

Abstract
Hepatitis C virus (HCV) orchestrates the different stages of its life cycle in time and space through the sequential participation of HCV proteins and cellular machineries; hence, these represent tractable molecular host targets for HCV elimination by combination therapies. We recently identified multifunctional Y-box-binding protein 1 (YB-1 or YBX1) as an interacting partner of NS3/4A protein and HCV genomic RNA that negatively regulates the equilibrium between viral translation/replication and particle production. To identify novel host factors that regulate the production of infectious particles, we elucidated the YB-1 interactome in human hepatoma cells by a quantitative mass spectrometry approach. We identified 71 YB-1-associated proteins that included previously reported HCV regulators DDX3, heterogeneous nuclear RNP A1, and ILF2. Of the potential YB-1 interactors, 26 proteins significantly modulated HCV replication in a gene-silencing screening. Following extensive interaction and functional validation, we identified three YB-1 partners, C1QBP, LARP-1, and IGF2BP2, that redistribute to the surface of core-containing lipid droplets in HCV JFH-1-expressing cells, similarly to YB-1 and DDX6. Importantly, knockdown of these proteins stimulated the release and/or egress of HCV particles without affecting virus assembly, suggesting a functional YB-1 protein complex that negatively regulates virus production. Furthermore, a JFH-1 strain with the NS3 Q221L mutation, which promotes virus production, was less sensitive to this negative regulation, suggesting that this HCV-specific YB-1 protein complex modulates an NS3-dependent step in virus production. Overall, our data support a model in which HCV hijacks host cell machinery containing numerous RNA-binding proteins to control the equilibrium between viral RNA replication and NS3-dependent late steps in particle production.
INTRODUCTION
Almost 200 million individuals worldwide are infected with the hepatitis C virus (HCV), a member of the genus Hepacivirus in the family Flaviviridae (1). Chronic infection often leads to progressive fibrosis, cirrhosis, hepatocellular carcinoma, and eventually death (2). While the efficacy of current treatments has significantly improved with the inclusion of HCV NS3 protease inhibitors in the new standard of care, this therapy has serious adverse side effects and the sustained virological response rates are still not optimal for infected populations (3). This unmet medical need is currently being addressed by the pharmaceutical industry through the development of novel classes of direct-acting antivirals (e.g., NS5B and NS5A inhibitors) and of host-targeted antivirals (HTAs) that inhibit host factors (e.g., miR-122, cyclophilin A) indispensable for the HCV life cycle (4–7). This highlights that resolving HCV and host factor physical and functional networks will help to identify novel molecular targets for the development of novel HTAs while providing an inexhaustible source of fundamental knowledge.
Following the entry of HCV into the target cell, the viral RNA (vRNA) genome is translated into a unique viral polyprotein precursor, which is further processed by cellular and viral proteases to generate 10 mature viral proteins (core, E1, E2, p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B). HCV infection is induced partly through NS4B's drastic rearrangement of cytoplasmic membranes, forming the so-called “membranous web,” where the viral RNA is replicated by HCV RNA polymerase NS5B (1, 8). Replication complex formation, integrity, and activity also depend on NS5A, NS3, and numerous host factors (e.g., PI4KIIIα, cyclophilin A) (4, 9–17). Until recently, mechanistic details of HCV particle assembly and egress were almost nonexistent because of the lack of experimental systems with which to explore the complete HCV infectious life cycle. In 2005, a breakthrough was achieved with the discovery of the JFH-1 strain (isolated from a Japanese patient with fulminant hepatitis) as the first HCV clone that robustly produces infectious viral particles in cell culture (18, 19). The extensive in vitro use of this strain, as well as various genetically engineered intergenotypic chimeric or adapted HCV clones, established the molecular basis of the study of particle production, which nevertheless remains poorly understood. HCV assembly is believed to be initiated by the targeting of the capsid protein core to the lipid droplet (LD), a cellular organelle involved in the storage of neutral lipids (20–22). Assembling capsids are most likely transferred to the endoplasmic reticulum, where they bud and acquire viral envelope proteins E1 and E2, and then use the very-low-density lipoprotein maturation and secretory pathway to exit from the cell (23). Each step in viral particle production seems to be tightly regulated in time and space, since all viral proteins (except NS5B), as well as numerous host factors (e.g., ApoE, ApoB, DGAT-1, MTP, annexin A2), have been shown to play crucial roles in this process (18, 23–39).
Very few mechanistic details of how HCV controls the transitions between the different stages of its life cycle in time and space are known. This is further complicated by the fact that several HCV proteins are multifunctional, an efficient way for the virus to condense the genetic information necessary for a complete and productive life cycle. For instance, the NS3 protein has multiple functions in polyprotein processing, in RNA replication (through its helicase and ATPase activities), and in particle assembly independently of its enzymatic properties (28, 40). The latter role of NS3 remains misunderstood, since no assembly-defective NS3 mutant has been clearly identified and reported for genotype 2a HCV. Nevertheless, the Q221L mutation within the helicase domain has been shown to increase virus production from a wild-type clone and also to rescue the assembly of various defective clones (28, 41, 42). Notably, several reports suggest that NS3, through its interaction with core multimers (43), may bridge assembling capsids to NS2, which controls their egress from LDs toward the secretory pathway (35–38, 41). The switch between the NS3 replication and assembly activities, which obviously do not occur at the same time point in the life cycle, may be controlled through the dynamic interactions with host factors. This model is supported by recent data from our group indicating that Y-box-binding protein 1 (YB-1 or YBX1) acts as a novel partner of NS3 and vRNA that modulates the equilibrium between HCV RNA replication and the production of infectious particles (44). Indeed, the knockdown of YB-1 gene expression compromised vRNA replication and drastically stimulated virus release by impeding negative regulation. Importantly, a subset of YB-1 protein redistributed to the surface of core-containing LDs in HCV-infected cells, where virus assembly occurs. Thus, the physical hijacking of YB-1 seems to be tightly linked to its functions in the HCV life cycle.
Other host factors, including RNA-binding proteins such as proteins involved in micro-RNA biogenesis and some DEAD box proteins (DDX3, DDX6), have been reported to be co-opted by HCV to the LDs similarly to YB-1, suggesting their involvement in a shared viral regulatory process as part of a common host machinery (45–49). However, the involvement of these host factors in the regulation of late stages of the HCV life cycle was never fully investigated or compared together, and differences between the approaches used (HCV clones and genotypes, methodologies) preclude any conclusion regarding a common functional relationship.
To gain further insight into the role of a functional ribonucleoprotein (RNP) complex during HCV infection, we elucidated the YB-1 interactome within the human hepatoma Huh7-derived cell line. A proteomic approach combining immunoprecipitation of YB-1 protein complexes coupled with mass spectrometry (MS) identified 71 potential YB-1 partners. A gene-silencing study of YB-1 partner hits revealed a strong enrichment of modulators of HCV RNA replication with the identification of 26 host cofactors. Following the extensive interaction and functional validation, we demonstrated a restrictive set of YB-1 interactors (C1QBP, LARP-1, and IGF2BP2) that not only redistribute to the surface of the LDs in JFH-1-expressing cells similarly to YB-1, DDX3, and DDX6 but also negatively regulate particle production, suggesting that they are part of the same functional YB-1 protein complex. Furthermore, we showed that the JFH-1 infectious strain with the single amino acid mutation Q221L in the NS3 protein was less sensitive than wild-type JFH-1 to the negative regulation of this YB-1 protein complex, further suggesting a role in the modulation of NS3-dependent steps in HCV particle production. Overall, this study reconciles data from previous independent reports on important HCV-host interactions and further defines a host YB-1 RNP complex that is hijacked by HCV to regulate late stages of its life cycle.
MATERIALS AND METHODS
Plasmids.
The construction of plasmids pCDNA3.1-Flag-MCS, pcDNA3.1-Flag-YB-1, and pEF/JFH-1-Rz/Neo and the corresponding Δ153-167 core mutant was previously described (44, 50, 51). For the construction of NS3 Q221L and NS2 Q199R mutants (Q1251L and Q1012R of the JFH-1 polyprotein), we performed a two-step recombinant PCR with Platinum Pfx DNA polymerase (Invitrogen) involving four oligonucleotides and the pEF/JFH-1-Rz/Neo plasmid as the template. The internal primers were designed to generate the desired point mutation in the NS3 and NS2 coding sequences. The final mutated NS2 and NS3 PCR products were cloned into the ClaI and BsiWI/SanDI cassettes of pEF/JFH1-Rz/Neo, respectively. The J6/JFH(p7-Rluc2A) plasmid encoding the Rluc reporter HCV (39) was a kind gift from Charles Rice (Rockefeller University) with the authorization of Apath LLC. An enhanced yellow fluorescent protein (eYFP)–YB-1 expression vector was generated by the in-frame ligation of a YB-1 coding sequence PCR product into the BamHI cassette of pcDNA3.1-Hygro-eYFP-MCS (51).
Cell lines, antibodies, and reagents.
The 293T, HeLa, and Huh7.5 cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, and 1% nonessential amino acids (all from Wisent). Cells were transfected with linear (25-kDa) polyethylenimine (PEI; Polysciences) (51) or Lipofectamine 2000 (Invitrogen) as described by the manufacturer. Huh7 cells stably expressing a reporter Con1 subgenomic replicon (Huh7-Con1-Fluc) (52) were a kind gift from Ralf Bartenschlager (University of Heidelberg) and were maintained in complete DMEM with 500 μg/ml G418 (Multicell). Huh7.5 cells stably expressing JFH-1 were generated following cell transfection with pEF/JFH-1-Rz/Neo and selection with 500 μg/ml G418 for 2 weeks. All of the primary antibodies used in this study are listed in Table S1 in the supplemental material with the corresponding dilutions for Western blotting and confocal microscopy analyses. Secondary antibodies coupled with horseradish peroxidase and Alexa Fluor were purchased from Bio-Rad and Invitrogen, respectively.
Immunoprecipitation and liquid chromatography (LC)-MS analysis.
For YB-1 interactome analysis, PEI-transfected cells from four confluent 150-mm dishes for each condition were used. Anti-Flag immunoprecipitations were performed exactly as described before (44), except that a 1-h preclearing step with 50 μl of a 50:50 slurry of IgG-Sepharose (GE Healthcare) was included to prevent nonspecific binding with the anti-Flag resin. Immune complexes were collected following two successive elutions with 100 μl of 0.5 M ammonium hydroxide (pH 11.7) at 4°C. Pooled eluates were evaporated on a SpeedVac, stored at −80°C, and further processed for quantitative MS (qMS) analysis. Samples were resuspended in 50 μl of 50 mM ammonium bicarbonate. Tris(2-carboxyethyl)phosphine was added to protein samples to a final concentration of 5 mM. Samples were incubated at 37°C with shaking at 650 rpm for 30 min. Thirty microliters of chloroacetamide at 55 mM was added, and samples were incubated at 37°C with shaking at 650 rpm for 30 min. One microgram of trypsin was added, and samples were digested overnight at 37°C. Samples were evaporated in a SpeedVac and resolubilized in 50 μl of 5% acetonitrile (ACN)–0.2% formic acid (FA). Twenty microliters of each sample was injected into a C18 precolumn (0.3 mm [inside diameter {i.d.}] by 5 mm), and peptides were separated on a C18 analytical column (150 μm [i.d.] by 100 mm) with an Eksigent nanoLC-2D system. A 56-min linear gradient of 10 to 60% B (A, 0.2% FA; B, ACN–0.2% FA) was used to elute peptides at a flow rate of 600 nl/min.
The LC system was coupled with an LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher). Each full MS spectrum was followed by six tandem MS (MS/MS) spectra on the most abundant multiply charged ions. MS/MS experiments were performed by collision-induced dissociation in the linear ion trap. The data were processed with the Mascot 2.2 (Matrix Science) search engine. The database used was IPI human (182,394 sequences).
Comparison of peptide abundance across different sample sets was achieved by label-free quantitative proteomics. Briefly, raw (.raw) data files from the Xcalibur software were converted into peptide map files representing all ions according to their corresponding m/z values, retention times, intensities, and charge states. Intensity values above a threshold of typically 10,000 counts were considered for further analysis. Peptide abundances were assessed by using apex intensity values. Clustering of peptide maps was performed on identified peptides by hierarchical clustering with tolerances of 15 ppm and 1 min for peptide mass and retention time, respectively. Normalization of retention time was then performed on the initial peptide cluster list by a dynamic and nonlinear correction that confines the retention time distribution to less than 0.1 min (<0.3% relative standard deviation [SD]) on average. For each LC-MS run, we normalize the peptide ratios so that the median of their logarithms was zero, to account for unequal protein amounts across conditions. Protein ratios reflecting their relative abundances were calculated as the median of all of the peptide ratios, while minimizing the effect of outliers. According to the relative protein abundance, we calculated an enrichment ratio for each identified protein (>1, enriched under the Flag–YB-1 condition; < −1, more abundant under the Flag-multi-cloning site [MCS] condition). A protein was considered significantly more abundant in the Flag–YB-1 immunoprecipitate and hence identified as a potential YB-1 partner when its fold enrichment factor was above a threshold of 3, according to the Gaussian distribution of the enrichment ratios for each cell line. We selected 10 and 18% of the most enriched proteins in the Flag–YB-1 immunoprecipitates from the Huh7.5 and Huh7 cell lines, respectively. As potential nonspecific RNA-dependent contaminant, ribosomal proteins were systematically excluded from the candidate list. The final YB-1 partner hit list was obtained from two independent qMS analyses of the YB-1 interactome in the Huh7 and Huh7.5 cell lines.
Lentivirus production and transduction.
293T cells were transfected with PEI by using plasmids pRSV-REV, pMD2-VSVG, and pMDLg/pRRE (a kind gift from Didier Trono) and short hairpin RNA (shRNA)-encoding plasmid pLKO.1-puro (TRC 1 generation; Sigma-Aldrich). The shRNAs used for the silencing of target genes are listed in Table S1 in the supplemental material. Two days later, cell culture media were collected and cleared through a 0.45-μm filter. Lentivirus preparations were titrated by colony assays in HeLa cells. Briefly, 4,000 cells were seeded into the wells of 96-well plates and transduced the day after with shRNA-expressing lentiviruses diluted 1:400 or 1:10,000 in the presence of 8 μg/ml Polybrene (Sigma-Aldrich). Infected cells were selected with 1 μg/ml puromycin (Wisent). At 7 days postransduction, cells were washed with phosphate-buffered saline (PBS) and colored with 1.25% crystal violet–25% ethanol. Colonies were counted, and viral titers (in PFU/ml) were calculated with the following formula: Number of colonies × Dilution × 12. For all RNA interference (RNAi) assays, cells were plated in the presence of 4 μg/ml Polybrene and lentiviruses at a multiplicity of infection (MOI) of 10. Sixteen to 24 h later, the culture medium was changed and puromycin was added at a final concentration of 3 μg/ml. The following day, cells were transfected with plasmids of interest. Two to five shRNAs were tested for each gene of interest, and their respective knockdown efficiencies were assessed by Western blotting with extracts from cells collected 4 to 5 days postransduction.
Luciferase assays.
Cells grown in white 96-well plates were washed once with 150 μl PBS. For firefly luciferase (Fluc) assays, 50 μl PBS and 50 μl of 2× luciferase buffer (100 mM Trizma acetate, 20 mM magnesium acetate, 2 mM EGTA, 1% Brij 58, 0.7% β-mercaptoethanol, 3.6 mM ATP, 45 μg/ml d-luciferin, pH 7.9) were added to the cells. Cells were incubated for 15 min at room temperature in the dark. For Renilla luciferase (Rluc) assays, 50 μl of PBS and 50 μl of 2 mM EDTA (pH 8)–5 μM coelenterazin (Nanolight) were added to the washed cells. Fluc and Rluc activities were measured with a MicroBeta JET luminescence counter (PerkinElmer).
Cytotoxicity assays.
For 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assays, cells were cultured in transparent 96-well plates. Twenty microliters of MTT stock solution (5 mg/ml in PBS) was added to the cells, which were incubated for 1 h at 37°C in the dark. Following the removal of the medium, cells were incubated at room temperature for 10 min with 150 μl of dimethyl sulfoxide containing 2 mM glycine, pH 11. Absorbance at 570 nm was read with a reference at 650 nm with a GENios Plus apparatus (Tecan).
For alamarBlue assays in the RNAi screening, cells were cultured in black 96-well plates. Ten microliters of alamarBlue reagent (Invitrogen; diluted 1:4 in PBS) was added to the cells, and following a 3-h incubation at 37°C, fluorescence at 595 nm (excitation wavelength, 531 nm) was measured with an EnVision plate reader (PerkinElmer). A control plate with medium only (no cells) or alamarBlue only was used to determine the background that was subtracted from the fluorescence value.
RNAi screening.
Five shRNA-encoding pLKO.1-puro plasmids per gene of the MS hits were cherry-picked from the MISSION TRC shRNA lentiviral human library (Sigma-Aldrich) at the Institut de Recherche en Immunologie et en Cancérologie (IRIC) High-Throughput Screening Core Facility. Lentivirus production and transduction were carried out with a Biomek FX (Beckman Coulter) enclosed in a class II cabinet. Twenty thousand 293T cells were seeded into the wells of 96-well plates and transfected the day after with the selected shRNA plasmids and MISSION lentiviral packaging mixture (SHP001; Sigma-Aldrich) with TransIT reagent (Mirus). Lentivirus-containing cell supernatants were collected at 48 and 72 h posttransfection and pooled. The average titer of the lentiviral production was determined by titrating random preparations from each plate as described above. For RNAi screening, 5,000 Huh7-Con1-Fluc cells were seeded into the wells of 96-well plates in the presence of 4 μg/ml Polybrene and lentiviruses were added to the cells with a Biomek FX at an MOI of 10 (see Fig. 1D for the setup used). A series of internal quality controls that included individually produced and titrated lentivirus encoding nontarget shRNA (shNT) and shRNA silencing YB-1 expression were added to each plate (four wells per shRNA). Treatment with the HCV inhibitor BILN 2061 (53) was also included (final concentration, 1 μM) as a positive control for total disruption of HCV replication. Finally, each plate contained wells with nontransduced cells in order to control for potential undesirable effects of lentiviral infection on HCV replication and cell viability. Four days postransduction, Fluc and alamarBlue assays were performed exactly as described above. This RNAi screening was performed in three independent experiments. The Fluc and alamarBlue signal levels determined with the shNT control in each culture plate were arbitrarily set to 1. For each assay, the mean values and corresponding SDs from three independent RNAi screenings were calculated. A tested protein was considered a hit when one or two corresponding shRNAs diminished Fluc activity (i.e., HCV Con1 RNA replication) by more than 70 or 50%, respectively, without significantly affecting cell viability (alamarBlue signal, >75%).
Fig 1.

A combined MS-RNAi screening approach identifies new HCV replication regulators. (A) Decision tree for the identification of YB-1 protein partners that regulate HCV particle production. (B) Huh7 cells were transfected with Flag–YB-1-expressing or control vectors. At 48 h posttransfection, cell extracts were prepared and subjected to immunoprecipitation (IP) with anti-Flag antibody-coupled resin. Cell extracts were analyzed by Western blotting with anti-YB-1, anti-Flag, and anti-actin antibodies. The content of the immunoprecipitates was analyzed by silver staining, attesting to the specificity of the immunoprecipitation and of the copurification of YB-1 partners. (C) Schematic representation of the reporter genotype 1b HCV subgenomic replicon stably expressed by the Huh7-Con1-Fluc cell line. This replicon expresses the Fluc and G418 resistance product in a bicistronic manner under the control of the HCV internal ribosome entry site (IRES), as well as the NS3-NS5B replication unit downstream encephalomyocarditis virus (EMCV) IRES. With the absence of structural proteins, this virus does produce particles and replicates only within the cell. (D) Plate map and assay design used for RNAi screening. Huh7-Con1-Fluc cells were seeded into the wells of 96-well plates and transduced with shRNA-expressing lentiviruses. The right and left columns were used exclusively for controls. Fluc and alamarBlue assays were performed 4 days postransduction. (E) TIGR (The Institute for Genomic Research) MultiExperimentViewer (TMeV) representation of the selected shRNA hits. Each column represents the results of the RNA screening for one shRNA hit with the indicated targeted protein at the top.
HCV production, purification, and infection.
Huh7.5 cells were transfected with plasmid pEF/JFH-1-Rz/Neo by using Lipofectamine 2000. At 3 days posttransfection, cells and culture media were collected. Cells were washed twice with PBS and lysed in 10 mM Tris-HCl–100 mM NaCl–0.5% Triton X-100 (pH 7.6) with protease inhibitors (Roche). Cell extracts were analyzed by Western blotting. Virus-containing culture medium was cleared through a 0.45-μm filter. For virus purification, 8 ml was deposited onto a 20% sucrose cushion (in PBS). Samples were ultracentrifuged at 35,000 rpm for 4 h at 4°C with an SW41 rotor (Beckman). Pelleted viral particles were resuspended in 1× SDS loading buffer, heated for 5 min at 95°C, and further analyzed by Western blotting. For viral infection, 100,000 Huh7.5 cells were seeded into the wells of six-well plates the day prior to infection. Medium was removed, and 1 ml of cleared virus-containing sample was added to the cells. Following incubation for 4 to 6 h at 37°C, the culture medium was replaced with fresh DMEM. At 3 days postinfection, cells were collected and total RNA was purified with the RNeasy Minikit (Qiagen). HCV RNA content was analyzed by quantitative real-time PCR (qRT-PCR) by the IRIC Genomic Core Facility.
For intracellular infectivity assays, washed cells were pelleted, resuspended in 1 ml of complete DMEM, and then subjected to four successive freeze-thaw cycles. Lysed cells were centrifuged at 10,000 rpm for 5 min at room temperature, and supernatants were used to infect naive Huh7.5 cells. An aliquot of cell extract was kept for total RNA extraction with TRIzol LS (Invitrogen) according to the manufacturer's instructions. Relative HCV RNA amounts were determined by qRT-PCR at the IRIC Genomic Core Facility.
For J6/JFH-1(p7-Rluc2A) virus production, the DNA template used for in vitro transcription (a kind gift from Charles Rice) was linearized by XbaI digestion and then purified by phenol-chloroform extraction and ethanol precipitation. J6/JFH-1(p7-Rluc2A) RNA was produced with the MEGAscript T7 in vitro transcription kit (Ambion) according to the manufacturer's instructions and purified with the RNeasy Minikit (Qiagen) by the RNA cleanup protocol. Huh7.5 cells resuspended in CytoMix buffer were electroporated with the J6/JFH-1(p7-Rluc2A) RNA exactly as described before (54), with a GenePulser Xcell apparatus (Bio-Rad). Cells were plated in a 150-mm cell culture dish. Cell medium was replaced at 24 h postelectroporation. The day after, cells were trypsinized and one-fifth of the cell preparation was replated on a 150-mm dish. Four days later, virus-containing culture medium was collected, cleared through a 0.45-μm filter and stored at −80°C. For infection, 75 μl of this virus preparation was added to 4,000 to 5,000 Huh7.5 cells that had been plated into the wells of 96-well plates the day before. Four to 6 h later, the culture medium was replaced. Three days postinfection, Rluc assays were performed as described above.
qRT-PCR.
For reverse transcription with total RNA from JFH-1-transfected cells, we used the QuantiTect RT kit (Qiagen) in order to avoid a contaminating DNA signal from the pEF/JFH-1/Rz plasmid. Other reverse transcription and all real-time PCRs were performed at the IRIC Genomic Core Facility with TaqMan-based assays. For HCV RNA detection, we used primers CATGGCGTTAGTATGAGTGTCG and GGTTCCGCAGACCACTATG and TaqMan-labeled probe CAGCCTCC (probe 75; no. 04688988001 from the Roche Universal Probe Library).
Confocal microscopy analyses.
Cells were grown on a coverslip in six-well plates, fixed with 4% paraformaldehyde (in PBS) for 20 min at room temperature, permeabilized with 0.2% Triton X-100 (in PBS) for 15 min at room temperature, and then stored a 4°C in PBS. Blocking was performed in PBS with 10% normal goat serum–5% bovine serum albumin (BSA)–0.02% sodium azide for 45 min at room temperature. Following three rapid washes, cells were labeled for 2 h at room temperature with primary antibodies diluted in 5% BSA–0.02% sodium azide–PBS as indicated in Table S1 in the supplemental material. Slides were washed three times in PBS and then probed with Alexa Fluor 488-, 594-, 647-, or 402-conjugated secondary antibodies (Invitrogen) diluted 1:500 in 5% BSA–0.02% sodium azide–PBS for 1 h in the dark. Cells were extensively washed and incubated with Hoechst dye (Invitrogen) at a final concentration of 1 μg/ml in PBS for 10 min. Following three rapid washes, a 1-h incubation was performed with HCS LipidTOX Deep Red (Invitrogen) diluted 1:1,000 in PBS to label the LDs in some experiments. Immediately afterward, the slides were mounted with 1,4-diazabicyclo[2.2.2]octane (Sigma-Aldrich) as an antifading agent. Labeled cells were then examined by confocal laser scanning microscopy with an LSM510 microscope (Zeiss) at the IRIC Bio-Imaging Core Facility. Colocalization percentages were determined with the MetaMorph image analysis software (Molecular Devices).
LD flotation assays.
In some experiments, Huh7.5 cells were treated with oleic acid, which was prepared exactly as previously described (55). Briefly, 0.5 g of fatty-acid-free BSA (catalog number A6003; Sigma-Aldrich) was dissolved in 3.6 ml of 0.1 M Tris-Cl, pH 8. A 12.6-mg sample of oleic acid (catalog number O1383; Sigma-Aldrich) was transferred into clean, fatty-acid free BSA (catalog number A6003; Sigma-Aldrich) and then diluted in 3.6 ml of Tris-BSA buffer by gently shaking the solution until it was clear (oleic acid stock at 12.38 mM). Cells were treated with 200 μM oleic acid at 37°C for 16 h before the assay, washed twice in PBS, collected in PBS–5 mM EDTA, pelleted, resuspended in 1.3 ml of HLM buffer (20 mM Tris, 1 mM EDTA [pH 7.4], protease inhibitors), and incubated for 10 min on ice. Cells were subjected to eight gentle strokes with a Dounce homogenizer. The nuclei were removed by centrifugation at 1,000 × g for 10 min at 4°C. Resulting cell homogenates (1 ml) were mixed with 0.5 volume of HLM buffer containing protease inhibitors and 60% (wt/vol) sucrose and deposited at the bottom of an ultracentrifuge tube. Five milliliters of HLM–5% (wt/vol) sucrose, followed by 5 ml of HLM, was gently added above the cell extract. The discontinuous sucrose gradients were centrifuged at 13,000 rpm with an SW41 rotor (Beckman Coulter) for 30 min at 4°C and then stopped with no brake. Three hundred to 500 μl of the floating LD fraction was collected, diluted in SDS-containing loading buffer, heated at 95°C for 5 min, resolved by SDS-PAGE, and finally analyzed by Western blotting.
Statistical analysis.
Statistical analysis was performed with the Office Excel 2010 software. All Student t tests were two tailed, and P values of <0.05 were considered significant.
RESULTS
Elucidation and functional analysis of the YB-1 interactome in HCV-expressing cells.
In order to identify novel regulators of the HCV replication and particle production equilibrium and to better understand the function of YB-1-containing RNP complexes during viral particle generation, we investigated its composition in hepatoma cell lines Huh7 and Huh7.5, which are permissive to HCV infection. Cells were transfected with plasmids encoding Flag-tagged YB-1 or the control vector (Fig. 1). Flag–YB-1 was expressed at subendogenous levels in order to reduce off-target and undesirable interactions (Fig. 1B). YB-1-associated proteins were immunopurified with anti-Flag antibodies. Western blotting and silver staining analyses of the corresponding proteins showed that Flag–YB-1 was purified as the predominant protein (Fig. 1B). Numerous YB-1 partners were specifically copurified, as evidenced by the silver-stained bands present in the Flag–YB-1 immunoprecipitation and absent from the negative-control condition. The protein profiles of these eluates were compared by a label-free qMS approach to determine changes in protein abundance between immunoprecipitates and the negative control. A detected protein was considered a YB-1 partner hit when it was enriched in the Flag–YB-1 condition by at least 3-fold compared to the Flag-MCS negative control (see Materials and Methods). The combined qMS analysis of the two preparations of Huh7 and Huh7.5 cells identified 71 potential YB-1 interactors, and most of them were RNA-binding proteins, including several heterogeneous nuclear RNPs (hnRNPs) and DEAD box (DDX/DHX) proteins (Fig. 1A; see Table S2 in the supplemental material). Interestingly, the newly identified YB-1 partner G3BP (or G3BP1) was previously shown to be required for the HCV life cycle (56, 57). YB-1 also associated with other RNA-binding proteins that were reported to influence the replication of other RNA viruses (see Table S4). For instance, the double-stranded-RNA-binding protein Staufen1 (STAU1) was shown to influence the life cycles of the human immunodeficiency and influenza viruses (58, 59). Finally, the YB-1 interactome also included membrane-associated dynamin 2 (DNM2) and apolipoprotein B (APOB), which are important for HCV entry and assembly, respectively (25, 60).
To determine if these putative YB-1 partners are involved in HCV RNA replication, we performed a targeted RNAi screening of the Huh7 cell line, which contains the reporter Con1 subgenomic replicon (Fig. 1C). This viral genome encodes a bicistronic Fluc whose expression levels strictly depend on RNA replication (44, 52). For each YB-1 partner hit, five different shRNA-expressing lentiviruses were tested individually by transduction of Huh7-Con1-Fluc cells. Four days postransduction, Fluc activity was measured to monitor the effects of gene knockdown on HCV RNA replication (Fig. 1D; see Table S3 in the supplemental material). An alamarBlue assay was performed as a counterscreening to evaluate the possible cytotoxicity induced by the shRNAs tested. Gene hits were selected as described in Materials and Methods, while taking into account undesirable cytotoxic effects, as well as the number and antiviral efficiency of shRNAs. This RNAi screening approach successfully identified 26 YB-1 partner hits whose gene knockdown significantly decreased HCV RNA replication (Fig. 1A and E).
We next confirmed the interaction between YB-1 and the identified partner hits by Flag coimmunoprecipitation assays and immunoblot assay detection in Huh7.5 cells that stably expressed the infectious molecular clone JFH-1 (Fig. 2A and B). YB-1 interactors could be specifically coimmunoprecipitated with Flag–YB-1. As a negative control, we did not detect any off-target interaction with Flag-eYFP. In reciprocal coimmunoprecipitation experiments, we showed that endogenous proteins DDX3, DDX6, IGF2BP2, LARP-1, ILF2, RHA, PABPN1, and ILF3 all individually interacted with endogenous YB-1 in uninfected cells (Fig. 2C). Other YB-1 partners, such as DDX6, IGF2BP2, LARP-1, and ILF3, were often detected by the immunoprecipitation of these endogenous proteins, suggesting pre-existing YB-1 RNP complexes (Fig. 2C). The YB-1 interactors (15 proteins) were then assessed for involvement in HCV RNA replication. Two to five shRNA-expressing lentiviruses per gene were individually tested for silencing activity (see Table S1 in the supplemental material). For each gene, the shRNA sequence with the best silencing efficiency was first used for transduction of Huh7-Con1-Fluc (Fig. 3A to K). Luciferase assays performed at 3 and 4 days postransduction showed that the knockdown of YB-1 partners significantly decreased HCV replication over time compared to the control condition with an shNT (Fig. 3L). Interestingly, the validated hits included the previously reported HCV regulators DDX3, DDX5, DDX6, and DHX9, suggesting that their functions during the HCV life cycle might be linked to those of YB-1 (45, 46, 48, 49, 61, 62). As controls, treatment with the NS3 protease inhibitor BILN 2061 completely abrogated Con1 replication, while decreased YB-1 expression by gene knockdown (YB-1 shRNA) inhibited HCV RNA replication by almost 80%. Parallel cell viability assays confirmed that none of the shRNAs, with the exception of shhnRNPG-T, induced apparent cytotoxicity. The dependency of HCV on the validated YB-1 interactors was then assessed following the infection of Huh7.5 cells with the genotype 2a Rluc reporter virus J6/JFH-1(p7-Rluc2A) (39) (Fig. 3L). In the context of a full infectious life cycle, we observed a more restrictive set of YB-1 interactors, i.e., DDX3, DDX28, IGF2BP2, LARP1, and ZC3HAV1, that significantly decreased HCV replication, although to a lesser extent than the Con1 replicon. In addition, the dependency of HCV on some host factors such as DHX9 and PABPN1 might be genotype specific since the decreased expression inhibited HCV 1b Con1 without affecting HCV 2a J6/JFH replication. Overall, our combined MS-RNAi approach identified previously reported and novel proteins as part of a YB-1-containing protein complex that contributes to the HCV replicative life cycle.
Fig 2.

Interaction and validation of YB-1 partner hits. (A, B) Huh7.5 cells stably expressing JFH-1 were transfected with the indicated Flag vectors. At 48 h posttransfection, cell extracts were prepared and subjected to immunoprecipitation (IP) with anti-Flag antibody-coupled resin. Resulting eluates, as well as cell extracts, were analyzed by Western blotting with anti-Flag, anti-NS3, anti-DDX3, anti-DDX6, anti-C1QBP, anti-IGF2BP2, anti-LARP1, anti-ILF2, anti-ILF3, anti-PABPN1, anti-DHX9, and anti-actin antibodies. Stau1 and IGF2BP3 MS hits were also detected in the Flag–YB-1 immunoprecipitate (data not shown). (C) Immunoprecipitation of endogenous proteins YB-1, DDX3, DDX6, IGF2BP2, LARP-1, ILF2, RHA, PABPN1, and ILF3, followed by Western blotting detection of endogenous YB-1 (Reciprocal Co-Ip) and Western blotting detection of other endogenous proteins of the YB-1 RNP complex, i.e., DDX3, DDX6, IGF2BP2, LARP-1, ILF2, RHA, PABPN1, and ILF3. Immunoprecipitation of FLAG protein and endogenous polymerase II (POLII) was used as a negative control. Ctrl −, negative control; Ab, antibody; arrow, specific protein; *, heavy chain of immunoglobulin; **, nonspecific protein.
Fig 3.

Assessment of the silencing efficiency of shRNAs and functional validation of YB-1 partner hits. (A to K) Two to 5 shRNAs per hit were selected from the TRC shRNA lentiviral library (Sigma-Aldrich) of the IRIC High-Throughput Screening Core Facility. ShRNA-expressing lentiviruses were produced in 293T cells and used for the transduction of Huh7, Huh7.5, or 293T cells. The following day, puromycin was added to the cells. Seventy-two hours later, cells were collected and lysed and their contents were analyzed for the indicated host factors by Western blotting with antibodies against YBX2 (A), C1QBP (B), DDX5 (C), DDX6 (D), DDX21 (E), DDX28 (F), DHX30 (G), hnRNPG-T (H), IGF2BP2 (I), PABPN1 and LARP1 (J), and ZC3HAV1 (K) indicated in Table S1 in the supplemental material. The shRNAs in bold are those that were selected for further experiments. (L) Huh7-Con1-Fluc cells were transduced with shRNA-expressing lentiviruses at an MOI of 5. Fluc and MTT assays were performed 3 and 4 days postransduction as readouts of HCV replication and cell viability, respectively. Huh7.5 cells were transduced with shRNA-expressing lentivirus and infected the day after with J6/JFH(p7-Rluc2A) reporter HCVs. The levels of HCV infection were monitored by measuring Rluc activity at 3 days postinfection. As a control for virus replication, cells were treated with 1 μM HCV protease inhibitor BILN 2061. Nontransduced cells control for potential off-target effects of the lentiviruses on cell viability and HCV replication. Statistically significant differences are indicated. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared to the shNT-expressing cell condition).
Host cofactors C1QBP, IGF2BP2, and LARP1 redistribute to LDs upon HCV infection.
To identify YB-1 interactors that regulate the late stages of the HCV life cycle, we next hypothesized that proteins would redistribute to the surface of core-containing LDs similarly to YB-1 (44) upon HCV infection. To test this, JFH-1-expressing cells were probed for HCV proteins, LDs, and YB-1 interactors and then examined by confocal laser scanning microscopy (Fig. 4). As a positive control, YB-1 was shown to redistribute to this organelle only in HCV-containing cells (Fig. 4A). We analyzed the cellular distribution of 19 YB-1 partner hits in normal and JFH-1-containing Huh7.5 cells. None of the hits tested colocalized with the LDs in uninfected cells (Fig. 4A to G, white arrows; data not shown), and most proteins, such as hnRNPG-T and DHX9, retained a normal distribution in JFH-1-expressing cells (Fig. 4G; data not shown) (44). Strikingly, among the validated YB-1 interactors, the three host cofactors C1QBP, LARP1, and IGF2BP2, as well as previously reported DDX3 and DDX6 (45, 46, 48, 49), all localized to the surface of core-containing LDs in JFH-1 cells, similarly to YB-1 (Fig. 4B to F, cells indicated by yellow arrows). In contrast, these proteins harbored an unaltered cellular distribution in uninfected cells (Fig. 4B to F, cells indicated by white arrows). In addition, triple colocalizations of eYFP-tagged YB-1, core, and IGF2BP2 or DDX3 were observed at the LDs (Fig. 5), strongly suggesting that HCV remodels the distribution of these host factors within a unique virus-host protein complex. We then confirmed the relocalization phenotype by a biochemical approach based on an LD flotation assay. In order to increase the size and number of LDs and hence facilitate their purification (D.L and L.C.C, unpublished results) (55), we treated cells with oleic acid in complex with BSA prior to cell homogenization and fractionation. As shown in Fig. 4H, we specifically detected core, NS3, YB-1, DDX3, DDX6, C1QBP, and IGF2BP2 in the LD fraction of JFH-1-expressing cells. Importantly, these proteins were not observed in preparations of uninfected cells, consistently with the confocal microscopy analysis. As a negative control, the confirmed YB-1 partner hnRNPG-T, whose distribution remains unaffected in JFH-1-expressing cells (Fig. 4G), was absent from the LD fraction. To a lesser extent, YB-1, DDX3, DDX6, and IGF2BP2 were also detected in the LD fraction of infected cells not treated with oleic acid (see Fig. 8A; data not shown). Moreover, HCV-induced redistribution of these host cofactors to LDs is dependent on core expression, similarly to YB-1 (44). Indeed, IGF2BP2 and DDX3 could not be detected in the LD fraction of cells that expressed the core-mutated clone JFH-1 Δ153-167 (L.C.C and D.L., unpublished results). Overall, our data acquired by confocal microscopy and confirmed by a cytoplasmic-fractionation approach strongly suggest that previously reported proteins YB-1, DDX3, and DDX6, as well as newly identified host cofactors C1QBP, LARP1, and IGF2BP2, all redistributed to the LDs as part of a unique virus-host YB-1 protein complex during JFH-1 infection.
Fig 4.

HCV induces the relocalization of DDX3, DDX6, IGF2BP2, LARP1, and C1QBP to core-containing LDs. (A to G) JFH-1-expressing Huh7.5 cells were fixed and probed with antibodies that recognize the indicated host and viral proteins. Nuclei and LDs were stained with Hoechst dye and LipidTOX, respectively. Unmerged and merged higher-magnification images of the boxed areas are shown. Yellow and white arrows indicate JFH-1-expressing cells and uninfected cells, respectively. (H) Huh7.5 cells were transfected with JFH-1 and the control vector. Five days later, cells were treated with 200 μM BSA-complexed oleic acid. Sixteen hours later, cells were collected and LDs were purified with a sucrose-based flotation assay. Aliquots of lysed cells and the LD fraction were analyzed by Western blotting with anti-ADRP, anti-core, anti-NS3, anti-YB-1, anti-DDX3, anti-DDX6, anti-IGF2BP2, anti-C1QBP, anti-hnRNG-T, and anti-actin antibodies.
Fig 5.

DDX3 and IGF2BP2 are corecruited with YB-1 to core-containing LDs. Huh7.5 cells, which stably express JFH-1, were transfected with eYFP-tagged YB-1, fixed 2 days later, and then probed with antibodies directed against core and DDX3 (A) or core and IGF2BP2 (B). Nuclei in panel A were stained with Hoechst. YB-1 was detected via the intrinsic fluorescence emitted by its eYFP tag.
Fig 8.

YB-1 knockdown decreases the association of NS3, DDX3, DDX6, and IGF2BP2 with LDs. Huh7.5 cells transduced with shRNA-expressing lentiviruses were transfected with a JFH-1-expressing plasmid. (A) Three days later, cells were collected and LDs were purified with a sucrose-based flotation assay. The LD fractions were analyzed by Western blotting with anti-ADRP, anti-core, anti-NS3, anti-DDX3, anti-DDX6, and anti-IGF2BP2 antibodies. (B) Cells were treated as in panel A, fixed, and probed with anti-NS3 and anti-core antibodies. Nuclei and LDs were stained with Hoechst dye and LipidTOX, respectively. Cells were analyzed by laser scanning microscopy. (C) Percentages of colocalization of NS3 or core with LDs were calculated for each condition with MetaMorph image analysis software and plotted.
YB-1, DDX6, C1QBP, IGF2BP2, and LARP1 restrain late steps in HCV particle production.
The assembly and release of HCV particles rely on the LD cell compartment (20–22), and thus, our data strongly suggest that redistributed host protein complexes regulate the production of infectious viral particles similarly to YB-1. To test this, virion-containing culture media were purified by ultracentrifugation from cells with the different LD-associated cofactors knocked down and then used to infect naive Huh7.5 cells. As a positive control, the knockdown of YB-1 reproducibly increased both the detection of extracellular core in purified particles and supernatant-associated infectivity (Fig. 6A and B), as reported in our previous study (44). Consistently, the overexpression of Flag-tagged YB-1 resulted in a marked decrease in extracellular infectivity (Fig. 7B, right side, extracellular). More importantly, when YB-1 partner DDX6, C1QBP, IGF2BP2, and LARP1 levels were individually decreased by gene silencing in JFH-1-transfected cells, we observed the same phenotype as with YB-1 with enhanced levels of released core and hence HCV particle production with increased supernatant-associated infectivity (Fig. 6). Unexpectedly, DDX3 knockdown did not significantly influence core release and supernatant infectivity, although this host factor redistributed to LDs upon HCV infection. Hence, our results identified DDX6, C1QBP, IGF2BP2, and LARP1 as novel host cofactors, which, similarly to YB-1, restrain the production of HCV infectious particles.
Fig 6.
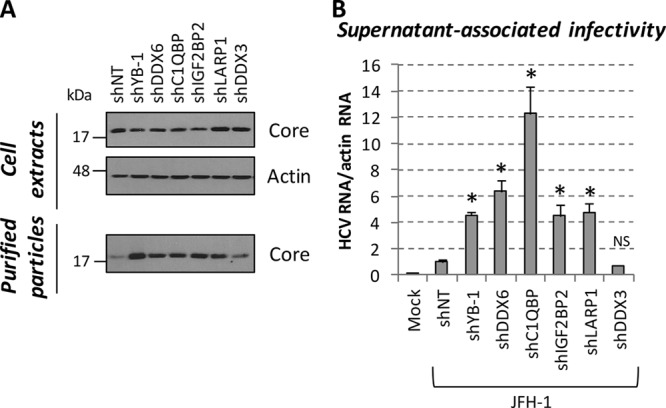
Knockdown of YB-1 partners significantly stimulates the production of infectious viral particles. Huh7.5 cells transduced with shRNA-expressing lentiviruses were transfected with a JFH-1-expressing plasmid. (A) Three days posttransfection, cells were collected and lysed while released viral particles were purified from culture medium. Cell extracts and lysed viruses were analyzed for their core contents by Western blotting. (B) Virus-containing culture media from panel A were used to infect naive Huh7.5 cells. Three days postinfection, cells were collected and analyzed for their HCV RNA content by qRT-PCR. HCV RNA levels were normalized to actin RNA content and arbitrarily set to 1 for the JFH-1-plus-shNT condition. Statistically significant differences are indicated. *, P < 0.05; NS, not significant (compared to the shNT-expressing cell condition).
Fig 7.
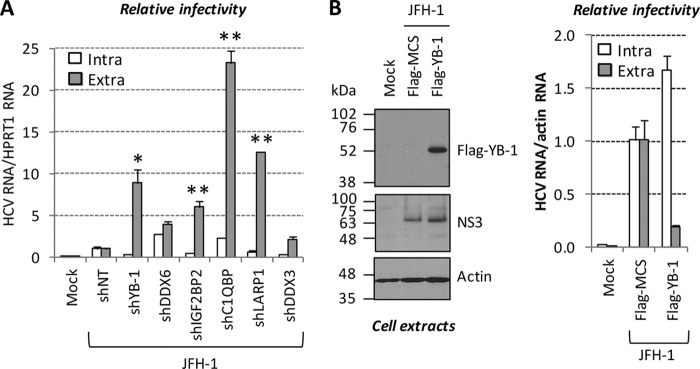
YB-1 RNP modulates the release and egress of HCV particles but not their assembly. Huh7.5 cells transduced with shRNA-expressing lentiviruses were transfected with a JFH-1-expressing plasmid. (A) Three days posttransfection, cells and culture media were collected. Intracellular viruses were prepared by freeze-thaw-based cell lysis. Virus-containing culture media (extracellular) and cell extracts (intracellular) were used to infect naive Huh7.5 cells. Three days postinfection, cells were collected and analyzed for their HCV RNA content by qRT-PCR. HCV RNA levels for intracellular and extracellular infectivities were normalized to HPRT1 RNA content and arbitrarily set to 1 for each JFH-1-plus-shNT condition. Statistically significant differences are indicated. *, P < 0.05; **, P < 0.01 (compared to the corresponding intracellular infectivity condition). (B) Huh7.5 cells were cotransfected with JFH-1- and Flag–YB-1-expressing plasmids. Three days later, an aliquot of transfected cells was analyzed for its Flag–YB-1 and HCV contents by Western blotting with anti-Flag and anti-NS3 antibodies (left side). Extracellular and intracellular viruses (prepared as in panel A) were used for infection. Infected cells were analyzed as in panel A, except that actin RNA was used for normalization (lower right side).
In order to determine if this phenotype is due to an increase in either assembly or egress, we analyzed both extracellular and intracellular infectivity in knockdown cells (Fig. 7). Intracellular infectivity reflects the amounts of assembled (but not released) viral particles inside the cell. If YB-1 partners influence virus formation, parallel upregulation of both extra- and intracellular infectivities is expected following gene knockdown. If, on the other hand, these host cofactors instead influence the egress of HCV but not particle assembly, only extracellular infectivity will be enhanced. As shown in Fig. 7A, YB-1 knockdown increased extracellular infectivity, while intracellular infectivity was reduced instead. This was attributable to the fact that YB-1 knockdown affects cytoplasmic vRNA levels because of its role in RNA replication. Consistently, the overexpression of Flag-tagged YB-1 decreased extracellular infectivity and slightly increased intracellular infectivity (Fig. 7B). Thus, both experiments strongly suggest that YB-1 negatively regulates particle production at the level of egress and/or release but not during virion assembly per se. As expected, the knockdown of DDX6, IGF2BP2, C1QBP, and LARP1 resulted in enhanced extracellular infectivity (Fig. 7A). Strikingly, intracellular infectivity was not increased by the gene silencing of IGF2BP2 and LARP1 and was weakly increased by C1QBP and DDX6 knockdown. Altogether, our results demonstrate that YB-1, DDX6, IGF2BP2, LARP1, and C1QBP negatively regulate the release of preformed viruses by infected cells and restrain HCV particle production.
YB-1-dependent localization of its partners and NS3 to LDs in HCV-infected cells.
We previously showed that YB-1 associates with the NS3 protein (44), which contributes to virus progeny (28, 40, 41; unpublished results). This strongly suggests that the YB-1-associated protein complex and the NS3 protein are functionally linked during the late stages of the HCV life cycle. Given that both NS3 and YB-1 are associated with LDs in a core-dependent manner during the late stages of the HCV life cycle (21, 44), we investigated a potential contribution of YB-1 to the cellular localization of NS3 and core. LDs were purified from JFH-1 cells previously transduced with control shNT- or YB-1 shRNA-expressing lentivirus. Western blot analysis (Fig. 8A) showed that both core and NS3 are specifically detected in the LD fraction from shNT/JFH-1 control cells. Strikingly, when YB-1 expression was decreased in knockdown cells, significantly lower NS3 levels were detected in the LD fraction, in contrast to core levels, which remained unaffected. Similar levels of the LD-associated control adipose differentiation-related protein (ADRP) were detected under the different conditions, attesting to the quality of the LD preparation. Consistently, immunofluorescence analysis of JFH-1-expressing Huh7.5 cells showed that YB-1 knockdown significantly decreased NS3 colocalization with LDs without, however, affecting core association with LDs (Fig. 8B and C). Strikingly, YB-1 knockdown also significantly decreased the levels of the IGF2BP2, DDX6, and DDX3 proteins associated with LDs (Fig. 8A). These results show that YB-1 is required for proper localization of NS3 and its partners to LDs and further pinpoint the key role of YB-1 as an organizer of this co-opted host machinery. Altogether, the data provide strong evidence of a YB-1 RNP host protein complex that is hijacked by HCV to regulate NS3-dependent stages of the HCV life cycle.
NS3 Q221L mutant virus overcomes the regulation of particle production by YB-1 RNP complex.
As YB-1 associates with NS3 and influences its localization, we explored a functional link between the YB-1-asssociated protein complex and NS3 during HCV particle production. We took advantage of the Q221L mutation in the NS3 helicase domain, which increases the production of infectious particles and can rescue defects in the late stages of the HCV life cycle (28, 41, 42). We investigated whether this mutant virus is able to evade the negative regulation imposed by the YB-1 protein complex that would account for its “overassembling” ability. The NS3 Q221L mutant virus was shown to produce normal amounts of viral protein (Fig. 9A, top) while releasing more infectious viral particles than wild-type JFH-1, with a 35-fold increase in extracellular infectivity (Fig. 9A, bottom). An infectious JFH-1 clone mutated in the C-terminal domain of NS2 (NS2 Q199R), which was also reported to “overassemble” (33), was included in our study and showed a 5-fold increase in extracellular infectivity (Fig. 9A). The clones defective in replication (NS5B GND) and assembly (Core Δ153-167) were used as negative controls that lack infectious-particle production (Fig. 9A). The supernatant-associated infectivities of the wild type and the NS2 Q199R and NS3 Q221L JFH-1 clones were then determined in cells by the knockdown of each protein of a YB-1 protein complex (DDX6, C1QBP, IGF2BP2, and LARP1). The expression of these different host factors was successfully reduced by gene silencing in JFH-1-expressing cells (Fig. 9B). In order to compare the effects of these protein knockdowns on the wild type and the NS3 Q221L and NS2 Q199R JFH-1 clones, the relative supernatant-associated infectivities of shNT-transduced control cells expressing each JFH-1 construct were used as individual references to normalize the HCV RNA/actin RNA ratio (Fig. 9C). The knockdown of each host factor increased the extracellular infectivity of wild-type JFH-1 as previously shown in Fig. 6 and 7 as a result of the enhanced release of HCV particles (Fig. 9C, black bar). Very strikingly, the yields of extracellular infectious particles produced from cells expressing NS3 Q221L JFH-1 were substantially less affected by the knockdown of each host cofactor and YB-1 compared to the increased infectivity of wild-type JFH-1. In contrast, the relative infectivity of NS2 Q199R JFH-1 was similar to that of the wild type in the various protein knockdown cells, strongly emphasizing that the negative-regulation phenotype is dependent on the function of NS3 in virion production. The resistance of the NS3 Q221L clone to YB-1 and associated host factors in knockdown cells was not due to a loss of YB-1–NS3 interaction, since both the wild-type and Q221L mutant NS3 proteins were detected to the same extent following immunoprecipitation with anti-YB-1 antibodies (Fig. 9D). Since the NS3 Q221L JFH-1 mutant circumvents YB-1-dependent negative regulation upon particle production, the data support a functional host machinery that controls the NS3-dependent late stages of the virus life cycle. The observations that YB-1, DDX6, C1QBP, IGF2BP2, and LARP1 interact together, redistribute at LDs, and induce similar phenotypes in various JFH-1 clones upon the knockdown of individual genes provide additional evidence that these cofactors regulate the HCV life cycle as part of the same protein complex.
Fig 9.

The virus production of the JFH-1 NS3 Q221L mutant is partially unresponsive to the knockdown of YB-1 RNP factors. (A) Huh7.5 cells were transfected with constructs encoding wild-type JFH-1 and mutant JFH-1 clones. Three days posttransfection, cells and released viral particles were collected. Cells were lysed and analyzed for their NS3, core, and actin contents by Western blotting (top). Virus-containing culture media from panel A were used to infect naive Huh7.5 cells. Three days postinfection, cells were collected and analyzed for their HCV RNA content by qRT-PCR (bottom). HCV RNA levels were normalized to actin RNA content and arbitrarily set to 1 for the wild-type (wt) JFH-1 condition. Statistically significant differences are indicated. *, P < 0.01; **, P < 0.0001 (compared to wild-type JFH-1). (B) Huh7.5 cells transduced with shRNA-expressing lentiviruses were transfected with a wild-type, NS2 Q199R, or NS3 Q221L JFH-1-expressing plasmid. Three days posttransfection, cells and culture media were collected. Cells expressing wild-type JFH-1 were lysed and analyzed for knockdown efficiency and core expression by Western blotting with anti-YB-1, anti-DDX6, anti-C1QBP, anti-IGF2BP2, anti-LARP1, anti-core, and anti-actin antibodies. (C) Virus-containing culture media were used to infect naive Huh7.5 cells. Three days postinfection, cells were collected and analyzed for their HCV RNA content by qRT-PCR. HCV RNA levels were normalized to actin RNA content. In order to separately compare the effects of the shRNAs on the JFH-1 clones, the relative HCV infectivity under shNT conditions for each JFH-1 construct was used as an individual reference and arbitrarily set to 1. (D) Huh7.5 cells were transfected with wild-type and NS3 Q221L JFH-1-expressing plasmids. Three days posttransfection, cell extracts were prepared and subjected to immunoprecipitation (IP) with antibodies directed against YB-1. Anti-hemagglutinin antibody immunoprecipitation was used as a control to monitor for nonspecific binding of lysate protein to the antibodies and/or the resin. Resulting eluates (right side) and cell extracts (left side) were analyzed by Western blotting with anti-YB-1, anti-NS3, anti-IGF2BP2, and anti-actin antibodies.
DISCUSSION
In this study, we combined an experimental approach based on MS and RNAi gene-silencing screening of YB-1 interactors to identify novel regulators of HCV infection. By first elucidating the YB-1 interactome in liver cells permissive to HCV infection, we found mostly RNA-binding proteins. This was not surprising, considering that YB-1 is frequently found in MS analyses of RNPs (63–68). We then challenged the YB-1 partner hits for a potential role in vRNA replication in HCV replicon-containing cells. Among the YB-1 partners that were identified as HCV cofactors by RNAi screening, several proteins were previously reported, such as ILF2, ILF3, DDX3, DDX6, hnRNPA1, and DHX9 (45, 46, 48, 49, 61, 62, 69, 70), suggesting that these proteins could be part of common functional virus-host RNP complexes. Hence, the data described in these studies could be reconciled with a proposed YB-1-containing RNP complex (44). Unfortunately, by using gene ontology analysis (DAVID) and interaction networking (Ingenuity Pathways Analysis) software resources, we were unable to highlight a clearly delineated cellular pathway that is co-opted by HCV, because most YB-1 interactors are directly or indirectly interconnected (data not shown). This lack of “networking resolution” probably originates from the very nature of RNA-binding proteins that are multifunctional. The function and RNA ligand specificity of a given RNA-binding protein are accepted to be often conferred by its RNP composition and its posttranslational modification pattern. Nevertheless, the identification of YB-1 partners in the proposed RNP complex could not point to any established role in DNA repair, splicing, transcription, or translation. Overall, this underscores the as-yet-unmet need for both conditional and spatiotemporal networking software that will take into account changes in protein composition and stoichiometry and in the cellular localization of interaction networks as a function of specific stimuli (virus infection, cytokine, pathogen-associated molecular pattern, neurotransmitter, etc.). Future studies based on qMS approaches will explore putative HCV-induced changes in YB-1 RNP component stoichiometry. Since the majority of the interactions were detected in uninfected Huh7.5 and 293T cells (Fig. 2C; L.L.C. and D.L., unpublished data), the putative proteomic remodeling of YB-1 RNP and its redistribution most likely occur on pre-existing complexes. Interestingly, our study has identified YB-1-associated cofactors that were previously described as important regulators of several RNA viruses. The reported viral functions of these cellular cofactors are compiled in Table S4 in the supplemental material. For instance, a role for YB-1 has been described upon infection with influenza virus and dengue virus, a member of the Flaviviridae family, like HCV (71, 72). Additionally, human immunodeficiency virus type 1 co-opts DDX3, DDX6, and hnRNP A1 during the late stages of its life cycle (73–75). Whether all of these viral functions involve interactions with YB-1 remains unclear, but from these studies emerges a potential panviral role for host macromolecular complexes composed of RNA-binding proteins, including YB-1 and others, such as DDXs, hnRNPs, and IGF2BPs. Future qMS and RNAi screening studies are required to confirm if RNA viruses besides HCV functionally co-opt a YB-1 RNP complex and whether its composition, stoichiometry, and cellular localization changes are panviral or rather specific to a given virus.
Most importantly, the cellular localization analysis of the validated YB-1 interactors revealed a selective set of proteins (DDX6, IGF2BP2, LARP1, and C1QBP) that redistributed to the surface of core-containing LDs upon HCV infection (Fig. 4). In fact, the distribution of the majority of the YB-1 partners studied is unaltered (Fig. 4G; data not shown), suggesting that they have a major role in vRNA replication and not in the regulation of particle production. However, we cannot exclude the possibility of the sporadic redistribution of some YB-1 partners below the detection limit of our assays. Unfortunately, the assay for quantification of particle production was not suitable for gene-silencing screening of the 71 YB-1 partner hits, and since the selection of cofactors was based on a Con1 RNA replication assay, we cannot rule out the possibility that we missed some YB-1 partners that modulate particle production but not replication. Nevertheless, the knockdown of LD-associated YB-1 partners confirmed that these host cofactors (with the exception of DDX3), similarly to YB-1, regulate the equilibrium between vRNA replication and the production of infectious particles. Thus, these proteins interact with YB-1; redistribute in a YB-1-dependent fashion, like NS3, to LDs during infection; and have the same functions during the HCV life cycle, strongly suggesting that they are part of the same virus host RNP complex hijacked from the replicative site by the assembling capsids.
The present study also provides important new information about the mechanism of action of the YB-1 RNP complex during the late stages of the HCV life cycle. First, core release experiments demonstrated that this host complex regulates the yield of released virus particles and apparently does not modulate their specific infectivity per se. Indeed, intracellular infectivity, which measures the cytoplasmic amounts of assembled and enveloped particles, did not depend on YB-1, LARP1, and IGF2BP2 expression levels. These data strongly suggest that the hijacked YB-1 RNP complex does not influence core multimerization and envelope covering but rather acts at a later stage during the egress and/or release of assembled viruses. This conclusion is further supported by the fact that core oligomerization is not modulated by YB-1, as demonstrated by comparable bioluminescence resonance energy transfer (BRET) signal of oligomeric core in YB-1 knockdown and control cells (L.C.C and D.L., unpublished data). Our data are also in line with recent findings providing evidence of a late role of several LD-associated proteins in HCV infection. As an example, core participates in virus egress from the cell by interacting with the endocytic pathway machinery (76), despite being associated mainly with LDs in infected cells. This might be achieved via the association of core with the μ subunit of clathrin adaptor protein complex 2, AP2M1, the knockdown of whose expression led to an accumulation of capsids at LDs and a loss of virus egress through the trans-Golgi network (77). Consistently, treatment of cells with the secretion inhibitor brefeldin A disrupts virus egress and release but not assembly, leading to the accumulation of NS3 and core at the LD surface (25, 41, 78). Moreover, our results are reminiscent of those obtained with host cofactor CHMP4B, which is localized at the LD surface in infected cells but has functions in particle production downstream of virus assembly (79). Hence, it is conceivable that assembling capsids hijack a limiting YB-1 RNP complex from the replicative site to control excessive downstream processes such as egress and release.
NS3 is involved in the transit of partially assembled capsids from the LD surface toward the secretory pathway compartment, a process visualized by the microtubule-dependent movement of core-containing puncta (41). Indeed, the Q221L mutation in the NS3 helicase domain restores defects in this process caused by mutations in the N-terminal domain of the NS2 protein (with functions in assembly distinct from those of its C terminus, which includes Q199). Considering that NS3 Q221L mutant virus release is less sensitive to the negative regulation of particle production imposed by YB-1 and newly identified host cofactors (Fig. 9), we propose that a hijacked YB-1 RNP host machinery restrains the NS3-dependent steps in particle production. This conclusion is also supported by the following evidence. First, YB-1 and NS3 interact with one another and colocalize at the LD surface (44). Second, NS3 association with LDs is diminished following YB-1 expression knockdown in virus-producing cells (Fig. 8), a result that is in contrast to that obtained by brefeldin A treatment. This suggests that under conditions where the YB-1 RNP complex is limiting at LDs, such as in a putative reduced exit from the vRNA replicative site, NS3-dependent egress of capsids is more rapid and particle production is more efficient. This model could be challenged in live-cell imaging experiments to assess NS3 cytoplasmic dynamics. As we could not demonstrate a reduced interaction between NS3 Q221L and YB-1 (see below), it remains unknown how this NS3 JFH-1 mutant is able to circumvent the YB-1 RNP complex-mediated negative regulation of particle production, which clearly could account for its “overassembling” properties. This mutation was shown to increase HCV intracellular infectivity, demonstrating a role for NS3 in the modulation of virus assembly (virus envelopment, core multimerization). However, we cannot rule out the possibility that NS3 has an additional function at a later time point in virus generation (i.e., egress and/or release), which would be under the control of a hijacked YB-1 RNP complex. The Q221L mutation is located at the surface of the helicase domain and does not affect NS3 enzymatic activities (28). Hence, it was proposed that NS3 would play its role in assembly through interaction with host cofactors, in support of YB-1 RNP-mediated regulation of particle production via an interaction with NS3. Unfortunately, this association was not significantly affected by the introduction of the NS3 Q221L mutation into JFH-1, as demonstrated by coimmunoprecipitation experiments (Fig. 9D), and hence, this cannot account for its phenotype. Additionally, BRET assays showed that YB-1 knockdown does not modulate NS3-core interaction (L.C.C and D.L., unpublished data), suggesting that YB-1 and core binding sites on the NS3 protein in the same hijacked RNP complex are potentially mutually exclusive. In addition to membranes, NS3 associates with tubulin similarly to YB-1, which also promotes tubulin nucleation in vitro (80–82). Hence, one can speculate that YB-1 controls the dynamics of the cytoskeleton in the vicinity of LDs, its association with NS3, and the microtubule-dependent exit of capsids from the LD compartment (41). Finally, YB-1 can be secreted by mesangial and monocytic cells in a nonclassical manner after an inflammatory challenge (83). Thus, HCV release might be regulated by the as-yet-unidentified cell machinery that controls YB-1 secretion. Additional studies are required to further explore the molecular mechanisms that underlie the negative regulation of NS3-dependent late steps in infectious particle production by a hijacked YB-1 RNP complex.
Interestingly, YB-1 and several of its identified interactors are overexpressed in hepatocellular carcinoma (often induced by HCV infection) and other cancer tissues (see Table S4 in the supplemental material). In particular, the high expression of proteins such as YB-1, C1QBP, and hnRNP A1 correlates with cancer aggressiveness and progression and is often associated with a poor survival prognosis (84–88). HCV can induce in vitro the epithelial mesenchymal transition of primary hepatocytes believed to be required for hepatocellular carcinoma metastasis (89). This important event in disease progression might be achieved by the co-opting of YB-1 RNP in chronic HCV infection. Indeed, YB-1 promotes the epithelial mesenchymal transition accompanied by enhanced metastatic potential in breast epithelial cells by stimulating the cap-independent translation of a specific set of genes (90).
In conclusion, by taking advantage of a combined approach based on qMS and RNAi screening, we have identified novel YB-1-associated cofactors of HCV replication. Further characterization of the selective set of YB-1 partners DDX6, IGF2BP2, LARP1, and C1QBP unraveled an RNP complex hijacked by the assembling capsids to the LD compartment, which restrains the NS3-dependent steps in virus release and/or egress. We propose that YB-1 and newly identified host cofactors, as part of a virus-host RNP complex, are critical for harmonizing the different stages of the HCV life cycle in time and space and might represent promising targets for the development of novel host-targeting antivirals.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jake Liang for plasmid pEF/JFH1-Rz/Neo; Takaji Wakita (National Institute of Infectious Diseases, Tokyo), Charles Rice (Rockefeller University), and Apath LLC for the use of JFH-1, for Huh7.5 cells, and for the J6-JFH-1(p7-Rluc2a) reporter virus; Ralf Bartenschlager (University of Heidelberg) for anti-NS5A antibodies and the luciferase-encoding Con1b subgenomic replicon; Darius Moradpour (University of Lausanne) for anti-NS5B antibodies; and Didier Trono (École Polytechnique Fédérale de Lausanne) for the lentiviral packaging plasmids. We are grateful to Marie-Ève Racine for construction of the eYFP–YB-1 expression plasmid; to the IRIC High-Throughput Screening Core Facility, Jean Duchaine, Karine Audette, and Julie Lafontaine for lentivirus production and transduction during RNAi screening; the IRIC Bio-Imagery Core Facility and Christian Charbonneau for confocal microscopy; and the IRIC Genomics Core Facility, Raphaëlle Lambert, Caroline Paradis, and Marianne Arteau for sequencing and real-time PCR. We thank Bridget Gagné and Margarita Zayas for critical reading of the manuscript.
This work was supported by an operating grant from the Canadian Institutes of Health Research (MOP-115058) to D.L. and L.C.-C. and by the Novartis/Canadian Liver Foundation Hepatology Research Chair to D.L. P.T. holds a Canada Research Chair in Proteomics and Bioanalytical Spectrometry. IRIC is supported in part by the Canadian Center of Excellence in Commercialization and Research (CECR), the Canadian Foundation for Innovation (CFI), and the Fonds de Recherche du Québec en Santé (FRQS).
Footnotes
Published ahead of print 28 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01474-13.
REFERENCES
- 1.Bartenschlager R, Cosset FL, Lohmann V. 2010. Hepatitis C virus replication cycle. J. Hepatol. 53:583–585 [DOI] [PubMed] [Google Scholar]
- 2.Tsai WL, Chung RT. 2010. Viral hepatocarcinogenesis. Oncogene 29:2309–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chatel-Chaix L, Baril M, Lamarre D. 2010. Hepatitis C virus NS3/4A protease inhibitors: a light at the end of the tunnel. Viruses 2:1752–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chatel-Chaix L, Germain MA, Gotte M, Lamarre D. 2012. Direct-acting and host-targeting HCV inhibitors: current and future directions. Curr. Opin. Virol. 2:588–598 [DOI] [PubMed] [Google Scholar]
- 5.Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, Serrano-Wu MH, Langley DR, Sun JH, O'Boyle DR, II, Lemm JA, Wang C, Knipe JO, Chien C, Colonno RJ, Grasela DM, Meanwell NA, Hamann LG. 2010. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465:96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Orum H. 2010. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327:198–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flisiak R, Feinman SV, Jablkowski M, Horban A, Kryczka W, Pawlowska M, Heathcote JE, Mazzella G, Vandelli C, Nicolas-Metral V, Grosgurin P, Liz JS, Scalfaro P, Porchet H, Crabbe R. 2009. The cyclophilin inhibitor Debio 025 combined with PEG IFNalpha2a significantly reduces viral load in treatment-naive hepatitis C patients. Hepatology 49:1460–1468 [DOI] [PubMed] [Google Scholar]
- 8.Rice CM. 2011. New insights into HCV replication: potential antiviral targets. Top. Antivir. Med. 19:117–120 [PMC free article] [PubMed] [Google Scholar]
- 9.Chatterji U, Bobardt M, Selvarajah S, Yang F, Tang H, Sakamoto N, Vuagniaux G, Parkinson T, Gallay P. 2009. The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. J. Biol. Chem. 284:16998–17005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Z, Yang F, Robotham JM, Tang H. 2009. Critical role of cyclophilin A and its prolyl-peptidyl isomerase activity in the structure and function of the hepatitis C virus replication complex. J. Virol. 83:6554–6565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berger KL, Cooper JD, Heaton NS, Yoon R, Oakland TE, Jordan TX, Mateu G, Grakoui A, Randall G. 2009. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl. Acad. Sci. U. S. A. 106:7577–7582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berger KL, Kelly SM, Jordan TX, Tartell MA, Randall G. 2011. Hepatitis C virus stimulates the phosphatidylinositol 4-kinase III alpha-dependent phosphatidylinositol 4-phosphate production that is essential for its replication. J. Virol. 85:8870–8883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaul A, Stauffer S, Berger C, Pertel T, Schmitt J, Kallis S, Zayas M, Lohmann V, Luban J, Bartenschlager R. 2009. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 5:e1000546. 10.1371/journal.ppat.1000546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim YS, Hwang SB. 2011. Hepatitis C virus NS5A protein interacts with phosphatidylinositol 4-kinase type IIIalpha and regulates viral propagation. J. Biol. Chem. 286:11290–11298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reiss S, Rebhan I, Backes P, Romero-Brey I, Erfle H, Matula P, Kaderali L, Poenisch M, Blankenburg H, Hiet MS, Longerich T, Diehl S, Ramirez F, Balla T, Rohr K, Kaul A, Buhler S, Pepperkok R, Lengauer T, Albrecht M, Eils R, Schirmacher P, Lohmann V, Bartenschlager R. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9:32–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tai AW, Salloum S. 2011. The role of the phosphatidylinositol 4-kinase PI4KA in hepatitis C virus-induced host membrane rearrangement. PLoS One 6:e26300. 10.1371/journal.pone.0026300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bianco A, Reghellin V, Donnici L, Fenu S, Alvarez R, Baruffa C, Peri F, Pagani M, Abrignani S, Neddermann P, De Francesco R. 2012. Metabolism of phosphatidylinositol 4-kinase IIIalpha-dependent PI4P is subverted by HCV and is targeted by a 4-anilino quinazoline with antiviral activity. PLoS Pathog. 8:e1002576. 10.1371/journal.ppat.1002576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626 [DOI] [PubMed] [Google Scholar]
- 19.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boulant S, Targett-Adams P, McLauchlan J. 2007. Disrupting the association of hepatitis C virus core protein with lipid droplets correlates with a loss in production of infectious virus. J. Gen. Virol. 88:2204–2213 [DOI] [PubMed] [Google Scholar]
- 21.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9:1089–1097 [DOI] [PubMed] [Google Scholar]
- 22.Shavinskaya A, Boulant S, Penin F, McLauchlan J, Bartenschlager R. 2007. The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly. J. Biol. Chem. 282:37158–37169 [DOI] [PubMed] [Google Scholar]
- 23.Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M, Jr, Ye J. 2007. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. U. S. A. 104:5848–5853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herker E, Harris C, Hernandez C, Carpentier A, Kaehlcke K, Rosenberg AR, Farese RV, Jr, Ott M. 2010. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat. Med. 16:1295–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gastaminza P, Cheng G, Wieland S, Zhong J, Liao W, Chisari FV. 2008. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 82:2120–2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang KS, Jiang J, Cai Z, Luo G. 2007. Human apolipoprotein E is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 81:13783–13793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Backes P, Quinkert D, Reiss S, Binder M, Zayas M, Rescher U, Gerke V, Bartenschlager R, Lohmann V. 2010. Role of annexin A2 in the production of infectious hepatitis C virus particles. J. Virol. 84:5775–5789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma Y, Yates J, Liang Y, Lemon SM, Yi M. 2008. NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J. Virol. 82:7624–7639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Appel N, Schaller T, Penin F, Bartenschlager R. 2006. From structure to function: new insights into hepatitis C virus RNA replication. J. Biol. Chem. 281:9833–9836 [DOI] [PubMed] [Google Scholar]
- 30.Masaki T, Suzuki R, Murakami K, Aizaki H, Ishii K, Murayama A, Date T, Matsuura Y, Miyamura T, Wakita T, Suzuki T. 2008. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 82:7964–7976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tellinghuisen TL, Foss KL, Treadaway J. 2008. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 4:e1000032. 10.1371/journal.ppat.1000032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones DM, Patel AH, Targett-Adams P, McLauchlan J. 2009. The hepatitis C virus NS4B protein can trans-complement viral RNA replication and modulates production of infectious virus. J. Virol. 83:2163–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Russell RS, Meunier JC, Takikawa S, Faulk K, Engle RE, Bukh J, Purcell RH, Emerson SU. 2008. Advantages of a single-cycle production assay to study cell culture-adaptive mutations of hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 105:4370–4375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yi M, Ma Y, Yates J, Lemon SM. 2009. Trans-complementation of an NS2 defect in a late step in hepatitis C virus (HCV) particle assembly and maturation. PLoS Pathog. 5:e1000403. 10.1371/journal.ppat.1000403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stapleford KA, Lindenbach BD. 2011. Hepatitis C virus NS2 coordinates virus particle assembly through physical interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J. Virol. 85:1706–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jirasko V, Montserret R, Lee JY, Gouttenoire J, Moradpour D, Penin F, Bartenschlager R. 2010. Structural and functional studies of nonstructural protein 2 of the hepatitis C virus reveal its key role as organizer of virion assembly. PLoS Pathog. 6:e1001233. 10.1371/journal.ppat.1001233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma Y, Anantpadma M, Timpe JM, Shanmugam S, Singh SM, Lemon SM, Yi M. 2011. Hepatitis C virus NS2 protein serves as a scaffold for virus assembly by interacting with both structural and nonstructural proteins. J. Virol. 85:86–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coller KE, Heaton NS, Berger KL, Cooper JD, Saunders JL, Randall G. 2012. Molecular determinants and dynamics of hepatitis C virus secretion. PLoS Pathog. 8:e1002466. 10.1371/journal.ppat.1002466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones CT, Murray CL, Eastman DK, Tassello J, Rice CM. 2007. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 81:8374–8383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones DM, Atoom AM, Zhang X, Kottilil S, Russell RS. 2011. A genetic interaction between the core and NS3 proteins of hepatitis C virus is essential for production of infectious virus. J. Virol. 85:12351–12361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Counihan NA, Rawlinson SM, Lindenbach BD. 2011. Trafficking of hepatitis C virus core protein during virus particle assembly. PLoS Pathog. 7:e1002302. 10.1371/journal.ppat.1002302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phan T, Kohlway A, Dimberu P, Pyle AM, Lindenbach BD. 2011. The acidic domain of hepatitis C virus NS4A contributes to RNA replication and virus particle assembly. J. Virol. 85:1193–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mousseau G, Kota S, Takahashi V, Frick DN, Strosberg AD. 2011. Dimerization-driven interaction of hepatitis C virus core protein with NS3 helicase. J. Gen. Virol. 92:101–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chatel-Chaix L, Melancon P, Racine ME, Baril M, Lamarre D. 2011. Y-box-binding protein 1 interacts with hepatitis C virus NS3/4A and influences the equilibrium between viral RNA replication and infectious particle production. J. Virol. 85:11022–11037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scheller N, Mina LB, Galao RP, Chari A, Gimenez-Barcons M, Noueiry A, Fischer U, Meyerhans A, Diez J. 2009. Translation and replication of hepatitis C virus genomic RNA depends on ancient cellular proteins that control mRNA fates. Proc. Natl. Acad. Sci. U. S. A. 106:13517–13522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jangra RK, Yi M, Lemon SM. 2010. DDX6 (Rck/p54) is required for efficient hepatitis C virus replication but not for internal ribosome entry site-directed translation. J. Virol. 84:6810–6824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ariumi Y, Kuroki M, Kushima Y, Osugi K, Hijikata M, Maki M, Ikeda M, Kato N. 2011. Hepatitis C virus hijacks P-body and stress granule components around lipid droplets. J. Virol. 85:6882–6892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Angus AG, Dalrymple D, Boulant S, McGivern DR, Clayton RF, Scott MJ, Adair R, Graham S, Owsianka AM, Targett-Adams P, Li K, Wakita T, McLauchlan J, Lemon SM, Patel AH. 2010. Requirement of cellular DDX3 for hepatitis C virus replication is unrelated to its interaction with the viral core protein. J. Gen. Virol. 91:122–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ariumi Y, Kuroki M, Abe K, Dansako H, Ikeda M, Wakita T, Kato N. 2007. DDX3 DEAD-box RNA helicase is required for hepatitis C virus RNA replication. J. Virol. 81:13922–13926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kato T, Matsumura T, Heller T, Saito S, Sapp RK, Murthy K, Wakita T, Liang TJ. 2007. Production of infectious hepatitis C virus of various genotypes in cell cultures. J. Virol. 81:4405–4411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baril M, Racine ME, Penin F, Lamarre D. 2009. MAVS dimer is a crucial signaling component of innate immunity and the target of hepatitis C virus NS3/4A protease. J. Virol. 83:1299–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113 [DOI] [PubMed] [Google Scholar]
- 53.Lamarre D, Anderson PC, Bailey M, Beaulieu P, Bolger G, Bonneau P, Bos M, Cameron DR, Cartier M, Cordingley MG, Faucher AM, Goudreau N, Kawai SH, Kukolj G, Lagace L, LaPlante SR, Narjes H, Poupart MA, Rancourt J, Sentjens RE, St George R, Simoneau B, Steinmann G, Thibeault D, Tsantrizos YS, Weldon SM, Yong CL, Llinas-Brunet M. 2003. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 426:186–189 [DOI] [PubMed] [Google Scholar]
- 54.Kato T, Date T, Murayama A, Morikawa K, Akazawa D, Wakita T. 2006. Cell culture and infection system for hepatitis C virus. Nat. Protoc. 1:2334–2339 [DOI] [PubMed] [Google Scholar]
- 55.Brasaemle DL, Wolins NE. 2006. Isolation of lipid droplets from cells by density gradient centrifugation. Curr. Protoc. Cell Biol. Chapter 3:Unit 3.15. [DOI] [PubMed] [Google Scholar]
- 56.Garaigorta U, Heim MH, Boyd B, Wieland S, Chisari FV. 2012. Hepatitis C virus (HCV) induces formation of stress granules whose proteins regulate HCV RNA replication and virus assembly and egress. J. Virol. 86:11043–11056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yi Z, Fang C, Pan T, Wang J, Yang P, Yuan Z. 2006. Subproteomic study of hepatitis C virus replicon reveals Ras-GTPase-activating protein binding protein 1 as potential HCV RC component. Biochem. Biophys. Res. Commun. 350:174–178 [DOI] [PubMed] [Google Scholar]
- 58.Chatel-Chaix L, Abrahamyan L, Frechina C, Mouland AJ, DesGroseillers L. 2007. The host protein Staufen1 participates in human immunodeficiency virus type 1 assembly in live cells by influencing pr55Gag multimerization. J. Virol. 81:6216–6230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Lucas S, Peredo J, Marion RM, Sanchez C, Ortin J. 2010. Human Staufen1 protein interacts with influenza virus ribonucleoproteins and is required for efficient virus multiplication. J. Virol. 84:7603–7612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu S, Kuo W, Yang W, Liu W, Gibson GA, Dorko K, Watkins SC, Strom SC, Wang T. 2010. The second extracellular loop dictates Occludin-mediated HCV entry. Virology 407:160–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goh PY, Tan YJ, Lim SP, Tan YH, Lim SG, Fuller-Pace F, Hong W. 2004. Cellular RNA helicase p68 relocalization and interaction with the hepatitis C virus (HCV) NS5B protein and the potential role of p68 in HCV RNA replication. J. Virol. 78:5288–5298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Isken O, Baroth M, Grassmann CW, Weinlich S, Ostareck DH, Ostareck-Lederer A, Behrens SE. 2007. Nuclear factors are involved in hepatitis C virus RNA replication. RNA 13:1675–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kozak SL, Marin M, Rose KM, Bystrom C, Kabat D. 2006. The anti-HIV-1 editing enzyme APOBEC3G binds HIV-1 RNA and messenger RNAs that shuttle between polysomes and stress granules. J. Biol. Chem. 281:29105–29119 [DOI] [PubMed] [Google Scholar]
- 64.Onishi H, Kino Y, Morita T, Futai E, Sasagawa N, Ishiura S. 2008. MBNL1 associates with YB-1 in cytoplasmic stress granules. J. Neurosci. Res. 86:1994–2002 [DOI] [PubMed] [Google Scholar]
- 65.Vashist S, Urena L, Chaudhry Y, Goodfellow I. 2012. Identification of RNA-protein interaction networks involved in the norovirus life cycle. J. Virol. 86:11977–11990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jønson L, Vikesaa J, Krogh A, Nielsen LK, Hansen T, Borup R, Johnsen AH, Christiansen J, Nielsen FC. 2007. Molecular composition of IMP1 ribonucleoprotein granules. Mol. Cell. Proteomics 6:798–811 [DOI] [PubMed] [Google Scholar]
- 67.Maher-Laporte M, Berthiaume F, Moreau M, Julien LA, Lapointe G, Mourez M, DesGroseillers L. 2010. Molecular composition of staufen2-containing ribonucleoproteins in embryonic rat brain. PLoS One 5:e11350. 10.1371/journal.pone.0011350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Höck J, Weinmann L, Ender C, Rüdel S, Kremmer E, Raabe M, Urlaub H, Meister G. 2007. Proteomic and functional analysis of Argonaute-containing mRNA-protein complexes in human cells. EMBO Rep. 8:1052–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim CS, Seol SK, Song OK, Park JH, Jang SK. 2007. An RNA-binding protein, hnRNP A1, and a scaffold protein, septin 6, facilitate hepatitis C virus replication. J. Virol. 81:3852–3865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Upadhyay A, Dixit U, Manvar D, Chaturvedi N, Pandey VN. 2013. Affinity capture and identification of host cell factors associated with hepatitis C virus (+) strand subgenomic RNA. Mol. Cell. Proteomics 12:1539–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kawaguchi A, Matsumoto K, Nagata K. 2012. YB-1 functions as a porter to lead influenza virus ribonucleoprotein complexes to microtubules. J. Virol. 86:11086–11095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Paranjape SM, Harris E. 2007. Y box-binding protein-1 binds to the dengue virus 3′-untranslated region and mediates antiviral effects. J. Biol. Chem. 282:30497–30508 [DOI] [PubMed] [Google Scholar]
- 73.Reed JC, Molter B, Geary CD, McNevin J, McElrath J, Giri S, Klein KC, Lingappa JR. 2012. HIV-1 Gag co-opts a cellular complex containing DDX6, a helicase that facilitates capsid assembly. J. Cell Biol. 198:439–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Monette A, Ajamian L, Lopez-Lastra M, Mouland AJ. 2009. Human immunodeficiency virus type 1 (HIV-1) induces the cytoplasmic retention of heterogeneous nuclear ribonucleoprotein A1 by disrupting nuclear import: implications for HIV-1 gene expression. J. Biol. Chem. 284:31350–31362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yedavalli VS, Neuveut C, Chi YH, Kleiman L, Jeang KT. 2004. Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell 119:381–392 [DOI] [PubMed] [Google Scholar]
- 76.Lai CK, Jeng KS, Machida K, Lai MM. 2010. Hepatitis C virus egress and release depend on endosomal trafficking of core protein. J. Virol. 84:11590–11598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Neveu G, Barouch-Bentov R, Ziv-Av A, Gerber D, Jacob Y, Einav S. 2012. Identification and targeting of an interaction between a tyrosine motif within hepatitis C virus core protein and AP2M1 essential for viral assembly. PLoS Pathog. 8:e1002845. 10.1371/journal.ppat.1002845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Matto M, Sklan EH, David N, Melamed-Book N, Casanova JE, Glenn JS, Aroeti B. 2011. Role for ADP ribosylation factor 1 in the regulation of hepatitis C virus replication. J. Virol. 85:946–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ariumi Y, Kuroki M, Maki M, Ikeda M, Dansako H, Wakita T, Kato N. 2011. The ESCRT system is required for hepatitis C virus production. PLoS One 6:e14517. 10.1371/journal.pone.0014517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chernov KG, Curmi PA, Hamon L, Mechulam A, Ovchinnikov LP, Pastre D. 2008. Atomic force microscopy reveals binding of mRNA to microtubules mediated by two major mRNP proteins YB-1 and PABP. FEBS Lett. 582:2875–2881 [DOI] [PubMed] [Google Scholar]
- 81.Chernov KG, Mechulam A, Popova NV, Pastre D, Nadezhdina ES, Skabkina OV, Shanina NA, Vasiliev VD, Tarrade A, Melki J, Joshi V, Baconnais S, Toma F, Ovchinnikov LP, Curmi PA. 2008. YB-1 promotes microtubule assembly in vitro through interaction with tubulin and microtubules. BMC Biochem. 9:23. 10.1186/1471-2091-9-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lai CK, Jeng KS, Machida K, Lai MM. 2008. Association of hepatitis C virus replication complexes with microtubules and actin filaments is dependent on the interaction of NS3 and NS5A. J. Virol. 82:8838–8848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Frye BC, Halfter S, Djudjaj S, Muehlenberg P, Weber S, Raffetseder U, En-Nia A, Knott H, Baron JM, Dooley S, Bernhagen J, Mertens PR. 2009. Y-box protein-1 is actively secreted through a non-classical pathway and acts as an extracellular mitogen. EMBO Rep. 10:783–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen YB, Jiang CT, Zhang GQ, Wang JS, Pang D. 2009. Increased expression of hyaluronic acid binding protein 1 is correlated with poor prognosis in patients with breast cancer. J. Surg. Oncol. 100:382–386 [DOI] [PubMed] [Google Scholar]
- 85.Yasen M, Kajino K, Kano S, Tobita H, Yamamoto J, Uchiumi T, Kon S, Maeda M, Obulhasim G, Arii S, Hino O. 2005. The up-regulation of Y-box binding proteins (DNA binding protein A and Y-box binding protein-1) as prognostic markers of hepatocellular carcinoma. Clin. Cancer Res. 11:7354–7361 [DOI] [PubMed] [Google Scholar]
- 86.Zhou ZJ, Dai Z, Zhou SL, Fu XT, Zhao YM, Shi YH, Zhou J, Fan J. 2013. Overexpression of HnRNP A1 promotes tumor invasion through regulating CD44v6 and indicates poor prognosis for hepatocellular carcinoma. Int. J. Cancer 132:1080–1089 [DOI] [PubMed] [Google Scholar]
- 87.Maciejczyk A, Szelachowska J, Ekiert M, Matkowski R, Halon A, Lage H, Surowiak P. 2012. Elevated nuclear YB1 expression is associated with poor survival of patients with early breast cancer. Anticancer Res. 32:3177–3184 [PubMed] [Google Scholar]
- 88.Wu Y, Yamada S, Izumi H, Li Z, Shimajiri S, Wang KY, Liu YP, Kohno K, Sasaguri Y. 2012. Strong YB-1 expression is associated with liver metastasis progression and predicts shorter disease-free survival in advanced gastric cancer. J. Surg. Oncol. 105:724–730 [DOI] [PubMed] [Google Scholar]
- 89.Bose SK, Meyer K, Di Bisceglie AM, Ray RB, Ray R. 2012. Hepatitis C virus induces epithelial-mesenchymal transition in primary human hepatocytes. J. Virol. 86:13621–13628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Evdokimova V, Tognon C, Ng T, Ruzanov P, Melnyk N, Fink D, Sorokin A, Ovchinnikov LP, Davicioni E, Triche TJ, Sorensen PH. 2009. Translational activation of snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial-mesenchymal transition. Cancer Cell 15:402–415 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.