

Abstract
The envelope glycoprotein (Env) is the sole antigenic feature on the surface of HIV and the target for the humoral immune system. Soluble, uncleaved gp140 Env constructs truncated at the transmembrane domain are being investigated intensively as potential vaccine immunogens by many groups, and understanding their structural properties is essential. We used hydrogen/deuterium-exchange mass spectrometry and small-angle X-ray scattering to probe structural order in a panel of commonly used gp140 constructs and matched gp120 monomers. We observed that oligomeric forms of uncleaved gp140, generally presumed to be trimeric, contain a protease-resistant form of gp41 akin to the postfusion, helical bundle conformation and appear to lack specific interactions between gp120 and gp41. In contrast, the monomeric form of gp140 shows significant stabilization of the gp120 inner domain imparted by the gp41 region, demonstrating excellent agreement with past mutagenesis studies. Moreover, the gp140 monomers respond to CD4 binding in manner that is consistent with the initial stages of Env activation: CD4 binding induces structural ordering throughout gp120 while loosening its association with gp41. The results indicate that uncleaved gp140 oligomers do not represent an authentic prefusion form of Env, whereas gp140 monomers isolated from the same glycoprotein preparations in many ways exhibit function and internal structural order that are consistent with expectations for certain aspects of native Env. gp140 monomers may thus be a useful reagent for advancing structural and functional studies.
INTRODUCTION
The envelope glycoprotein (Env) is the sole target of neutralizing antibodies against HIV. Env is expressed as a single polypeptide (gp160), which oligomerizes into a trimer, is extensively glycosylated, and is proteolytically cleaved to produce the gp120 surface subunit containing the receptor binding sites and the membrane-anchored gp41 subunit. Engagement of the primary receptor, CD4, leads to conformational changes that expose elements necessary for binding the coreceptor. These receptor interactions trigger a series of conformational changes in Env that catalyze host-viral membrane fusion, with gp41 ultimately forming a stable six helix bundle in the postfusion state (1–3). Crystal structures have been determined for gp120 core in complex with stabilizing ligands such as CD4 or antibody Fabs (4–8); however, these generally have variable loops truncated and are heavily deglycosylated. The core of gp120 consists of an outer domain that contains the majority of glycosylation sites, an inner domain composed of three structural layers plus a β-sandwich motif, and a bridging sheet formed by strands from the inner and outer domains. Cryo-electron microscopy (cryo-EM) structures of both viral surface Env, detergent-solubilized trimer, and stabilized, soluble trimeric constructs have suggested an intimate association between gp120 and gp41 (9–12), and numerous biochemical studies have implicated residues involved in this interaction (13–20), but the details of the gp120-gp41 interaction remain unknown. To date, the only high-resolution structures available for gp41 are of the postfusion conformation, in which both heptad repeat (HR) regions are bound into the stable 6-helix bundle (1, 2, 21), whereas the prefusion form is known to be structurally distinct and yet remains largely uncharacterized (22).
A common approach to studying Env has been to express gp120, together with the ectodomain portion of gp41, to yield a soluble Env construct termed “gp140.” One drawback to this approach is that the interactions needed to maintain the trimer structure are relatively labile, and shedding of gp120 has been observed (23). Multiple approaches have been taken to stabilize gp140 constructs including (i) furin site modification, yielding uncleaved gp140 constructs (24, 25), (ii) addition of C-terminal trimerization motifs (26–28), (iii) introduction of intersubunit disulfide bonds (15, 22), (iv) mutations to favor the prefusion conformation (SOSIP) (23), (v) the use of directed evolution to select Env constructs with increased stability (29), and (vi) the inclusion of the transmembrane region in Env constructs (9–11). Several of these gp140 constructs have been shown to elicit stronger immune responses than gp120, but in all cases thus far the improvement was relatively modest (24, 28, 30–33). In order to understand the structural basis for the observed immune responses to gp140 oligomers, as well as to understand how to improve immunogen design, it is necessary to characterize structure and presentation of key epitopes in these constructs.
Here we apply hydrogen/deuterium (H/D)-exchange with mass spectrometry (HDX-MS) to characterize several full-length, glycosylated gp140 constructs and matched gp120 monomers. HDX-MS probes backbone amide hydrogen bonding and hence is a sensitive means of mapping stable secondary structure, identifying regions of structural order and disorder and mapping protein-ligand interfaces. In addition, it can detect sequence-specific changes in conformational dynamics or allosteric effects induced by ligand binding (34) that low-to-moderate resolution structural analysis approaches, such as by electron microscopy, are still unable to resolve. We have previously used this method to show that full-length, monomeric gp120 is distinct from the receptor-bound structure determined for the truncated gp120 core by crystallography (8, 35). Similar differences between a minimal gp120 core in an unliganded and CD4-bound state were also examined by others using HDX-MS (36). Here, we extended our analysis to probe the structural features of uncleaved gp140 constructs, which should have all the elements of the Env ectodomain present and enable us to examine structural properties of the gp140 trimers and to probe the gp120-gp41 interface. In addition, we used small-angle X-ray scattering (SAXS) to compare the low-resolution solution structures of monomeric gp120 and uncleaved, monomeric gp140 in order to localize the relative position of the gp41 subunit (37). We found that in many respects the uncleaved gp140 monomer behaves and shows local structural ordering of gp120-gp41 interactive regions consistent with sites that have been mapped through mutagenesis studies in functional Env. In contrast, the oligomeric forms of uncleaved gp140s that have been the focus of immunogen studies exhibit prominent, non-native structural features and lack a functional response to CD4 that would be expected for authentic Env trimers.
MATERIALS AND METHODS
Sample preparation.
Soluble gp120 and gp140 constructs of isolates SF162 and QH0692 were expressed and purified from HEK293 cells (25). Glycoprotein fractions were resolved by size exclusion (Superdex 200) in phosphate-buffered saline (20 mM sodium phosphate [pH 7.4], 150 mM NaCl, 0.02% sodium azide, 1 mM EDTA). Monomeric gp120 and gp140 constructs were repurified by size exclusion immediately prior to SAXS to minimize any dimer contamination. 92UG37 gp120 and gp140 were expressed and purified as described previously (28). Soluble two-domain CD4 (sCD4) (38) was obtained from the National Institutes of Health (NIH) AIDS reagents program. Proteins were concentrated using stirred pressure cells (Amicon 30-kDa cutoff; Millipore, Billerica, MA). Sample purity was assessed with reducing and nonreducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and blue native-polyacrylamide gel electrophoresis (BN-PAGE) (Fig. 1). The complex with CD4 was formed by overnight incubation with a 3-fold molar excess of sCD4 at 4°C.
Fig 1.
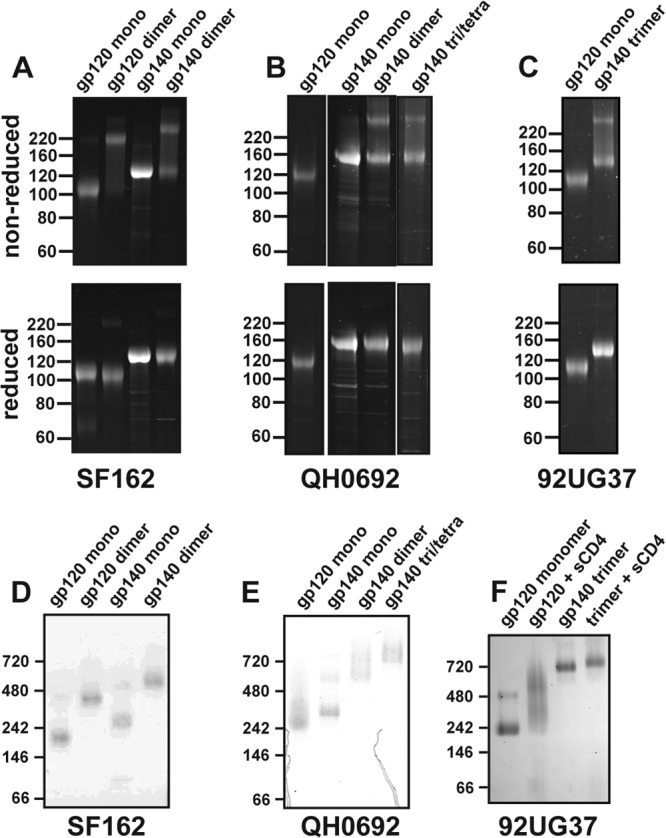
SDS-PAGE and BN-PAGE analysis of gp140 constructs. (A to C) SDS-PAGE gels under nonreducing and reducing conditions for SF162 (A), QH0692 (B), and 92UG37 (C) constructs. In gp140 dimer and trimer materials, bands corresponding to disulfide bonded protomers are evident. Gels were stained with SYPRO Ruby (Invitrogen, Carlsbad, CA). (D to F) BN-PAGE gels are shown for the respective constructs.
Hydrogen/deuterium-exchange mass spectrometry.
A total of 30 to 60 pmol (ca. 3 to 7 μg) of protein (per time point) was incubated in deuterated buffer (20 mM Na3PO4 (pH 7.4), 150 mM NaCl, 0.02% sodium azide, 1 mM EDTA, 85% D2O) at 22°C, with time points ranging from 3 s up to 20 h. Samples were then added to an equal volume (100 μl) of ice-cold quench solution (1 M Tris (2-carboxyethyl) phosphine [TCEP], 0.02% formic acid) with 10 μl of a 3-mg/ml pepsin solution in 100 mM Na3PO4 (pH 5.0) for a final pH of 2.5. After 5 min of pepsin digestion on ice, samples were flash frozen in liquid nitrogen and stored at −80°C until analysis. A fully deglycosylated sample was prepared by identical pepsin digestion, neutralization by the addition of 10 M NaOH (final pH 7.5), treatment with 0.5 mU of PNGase F (Prozyme, Hayward, CA) at 37°C for 12 h, and acidification with 5% formic acid. A “zero” time point to account for deuterium exchange during pepsin digestion was obtained by mixing the deuterated buffer with the quench solution prior to adding the protein. A fully deuterated sample was prepared by denaturation (15 min at 85°C in 3 M guanidinium-hydrochloride [GndHCl] and 100 mM dithiothreitol [DTT]), followed by deuteration in the same buffer as for the other samples for 1 h at 85°C. For many of the gp41 fragments, the signal with the fully deuterated sample was very weak or absent. For these fragments, the mass shift for each region plateaus at a point consistent with the expected level of complete deuteration based on the number of amides and the 15 to 20% back-exchange observed in our analysis. Therefore, the position of the plateau was considered the fully deuterated position for the gp41 fragments. Pressure-induced unfolding was attempted with a Barocycler 2320 (Pressure Biosciences, South Easton, MA), applying 10 cycles of pressure at 35,000 lb/in2 (for 30 s each cycle) at 4°C and immediate pepsin digestion.
Frozen samples were thawed on ice for 5 min and manually injected onto a Waters BEH 1.7 trap column (1.2 by 5 mm; Waters) flowing 0.1% trifluoroacetic acid (TFA) at 100 μl/min. After 3 min of washing, the peptides were resolved over a Hypersil 2.1-μm C18 column (1 by 50 mm; Thermo Scientific) using a gradient of 15 to 40% solvent B for 8 min (solvent A: 0.05% TFA, 5% acetonitrile [ACN]; solvent B: 0.05% TFA, 80% ACN). Injections of 10% formic acid, 80% methanol, 2:1 isopropanol-ACN, and 80% ACN were used to wash the syringe, loop, and trap column between injections, whereas two rapid (10 to 100% solvent B for 30 s) gradients were used to clean the resolving column (39) and minimize carryover (<5%). Eluting peptides were analyzed with a Waters Synapt Q-TOF mass spectrometer with source and desolvation temperatures of 80 and 150°C, respectively. The percent exchange for each fragment was calculated as described previously (35). Peptic peptides were identified by exact mass and tandem mass spectrometry (MS/MS) spectra. Identification of glycopeptides was aided by MS/MS spectra of the fully deglycosylated sample.
Light scattering.
Dynamic and static light scattering measurements were performed at 20°C on a Dynapro Nanostar (Wyatt Technology, Santa Barbara, CA). A total of 30 acquisitions of 5 s were collected at a range of concentrations (0.1 to 1 mg/ml). The data were fit using dynamics analysis software (Wyatt Technology), assuming a spherical model. Theoretical molecular masses were calculated from the molecular mass of the polypeptide plus the mass of the glycans using Man8 (1.7 kDa) for the high-mannose type and fucosylated monosialylated biantennary (2.1 kDa) for the complex type. Based on the current MS data, together with previous site-specific glycosylation studies (40), SF162 gp120 contains 10 high-mannose and 11 complex type glycans, QH0692 gp120 contains 11 high-mannose and 14 complex type glycans, and 92UG37 contains 9 high-mannose and 15 complex type glycans. To our knowledge, the glycan types in gp41 have yet to be determined and for present calculations have been assumed to contain 2 high-mannose and 2 complex type glycans.
SAXS.
Small-angle X-ray scattering (SAXS) measurements were conducted on Beam Line 4-2 at the Stanford Synchrotron Radiation Laboratory (41); the details of the collection protocol have been described previously (35). All samples were collected over a broad concentration range (0.25 to 3 mg/ml) at 15°C. The data sets at the highest concentration free of self-association or interparticle interference, as reflected by the linearity of the Guinier regions (QRg < 1.3), were used for further analysis. SAXS patterns were analyzed with the ATSAS software suite (42–44).
Molecular masses were calculated for each sample using the zero angle scattering intensity [I(0)] relative to water or RNase A as a standard (45), with adjustments for carbohydrate content as described previously (46). The particle distance distribution function [P(r)] plot generated from GNOM (44) was used for ab initio shape reconstruction using DAMMIN v5.3i (47). The bead models were aligned using SUPCOMB13 with enantiomers considered (48) and spatially filtered using DAMFILT (49). The resulting models were converted to a volume envelope using the program pdb2vol within the SITUS2.2 package (50).
RESULTS
The dominant oligomeric form of uncleaved gp140s is a dimer rather than a trimer.
Expression of gp140 constructs from two clade B isolates (SF162 and QH0692) using expression systems and purification approaches identical to previous studies of uncleaved gp140 glycoprotein (25) yielded several oligomeric forms that were isolated by size-exclusion chromatography. Both isolates contained a monomeric fraction, a higher-order oligomeric species that was the major population, and larger oligomers that eluted near the void volume in size exclusion chromatography. Static light scattering (SLS) and zero-angle intensity from SAXS were used to assess the oligomeric state of each species since these methods have been shown to be accurate for such highly glycosylated proteins (35, 46). In addition, dimeric and monomeric gp120 were examined to validate the methods for molecular mass determination (Table 1). For both isolates, the predominant gp140 species corresponded to a dimer. For QH0692, a higher-order oligomer was also purified to apparent homogeneity (as evaluated by size-exclusion chromatography); however, the calculated molecular mass was ambiguous, with a value somewhere between that expected for a trimer and tetramer. A small dimeric contaminant within the QH0692 gp120 and gp140 monomers was observed by BN-PAGE and likely accounts for the slightly larger than expected molecular mass (Fig. 1). The radius of hydration from dynamic light scattering (DLS) is consistent for both the monomers and the dimers between the two isolates (Table 1). Nonreducing SDS-PAGE revealed that gp120 dimers, gp140 dimers, and larger oligomers contain substantial populations with intermolecular disulfide bonds, although the extent of dimerization varies slightly between isolates (Fig. 1A and B).
Table 1.
Molecular mass calculations
| Isolate and glycoprotein | Mass (kDa)a |
I(0) (kDa)b |
Rh (Å) (mean ± SD)c | |||
|---|---|---|---|---|---|---|
| Expected | SLS | DLS (mean ± SD) | Water | Std | ||
| SF162 | ||||||
| gp120 monomer | 92.2 | 91.9 | 123 ± 2 | 89.4 | 91.8 | 46.5 ± 0.4 |
| gp120 dimer | 184.4 | 152.8 | 204 ± 7 | 145.1 | 149.1 | 57.8 ± 0.9 |
| gp140 monomer | 119.9 | 128.6 | 158 ± 4 | 110.8 | 123.0 | 51.8 ± 0.5 |
| gp140 dimer | 239.8 | 271.4 | 381 ± 10 | 237.6 | 263.7 | 75.4 ± 0.9 |
| QH0692 | ||||||
| gp120 monomer | 100.4 | 117.5 | 171 ± 2 | ND | ND | 53.6 ± 0.6 |
| gp140 monomer | 128.2 | 175.8 | 192 ± 3 | ND | ND | 56.3 ± 0.4 |
| gp140 dimer | 256.4 | 275.8 | 368 ± 16 | ND | ND | 74.4 ± 1.3 |
| gp140 tri/tetramer | 384.6/512.8 | 491.1 | 632 ± 14 | ND | ND | 93.7 ± 0.9 |
| 92UG37 | ||||||
| gp120 monomer | 101.4 | 113.3 | 139 ± 2 | ND | ND | 49.1 ± 0.4 |
| gp140 trimer | 399.2 | 506.6 | 545 ± 13 | ND | ND | 87.9 ± 0.9 |
The theoretical molecular mass calculations (“Expected”) are described in Materials and Methods. For static light scattering (SLS), the molecular mass was estimated from static forward scattering. Uncertainty in this measurement is primarily dependent on the uncertainty in concentration determination, expected to be <15%. For dynamic light scattering (DLS), molecular mass estimations were obtained from DLS, assuming a spherical model.
For the I(0) values, the molecular mass estimations were derived from SAXS using zero angle scattering either relative to the intensity of water or to the intensity of an internal protein standard (Std). ND, not determined.
The radius of hydration (Rh) was obtained from DLS, assuming a spherical model.
Monomeric gp140 shows a stable gp120-gp41 interface.
As a first step, we compared the H/D-exchange profiles of monomeric gp140 and monomeric gp120 from the same isolate and expression system to examine the effects of the gp41 interaction. Since isolate-specific variations in conformational dynamics are often significant (51), we focused the analysis on comparing gp120/gp140 constructs for the same isolate. Peptic fragments of monomeric gp140 (SF162) were very similar to those seen with monomeric gp120, with only a slight loss in coverage due to coeluting overlapping peptides, resulting in 71% coverage of the gp120 region. A total of 36 peptic fragments from the gp41 region were observed, yielding coverage of most of gp41 except for the highly glycosylated gp41 loop and the N-terminal half of HR2 (see Fig. S1 in the supplemental material). Glycosylation patterns on the observable glycopeptides revealed no major differences between gp120 and gp140, with the exception of fragment 224-259 (HXB2 numbering), which had significantly larger glycoforms of high-mannose glycans in monomeric gp140 (Fig. 2). Unlike gp120, monomeric gp140 did not exhibit observable O-linked glycosylation at T499 (35).
Fig 2.
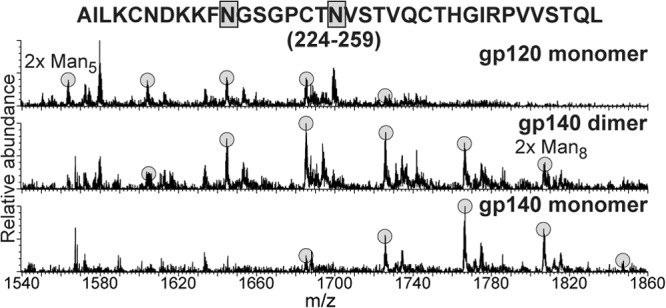
Glycoform distributions at positions 234 and 241 in gp120, gp140 dimer, and gp140 monomer. The peptic 224-259 fragment was found to contain two high-mannose type N-linked glycans. Each glycoform resulting from different degrees of mannose trimming is highlighted.
Comparison of the SF162 monomeric gp120 to monomeric gp140 revealed several regions with significantly increased protection imparted by the gp41 domain (Fig. 3A). For example, the WVTVY segment (segment 35-39, HXB2 numbering) at the N-terminal extension of gp120 fully exchanged within seconds in gp120 but was protected even at 20 h in monomeric gp140. Similarly, fragments spanning residues 40 to 83 (HXB2 numbering) in layer 1 of the inner domain were in rapid exchange in gp120 but were significantly protected in monomeric gp140. Residues 105 to 111 in layer 2 of the inner domain were somewhat more protected in gp140. Based on the comparison of the 484-501 (in gp120) and 484-503 (in gp140) segments, protection also increased at the C-terminal end of gp120 immediately preceding the furin cleavage site. In contrast, the variable loops, the outer domain, and the bridging sheet elements did not appear to be affected by the presence or interaction with gp41. Within gp41, the fusion peptide-proximal region (FPPR) and the majority of HR1 with part of the downstream gp41 loop region showed moderate protection, whereas the N-terminal portion of gp41 (fusion peptide) and all of HR2 past residue 646 were in very rapid exchange. The membrane-proximal external region (MPER) was also in rapid exchange, with the exception of slight protection at the far C terminus.
Fig 3.

Butterfly plots comparing the H/D-exchange profiles of monomeric gp120 and gp140. The upper plot shows the exchange profiles of all observable fragments for gp120 (top) and monomeric gp140 (bottom) for isolates SF162 (A) and QH0692 (B). The percent exchange is shown each fragment at the midpoint in primary sequence. For example the exchange profile of fragment 104-110 will be plotted at position 107. The positions of the variable loops and heptad repeats are highlighted in gray. The difference plots under each butterfly plot show the difference in percent exchange at each time point. The exchange plots of each individual fragment are shown in Fig. S1 and S2 in the supplemental material. (C) Differences in exchange profiles between gp120 and monomeric gp140 are shown on the core structure of gp120 (hybrid of PDB 2NY7 and 3JWD as described previously [35]). Regions are colored from purple (more protected/more ordered) to red (less protected/more disordered relative to gp120).
In contrast to SF162, the peptic coverage of QH0692 constructs was relatively poor, with only 38% coverage of the gp120 region. This is due to the additional N-linked glycosylation sites and sequence variation, resulting in a loss of peptic cleavage sites (see Fig. S2 in the supplemental material), along with an overall weaker signal due to the limited sample available. However, nearly all of the same gp41 fragments could be observed with QH0692 monomeric gp140. The major differences between gp120 and monomeric gp140 are consistent with the results from SF162 (Fig. 3B). One detectable difference was slight protection observed at the center of V2 within QH0692 gp140 monomer at residues 173 to 176 (HXB2 numbering). Due to the limited sequence coverage of gp120, it is possible that differences may exist in the QH0692 data that are distinct from SF162. For example, since fragments corresponding to the C-terminal end of gp120 were not observed in QH0692 gp140 monomer, the effects beyond residue 502, could not be analyzed for the QH0692 constructs. Accepting this caveat, nearly every region that was available for comparison (including in gp41, as described below) showed similar patterns of protection in QH0692 and SF162 (Fig. 3).
Throughout the course of H/D-exchange studies, we noticed that the fully deuterated gp140 monomer sample, prepared by denaturation in guanidine and DTT, led to the loss of nearly all of the gp41 fragments, without affecting the signal for any of the gp120 fragments. BN-PAGE was used to assess the effects of denaturation on monomeric gp140. Even mild destabilization (10 min of incubation with either 4 M urea or at 50°C) resulted in efficient oligomerization, yielding a discrete oligomer rather than a nonspecific aggregate (Fig. 4). We speculate that the denaturation step causes gp41 to transition into a stable conformation that is highly protease resistant under HDX-MS quench conditions (pH 2.5 and 0°C). Interestingly, the addition of sCD4 had a similar effect, resulting in some dimerization of monomeric gp140.
Fig 4.
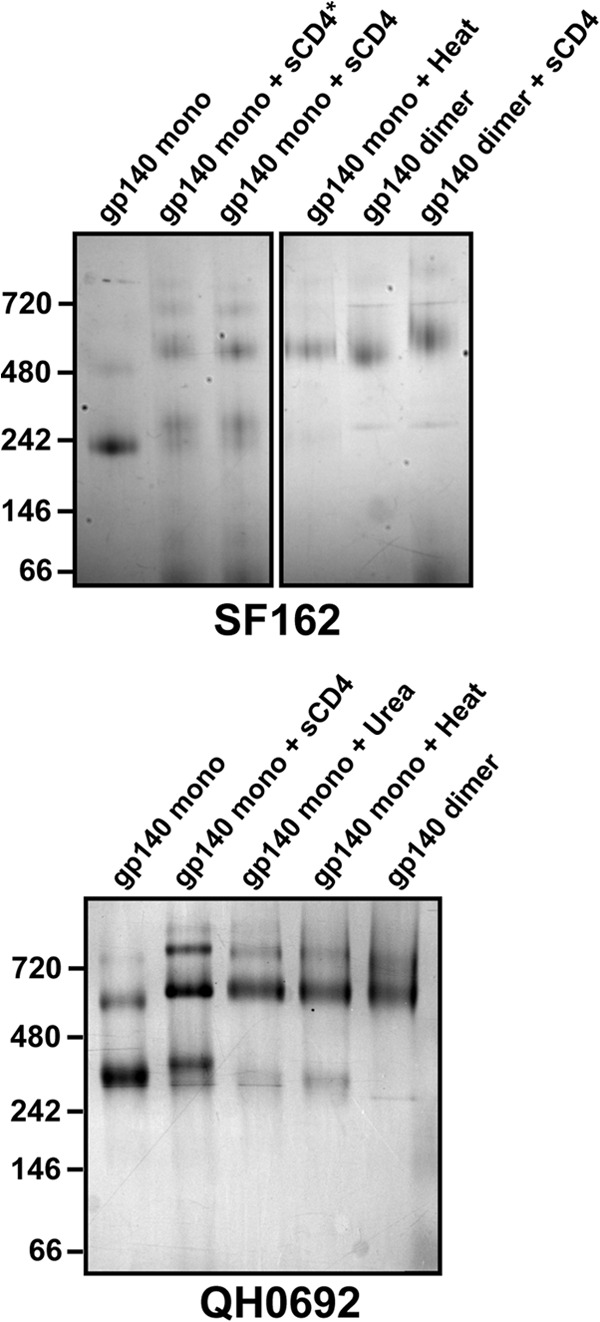
Dimerization upon destabilization of monomeric gp140. BN-PAGE analysis of monomeric gp140 SF162 (top) and QH0692 (bottom) after sCD4 binding, chemical perturbation (4 M Urea for 10 min), or heat denaturation (50°C for 10 min). *, gp140 was incubated with sCD4 for 2 h, rather than added immediately before BN-PAGE.
CD4 binding alters the gp120-gp41 interaction in monomeric gp140.
Since sCD4 binding had large-scale effects on monomeric gp140, we sought to use HDX-MS to investigate specific changes induced by CD4 binding. Identical experiments were performed side-by-side with monomeric gp120 to assess any unique changes occurring in gp140. The changes in the exchange profile for the gp120-sCD4 complex reveal increased protection at the inner domain, the bridging sheet, and the CD4 binding loop (Fig. 5A and D). This pattern of changes is very consistent with our previous observations for monomeric gp120 from CHO cells (35). In the case of monomeric gp140, the changes at the CD4 binding loop and bridging sheet are nearly identical to those seen in gp120 (Fig. 2B). On the other hand, the N-terminal extension and C terminus of gp120 become less protected upon sCD4 binding (Fig. 5F and H), and very little change was observed within layer 1 of the inner domain (Fig. 5G). Very similar effects were seen for the CD4 complex with the QH0692 gp140 monomer (Fig. 5E), with an additional loss of protection at center of V2.
Fig 5.

Effect of CD4 binding to gp120 and gp140 monomers as monitored by H/D exchange. (A, B, D, and E) Butterfly plots for monomeric gp120 with or without sCD4 (A and D) and gp140 monomer with or without sCD4 (B and E). Plot descriptions are described in Fig. 3. (C) Changes in monomeric gp140 upon sCD4 binding shown on the CD4-bound core crystal structure (PDB 3JWD). Regions are colored from purple (more protected/more ordered) to red (less protected/more disordered relative to gp120). The position of CD4 is illustrated as a transparent yellow surface. (F to I) Individual deuterium uptake profiles are shown for peptides (isolate SF162) from the N-terminal extension (F), layer 1 of the gp120 inner domain (G), the C-terminal end of gp120 (H), and HR1 from gp41 (I). Numbering in parentheses refers to HXB2 numbering.
Most notably there were several large changes in the gp140 monomer, observed within the gp41 region in response to CD4 binding. In both SF162 and QH0692 isolates, the largest effect was within HR1, where the N-terminal half of HR1 (residues 548 to 581, HXB2 numbering) became significantly less protected upon CD4 binding (Fig. 5I). Other effects in gp41 were more varied between the two isolates. In SF162 the C-terminal portion of HR1 (residues 582 to 602, HXB2 numbering) appeared to be unaffected by CD4 binding, but increased protection was observed at this same region in QH0692. The center of HR2 (residues 642 to 645, HXB2 numbering) also responded differently to CD4 binding in the two isolates, becoming slightly less protected in SF162, but more protected in QH0692. Lastly, the C-terminal end of the MPER region also showed a moderate increase in protection with CD4 binding in QH0602 but showed no change in SF162.
CD4 binding results in some degree of oligomerization with both gp140 monomers (Fig. 4), potentially complicating the HDX-MS analysis of the bound form. Based on the decreased signal intensity for the gp41 fragments (while gp120 fragments showed similar intensities), it appears that some of the gp41 in the population forms a pepsin-resistant oligomer similar to that observed in preparation of fully deuterated samples that had denaturant present. Since the oligomerized population is pepsin resistant, all of the gp41-derived fragments observed by HDX-MS reflect the changes that occur within the gp41 region of the monomeric subpopulation of the gp140-sCD4 complex.
Structural model of monomeric gp140 from SAXS.
Since monomeric gp140 has not yet been studied even by low-resolution structural techniques, we sought to compare the large-scale organization of gp140 and gp120 monomers by SAXS. We obtained SAXS data for freshly gel filtration-purified constructs, ensuring the samples were free of oligomer contamination. The Guinier region showed good linearity for both data sets (Fig. 6A and B), with no dependence on protein concentration up to 3 mg/ml. Since the data were free of concentration-dependent effects and the correct molecular mass was calculated from the scattering at zero angle [I(0)], the data was deemed to be suitable for structural interpretation (52). The radii of gyration (Rg) obtained from Guinier analysis were 42.3 ± 0.1 Å and 37.2 ± 0.1 Å for the gp140 and gp120 monomers, respectively. Similarly, the maximal dimension (Dmax) was larger for gp140 (160 Å versus 130 Å) (Fig. 6C).
Fig 6.

SAXS of monomeric gp120 and gp140 SF162 suggests gp41 is positioned alongside the gp120 inner domain. (A and B) Experimental SAXS patterns (black) measured for gp120 (A) and gp140 (B) monomers compared to fits obtained from ab initio reconstructions using DAMMIN (red) (47). Insets show the linearity of the Guinier plots. (C) Distance distribution [P(r)] plots for gp120 (red) and gp140 (blue) obtained from GNOM (47). (D and E) Shape reconstructions for monomeric gp120 (D) and monomeric gp140 (E) are shown with the structural model (PDB 3JWD and 2B4C to show approximate locations for N/C-terminal extensions and V3) docked into the SAXS envelope; gp120 core residues that have been identified as playing a role in association with gp41 are shown in space-filling balls and colored as C-terminal extenions (purple), β-sheet “platform” (yellow), layer 1 (magenta), and layer 2 (red) (60). (F) Comparison of SAXS reconstructions for gp120 (blue) and gp140 monomers (gray) reveals additional density in the gp140 monomer localized around the putative gp41 interactive face of gp120 (box). (G and H) Comparison of a gp140 monomer SAXS model with cryo-EM reconstruction for the Env trimer (gray, EMD-5019 [9]) shows good agreement, suggesting that the protomer retains large-scale organization similar to what exists in the membrane-presented Env trimer.
Ab initio shape reconstructions were performed for monomeric gp140 and gp120 to visualize and compare the overall shape envelopes and to help localize the portions attributable to gp41. The resulting models were consistent with the scattering data with chi values of 1.224 ± 0.001 (gp120) and 1.300 ± 0.007 (gp140). Normalized spatial discrepancy between the individual models indicated relatively good convergence among the 12 individual models obtained for each data set (0.676 ± 0.010 and 0.683 ± 0.018). Based on this low-resolution shape reconstruction, monomeric gp140 is slightly larger than gp120 but with a similar morphology (Fig. 6D and E). Superpositioning of the shape envelopes of gp140 monomer and gp120 monomer indicates that gp140 shows a bulkier base and a notable hip of density alongside the gp120 core (Fig. 6F). Based on previous interpretation of the crown and basal lobes using existing gp120 core crystal structures (5, 8, 35), the variable loops are also positioned at the crown (top) portion of the envelope in the gp140 monomer. From this comparison the additional density in the gp140 reconstruction that we attribute to gp41 appears to be located adjacent to layers 1 and 2 and the β-sheet “platform” of the gp120 inner domain (Fig. 6E and F), in excellent agreement with regions observed to be more ordered in gp140 versus gp120 (Fig. 3C).
The gp140 monomer SAXS model was docked into the low-resolution structure for the Env trimer presented on the surface of virus particles that was previously determined by cryo-electron tomography (9) and was observed to approximate one of the three blades of the propeller-shaped trimer spike (Fig. 6G and H), although the V1/V2 loops in the trimer are likely drawn down toward the central axis, where they are believed to be involved in intersubunit interactions (9, 10). It appears that although it lacks trimeric contacts, the gp140 monomer retains the approximate shape and large-scale organization of a protomer in the Env trimer.
gp120-gp41 interactions are absent in oligomeric gp140s.
The H/D-exchange profile of the oligomeric gp140s was examined to investigate the nature of this material that is often presumed to be representative of trimeric Env ectodomains, though, as we describe above, they are in fact predominantly dimers of gp140. The coverage of the gp120 region was identical to gp140 monomers for both SF162 and QH0692 isolates, but nearly all coverage of gp41 in the gp140 dimers was lost (Fig. 7), with the exception of a weak signal for peptides within the loop region between HR1 and HR2 (residues 593 to 602 and 593 to 604, HXB2 numbering) and two very large, late-eluting species corresponding to large fragments of gp41 spanning from the fusion peptide to the end of HR1 (residues 484 to 602 and 519 to 602, HXB2 numbering) (see Fig. S1 and S2 in the supplemental material). This pattern of proteolysis is consistent with HR1 existing in a stable, protease-resistant form, with some protease accessibility at the gp41 loop bounded by HR1 and HR2, i.e., an organization consistent with a helical bundle organization for gp41. Inclusion of 3 M GndHCl, 6 M urea, or 20% acetonitrile in the quench solution and high-pressure treatment (35kpsi) to unfold gp41 failed enhance proteolytic cleavage, suggesting that the gp41 region was indeed in a highly stable state. Under identical experimental conditions, the gp140 monomers, which are identical in primary sequence and chemical composition although conformationally distinct, did yield extensive peptic digestion of these same regions, suggesting that the experimental conditions (e.g., quench conditions) were not responsible for the protease resistance of the gp140 dimer material. Instead, this appears to be a preexisting trait of the dimeric constructs.
Fig 7.
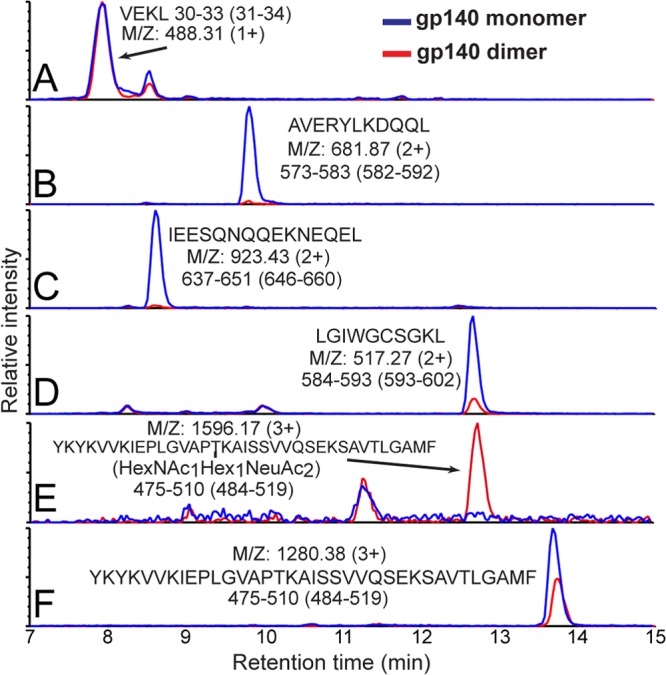
Ion-extracted traces for various peptic fragments. The intensities for several peptides from monomeric (blue) and dimeric (red) gp140 (SF162) after pepsin digestion are shown. The residue numbers for each segment are shown below the amino acid sequence, with HXB2 numbering in parentheses. Although gp120 segment intensities (see, for example, section A) were nearly identical in various constructs, as shown in sections B to F, gp41 fragments in dimeric gp140 were not observed except for a moderate signal for LGIWGCSGKL (section D). A large fragment from the C terminus of gp120 was observed bearing an O-linked glycan at T499 only in dimeric gp140 (sections E and F).
Unlike with the gp140 monomers, the gp140 dimers showed very little difference in their exchange profile for gp120 regions relative to isolated gp120 monomers. The comparison reveals minor protection at the N-terminal extension of gp120, within the V2 loop, and within the SF162 data set, a minor increase in protection in layer 2 of the gp120 inner domain (residues 105 to 111) (Fig. 8A and B). The same pattern of differences was observed when we compared the gp120 monomer to gp120 dimers (Fig. 8C), indicating that gp140 dimers are very similar to gp120 dimers. Unlike monomeric gp140, dimeric gp140 shows very few effects resulting from inclusion of the gp41 region. The exchange profile of the N-terminal extension in gp140 monomers shows significant protection, indicating local ordering, whereas both gp120 dimers and gp140 dimers are only marginally protected (Fig. 8E). Likewise, for layer 1 of gp120 and the C-terminal end of gp120, which show a complex pattern of protection in gp140 monomers, in the dimers no measurable protection is observed (Fig. 8F and G). Lastly, unlike with monomeric gp140 that showed significant protection within the gp41 loop region, the loop fragments in gp140 dimers showed essentially no protection in both SF162 and QH0692 isolates (Fig. 8H). We further note that the higher-order (trimer/tetramer) oligomeric form of QH0692 gp140 exhibited the same overall trend as the gp140 dimers (Fig. 8D). These comparisons indicate that among the constructs, only the monomeric gp140 contains a stable gp120-gp41 interface.
Fig 8.

H/D-exchange profiles of monomeric gp120 and oligomeric gp140s reveals no gp120-gp41 interactions in the oligomers. Butterfly plots for SF162 monomeric gp120 versus dimeric gp140 (A), QH0692 monomeric gp120 versus dimeric gp140 (B), SF162 monomeric gp120 versus dimeric gp120 (C), and QH0692 monomeric gp120 versus trimeric/tetrameric gp140 (D) are shown. An explanation for data representation in these types of plots is provided in the legend for Fig. 3. (E to H) Deuterium-uptake plots of selected peptides from both monomeric and dimeric gp120 and gp140 (SF162) constructs; numbering in parentheses refers to HXB2 numbering.
Foldon-stabilized, trimeric gp140 shows no evidence of gp120-gp41 interactions with a T4 fibritin foldon trimerization motif.
In order to investigate a trimeric Env construct that has been suggested as a prospective immunogen candidate, we next applied H/D-exchange to study gp140 trimers (isolate 92UG37) (28). BN-PAGE suggested that the proteins were homogeneous (Fig. 1C), although nonreducing SDS revealed the presence of some disulfide-bonded dimer, akin to that seen with the previous gp140 dimers (Fig. 1F). Thus, some form of heterogeneity among protomers in the population exists. It is plausible that within a portion of the trimers, two of the protomeric units may be disulfide bonded, whereas the remaining protomer is only noncovalently associated. The slightly larger than expected molecular mass by light scattering may result from an underestimate of the theoretical molecular mass, since several glycans may be larger than expected based on our examination of the other gp140 constructs (Table 1). The sequence coverage of the gp120 portions of the 92UG37 gp140 trimer was reasonable (61%), but nearly all gp41 fragments were absent. Just as with the protease-resistant gp140 dimers described above, the only observable peptides from this region were segment 593-602, corresponding to the segment in the gp41 loop region, and two very large fragments spanning from the end of gp120 all the way to the end of HR1 (see Fig. S3 in the supplemental material). A comparison of the exchange profile of monomeric gp120 to trimeric gp140 strongly resembles the results obtained with the gp140 dimers. Namely, aside from a moderate increase in protection at the N-terminal extension, the two have essentially the same exchange profile (Fig. 9A). A fragment from gp120 layer 1, predicted to be a key part of the gp120-gp41 interactive face (6, 8), served as a useful diagnostic peptide for this comparison (Fig. 9B). Monomeric gp140s (both SF162 and QH0692) showed a significant increase in protection at this region, but the 92UG37 gp120 monomer and gp140 trimer show exactly the same exchange profile, indicating that, as in the gp140 dimers, the foldon-stabilized gp140 trimer lacks stabilizing interactions between gp120 and gp41 regions.
Fig 9.

Uncleaved gp140 trimer from isolate 92UG37 does not exhibit functional gp120-gp41 interactions. (A) Plots comparing the exchange profiles of 92UG37 monomeric gp120 versus foldon-stabilized, trimeric gp140. Individual exchange plots are shown for a fragment from gp120 layer 1 (B) and the large fragment spanning the C-terminal end of gp120 to the end of HR1 (C). Numbering in parentheses refers to HXB2 numbering. Plots comparing the exchange profile with or without sCD4 are shown for monomeric gp120 (D) and trimeric gp140 (E) are also shown. Individual exchange plots for each fragment are shown in Fig. S3 in the supplemental material.
To further assess the functional state of this gp140 trimer, we examined the effect of CD4 binding to trimeric gp140, side-by-side in comparison to monomeric gp120. For both constructs, the changes parallel what is observed with gp120 monomers from SF162 and QH0692, namely, CD4 binding results in stabilization of the CD4 binding loop, gp120 inner domain, and elements of the bridging sheet (Fig. 9D and E). The extent of stabilization imposed by CD4 is likewise similar for monomeric gp120 and trimeric gp140. A slight decrease in protection within the V2 loop is present in both comparisons. The large gp41 derived peptide covering HR1 reveals that CD4 binding had no effect on gp41 in the gp140 trimer (Fig. 9C).
DISCUSSION
Uncleaved gp140 oligomers are largely non-native in structure.
Uncleaved forms of gp140 have been widely investigated as prospective vaccine candidates with the expectation that compared to isolated gp120 subunits, the inclusion of gp41 and resulting oligomerization of gp140 protomers would more closely mimic native Env on virus and produce a more effective neutralizing immune response. The uncleaved gp140 constructs investigated thus far, however, have only demonstrated a moderate improvement in immunogenicity over gp120 (24, 28, 30, 31, 33). The structural analysis provided here suggests possible structural explanations for the muted immune responses to some of the oligomeric, uncleaved soluble gp140s.
Based upon our biophysical characterization, the oligomeric forms of the two clade B, uncleaved gp140s are predominantly dimers, along with monomers and some portion of higher-order oligomers (Table 1). This is consistent with previous studies revealing uncleaved gp140 constructs to be a mixture of oligomers that vary in relative abundance and size between isolates (53, 54). We note that the uncleaved gp140s examined here were expressed and purified by methods identical to those previously described and in widespread use (25, 28). Obtaining accurate molecular mass measurements for glycoproteins is not trivial (46), and it is plausible that the oligomers that are presumed to be trimers based on native PAGE or gel filtration chromatography in many cases are in fact dimerized gp140.
Both the clade B dimers and the clade C trimer forms of uncleaved gp140 contain a significant subpopulation exhibiting interprotomer disulfide bonds, which are also present in gp120 dimers, and thought to be nonphysiological (55, 56). Therefore, even by this biochemical assay, heterogeneity among uncleaved gp140 oligomers is evident. The gp140 dimers and higher-order oligomers exhibit striking similarities in the H/D-exchange profiles to dimeric gp120 (Fig. 8A and B). They show only a slight increase in protection within the N-terminal extension and V2 relative to gp120 monomer. Both V1/V2 and the N-terminal extension regions have been shown to be involved in aberrant gp120 dimerization (35, 56, 57). A small degree of increased protection was also seen in layer 2 of the inner domain (HEDIISL), which is also likely to be involved in the oligomeric interface since mutations within this region have been reported to hinder dimerization (57). Even the trimerization motif-stabilized, uncleaved clade C (92UG37) gp140, which indeed bears the molecular mass of a trimer, showed the same pattern in its HDX-MS profile (Fig. 9A), suggesting that it adopts a structure with characteristics very similar to what is observed with the dimerized gp120 and gp140s, rather than exhibiting traits expected for a native gp120-gp41 interface. Furthermore, CD4 binding to this gp140 trimer had no effect on the gp41 domain, which is consistent with it lacking gp41-gp120 functional interactions.
Both gp140 dimers (clade B, SF162 and QH0692) and trimers (clade C, 92UG37) were found to be protease resistant throughout most of gp41, including the entirety of HR1. We infer that this domain has likely formed extended helices that may gain significant protection from proteolysis by involvement in a helical bundle structure (1, 2). The stable helices may extend beyond the HR domains proper, as suggested in a recent structure of gp41 that included the MPER and FPPR (3). It is also plausible that the HR2 region, which has previously been shown to form dimeric helical bundle interactions when the residue 630-683 segment was examined in isolation, may be involved in a helical bundle formation in the gp140 oligomers (58). In a recent study we observed that influenza virus hemagglutinin, another type 1 fusion protein, also becomes pepsin resistant only after it has adopted the postfusion helical bundle form (unpublished observations). The only observable, small fragment of gp41 in the gp140 oligomer samples that was released by proteolysis was from gp41 the loop region, directly C terminal to HR1. This segment exhibited very rapid solvent exchange, suggesting an organization that is consistent with the postfusion gp41 6-helix bundle NMR structure (21). Previous antibody binding assays suggested that a structure other than the 6-helix bundle was adopted by these same 92UG37 uncleaved gp140 trimers (59); therefore, the exact nature of the protease-resistant form of gp41 remains unknown.
Taken together, the current data indicate that the uncleaved gp140 trimers and dimers are composed of a stable, post-fusion-like gp41 with gp120 associated solely by the covalent link that exists in the absence of gp120-gp41 cleavage (Fig. 10A). A similar description of uncleaved gp140 trimers has previously been proposed and motivated efforts to stabilize Env in a more native conformation (15). If the uncleaved gp140 oligomers used for previous studies embodied nonfunctional gp41 subunits with loosely tethered gp120 subunits, as our data suggest, then it provides a structural rationale for why dramatic, qualitative improvements in immunogenicity over monomeric gp120 were not achieved. The moderate improvement may have arisen from enhanced immune response to the multivalent presentation of gp120s rather than as a result of the gp140 constructs authentically mimicking the native Env trimer.
Fig 10.
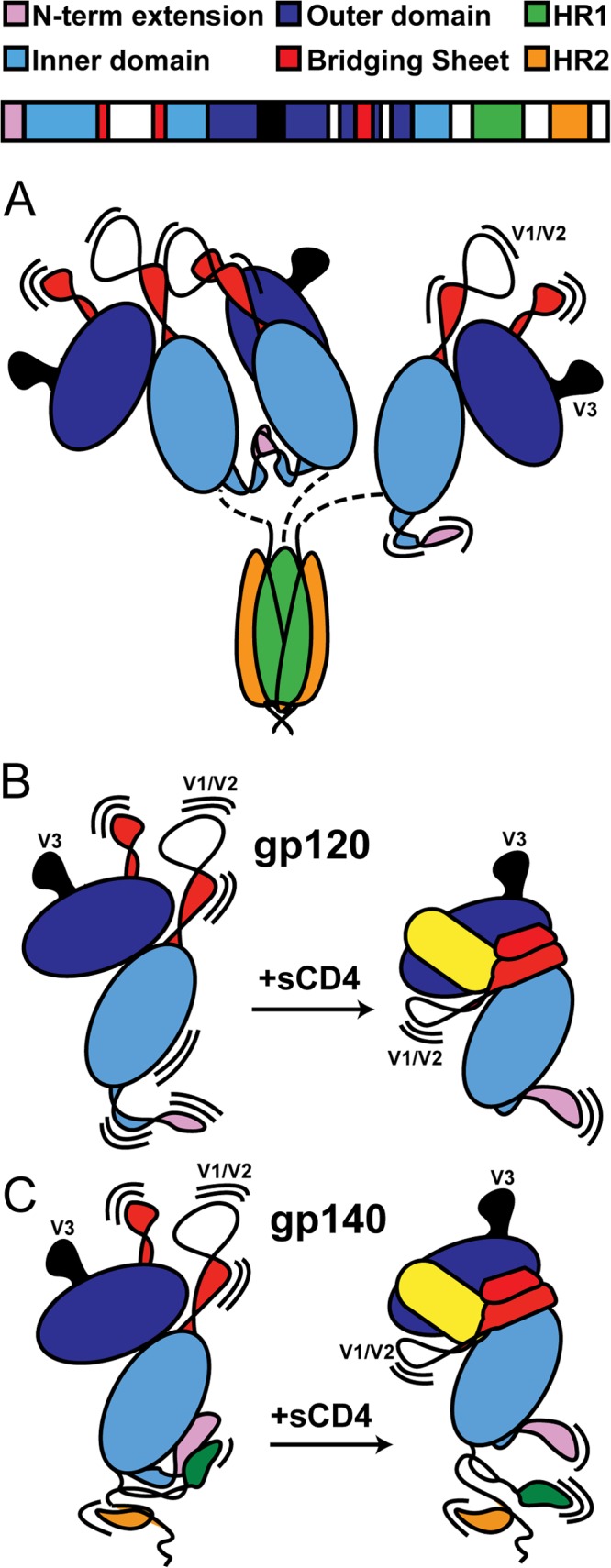
Structural models of various Env constructs. (A) gp140 trimers (and dimers) show no evidence of gp41/gp120 interactions. Instead, gp41 adopts a post-fusion-like state, while a portion of gp120s form dimers, presumably through contacts in V1/V2 and the N-terminal extension. (B) With monomeric gp120, CD4 (shown in yellow) binding stabilizes the gp120 inner domain, along with the elements that form the bridging sheet. (C) With monomeric gp140, CD4 has a similar effect on the bridging sheet but at the same time weakens the interface between gp120 and gp41, which involves the N- and C-terminal extensions of gp120 and HR1 of gp41. A color-coded sequence of gp140 is shown at the top, and the structural regions of interest are annotated in Fig. S1 to S3 in the supplemental material.
Interactions between gp41 and gp120 in monomeric gp140 protomers.
In contrast to the observations with gp140 oligomers, our data indicate that monomeric gp140 exhibits features and gp41-related ordering in gp120 that suggest it may exist in a relatively native-like functional state. The presence of gp41 was found to impart a significant degree of stabilization to regions of gp120 that have been identified as mediating gp120-gp41 association in Env (6, 13, 15, 60). Specifically, the regions showing increased deuterium-exchange protection are the N and C termini of gp120, along with layers 1 and 2 of the inner domain (Fig. 3C) in the region often referred to as the “gp41 interactive face” (6). In addition, the glycosylation profile on layer 2 of the inner domain shows less mannose trimming on monomeric gp140 than gp120, a finding consistent with increased protection at this site (Fig. 2). On this same note, the lack of O-glycosylation at position T499 in gp140 monomers (but not gp140 dimers) is consistent with the increased protection of the C-terminal end of gp120 (Fig. 7). Low-resolution shape reconstructions from SAXS show an enlarged bulge positioned along the gp120 core (Fig. 6D to F), paralleling the same surface implicated by HDX-MS and overlapping the sites that show differential glycosylation in monomeric gp140 and gp120. Although these reconstructions are resolution limited, they show consistency with the major expectations for gp120-gp41 association and also consistency with general morphology observed in Env trimers (Fig. 6G and H) (9).
In terms of structural order in the gp41 region, unlike the profile measured for gp140 oligomers, the H/D-exchange profile of gp41 in monomeric gp140 may resemble key aspects of the prefusion form of the Env protomer. The complex protection pattern, including portions of the gp41 loop region, and lack of protection in the C-terminal half of HR2 are inconsistent with known gp41-related helical bundle structures (1, 3) (Fig. 3), since the helical bundle organization would be expected to yield significant deuterium-exchange protection of backbone amide groups due to hydrogen bonding throughout the entirety of HR1 and HR2. Based on the observed protection profile in gp41 and the response to CD4 binding, the data suggest that the gp120-gp41 interface involves the majority of HR1 (Fig. 3 and 10C). The FPPR, the N-terminal portion of the gp41 loop region, and some of the N-terminal half of HR2 also show some evidence of being ordered due to interactions with gp120. This is consistent with mutagenesis studies of gp41 that have observed large effects on the gp120-gp41 association arising from mutations at the C-terminal portion of HR1 (20) and within FPPR (14, 19). Several studies have also implicated portions of the gp41 loop region as a site of interaction with gp120 (16–18); however, due to its high glycan content, the latter part of this region could not be tracked by the current HDX-MS analysis.
It may seem surprising that the fusion peptide segment itself was highly accessible to exchange in gp140 monomers. However, we note that in Env prior to cleavage, it would be necessary for this segment to be surface accessible for furin processing. Based upon comparisons to an analogous type 1 viral fusion protein, influenza virus hemagglutinin, in cleaved and uncleaved forms, one expects that only after cleavage would the fusion peptide sequestered into a protected cavity (61, 62). In the uncleaved hemagglutinin HA0 trimer, the fusion peptide segment is presented as a surface accessible loop, but cleavage allows the peptide to reorganize and become buried at an intersubunit interface without producing major structural changes to the rest of the trimer structure. A comparison of detergent-solubilized uncleaved gp160 trimers (10, 11) and native Env trimers on virions (9) suggests that in the context of a trimer, cleavage does not dramatically alter the overall organization; however, proteolytic processing has been reported to have measurable effects on the antigenic profile of conformational flexibility in Env (63–65). Indeed, furin cleavage and intersubunit contacts within native trimers are expected to exert an influence on gp120-gp41 interactions, potentially affecting the response to CD4 binding. Although it is clear the monomer cannot entirely mimic the protomer structure in the intact, cleaved trimer, due to the lack of quaternary contacts and proteolytic processing, it appears that the uncleaved gp140 monomers otherwise possess a gp120 and gp41 organization that retains many important features of the protomer in more complete assemblies. The current HDX-MS analysis complements existing cryo-EM reconstructions by providing relatively detailed sequence-specific structural information that is not available in the EM models.
It remains to be determined why the apparently functional gp140 monomer does not associate into native trimers. We posit that under the native pathway, Env trimers would fold and oligomerize while membrane anchored. Proper oligomerization may not be favored in constructs lacking the transmembrane anchor. This notion is supported by the recent higher resolution structural study of trimeric Env, using a larger Env construct that contained the transmembrane portion (9–11). Alternatively, with soluble constructs, portions of the MPER region may occlude hydrophobic patches on gp120 or gp41 that are necessary for proper trimerization.
gp140 monomers appear to adopt a meta-stable conformation that is destabilized by CD4 binding or nonspecific perturbation.
HIV Env, like other type I viral fusion proteins, is hypothesized to exist in a kinetically trapped, metastable conformation prior to triggering (66). Once triggered, the fusogenic subunit (gp41 in Env) converts to a highly stable, postfusion helical bundle structure that would draw the two membranes together to induce their fusion. In the constructs we examined here, monomeric gp140 also appears to exist in a meta-stable state. Slight perturbation by introduction of destabilizing denaturants or CD4 binding leads to conformational changes that lead to significant oligomerization and structural changes within gp41 (Fig. 4). This suggests that in gp140 monomers the gp41 subunit exists in a metastable state that, upon destabilization, is able to transition to a lower-energy organization with gp41 most likely associated in highly stable helical bundles.
CD4 induces large-scale changes in structural order in both gp120 and gp140 monomers. The comparison of exchange profiles with or without sCD4 for both SF162 and QH0692 gp120 monomers are consistent with our earlier studies with CHO expressed material (35), marked primarily by the formation of the bridging sheet, ordering of the CD4 binding site, and ordering of the inner domain (Fig. 10B). The response to CD4 in gp140 monomers shows similar changes; however, in gp140 monomers, much of the gp120 subunit's layer 1 is already ordered, and the N-terminal and C-terminal regions of gp120 in fact become less ordered upon CD4 binding, as if a structural constraint becomes released (Fig. 5C and 10C). These changes are consistent with earlier findings that monomeric gp140 has epitopes at these regions occluded by the interaction with gp41, and the occlusion is disrupted by CD4 binding (67). In contrast, gp41 in the oligomeric forms of gp140 has already adopted a highly stable conformation and exhibits no response to CD4 binding.
The effects of CD4 binding extend to the gp41 domain in gp140 monomers, demonstrating the allosteric coupling of the CD4 binding site to the gp120-gp41 interface. This binding event results in increased exchange within HR1, which is consistent with earlier findings that CD4 binding increases the accessibility of HR1 (68). This allosteric effect at HR1, we suspect, in an analogous context in Env would destabilize the prefusion conformation of gp41, representing an initial step in the activation of the gp41 fusion machinery (Fig. 10C). The changes observed throughout the inner domain upon CD4 binding agree closely with mutagenesis studies, implicating the layers of the inner domain in the allosteric mechanism of gp41 activation (60, 69). Thus, in contrast to the panel of oligomeric forms of uncleaved gp140 examined here, monomeric HIV-1 gp140 embodies several important features of gp120-gp41 association, making it a potentially useful soluble reagent for studying the structure and function of an Env protomer and for making comparisons between different isolates in lieu of well-behaved, native-like trimers. Modifications to gp140 constructs that destabilize helical bundle formation, for example, an isoleucine-to-proline helix breaking change introduced into the HR1 region (23) or changes that stabilize prefusion forms of the trimer (29) may rebalance the conformational equilibrium to disfavor the proteolytically resistant gp140 oligomer that dominates the types of constructs we examined and favor more native-like conformations in soluble Env constructs.
Supplementary Material
ACKNOWLEDGMENTS
We thank Tsutomu Matsui, Lester Carter, and the support staff at the Stanford Synchrotron Radiation Lightsource for assistance with SAXS data collection. SF162 and QH0692 gp120 and gp140 constructs were generously provided by Noah Sather, George Sellhorn, and Leonidas Stamatatos (Seattle Biomedical Research Institute). We are grateful to James Kovacs and Bing Chen (Boston Children's Hospital) for providing 92UG37 gp120 and gp140 constructs. The following reagent was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH: sCD4-183 from Pharmacia, Inc.
This study was supported by NIH grants F32-GM097805 (M.G.), R00-GM080352, and R01-GM099989 and by UAB CFAR grant (P30-AI027767) through the CNIHR program for new HIV investigators (K.K.L.). Data collection at SSRL was supported by grant P41-RR001209 from the National Center for Research Resources, a component of the NIH.
Footnotes
Published ahead of print 21 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01681-13.
REFERENCES
- 1.Lu M, Blacklow SC, Kim PS. 1995. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat. Struct. Biol. 2:1075–1082 [DOI] [PubMed] [Google Scholar]
- 2.Chan DC, Fass D, Berger JM, Kim PS. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263–273 [DOI] [PubMed] [Google Scholar]
- 3.Buzon V, Natrajan G, Schibli D, Campelo F, Kozlov MM, Weissenhorn W. 2010. Crystal structure of HIV-1 gp41 including both fusion peptide and membrane proximal external regions. PLoS Pathog. 6:e1000880. 10.1371/journal.ppat.1000880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou T, Xu L, Dey B, Hessell AJ, Van Ryk D, Xiang SH, Yang X, Zhang MY, Zwick MB, Arthos J, Burton DR, Dimitrov DS, Sodroski J, Wyatt R, Nabel GJ, Kwong PD. 2007. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature 445:732–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pancera M, Majeed S, Ban YE, Chen L, Huang CC, Kong L, Kwon YD, Stuckey J, Zhou T, Robinson JE, Schief WR, Sodroski J, Wyatt R, Kwong PD. 2010. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc. Natl. Acad. Sci. U. S. A. 107:1166–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou T, Georgiev I, Wu X, Yang Z-Y, Dai K, Finzi A, Do Kwon Y, Scheid J, Shi W, Xu L, Yang Y, Zhu J, Nussenzweig MC, Sodroski J, Shapiro L, Nabel GJ, Mascola JR, Kwong PD. 2010. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science 329:811–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kwon YD, Finzi A, Wu X, Dogo-Isonagie C, Lee LK, Moore LR, Schmidt SD, Stuckey J, Yang Y, Zhou T, Zhu J, Vicic DA, Debnath AK, Shapiro L, Bewley CA, Mascola JR, Sodroski JG, Kwong PD. 2012. Unliganded HIV-1 gp120 core structures assume the CD4-bound conformation with regulation by quaternary interactions and variable loops. Proc. Natl. Acad. Sci. U. S. A. 109:5663–5668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. 2008. Molecular architecture of native HIV-1 gp120 trimers. Nature 455:109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mao Y, Wang L, Gu C, Herschhorn A, Xiang SH, Haim H, Yang X, Sodroski J. 2012. Subunit organization of the membrane-bound HIV-1 envelope glycoprotein trimer. Nat. Struct. Mol. Biol. 19:893–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mao Y, Wang L, Gu C, Herschhorn A, Desormeaux A, Finzi A, Xiang SH, Sodroski JG. 2013. Molecular architecture of the uncleaved HIV-1 envelope glycoprotein trimer. Proc. Natl. Acad. Sci. U. S. A. 110:12438–12443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris A, Borgnia MJ, Shi D, Bartesaghi A, He H, Pejchal R, Kang YK, Depetris R, Marozsan AJ, Sanders RW, Klasse PJ, Milne JL, Wilson IA, Olson WC, Moore JP, Subramanian S. 2011. Trimeric HIV-1 glycoprotein gp140 immunogens and native HIV-1 envelope glycoproteins display the same closed and open quaternary molecular architectures. Proc. Natl. Acad. Sci. U. S. A. 108:11440–11445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Helseth E, Olshevsky U, Furman C, Sodroski J. 1991. Human immunodeficiency virus type 1 gp120 envelope glycoprotein regions important for association with the gp41 transmembrane glycoprotein. J. Virol. 65:2119–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao J, Bergeron L, Helseth E, Thali M, Repke H, Sodroski J. 1993. Effects of amino acid changes in the extracellular domain of the human immunodeficiency virus type 1 gp41 envelope glycoprotein. J. Virol. 67:2747–2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Binley JM, Sanders RW, Clas B, Schuelke N, Master A, Guo Y, Kajumo F, Anselma DJ, Maddon PJ, Olson WC, Moore JP. 2000. A recombinant human immunodeficiency virus type 1 envelope glycoprotein complex stabilized by an intermolecular disulfide bond between the gp120 and gp41 subunits is an antigenic mimic of the trimeric virion-associated structure. J. Virol. 74:627–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maerz AL, Drummer HE, Wilson KA, Poumbourios P. 2001. Functional analysis of the disulfide-bonded loop/chain reversal region of human immunodeficiency virus type 1 gp41 reveals a critical role in gp120-gp41 association. J. Virol. 75:6635–6644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.York J, Nunberg JH. 2004. Role of hydrophobic residues in the central ectodomain of gp41 in maintaining the association between human immunodeficiency virus type 1 envelope glycoprotein subunits gp120 and gp41. J. Virol. 78:4921–4926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacobs A, Sen J, Rong L, Caffrey M. 2005. Alanine scanning mutants of the HIV gp41 loop. J. Biol. Chem. 280:27284–27288 [DOI] [PubMed] [Google Scholar]
- 19.Bellamy-McIntyre AK, Lay CS, Baar S, Maerz AL, Talbo GH, Drummer HE, Poumbourios P. 2007. Functional links between the fusion peptide-proximal polar segment and membrane-proximal region of human immunodeficiency virus gp41 in distinct phases of membrane fusion. J. Biol. Chem. 282:23104–23116 [DOI] [PubMed] [Google Scholar]
- 20.Sen J, Yan T, Wang J, Rong L, Tao L, Caffrey M. 2010. Alanine scanning mutagenesis of HIV-1 gp41 heptad repeat 1: insight into the gp120-gp41 interaction. Biochemistry 49:5057–5065 [DOI] [PubMed] [Google Scholar]
- 21.Caffrey M, Cai M, Kaufman J, Stahl SJ, Wingfield PT, Covell DG, Gronenborn AM, Clore GM. 1998. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J. 17:4572–4584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mische CC, Yuan W, Strack B, Craig S, Farzan M, Sodroski J. 2005. An alternative conformation of the gp41 heptad repeat 1 region coiled coil exists in the human immunodeficiency virus (HIV-1) envelope glycoprotein precursor. Virology 338:133–143 [DOI] [PubMed] [Google Scholar]
- 23.Sanders RW, Vesanen M, Schuelke N, Master A, Schiffner L, Kalyanaraman R, Paluch M, Berkhout B, Maddon PJ, Olson WC, Lu M, Moore JP. 2002. Stabilization of the soluble, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. J. Virol. 76:8875–8889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang X, Wyatt R, Sodroski J. 2001. Improved elicitation of neutralizing antibodies against primary human immunodeficiency viruses by soluble stabilized envelope glycoprotein trimers. J. Virol. 75:1165–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sellhorn G, Caldwell Z, Mineart C, Stamatatos L. 2009. Improving the expression of recombinant soluble HIV envelope glycoproteins using pseudo-stable transient transfection. Vaccine 28:430–436 [DOI] [PubMed] [Google Scholar]
- 26.Yang X, Florin L, Farzan M, Kolchinsky P, Kwong PD, Sodroski J, Wyatt R. 2000. Modifications that stabilize human immunodeficiency virus envelope glycoprotein trimers in solution. J. Virol. 74:4746–4754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang X, Lee J, Mahony EM, Kwong PD, Wyatt R, Sodroski J. 2002. Highly stable trimers formed by human immunodeficiency virus type 1 envelope glycoproteins fused with the trimeric motif of T4 bacteriophage fibritin. J. Virol. 76:4634–4642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kovacs JM, Nkolola JP, Peng H, Cheung A, Perry J, Miller CA, Seaman MS, Barouch DH, Chen B. 2012. HIV-1 envelope trimer elicits more potent neutralizing antibody responses than monomeric gp120. Proc. Natl. Acad. Sci. U. S. A. 109:12111–12116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leaman DP, Zwick MB. 2013. Increased functional stability and homogeneity of viral envelope spikes through directed evolution. PLoS Pathog. 9:e1003184. 10.1371/journal.ppat.1003184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Earl PL, Sugiura W, Montefiori DC, Broder CC, Lee SA, Wild C, Lifson J, Moss B. 2001. Immunogenicity and protective efficacy of oligomeric human immunodeficiency virus type 1 gp140. J. Virol. 75:645–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Srivastava IK, Stamatatos L, Legg H, Kan E, Fong A, Coates SR, Leung L, Wininger M, Donnelly JJ, Ulmer JB, Barnett SW. 2002. Purification and characterization of oligomeric envelope glycoprotein from a primary R5 subtype B human immunodeficiency virus. J. Virol. 76:2835–2847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beddows S, Franti M, Dey AK, Kirschner M, Iyer SP, Fisch DC, Ketas T, Yuste E, Desrosiers RC, Klasse PJ, Maddon PJ, Olson WC, Moore JP. 2007. A comparative immunogenicity study in rabbits of disulfide-stabilized, proteolytically cleaved, soluble trimeric human immunodeficiency virus type 1 gp140, trimeric cleavage-defective gp140 and monomeric gp120. Virology 360:329–340 [DOI] [PubMed] [Google Scholar]
- 33.Du SX, Idiart RJ, Mariano EB, Chen H, Jiang P, Xu L, Ostrow KM, Wrin T, Phung P, Binley JM, Petropoulos CJ, Ballantyne JA, Whalen RG. 2009. Effect of trimerization motifs on quaternary structure, antigenicity, and immunogenicity of a noncleavable HIV-1 gp140 envelope glycoprotein. Virology 395:33–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marcsisin SR, Engen JR. 2010. Hydrogen exchange mass spectrometry: what is it and what can it tell us? Anal. Bioanal. Chem. 397:967–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guttman M, Kahn M, Garcia NK, Hu SL, Lee KK. 2012. Solution structure, conformational dynamics, and CD4-induced activation in full-length, glycosylated, monomeric HIV gp120. J. Virol. 86:8750–8764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kong L, Huang CC, Coales SJ, Molnar KS, Skinner J, Hamuro Y, Kwong PD. 2010. Local conformational stability of HIV-1 gp120 in unliganded and CD4-bound states as defined by amide hydrogen/deuterium exchange. J. Virol. 84:10311–10321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Putnam CD, Hammel M, Hura GL, Tainer JA. 2007. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 40:191–285 [DOI] [PubMed] [Google Scholar]
- 38.Garlick RL, Kirschner RJ, Eckenrode FM, Tarpley WG, Tomich CS. 1990. Escherichia coli expression, purification, and biological activity of a truncated soluble CD4. AIDS Res. Hum. Retrovir. 6:465–479 [DOI] [PubMed] [Google Scholar]
- 39.Fang J, Rand KD, Beuning PJ, Engen JR. 2011. False EX1 signatures caused by sample carryover during HX MS analyses. Int. J. Mass Spectrom. 302:19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leonard CK, Spellman MW, Riddle L, Harris RJ, Thomas JN, Gregory TJ. 1990. Assignment of intrachain disulfide bonds and characterization of potential glycosylation sites of the type 1 recombinant human immunodeficiency virus envelope glycoprotein (gp120) expressed in Chinese hamster ovary cells. J. Biol. Chem. 265:10373–10382 [PubMed] [Google Scholar]
- 41.Smolsky IL, Liu P, Niebuhr M, Ito K, Weiss TM, Tsuruta H. 2007. Biological small-angle X-ray scattering facility at the Stanford Synchrotron Radiation Laboratory. J. Appl. Crystallogr. 40:s453–s458 [Google Scholar]
- 42.Petoukhov MV, Svergun DI. 2007. Analysis of X-ray and neutron scattering from biomolecular solutions. Curr. Opin. Struct. Biol. 17:562–571 [DOI] [PubMed] [Google Scholar]
- 43.Svergun DI. 1991. Mathematical methods in small-angle scattering data analysis. J. Appl. Crystallogr. 24:485–492 [Google Scholar]
- 44.Svergun DI. 1992. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25:495–503 [Google Scholar]
- 45.Mylonas E, Svergun DI. 2007. Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. J. Appl. Crystallogr. 40:s245–s249 [Google Scholar]
- 46.Guttman M, Weinkam P, Sali A, Lee KK. 2013. All-atom ensemble modeling to analyze small-angle X-ray scattering of glycosylated proteins. Structure 21:321–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Svergun DI. 1999. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76:2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kozin MB, Svergun DI. 2001. Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34:33–41 [Google Scholar]
- 49.Volkov VV, Svergun DI. 2003. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36:860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wriggers W, Birmanns S. 2001. Using situs for flexible and rigid-body fitting of multiresolution single-molecule data. J. Struct. Biol. 133:193–202 [DOI] [PubMed] [Google Scholar]
- 51.Davenport TM, Guttman M, Guo W, Cleveland B, Kahn M, Hu SL, Lee KK. Isolate-specific differences in the conformational dynamics and antigenicity of HIV-1 gp120. J. Virol. 87:10855–10873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jacques DA, Trewhella J. 2010. Small-angle scattering for structural biology: expanding the frontier while avoiding the pitfalls. Protein Sci. 19:642–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Staropoli I, Chanel C, Girard M, Altmeyer R. 2000. Processing, stability, and receptor binding properties of oligomeric envelope glycoprotein from a primary HIV-1 isolate. J. Biol. Chem. 275:35137–35145 [DOI] [PubMed] [Google Scholar]
- 54.Jeffs SA, Goriup S, Kebble B, Crane D, Bolgiano B, Sattentau Q, Jones S, Holmes H. 2004. Expression and characterisation of recombinant oligomeric envelope glycoproteins derived from primary isolates of HIV-1. Vaccine 22:1032–1046 [DOI] [PubMed] [Google Scholar]
- 55.Beddows S, Schulke N, Kirschner M, Barnes K, Franti M, Michael E, Ketas T, Sanders RW, Maddon PJ, Olson WC, Moore JP. 2005. Evaluating the immunogenicity of a disulfide-stabilized, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. J. Virol. 79:8812–8827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Center RJ, Earl PL, Lebowitz J, Schuck P, Moss B. 2000. The human immunodeficiency virus type 1 gp120 V2 domain mediates gp41-independent intersubunit contacts. J. Virol. 74:4448–4455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Finzi A, Pacheco B, Zeng X, Kwon YD, Kwong PD, Sodroski J. 2010. Conformational characterization of aberrant disulfide-linked HIV-1 gp120 dimers secreted from overexpressing cells. J. Virol. Methods 168:155–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu J, Deng Y, Li Q, Dey A, Moore JP, Lu M. 2010. Role of a putative gp41 dimerization domain in human immunodeficiency virus type 1 membrane fusion. J. Virol. 84:201–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frey G, Chen J, Rits-Volloch S, Freeman MM, Zolla-Pazner S, Chen B. 2010. Distinct conformational states of HIV-1 gp41 are recognized by neutralizing and non-neutralizing antibodies. Nat. Struct. Mol. Biol. 17:1486–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Finzi A, Xiang SH, Pacheco B, Wang L, Haight J, Kassa A, Danek B, Pancera M, Kwong PD, Sodroski J. 2010. Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol. Cell 37:656–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen J, Lee KH, Steinhauer DA, Stevens DJ, Skehel JJ, Wiley DC. 1998. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 95:409–417 [DOI] [PubMed] [Google Scholar]
- 62.Wilson IA, Skehel JJ, Wiley DC. 1981. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature 289:336–373 [DOI] [PubMed] [Google Scholar]
- 63.Dey AK, David KB, Lu M, Moore JP. 2009. Biochemical and biophysical comparison of cleaved and uncleaved soluble, trimeric HIV-1 envelope glycoproteins. Virology 385:275–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chakrabarti BK, Pancera M, Phogat S, O'Dell S, McKee K, Guenaga J, Robinson J, Mascola J, Wyatt RT. 2011. HIV type 1 Env precursor cleavage state affects recognition by both neutralizing and nonneutralizing gp41 antibodies. AIDS Res. Hum. Retrovir. 27:877–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haim H, Salas I, Sodroski J. 2013. Proteolytic processing of the human immunodeficiency virus envelope glycoprotein precursor decreases conformational flexibility. J. Virol. 87:1884–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Carr CM, Chaudhry C, Kim PS. 1997. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc. Natl. Acad. Sci. U. S. A. 94:14306–14313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wyatt R, Desjardin E, Olshevsky U, Nixon C, Binley J, Olshevsky V, Sodroski J. 1997. Analysis of the interaction of the human immunodeficiency virus type 1 gp120 envelope glycoprotein with the gp41 transmembrane glycoprotein. J. Virol. 71:9722–9731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Si Z, Madani N, Cox JM, Chruma JJ, Klein JC, Schon A, Phan N, Wang L, Biorn AC, Cocklin S, Chaiken I, Freire E, Smith AB, III, Sodroski JG. 2004. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc. Natl. Acad. Sci. U. S. A. 101:5036–5041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Desormeaux A, Coutu M, Medjahed H, Pacheco B, Herschhorn A, Gu C, Xiang SH, Mao Y, Sodroski J, Finzi A. 2012. The highly conserved layer 3 component of the HIV-1 gp120 inner domain is critical for CD4-required conformational transitions. J. Virol. 87:2549–2562 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.