 5C). These results indicate that either the integrity of the 3′-end sequence itself or the lack of complementarity with the 5′ end is important for efficient virus replication.
Fig 5.
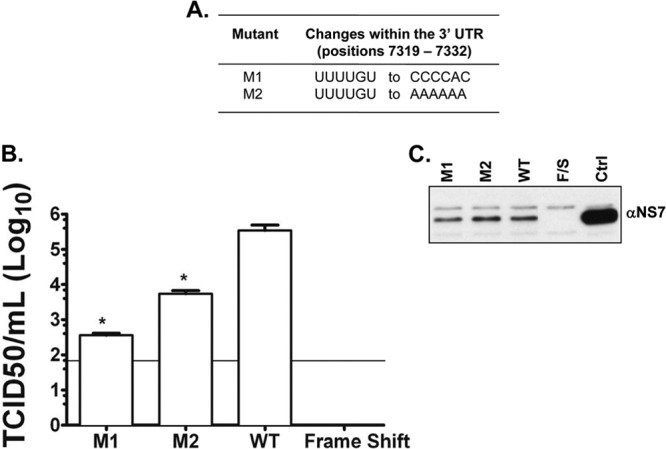
Nucleotide changes within the 3′ UTR of recovered viruses obtained from BSRT7 cells transfected with the in vitro-transcribed capped RNA pT7MNV-1:3′RZ infectious clone containing a 6-nt mutation. (A) The 6-nt changes within M1 and M2. (B) Virus recovered at 24 h posttransfection and total virus levels assayed by TCID50 in Raw264.7 cells. The limit of detection was 50 TCID50/ml. Transfections were performed in triplicate, and the average log titers are plotted, together with standard errors. Significance was also tested using one-way ANOVA compared to the wild type. *, P < 0.05. (C) Western blot analysis was carried out on lysates of transfected BSRT7 cells to assess the translation of NS7. Ctrl, total mock-transfected cell lysates. Frameshift (F/S) was also used as a recovery negative control. The data shown are representative of at least 3 independent experiments.
To further determine if the RNA-RNA interactions were necessary for viral replication, mutations within the 5′ CS predicted to reconstitute the RNA-RNA interactions were introduced into the wt MNV-1 cDNA. However, since the CS is located within a highly conserved coding region, nucleotide changes were not possible without alterations to the amino acid sequence of the NS1/2 protein. The wt 5′ CS ACAAGA (Asn-Lys-Lys) was changed to GTAGGAG (Asn-Arg-Arg) or ACAGGAG (Ser-Arg-Arg) and tested within the wt cDNA; these coding changes resulted in a significant reduction in viral recovery from a cDNA clone containing a wt 3′ end, M1, or M2 (data not shown), indicating that conservation of the amino acids is important for virus replication.
hnRNP A1 and PCBP2 interact with MNV-1 RNA during virus replication.
To validate that both hnRNP A1 and PCBP2 associate with MNV-1 RNA during authentic virus replication, an RNA coimmunoprecipitation assay was performed (Fig. 6A). Both PCBP2 and hnRNP1 were immunoprecipitated from MNV-1-infected Raw264.7 cell extracts; the copurified RNA was extracted from the immunoprecipitated complex and subjected to RT-PCR using MNV-1-specific primers. As expected, MNV-1 RNA was copurified with the viral RNA polymerase (NS7), but also with PCBP2 and hnRNP A1 (Fig. 6A). No viral RNA was amplified when an irrelevant purified rabbit IgG antibody (Fig. 6A, IgG) was used or when the assay was performed with noninfected cells. Taken together, these results confirm that hnRNP A1 and PCBP2 are associated with MNV-1 RNA during viral replication.
Fig 6.

hnRNP A1 and PCBP2 subcellular localization and interaction with the viral RNA during MNV-1 replication. (A) Antisera to MNV-1 NS7, hnRNP A1, and PCBP2 precipitated viral RNA from MNV-1-infected cells. Viral RNA was coimmunoprecipitated from MNV-1-infected Raw264.7 cell extracts using an antibody directed against viral polymerase (NS7) or anti-hnRNP A1 or anti-PCBP2 antibodies, respectively. Purified IgG (IgG) was used as a negative control. RNA was extracted from the immunoprecipitated complex or from an aliquot of the input lysate and subjected to RT-PCR using MNV-1-specific primers as detailed in Materials and Methods. The RT-PCR products were visualized on a 1% agarose gel. (B and C) Subcellular localization of hnRNP A1 (B) and PCBP2 (C) in mock-infected and MNV-1-infected Raw264.7 cells. Mock-infected Raw264.7 cells (top rows) or Raw264.7 cells infected with MNV-1 for 16 h at an MOI of 5 (bottom rows) were immunostained with an anti-hnRNP A1 (B) or an anti-PCBP2 (C) monoclonal antibody, followed by anti-mouse Alexa Fluor 488 (green) and DAPI (blue) staining. To identify the infected cells, the MNV-1 NS7 protein was immunostained with an anti-NS7 antibody, followed by anti-rabbit Alexa Fluor 594 (red) staining. The cells were observed using the Zeiss LSM 700 confocal microscope. The images depict single confocal slices taken from z-stacks. The colocalization coefficients of NS7 with hnRNP A1 and PCBP2 determined by ZEN 2010 software were 0.11/1 and 0.46/1, respectively. Enlarged views of the boxed areas are shown on the right. 3D, maximally projected z-stack. (D) Western blot of the hnRNP A1 protein in nuclear and cytoplasmic extracts from mock-infected (Mock) and MNV-1-infected (INF) Raw264.7 cells. The annexin II expression levels were used to show that cytosol and nuclear extracts were well separated. The actin expression levels were used as protein-loading controls. The data shown are representative of at least 3 independent experiments.
hnRNP A1 and PCBP2 are associated with the viral replication complex.
Given the association of both hnRNP A1 and PCBP2 with the MNV-1 genomic RNA in infected cells, it was important to determine their subcellular localization during MNV-1 infection. Analysis of the localization of both hnRNP A1 and PCBP2 after MNV-1 infection of Raw264.7 cells was performed using confocal microscopy. Immunostaining analysis showed that hnRNP A1 (green) is located mostly in the nuclei of noninfected Raw264.7 cells (Fig. 6B, top row). During MNV-1 infection, although hnRNP A1 remained predominantly nuclear, those cells expressing high levels of NS7 showed an increase of hnRNP A1 staining in the cytoplasm that correlated with a decrease in the nuclear hnRNP A1 levels (Fig. 6B, bottom row). The costaining of NS7 (red) and hnRNP A1 resulted in a degree of signal overlap in the perinuclear area, where the majority of the viral antigen or dsRNA has been found (45, 50) (Fig. 6B). The presence of hnRNP A1 in the nuclei and the cytoplasm of the infected cells was confirmed by the biochemical separation of nuclear and cytoplasmic fractions (Fig. 6D). Annexin II showed that the cytosol and nuclear fractions were well separated. Infection was demonstrated by the presence of NS7; the detection of NS7 in the nuclear fractions is due to the copurification of the replication complexes closely associated with the perinuclear membranes (50). In contrast, the distribution of PCBP2 in the noninfected Raw264.7 cells was detected throughout the nucleus and the cytoplasm, and it was mostly unaffected by MNV-1 infection (Fig. 6C, top row). Some degree of signal overlap of NS7 was also observed, with punctate foci of PCBP2 being found within the NS7-positive perinuclear area (Fig. 6C, bottom row).
Inhibition of expression of hnRNP A1 and PCBP2 results in the reduction of MNV1 replication.
The results presented above show that hnRNP A1 and PCBP2 stabilize the in vitro 5′-3′-end contacts of the MNV-1 genomic RNA and bind to the viral RNA in MNV-1 infected cells, suggesting that this association could have a role during viral replication. To confirm the role of hnRNP A1 and PCBP2 in the MNV-1 life cycle, BV2 cells were transfected with specific siRNAs directed toward hnRNP A1 or PCBP2, and the levels of both proteins were detected by Western blot assay. BV2 cells, previously shown to be highly permissive for MNV-1 infection (37, 45), were chosen for these assays, since they allow robust and reproducible siRNA knockdown of host factors in comparison with Raw264.7 cells, where transfection of either siRNAs or short hairpin RNA (shRNA)-expressing plasmids has been largely unsuccessful (45). Transfection of specific siRNAs resulted in a decrease in hnRNP A1 and PCBP2 levels (Fig. 7A and B, respectively) compared to cells transfected with an irrelevant siRNA.
Fig 7.

hnRNP A1 and PCBP2 siRNA affect virus replication. (A and B) BV-2 cells were transfected with an irrelevant siRNA or an siRNA specific for either hnRNP A1 (A1) (A) or PCBP2 (B) for 12 h. (C and D) The treated cells were then infected with MNV-1 at an MOI of 5 for 18 h, and RNA (C) and virus titer (D) determinations were performed by qRT-PCR or TCID50 assays, respectively. Detection of La protein was used as the loading control. Detection of NS7 indicates infection. The error bars represent the standard deviations from three independent experiments. ***, P < 0.001 for qRT-PCR, and **, P < 0.01 for TCID50 experiments by two-way ANOVA.
The consequences of hnRNP A1 and PCBP2 knockdown for MNV1 replication were evaluated by monitoring the levels of RNA and infectious virus produced. hnRNP A1 knockdown was found to significantly reduce MNV-1 RNA levels (Fig. 7C, left bar) in comparison to the RNA levels obtained from the cells treated with an irrelevant siRNA (Fig. 7C, right bar). Moreover, a reduction of genomic RNA production in PCBP2 knockdown cells was also observed (Fig. 7C, middle bar).
In agreement with the observed effect on RNA levels, cells transfected with hnRNP A1 or PCBP2 siRNA displayed reduced viral titers compared to the viral titers observed in the irrelevant-siRNA-treated cells both at high (Fig. 7D) and low (data not shown) MOI. The viability and growth of cells was unaffected by siRNA treatment (data not shown), and therefore, the observed decreased replication of MNV-1 in hnRNP A1 and PCBP2 siRNA-transfected cells was specific and not due to an indirect effect resulting in gross changes in cellular metabolism. Although the levels of RNA and the viral titer were not drastically reduced, these results taken together indicate that both hnRNP A1 and PCBP2 participate in the MNV-1 life cycle.
hnRNP A1 and PCBP2 induce MNV-1 RNA circularization.
The ability of hnRNP A1 and PCBP2 to bind to both the 5′ and the 3′ ends of the MNV-1 genomic RNA, as well as its association with the MNV-1 genomic RNA in infected cells, suggests that these proteins may have a role in the MNV-1 replicative cycle. Moreover, the coprecipitation of the 5′ end and the 3′ UTR in the presence of both proteins suggests that they could be involved in the MNV-1 RNA circularization. To support this idea, the protein-mediated changes of the MNV-1 RNA conformation were observed by EM. To avoid the secondary structures of single-stranded RNAs, RNA consisting of the single-stranded MNV-1 5′ end and 3′ UTR-flanking 933-bp double-stranded RNA of the luciferase gene were generated (Fig. 8A). After interacting with both hnRNP A1-His and His-PCBP2 proteins or unrelated BSA, the RNAs were observed by EM. In the absence of protein or in the presence of BSA, the RNA molecules appeared in linear conformation (Fig. 8B, left and middle). In the presence of both hnRNP A1-His and His-PCBP, circularization of the RNA molecules was observed (Fig. 8C, right), indicating that, as noted for other RNA viruses, MNV-1 genome circularization is at least partially mediated by cellular factors, including hnRNP A1 and PCBP (20, 25–27, 31, 49).
Fig 8.

MNV-1 RNA circularizes in the presence of hnRNP A1 and PCBP2. (A) Scheme of the structure of the RNA molecule used (see Materials and Methods). (+), positive sense RNA molecule; (−), antisense RNA molecule. (B) RNA electron micrographs. RNA molecules with 5′ ends and 3′ UTRs were incubated with BSA or hnRNP A1 and PCBP2 and observed under EM. (C) Amplification of a circular molecule. One hundred molecules were counted for each set, and the percentages of circular, pseudocircular, and linear RNA molecules are indicated.
DISCUSSION
The majority of the cis-acting signals required for the regulation of translation and viral replication of positive-strand RNA viruses, such as NoVs, are thought to be located in the 5′- and 3′-terminal regions of the viral genomes. Since both processes arise from the same molecule and depend on both terminal regions, long-distance interactions within the viral RNA genomes take place to ensure an efficient mechanism to guarantee the generation of the viral progeny (22, 23, 27, 31). These interactions can occur via direct RNA-RNA interactions (22, 23) or can be mediated by cellular proteins (20, 31).
We have previously demonstrated by in vitro coprecipitation assays that 5′-3′-end interactions occur in the NV genomic RNA in the presence of cellular proteins (33); however, to further determine if these interactions could have a role during NoV replication, the formation of 5′-3′-end interactions was analyzed in the RNA of MNV-1, the only NoV that replicates efficiently in tissue culture. By computational analysis, a 6-nt complementarity was predicted to occur between the 5′ end and the 3′ UTR of the MNV-1 genomic RNA. These sequences and the predicted 5′-3′ interactions are conserved across several MNV-1 strains, yet single-nucleotide synonymous variations within the 5′ CS were found, indicating the importance of amino acid conservation in this coding region. The 3′ CS was also highly conserved, showing that the sequences within the UTRs are involved in maintaining the cis-acting RNA structures involved in the viral life cycle (18, 45).
In this study, the in vitro coprecipitation assays showed that in silico-predicted RNA-RNA contact is required to allow the binding of cellular proteins that further stabilize the interaction. This is similar to our previously described analysis of NV RNA (33). Moreover, the disruption of the RNA-RNA interactions resulted in a reduction in complex formation, suggesting that both the integrity of the CS and the presence of cellular proteins are needed for the in vitro stabilization of the 5′-3′-end contacts of the MNV-1 genomic RNA.
In this study, two of the proteins directly involved in the stabilization of the 5′-3′ RNA-RNA interactions were identified as hnRNP A1 and PCBP2. hnRNP A1 is one of the most abundant and best-studied components of hnRNP complexes; together with other hnRNP proteins, hnRNP A1 packages nascent pre-mRNAs for processing in the nucleus (reviewed in references 46, 47, and 51) It is broadly distributed throughout the nucleoplasm and shuttles continuously between the nucleus and the cytoplasm (47), associates with poly(A) RNAs from both compartments, and is involved in several RNA metabolic processes, such as pre-mRNA splicing and trafficking. The N-terminal domain of hnRNP A1 contains two RNA recognition motifs (RRMs), which are known as unwinding protein 1 (U1), while the C-terminal region, particularly rich in glycine (52, 53), is involved in protein-protein interactions and also contributes to stable RNA binding (54). This protein facilitates the efficient annealing of cRNA or DNA molecules in vitro and the base-pairing interactions between small nuclear RNAs and pre-mRNAs (55, 56). In fact, sequence-specific RNA binding and RNA annealing are necessary for the ability of hnRNP A1 to regulate alternative splicing. The reannealing activity of hnRNP A1 is involved in the modulation of RNA secondary structures via its RNA helicase activity (57). This activity may be related to the roles that hnRNP A1 has in the translation and/or replication of numerous RNA viruses, such as mouse hepatitis virus (MHV) (49, 58), HCV (25, 59), dengue virus (60), enterovirus 71 (EV71) (61, 62), and Sindbis virus (61).
PCBP2, also known as hnRNP E2 and αCP2, is a shuttling protein involved in a remarkable array of biological processes, such as mRNA stability and the translational control of specific cellular mRNAs (27, 63–65). PCBP2 specifically interacts with nucleic acids containing tandem poly(C) motifs as a general mechanism for the stabilization of long-lived cellular mRNAs (66, 67). It stabilizes the α-globin and collagen 1α (22) mRNAs in vivo through its interaction with three pyrimidine-rich patches within their 3′ UTRs (68–70). PCBP2 is also implicated in the stabilization of viral RNAs and in the regulation of translation and/or replication and gene expression of several viruses, including poliovirus (30, 71–73), hepatitis A virus (HAV) (74), human papillomavirus (HPV) (75), vesicular stomatitis virus (VSV) (76), and HCV (27).
The precise binding sites of PCBP2 to the 3′ end of the MNV-1 genome have been determined; the most prominent PCBP2 binding site in the 3′ UTR has been mapped in the UUUUCUUU sequence present in the loop of the SL3 structure (nt 7350 to 7362) (14). An additional PCBP2 binding site within the 3′ UTR has been proposed, although not yet identified. In addition, two poly(C) sequences within the first 146 nt of the MNV-1 genomic RNA, located specifically in nt 24 to 38 and 110 to 121, correspond to potential PCBP2 binding sites (unpublished data). The interaction of hnRNP A1 within the 3′ UTR of the MNV-1 RNA has been recently reported, although no specific binding site, interaction with the 5′ end, or function of the interaction was described (45). In this work, hnRNP A1 was found to interact with both the 5′ and the 3′ ends of the MNV-1 genome. Although the precise binding sites were not determined, the specific AGAAG sequence responsible for the interaction of hnRNP A1 with the MHV RNA (49) is present within nt 67 to 71 of the MNV-1 genomic RNA, near the 5′ complementarity region (Fig. 1A). Downstream of this region, a UAGUGU sequence (nt 86 to 91) may also represent another putative hnRNP A1 binding site, since UAGAGU or the related sequence UAGGGU allows hnRNP A1 binding to its own pre-mRNA (77). Finally, an AU-rich element (nt 136 to 145) could also indicate another putative binding site (78, 79). It is interesting that both hnRNP A1 and PCBP2 are particularly involved in the circularization of viral RNAs and promote viral replication; hnRNP A1 and PTB are responsible for the MHV 5′-3′-end contacts (49), while PCBP2, PABP, and the viral protease-polymerase 3CD participate in the circularization of PV genomic RNA (31). Moreover, two PCBP2 molecules have recently been implicated in the circularization of HCV through a 5′- and 3′-UTR interaction (27).
The ability of PCBP2 and hnRNP A1 to specifically bind to both the 5′ and 3′ ends of the MNV-1 genomic RNA in infected cells, as well as the physical RNA-RNA interaction stimulated by these proteins, as observed by EM, provide evidence of a possible role during NoV genome circularization. It has been proposed that hnRNP A1 has the ability to stabilize RNA-RNA interactions either by a single molecule simultaneously binding to two different nucleic acid strands or by the dimerization of two hnRNP A1 molecules bound to two complementary strands (57). However, the requirement for both hnRNP A1 and PCBP2 to stabilize the RNA-RNA interactions of the MNV-1 genomic RNA suggests that both molecules bound near the CS could contribute to bring together both ends of the RNA molecules. It is worth noting that a physical interaction of PCBP2 and hnRNP A1 has not as yet been reported.
The communication between the 5′ m7GpppN cap and the poly(A) tail on eukaryotic mRNAs is mediated by cellular proteins and results in the synergistic enhancement of translation initiation, contributing to the control of mRNA expression in the eukaryotic cell (80–82). Moreover, long-range contacts that occur in the positive-strand RNA viral genomes often promote either RNA translation (83) or replication (31, 84) from a single molecule and can take place by RNA-RNA (22, 23) or RNA-protein (25–27, 31) interactions. Bringing the initiation sites for translation and RNA synthesis physically together also provides a strategy to recruit the ribosome or the replicase to the appropriate sites and to coordinate the two processes. The importance of the complementarity between the two ends of the MNV-1 genomic RNA, as well as the role of both hnRNP A1 and PCBP2 in virus production, was demonstrated in this work. In addition, the significant reduction in the recovery of infectious viruses containing mutations within the 3′-UTR CS that modify the interaction observed with the wild-type CS (M1 and M2) could be a consequence of the disruption of the 5′-3′-end interactions and/or the result of alterations in the structured regions within the 3′ UTR of the genomic RNA implicated in efficient viral replication. Unfortunately, due to the highly conserved amino acid sequence within the nonstructural proteins NS1/2, we were unable to provide additional evidence of the role of the CS sequence using reconstitution experiments, as nonsynonymous changes in this region were debilitating in the presence or absence of a mutated 3′ UTR.
The requirement for both hnRNP A1 and PCBP2 during MNV-1 replication was confirmed by the specific knockdown of each protein, which in both cases displayed comparable inhibitory effects on MNV-1 RNA and virus production, thus suggesting that both proteins are positive regulators required for efficient MNV-1 replication. However, although statistically significant and reproducible reduction in virus replication was observed, dramatic reduction in the RNA levels and viral titer was not achieved. One explanation of this observation could be the fact that proteins in the hnRNP A/B family are sometimes functionally interchangeable; therefore, the function of hnRNP A1 may be partially replaced by the presence of another hnRNP, and a more drastic inhibitory effect may be observed only after both proteins are knocked down (61). It is also worth noting that the levels of cellular RNA binding proteins are often increased in immortalized cells, and therefore, partial complementation due to overexpression of other cellular factors involved in genome circularization could occur. Further studies will require the use of primary cells to further delineate the functions of hnRNP A1 and PCBP2 in the MNV-1 life cycle.
hnRNP A1 and PCBP2 have been implicated in a number of functions related to viral gene expression, such as maintaining secondary and tertiary RNA structures, bringing the initiation sites for translation and RNA synthesis physically together, and promoting either internal ribosome entry site (IRES)-dependent or non-IRES-dependent translation. Moreover, they also have been implicated in the RNA replication and stability of several RNA viruses (27, 30, 85, 86); thus, it is possible that these proteins may have a role in promoting or coordinating MNV-1 translation and/or RNA replication. It has been suggested that binding of either PTB or PCBP2 to the 3′ polypyrimidine tract [p(Y)] within the MNV-1 genomic RNA has a repressive effect on virus translation, since increased levels of viral proteins were produced by viruses lacking this region (14); moreover, the function of PTB as a negative regulator of feline calicivirus (FCV) translation has been demonstrated (87). In addition to PCBP2 and hnRNP A1, numerous other proteins have been recently reported as being recruited to the ends of the MNV-1 genome (45) and may also be implicated in the formation of ribonucleoprotein complexes that participate in multiple aspects of the viral life cycle.
Taken together, these results suggest that hnRNP A1 and PCBP2 bind the 5′ end and the 3′ UTR of the MNV-1 genomic RNA and contribute to genome circularization and the virus life cycle. These data add NoVs to the growing list of RNA viruses in which genome circularization plays a role in the virus life cycle.
ACKNOWLEDGMENTS
We thank Carlos Sandoval and Laura Baltierra for cloning and expression of the hnRNP A1 gene and B. Semler for providing the pET22B:PCBP2 plasmid. We also thank Rosa M. del Angel and Juan Ludert for many helpful suggestions and critical comments on the manuscript.
This work was supported by grants 154767 from the Consejo Nacional de Ciencia y Tecnología, Mexico City, D.F., and ICYTDF/247 from the Instituto de Ciencia y Tecnología del Distrito Federal, Mexico City, D.F., to A.L.G.-E.; by a studentship from the Panamanian government to L.U.; and by a grant from the Wellcome Trust and the U.K. Biotechnology and Biological Sciences Research Council (BBSRC) to I.G. I.G. is a Wellcome Senior Fellow.
Footnotes
Published ahead of print 14 August 2013
REFERENCES
- 1.Lopman B, Zambon M, Brown DW. 2008. The evolution of norovirus, the “Gastric Flu.” PLoS Med. 5:e42. 10.1371/journal.pmed.0050042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patel MM, Widdowson M-A, Glass RI, Akazawa K, Vinje J, Parashar UD. 2008. Systematic literature review of role of noroviruses in sporadic gastroenteritis. Emerg. Infect. Dis. 14:1224–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Teunis PFM, Moe CL, Liu P, Miller SE, Lindesmith L, Baric RS, Le Pendu J, Calderon RL. 2008. Norwalk virus: how infectious is it? J. Med. Virol. 80:1468–1476 [DOI] [PubMed] [Google Scholar]
- 4.Clarke IN, Lambden PR. 1997. The molecular biology of caliciviruses. J. Gen. Virol. 78:291–301 [DOI] [PubMed] [Google Scholar]
- 5.McFadden N, Bailey D, Carrara G, Benson A, Chaudhry Y, Shortland A, Heeney J, Yarovinsky F, Simmonds P, Macdonald A, Goodfellow I. 2011. Norovirus regulation of the innate immune response and apoptosis occurs via the product of the alternative open reading frame 4. PLoS Pathog. 7:e1002413. 10.1371/journal.ppat.1002413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duizer E, Schwab KJ, Neill FH, Atmar RL, Koopmans MPG, Estes MK. 2004. Laboratory efforts to cultivate noroviruses. J. Gen. Virol. 85:79–87 [DOI] [PubMed] [Google Scholar]
- 7.Karst SM, Wobus CE, Lay M, Davidson J, Virgin HW. 2003. STAT1-dependent innate immunity to a Norwalk-like virus. Science 299:1575–1578 [DOI] [PubMed] [Google Scholar]
- 8.Guix S, Asanaka M, Katayama K, Crawford SE, Neill FH, Atmar RL, Estes MK. 2007. Norwalk virus RNA is infectious in mammalian cells. J. Virol. 81:12238–12248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaudhry Y, Nayak A, Bordeleau M-E, Tanaka J, Pelletier J, Belsham GJ, Roberts LO, Goodfellow IG. 2006. Caliciviruses differ in their functional requirements for eIF4F components. J. Biol. Chem. 281:25315–25325 [DOI] [PubMed] [Google Scholar]
- 10.Daughenbaugh KF, Fraser CS, Hershey JWB, Hardy ME. 2003. The genome-linked protein VPg of the Norwalk virus binds eIF3, suggesting its role in translation initiation complex recruitment. EMBO J. 22:2852–2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daughenbaugh KF, Wobus CE, Hardy ME. 2006. VPg of murine norovirus binds translation initiation factors in infected cells. Virol. J. 3:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodfellow I, Chaudhry Y, Gioldasi I, Gerondopoulos A, Natoni A, Labrie L, Laliberte JF, Roberts L. 2005. Calicivirus translation initiation requires an interaction between VPg and eIF4E. EMBO Rep. 6:968–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wobus CE, Thackray LB, Virgin HW. 2006. Murine norovirus: a model system to study norovirus biology and pathogenesis. J. Virol. 80:5104–5112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bailey D, Karakasiliotis I, Vashist S, Chung LMW, Rees J, McFadden N, Benson A, Yarovinsky F, Simmonds P, Goodfellow I. 2010. Functional analysis of RNA structures present at the 3′ extremity of the murine norovirus genome: the variable polypyrimidine tract plays a role in viral virulence. J. Virol. 84:2859–2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hardy ME, Estes MK. 1996. Completion of the Norwalk virus genome sequence. Virus Genes 12:287–290 [DOI] [PubMed] [Google Scholar]
- 16.Gutierrez-Escolano AL, Brito ZU, del Angel RM, Jiang X. 2000. Interaction of cellular proteins with the 5′ end of Norwalk virus genomic RNA. J. Virol. 74:8558–8562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gutierrez-Escolano AL, Vazquez-Ochoa M, Escobar-Herrera J, Hernandez-Acosta J. 2003. La, PTB, and PAB proteins bind to the 3′ untranslated region of Norwalk virus genomic RNA. Biochem. Biophys. Res. Commun. 311:759–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simmonds P, Karakasiliotis I, Bailey D, Chaudhry Y, Evans DJ, Goodfellow IG. 2008. Bioinformatic and functional analysis of RNA secondary structure elements among different genera of human and animal caliciviruses. Nucleic Acids Res. 36:2530–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Filomatori CV, Lodeiro MF, Alvarez DE, Samsa MM, Pietrasanta L, Gamarnik AV. 2006. A 5′ RNA element promotes dengue virus RNA synthesis on a circular genome. Genes Dev. 20:2238–2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gamarnik AV, Andino R. 1998. Switch from translation to RNA replication in a positive-stranded RNA virus. Genes Dev. 12:2293–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villordo SM, Alvarez DE, Gamarnik AV. 2010. A balance between circular and linear forms of the dengue virus genome is crucial for viral replication. RNA 16:2325–2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alvarez DE, Filomatori CV, Gamarnik AV. 2008. Functional analysis of dengue virus cyclization sequences located at the 5′ and 3′ UTRs. Virology 375:223–235 [DOI] [PubMed] [Google Scholar]
- 23.Alvarez DE, Lodeiro MF, Luduena SJ, Pietrasanta LI, Gamarnik AV. 2005. Long-range RNA-RNA interactions circularize the dengue virus genome. J. Virol. 79:6631–6643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheung P, Zhang M, Yuan J, Chau D, Yanagawa B, McManus B, Yang DC. 2002. Specific interactions of HeLa cell proteins with coxsackievirus B3 RNA: La autoantigen within the 5′ untranslated region binds differentially to multiple sites. Virus Res. 90:23–36 [DOI] [PubMed] [Google Scholar]
- 25.Isken O, Baroth M, Grassmann CW, Weinlich S, Ostareck DH, Ostareck-Lederer A, Behrens SE. 2007. Nuclear factors are involved in hepatitis C virus RNA replication. RNA 13:1675–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Isken O, Grassmann CW, Sarisky RT, Kann M, Zhang S, Grosse F, Kao PN, Behrens SE. 2003. Members of the NF90/NFAR protein group are involved in the life cycle of a positive-strand RNA virus. EMBO J. 22:5655–5665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang LY, Jeng KS, Lai MMC. 2011. Poly(C)-binding protein 2 interacts with sequences required for viral replication in the hepatitis C virus (HCV) 5′ untranslated region and directs HCV RNA replication through circularizing the viral genome. J. Virol. 85:7954–7964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bedard KM, Walter BL, Semler BL. 2004. Multimerization of poly(rC) binding protein 2 is required for translation initiation mediated by a viral IRES. RNA 10:1266–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blyn LB, Swiderek KM, Richards O, Stahl DC, Semler BL, Ehrenfeld E. 1996. Poly(rC) binding protein 2 binds to stem-loop IV of the poliovirus RNA 5′ noncoding region: identification by automated liquid chromatography tandem mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 93:11115–11120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blyn LB, Towner JS, Semler BL, Ehrenfeld E. 1997. Requirement of poly(rC) binding protein 2 for translation of poliovirus RNA. J. Virol. 71:6243–6246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herold J, Andino R. 2001. Poliovirus RNA replication requires genome circularization through a protein-protein bridge. Mol. Cell 7:581–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perera R, Daijogo S, Walter BL, Nguyen JHC, Semler BL. 2007. Cellular protein modification by poliovirus: the two faces of poly(rC)-binding protein. J. Virol. 81:8919–8932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sandoval-Jaime C, Gutierrez-Escolano AL. 2009. Cellular proteins mediate 5′-3′ end contacts of Norwalk virus genomic RNA. Virology 387:322–330 [DOI] [PubMed] [Google Scholar]
- 34.Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31:3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buchholz UJ, Finke S, Conzelmann KK. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 73:251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. 1990. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J. Neuroimmunol. 27:229–237 [DOI] [PubMed] [Google Scholar]
- 37.Cox C, Cao SB, Lu YA. 2009. Enhanced detection and study of murine norovirus-1 using a more efficient microglial cell line. Virol. J. 6:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bradford MM. 1976. Rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 39.Barton DJ, Flanegan JB. 1993. Coupled translation and replication of poliovirus RNA in vitro: synthesis of functional 3D polymerase and infectious virus. J. Virol. 67:822–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Solano-Gonzalez E, Burrola-Barraza E, Leon-Sicairos C, Avila-Gonzalez L, Gutierrez-Escolano L, Ortega-Lopez J, Arroyo R. 2007. The trichomonad cysteine proteinase TVCP4 transcript contains an iron-responsive element. FEBS Lett. 581:2919–2928 [DOI] [PubMed] [Google Scholar]
- 41.Timchenko LT, Iakova P, Welm AL, Cai ZJ, Timchenko NA. 2002. Calreticulin interacts with C/EBP alpha and C/EBP beta mRNAs and represses translation of C/EBP proteins. Mol. Cell. Biol. 22:7242–7257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yunus MA, Chung LM, Chaudhry Y, Bailey D, Goodfellow I. 2010. Development of an optimized RNA-based murine norovirus reverse genetics system. J. Virol. Methods 169:112–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chaudhry Y, Skinner MA, Goodfellow IG. 2007. Recovery of genetically defined murine norovirus in tissue culture by using a fowlpox virus expressing T7 RNA polymerase. J. Gen. Virol. 88:2091–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cancio-Lonches C, Yocupicio-Monroy M, Sandoval-Jaime C, Galvan-Mendoza I, Urena L, Vashist S, Goodfellow I, Salas-Benito J, Lorena Gutierrez-Escolano A. 2011. Nucleolin interacts with the feline calicivirus 3′ untranslated region and the protease-polymerase NS6 and NS7 proteins, playing a role in virus replication. J. Virol. 85:8056–8068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vashist S, Urena L, Chaudhry Y, Goodfellow I. 2012. Identification of RNA-protein interaction networks involved in the norovirus life cycle. J. Virol. 86:11977–11990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pinol-Roma S. 1997. HnRNP proteins and the nuclear export of mRNA. Semin. Cell Dev. Biol. 8:57–63 [DOI] [PubMed] [Google Scholar]
- 47.Pinol-Roma S, Dreyfuss G. 1992. Shuttling of pre-messenger-RNA binding-proteins between nucleus and cytoplasm. Nature 355:730–732 [DOI] [PubMed] [Google Scholar]
- 48.Polacek C, Friebe P, Harris E. 2009. Poly(A)-binding protein binds to the non-polyadenylated 3′ untranslated region of dengue virus and modulates translation efficiency. J. Gen. Virol. 90:687–692 [DOI] [PubMed] [Google Scholar]
- 49.Huang PY, Lai MMC. 2001. Heterogeneous nuclear ribonucleoprotein A1 binds to the 3′-untranslated region and mediates potential 5′-3′-end cross talks of mouse hepatitis virus RNA. J. Virol. 75:5009–5017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hyde JL, Sosnovtsev SV, Green KY, Wobus C, Virgin HW, Mackenzie JM. 2009. Mouse norovirus replication is associated with virus-induced vesicle clusters originating from membranes derived from the secretory pathway. J. Virol. 83:9709–9719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gorlach M, Burd CG, Portman DS, Dreyfuss G. 1993. The hnRNP proteins. Mole. Biol. Rep. 18:73–78 [DOI] [PubMed] [Google Scholar]
- 52.Buvoli M, Cobianchi F, Biamonti G, Riva S. 1990. Recombinant hnRNP protein a1 and its N-terminal domain show preferential affinity for oligodeoxynucleotides homologous to intron exon acceptor sites. Nucleic Acids Res. 18:6595–6600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Merrill BM, Stone KL, Cobianchi F, Wilson SH, Williams KR. 1988. Phenylalanines that are conserved among several RNA-binding proteins form part of a nucleic acid-binding pocket in the A1 heterogeneous nuclear ribonucleoprotein. J. Biol. Chem. 263:3307–3313 [PubMed] [Google Scholar]
- 54.Kumar A, Wilson SH. 1990. Studies of the strand-annealing activity of mammalian hnRNP complex protein-A1. Biochemistry 29:10717–10722 [DOI] [PubMed] [Google Scholar]
- 55.Mayeda A, Munroe SH, Xu RM, Krainer AR. 1998. Distinct functions of the closely related tandem RNA-recognition motifs of hnRNP A1. RNA 4:1111–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Munroe SH, Dong XF. 1992. Heterogeneous nuclear ribonucleoprotein-A1 catalyzes RNA-RNA annealing. Proc. Natl. Acad. Sci. U. S. A. 89:895–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cobianchi F, Biamonti G, Maconi M, Riva S. 1994. Human hnRNP protein A1: a model polypeptide for a structural and genetic investigation of a broad family of RNA-binding proteins. Genetica 94:101–114 [DOI] [PubMed] [Google Scholar]
- 58.Shi ST, Huang PY, Li HP, Lai MMC. 2000. Heterogeneous nuclear ribonucleoprotein A1 regulates RNA synthesis of a cytoplasmic virus. EMBO J. 19:4701–4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim CS, Seol SK, Song OK, Park JH, Jang SK. 2007. An RNA-binding protein, hnRNP A1, and a scaffold protein, septin 6, facilitate hepatitis C virus replication. J. Virol. 81:3852–3865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paranjape SM, Harris E. 2007. Y box-binding protein-1 binds to the dengue virus 3′-untranslated region and mediates antiviral effects. J. Biol. Chem. 282:30497–30508 [DOI] [PubMed] [Google Scholar]
- 61.Lin JY, Shih SR, Pan MJ, Li C, Lue CF, Stollar V, Li ML. 2009. hnRNP A1 interacts with the 5′ untranslated regions of enterovirus 71 and Sindbis virus RNA and is required for viral replication. J. Virol. 83:6106–6114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shih SR, Stollar V, Li ML. 2011. Host factors in enterovirus 71 replication. J. Virol. 85:9658–9666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berry AM, Flock KE, Loh HH, Ko JL. 2006. Molecular basis of cellular localization of poly C binding protein 1 in neuronal cells. Biochem. Biophys. Res. Commun. 349:1378–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Makeyev AV, Liebhaber SA. 2002. The poly(C)-binding proteins: a multiplicity of functions and a search for mechanisms. RNA 8:265–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ostareck-Lederer A, Ostareck DH, Hentze MW. 1998. Cytoplasmic regulatory functions of the KH-domain proteins hnRNPs K and E1/E2. Trends Biochem. Sci. 23:409–411 [DOI] [PubMed] [Google Scholar]
- 66.Du Z, Lee JK, Fenn S, Tjhen R, Stroud RM, James TL. 2007. X-ray crystallographic and NMR studies of protein-protein and protein-nucleic acid interactions involving the KH domains from human poly(C)-binding protein-2. RNA 13:1043–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Holcik M, Liebhaber SA. 1997. Four highly stable eukaryotic mRNAs assemble 3′ untranslated region RNA-protein complexes sharing cis and trans components. Proc. Natl. Acad. Sci. U. S. A. 94:2410–2414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kiledjian M, Wang X, Liebhaber SA. 1995. Identification of two KH domain proteins in the alpha-globin mRNP stability complex. EMBO J. 14:4357–4364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stefanovic B, Hellerbrand C, Holcik M, Briendl M, Aliebhaber S, Brenner DA. 1997. Posttranscriptional regulation of collagen alpha1(I) mRNA in hepatic stellate cells. Mol. Cell. Biol. 17:5201–5209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang X, Kiledjian M, Weiss IM, Liebhaber SA. 1995. Detection and characterization of a 3′ untranslated region ribonucleoprotein complex associated with human alpha-globin mRNA stability. Mol. Cell. Biol. 15:1769–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fitzgerald KD, Semler BL. 2011. Re-localization of cellular protein SRp20 during poliovirus infection: bridging a viral IRES to the host cell translation apparatus. PLoS Pathog. 7:e1002127. 10.1371/journal.ppat.1002127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Spear A, Sharma N, Flanegan JB. 2008. Protein-RNA tethering: the role of poly(C) binding protein 2 in poliovirus RNA replication. Virology 374:280–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Toyoda H, Franco D, Fujita K, Paul AV, Wimmer E. 2007. Replication of poliovirus requires binding of the poly(rC) binding protein to the cloverleaf as well as to the adjacent C-rich spacer sequence between the cloverleaf and the internal ribosomal entry site. J. Virol. 81:10017–10028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Graff J, Cha J, Blyn LB, Ehrenfeld E. 1998. Interaction of poly(rC) binding protein 2 with the 5′ noncoding region of hepatitis A virus RNA and its effects on translation. J. Virol. 72:9668–9675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Collier B, Goobar-Larsson L, Sokolowski M, Schwartz S. 1998. Translational inhibition in vitro of human papillomavirus type 16 L2 mRNA mediated through interaction with heterogenous ribonucleoprotein K and poly(rC)-binding proteins 1 and 2. J. Biol. Chem. 273:22648–22656 [DOI] [PubMed] [Google Scholar]
- 76.Dinh PX, Beura LK, Panda D, Das A, Pattnaik AK. 2011. Antagonistic effects of cellular poly(C) binding proteins on vesicular stomatitis virus gene expression. J. Virol. 85:9459–9471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chabot B, Blanchette M, Lapierre I, La Branche H. 1997. An intron element modulating 5′ splice site selection in the hnRNP A1 pre-mRNA interacts with hnRNP A1. Mol. Cell. Biol. 17:1776–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hamilton BJ, Nagy E, Malter JS, Arrick BA, Rigby WFC. 1993. Association of heterogeneous nuclear ribonucleoprotein-A1 and C proteins with reiterated AUUUA sequences. J. Biol. Chem. 268:8881–8887 [PubMed] [Google Scholar]
- 79.Hamilton BJN, Burns CM, Nichols RC, Rigby WFC. 1997. Modulation of AUUUA response element binding by heterogeneous nuclear ribonucleoprotein A1 in human T lymphocytes: the roles of cytoplasmic location, transcription, and phosphorylation. J. Biol. Chem. 272:28732–28741 [DOI] [PubMed] [Google Scholar]
- 80.Gallie DR. 1991. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev. 5:2108–2116 [DOI] [PubMed] [Google Scholar]
- 81.Preiss T, Hentze MW. 1998. Dual function of the messenger RNA cap structure in poly(A)-tail-promoted translation in yeast. Nature 392:516–520 [DOI] [PubMed] [Google Scholar]
- 82.Tarun SZ, Jr, Sachs AB. 1995. A common function for mRNA 5′ and 3′ ends in translation initiation in yeast. Genes Dev. 9:2997–3007 [DOI] [PubMed] [Google Scholar]
- 83.Guo L, Allen EM, Miller WA. 2001. Base-pairing between untranslated regions facilitates translation of uncapped, nonpolyadenylated viral RNA. Mol. Cell 7:1103–1109 [DOI] [PubMed] [Google Scholar]
- 84.Khromykh AA, Meka H, Guyatt KJ, Westaway EG. 2001. Essential role of cyclization sequences in flavivirus RNA replication. J. Virol. 75:6719–6728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Andino R, Rieckhof GE, Achacoso PL, Baltimore D. 1993. Poliovirus RNA synthesis utilizes an RNP complex formed around the 5′-end of viral RNA. EMBO J. 12:3587–3598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Murray KE, Roberts AW, Barton DJ. 2001. Poly(rC) binding proteins mediate poliovirus mRNA stability. RNA 7:1126–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Karakasiliotis I, Vashist S, Bailey D, Abente EJ, Green KY, Roberts LO, Sosnovtsev SV, Goodfellow IG. 2010. Polypyrimidine tract binding protein functions as a negative regulator of feline calicivirus translation. PLoS One 5:e9562. 10.1371/journal.pone.0009562 [DOI] [PMC free article] [PubMed] [Google Scholar]