

Abstract
All bacteria use the conserved Sec pathway to transport proteins across the cytoplasmic membrane, with the SecA ATPase playing a central role in the process. Mycobacteria are part of a small group of bacteria that have two SecA proteins: the canonical SecA (SecA1) and a second, specialized SecA (SecA2). The SecA2-dependent pathway exports a small subset of proteins and is required for Mycobacterium tuberculosis virulence. The mechanism by which SecA2 drives export of proteins across the cytoplasmic membrane remains poorly understood. Here we performed suppressor analysis on a dominant negative secA2 mutant (secA2 K129R) of the model mycobacterium Mycobacterium smegmatis to better understand the pathway used by SecA2 to export proteins. Two extragenic suppressor mutations were identified as mapping to the promoter region of secY, which encodes the central component of the canonical Sec export channel. These suppressor mutations increased secY expression, and this effect was sufficient to alleviate the secA2 K129R phenotype. We also discovered that the level of SecY protein was greatly diminished in the secA2 K129R mutant, but at least partially restored in the suppressors. Furthermore, the level of SecY in a suppressor strongly correlated with the degree of suppression. Our findings reveal a detrimental effect of SecA2 K129R on SecY, arguing for an integrated system in which SecA2 works with SecY and the canonical Sec translocase to export proteins.
INTRODUCTION
Bacteria use both conserved and specialized protein export systems to deliver proteins to the bacterial cell surface and to the extracellular environment. These exported proteins are important for critical cell functions, like nutrient acquisition, cell structure and, in the case of pathogens, virulence. All bacteria possess the conserved Sec protein export system, which performs the bulk of “housekeeping” protein export. More recently, it was discovered that mycobacteria and some Gram-positive bacteria possess a second, specialized Sec export system (1–7). This specialized protein export system is only partially understood, but in pathogens it is often required for virulence.
In the housekeeping Sec export system, the motor protein SecA plays a central role in exporting unfolded proteins through a membrane channel comprised of the integral membrane proteins SecY, SecE, and SecG, where SecY is the major component of the translocon (8). SecA is a cytoplasmic ATPase that provides energy for protein export through successive rounds of ATP binding and hydrolysis (9, 10). Proteins exported by the Sec system possess an N-terminal signal peptide, which is cleaved from the mature protein following export. Because many important proteins rely on the Sec system for export, many Sec proteins are essential, including SecA and SecY (1, 11). Bacteria containing a second, specialized Sec export system are characterized by the presence of two SecA proteins with distinct functions. The second SecA protein, called SecA2, also possesses ATPase activity (12, 13) and is generally nonessential (14).
In mycobacteria, SecA2 is responsible for the export of a small subset of proteins (15, 16) and is required for virulence of the pathogen Mycobacterium tuberculosis (16). The model organism Mycobacterium smegmatis also possesses a SecA2 system, which is functionally conserved with that of M. tuberculosis (1, 17). Studies with M. smegmatis have shown that SecA2-dependent proteins can contain N-terminal signal peptides indistinguishable from classical Sec signal peptides and that it is the mature domain of the protein that determines the requirement for SecA2 (18). However, the mechanism of SecA2-dependent protein export in mycobacteria is unknown. Some SecA2 export systems, referred to as SecA2-SecY2 systems, include a second distinct SecY protein (named SecY2) that is required for export of SecA2-dependent proteins. In these systems, SecY2 is presumed to function as the channel for protein translocation across the membrane (3, 4, 14). In mycobacteria, there is no SecY2 protein evident, making it unclear how SecA2-dependent proteins are exported in this system. One possibility is that the mycobacterial SecA2 system exports proteins through the SecYEG channel, sharing this channel with the housekeeping Sec export system; however, this model has not been proven.
To increase our understanding of SecA2 export and to identify additional components of the SecA2 system, we carried out a suppressor analysis of a secA2 mutant of M. smegmatis. For this purpose, we used a secA2 K129R mutant that encodes for a SecA2 protein with an amino acid substitution (K129R) in the Walker box, which is an amino acid motif important for ATP binding and hydrolysis. We previously demonstrated that this K129R substitution disrupts the ATPase activity of SecA2, rendering it nonfunctional (12, 17). In addition, SecA2 K129R has a dominant negative effect on wild-type SecA2 (17). This suggests that SecA2 K129R is still able to interact with its normal binding partners within the cell, but because it is nonfunctional, it interferes with the functions of these partners. Further, SecA2 K129R must disrupt an important process in the cell, because the phenotypes of the secA2 K129R allele are worse than those of a secA2 deletion mutant. These properties make the secA2 K129R allele a good starting point for suppressor analysis.
In the present study, we characterized two extragenic suppressor mutations of secA2 K129R, both of which are located in the promoter region of the only secY gene of mycobacteria. We also discovered that SecY protein levels are drastically reduced in the secA2 K129R mutant and that these suppressor mutations increase secY expression to partially restore SecY protein levels and suppress secA2 K129R phenotypes. Finally, we found that SecY levels were increased relative to the secA2 K129R strain in six additional extragenic suppressors. Taken together, these findings suggest that SecA2 K129R disrupts the housekeeping Sec export system, causing SecY degradation and thus the severe phenotypes of the secA2 K129R mutant. The data presented here argue for SecA2 working in concert with SecY and the housekeeping Sec pathway to export SecA2-dependent proteins.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
M. smegmatis strains used in this study are described in Table 1 and were grown at 37°C or 30°C in Middlebrook 7H9/7H10 or Mueller-Hinton medium. The secA2 K129R strain (NR178) has a severe growth defect on Mueller-Hinton agar (17) and was therefore propagated using Middlebrook 7H9/7H10 medium. To limit acquisition of suppressors when working with the secA2 K129R strain, starter cultures were generally grown at 30°C, but the actual experiments were performed at 37°C. Media were supplemented with 0.5% glycerol plus 0.2% glucose (7H9/7H10 medium only) and 0.1% Tween 80 (all media). The antibiotics kanamycin (20 μg/ml) and hygromycin B (50 μg/ml) were added as needed. When required, plasmids were introduced into M. smegmatis strains by electroporation (19). Escherichia coli strains were grown at 37°C in Luria-Bertani broth or on Luria-Bertani agar. The antibiotics kanamycin (40 μg/ml) and hygromycin B (150 μg/ml) were added as needed.
Table 1.
Mycobacterium smegmatis strains used in this study
| M. smegmatis strain | Description | Source |
|---|---|---|
| mc2155 | Wild type | 38 |
| NR116 | ΔsecA2 | 17 |
| NR158 | mc2155 + pMV306.kan, wild type + empty plasmid | 17 |
| NR160 | NR116 + pMV306.kan, ΔsecA2 + empty plasmid | 17 |
| NR172 | NR116 + pYA810, ΔsecA2 + secA2 complementing plasmid | 17 |
| NR178 | NR116 + pNR25, ΔsecA2 + secA2 K129R plasmid | 17 |
| NR236 | NR178 with extragenic suppressor mutation 4S | This work |
| NR151 | NR178 with extragenic suppressor mutation 24S | This work |
| NR154 | NR178 with extragenic suppressor mutation 29S | This work |
| NR155 | NR178 with extragenic suppressor mutation 33S | This work |
| NR156 | NR178 with extragenic suppressor mutation 33B | This work |
| NR230 | NR178 with extragenic suppressor mutation 1S | This work |
| NR234 | NR178 with extragenic suppressor mutation 3S | This work |
| NR248 | NR178 with extragenic suppressor mutation 10S | This work |
Suppressor collection.
The secA2 K129R strain encodes a SecA2 protein in which the lysine at position 129 is replaced with an arginine (amino acid numbering based on NCBI GenBank accession number AF287049 [1]). Suppressors of secA2 K129R were isolated by plating 38 independently grown cultures of the secA2 K129R strain onto Mueller-Hinton agar at 37°C. Spontaneous suppressors (i.e., colonies that grew on Mueller-Hinton agar) were obtained, and one small (S) and one large (B) suppressor colony was chosen from each independent culture.
Plasmid construction.
Plasmids used in this study are described in Table 2, and the primers are described in Table S1 of the supplemental material. In all cases, newly constructed plasmids were sequenced. To create secY′-′lacZ fusion plasmids, a region upstream of secY was PCR amplified from M. smegmatis genomic DNA of strains NR178 and NR236 (suppressor 4S) to generate wild-type and 4S secY promoter sequences, respectively. Each PCR product contained 379 bp upstream of secY along with 34 bp of secY coding sequence and engineered EcoRI restriction sites; the products were each cloned into pCR2.1-TOPO (Invitrogen), yielding plasmids pLL5 and pLL6. The EcoRI secY promoter fragments were cut from pLL5 and pLL6 and ligated into EcoRI-digested pCV125, yielding plasmids pLL11 and pLL8, which contained secY′-′lacZ translational fusions. pLL11 was mutated by site-directed mutagenesis (Stratagene QuikChange II) to recreate the mutation found upstream of secY in suppressor NR151 (24S), yielding plasmid pLL15.
Table 2.
Plasmids used in this study
| Plasmid | Genotype | Description | Source |
|---|---|---|---|
| pMV306.kan | aph int attPL5 ColE1 | Single-copy mycobacterial shuttle vector, integrates in mycobacteriophage L5 attB site | 39 |
| pMV361.kan | aph Phsp60 int attPL5 ColE1 | Single-copy mycobacterial shuttle vector with hsp60 promoter, integrates in mycobacteriophage L5 attB site | 39 |
| pYA810 | aph Phsp60-secA2 int attPL5 ColE1 | M. smegmatis secA2 in pMV361.kan | 15 |
| pNR25 | aph Phsp60-secA2K129R int attPL5 ColE1 | M. smegmatis secA2 K129R in pMV361.kan | 17 |
| pCR2.1-TOPO | aph bla ColE1 | TOPO TA cloning plasmid | Invitrogen |
| pLL5 | aph bla PsecY-secY′ ColE1 | M. smegmatis secY promoter and 34 bp of secY gene in pCR2.1-TOPO | This work |
| pLL6 | aph bla PsecY(4S)-secY′ ColE1 | NR236 (4S) secY promoter and 34 bp of secY gene in pCR2.1-TOPO | This work |
| pCV125 | hyg ′lacZ oriM ColE1 | Multicopy mycobacterial shuttle vector with promoterless lacZ gene | MedImmune |
| pLL11 | hyg PsecY-secY′-′lacZ oriM ColE1 | M. smegmatis secY translationally fused to lacZ in pCV125 | This work |
| pLL8 | hyg PsecY(4S)-secY′-′lacZ oriM ColE1 | NR236 (4S) secY translationally fused to lacZ in pCV125 | This work |
| pLL15 | hyg PsecY(24S)-secY′-′lacZ oriM ColE1 | NR151 (24S) secY translationally fused to lacZ in pCV125 | This work |
| pLL17 | aph bla secY ColE1 | M. smegmatis secY in pCR2.1-TOPO | This work |
| pLL19 | aph bla secY(4S) ColE1 | NR236 (4S) secY in pCR2.1-TOPO | This work |
| pYUB2063 | hyg bla int attPTweety ColE1 cosλ | Single-copy mycobacterial shuttle vector, integrates in mycobacteriophage Tweety attB site | W. R. Jacobs, Albert Einstein College of Medicine |
| pLL2 | hyg bla int attPTweety ColE1 | pYUB2063 with 3,299-bp PciI fragment removed, single-copy mycobacterial shuttle vector, integrates in mycobacteriophage Tweety attB site | This work |
| pLL21 | hyg bla secY int attPTweety ColE1 | M. smegmatis secY in pLL2 | This work |
| pLL23 | hyg bla secY(4S) int attPTweety ColE1 | NR236 (4S) secY in pLL2 | This work |
To construct integrating secY expression plasmids, plasmid pYUB2063 was first digested with PciI and religated to create a smaller plasmid of 5,198 bp, named pLL2. This plasmid integrates at the Tweety mycobacteriophage attB site in the M. smegmatis genome (20). The entire secY gene along with its upstream promoter was PCR amplified from NR178 and NR236 genomic DNA, with engineered NotI and EcoRV restriction sites for cloning purposes. Each PCR product was cloned into pCR2.1-TOPO (Invitrogen), yielding plasmids pLL17 and pLL19. The NotI-EcoRV secY fragments were cut from pLL17 and pLL19 and ligated into NotI-EcoRV-digested pLL2, yielding plasmids pLL21 and pLL23.
Azide sensitivity assays.
Cultures were plated for azide sensitivity as previously described (17), by mixing 200 μl of a saturated culture with 7H9 top agar and pouring over 7H10 agar plates (with Tween supplementation omitted) in three technical replicates. Discs soaked with 10 μl of 0.15 M sodium azide were added to the center of each plate. The diameter of the zone of inhibition was measured after 2 days and is presented as a percentage of the entire plate diameter, yielding the percent azide inhibition.
Whole genome and directed sequencing.
Genomic DNA was isolated (as described previously [21]) from the 4S suppressor strain (NR236) and submitted for whole-genome sequencing at the High-Throughput Sequencing Facility at the University of North Carolina at Chapel Hill. Sequencing was performed using Illumina GA II technology with 36-bp single-end reads. Reads were aligned to the M. smegmatis mc2155 reference genome (NCBI RefSeq accession number NC_008596.1) using SOAP (22) (with default parameters), resulting in an average sequence coverage of 47.5. Single nucleotide polymorphisms, insertions, and deletions were located using SOAP and BLAT (23) (with default parameters), resulting in identification of a total of 86 mutations. Mutations that had also been identified in other mc2155-derived strains sequenced in our laboratory were discarded as background mutations (i.e., mutations already present in the parent strain), leaving 15 mutations that appeared unique to the 4S strain. Following further confirmatory sequencing of PCR-amplified products (Eton Bioscience, Inc., or Genewiz, Inc.), all but one of these mutations were eliminated as either background or false positives.
5′ RLM-RACE.
RNA ligase-mediated rapid amplification of cDNA ends (RLM-RACE) was performed using the GeneRacer kit (Invitrogen) according to the manufacturer's instructions. Briefly, total RNA was isolated from strain NR172, treated with tobacco acid pyrophosphatase to remove 5′-triphosphates, and ligated to the GeneRacer RNA oligonucleotide. secY 5′ ends were reverse transcribed to cDNA by using a gene-specific primer located within the secY coding sequence (primer R1) (see Fig. 2B, below). 5′-end cDNA was then amplified using a nested PCR strategy involving a first round of PCR with a second gene-specific primer located within the secY coding sequence (primer R2) and the GeneRacer 5′ primer (homologous to the GeneRacer RNA oligonucleotide), followed by a second round of PCR with a primer located upstream of the secY coding sequence (primer R3) and the GeneRacer 5′ primer. PCR products were separated by agarose gel electrophoresis, individually gel purified, and cloned into pCR2.1-TOPO (Invitrogen). A minimum of 4 clones originating from each PCR product were submitted to Genewiz, Inc., for sequencing. Transcript 5′ ends were identified as the first nucleotide following the sequence of the GeneRacer RNA oligonucleotide. Nucleotide positions were numbered relative to the translational start site of secY (NCBI GenBank accession number ABK75688).
Fig 2.

4S and 24S mutations are located within the secY promoter. (A) Relative to the predicted translational start site of secY (indicated by start codon GTG), suppressor 4S (NR236) contains a 2-bp insertion between nucleotides −137 and −138. Suppressor 24S (NR151) contains a single nucleotide polymorphism at nucleotide −134. Suppressor mutations are boxed. Potential promoter −10 regions are shown in bold letters. The M. smegmatis −10 region consensus sequence is shown as a sequence logo representing the frequency at which each nucleotide occurs (the percentages are indicated beneath each nucleotide), as reported by Newton-Foot and Gey van Piteus (27). The drawing is not to scale. (B) Transcript 5′ ends identified upstream of secY. By using primer R3, two species of secY transcript 5′ ends were amplified (see the agarose gel inset) from RNA from the complemented secA2 strain (NR172). Relative to the predicted translational start site of secY (GTG), the longer species has a 5′ end at nucleotides −129 to −127, and the shorter species has a 5′ end at nucleotide −75. 5′-end nucleotides are boxed. Potential promoter −10 regions are shown in bold letters. Amplified 5′ end products are represented by wavy lines.
Quantitative RT-PCR.
Strains were grown in 7H9 broth to an optical density at 600 nm (OD600) of approximately 1.0, pelleted by centrifugation at 1,600 × g for 10 min, and flash-frozen. RNA was isolated using one of two protocols. For both protocols, bacteria were lysed in 1 ml 3:1 chloroform-methanol and then vortexed with 5 ml TRIzol and incubated for 10 min at room temperature. Phases were separated by centrifugation at 1,600 × g for 15 min at 4°C. For the first protocol, the upper phase was mixed with a 1× volume of isopropanol to precipitate overnight at 4°C. RNA was pelleted by centrifugation at 20,000 × g for 30 min at 4°C, washed twice with cold 70% ethanol, and resuspended in RNase-free water. For the second protocol, the upper phase was mixed with a 0.625× volume of 95% ethanol and column purified (Promega SV total RNA isolation system). All RNA samples were treated with DNase (Promega) and then column purified (Zymo DNA-free RNA kit). Following RNA isolation, quantitative reverse transcription-PCR (RT-PCR) was performed in triplicate technical replicates using 25 or 50 ng RNA in each reaction mixture. Products were reverse transcribed and amplified from total RNA using the Bioline SensiMix SYBR and fluorescein one-step kit and amplified from a DNA standard by using the Bioline SensiMix SYBR and fluorescein kit. For quantitative RT-PCR on secY, products were amplified from the 5′ ends of secY and rpoB gene sequences, the starting quantity of each transcript was calculated relative to DNA standards, and secY transcripts were normalized to rpoB transcripts (as a control) in each sample (24).
LacZ (β-galactosidase) activity assays.
LacZ activity assays were performed similar to those described previously (25). Each strain was grown in 7H9 broth to late-log phase or saturated phase, and 800 μl was pelleted in a microcentrifuge. Cell pellets were resuspended in 800 μl Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol), then lysed with 35 μl chloroform and 1 μl of 0.1% SDS, and vortexed for 30 s. A 160-μl volume of o-nitrophenyl-β-d-galactopyranoside (4 mg/ml in Z buffer) was added to each reaction mixture, and mixtures were incubated for 1 h at room temperature. Reactions were terminated by addition of 400 μl of 1 M Na2CO3. Debris was removed by centrifugation at 16,000 × g for 3 min, and the OD420 was read from the supernatant. LacZ activity (Miller units) was calculated by the following formula: (1,000 × OD420)/([reaction time in minutes] × [culture volume used in the reaction, in ml] × OD600).
Subcellular fractionation and Western blotting.
Each strain was grown in Mueller-Hinton broth to an OD600 of approximately 1.0. Subcellular fractions were prepared as previously described (15). Briefly, cell pellets were lysed by passage through a French press. Cell debris was removed by centrifugation at 1,600 × g for 30 min, and clarified whole-cell lysates (WCL; supernatants) were standardized for equal protein content by using the Pierce bicinchoninic acid protein assay. Clarified whole-cell lysates were centrifuged at 100,000 × g for 2 h to separate cell envelope (pellet) and soluble (supernatant) fractions. In some cases, 1% Igepal CA-630 was added during cell envelope resuspension to facilitate solubilization of SecY. WCL (loaded for equal protein content) or subcellular fractions (loaded for equivalent starting cell material to WCL samples) were boiled, separated by SDS-PAGE, and transferred to nitrocellulose membranes for Western blotting. SecY was detected using a rabbit anti-SecY polyclonal antiserum at a 1:150 dilution. Primary antibodies were detected using alkaline phosphatase-conjugated goat anti-rabbit IgG secondary antibodies, enhanced chemifluorescence substrate (GE Healthcare), and a Molecular Dynamics Storm 860 PhosphorImager or Syngene G:BOX machine.
SecY antiserum production.
Anti-SecY antiserum was produced for this work by Yenzym Antibodies, LLC, and was raised in rabbits to a peptide corresponding to the C-terminal 16 amino acids of M. smegmatis SecY (QIESQLMQRNYEGFLK) by using TiterMax Gold adjuvant. Affinity-purified antibody was used in this study.
RESULTS
Spontaneous suppressor mutations alleviate the severe phenotypes of the secA2 K129R allele.
Compared to wild-type M. smegmatis, a secA2 deletion mutant exhibits a moderate growth defect on rich agar medium, such as Mueller-Hinton agar, and increased sensitivity to azide (1). Azide is known to inhibit SecA ATPase activity in E. coli (26), and we showed previously that SecA1 in mycobacteria is also affected by azide (1). Both of these secA2 deletion phenotypes are complemented by adding the wild-type secA2 allele, expressed from the constitutive hsp60 promoter, into the secA2 deletion mutant (Fig. 1A) (17). However, introduction of the secA2 K129R allele, also expressed from the hsp60 promoter, into the secA2 deletion mutant fails to complement secA2 deletion phenotypes. SecA2 K129R is nonfunctional as a result of an amino acid substitution in the ATP binding Walker box. Moreover, the secA2 K129R allele exacerbates both the growth defect on Mueller-Hinton agar and sensitivity to azide phenotypes (i.e., secA2 K129R phenotypes are more severe than phenotypes of the secA2 deletion) (Fig. 1B) (17). As reported previously, the secA2 K129R allele is also dominant negative, exhibiting phenotypes even in the presence of a wild-type secA2 allele (17).
Fig 1.
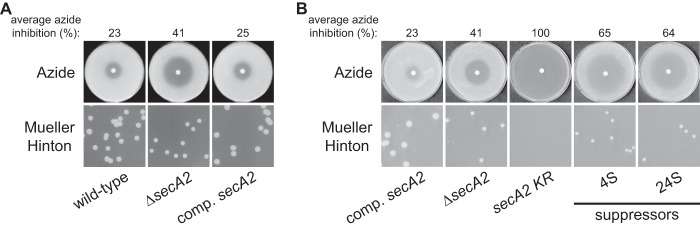
Azide sensitivity and Mueller-Hinton agar growth phenotypes. The indicated strains were plated for sensitivity to 10 μl of 0.15 M sodium azide for 2 days at 37°C and growth on Mueller-Hinton agar plates for 6 days at 37°C. Average azide inhibition was calculated by measuring the diameter of the zone of azide inhibition as a percentage of the plate diameter, and values are the means of three technical replicates. Results shown are representative of at least three independent experiments. Strains tested were the following: (A) wild type + empty plasmid (NR158), ΔsecA2 + empty plasmid (NR160), and complemented secA2 (NR172); (B) complemented secA2 (NR172), ΔsecA2 + empty plasmid (NR160), secA2 K129R (NR178), 4S (NR236), and 24S (NR151).
Suppressor mutations spontaneously arise in the secA2 K129R background and are easily identified by their ability to grow on Mueller-Hinton agar. To begin our suppressor analysis, we collected 63 suppressor mutants. All suppressors alleviated the severe phenotypes associated with the secA2 K129R allele, improving both growth on Mueller-Hinton agar and resistance to azide (Fig. 1B). Throughout this study, we routinely compared suppressor strains to the complemented secA2 strain (NR172) as a control, instead of the wild-type strain. By doing so, we could study the effect of the suppressor mutations across a series of strains that all express secA2 alleles from the same hsp60 promoter. In no case did suppressors rescue phenotypes to the level of the complemented secA2 strain. Instead, suppressor phenotypes more closely resembled the phenotypes of the secA2 deletion strain, suggesting that suppressor mutations overcome the detrimental effect of SecA2 K129R but do not restore SecA2-dependent export. Of the 63 suppressors isolated, 40 did not produce full-length SecA2 K129R protein (based on Western blotting [data not shown]), suggesting that these suppressors alleviate secA2 K129R phenotypes by either truncating the SecA2 K129R protein or preventing SecA2 K129R production. This is a category of suppressors that we expected to observe, but they were eliminated from further study as they seemed unlikely to help us identify proteins that work with SecA2 during export. Of the remaining 23 suppressors, 21 strains produced normal levels of full-length SecA2 K129R protein (including all strains examined in detail in the current study), and 2 strains produced full-length SecA2 K129R protein at a somewhat reduced level.
We next sequenced the secA2 K129R allele in the 23 suppressors producing SecA2 K129R protein. Of these 23 suppressors, 9 strains contained mutations in the secA2 K129R allele (intragenic), while the remaining 14 strains had no mutations in the secA2 K129R allele and were therefore extragenic suppressors. Because our goal was to identify proteins that work with SecA2 during export, we focused on characterizing extragenic suppressors.
Suppressors 4S and 24S contain mutations upstream of secY.
We chose a single extragenic suppressor with normal SecA2 K129R protein levels, identified as 4S (strain NR236), and performed whole-genome sequencing. Alignment of sequence reads to the M. smegmatis mc2155 reference genome resulted in average sequence coverage of 47.5, sufficient to identify mutations in the 4S genome. Each candidate suppressor mutation was PCR amplified and resequenced from both 4S and its parent secA2 K129R strain. Following this directed sequencing, false positives and background mutations were discarded, leaving only a single mutation uniquely present in suppressor 4S. This unique mutation was a 2-bp insertion located 137 bp upstream of the translational start site for the predicted M. smegmatis secY gene, msmeg_1483 (Fig. 2A). The msmeg_1483 open reading frame is the only secY homolog in M. smegmatis (43% identical and 63% similar at the amino acid level to the canonical E. coli SecY).
To determine whether any other extragenic suppressors in our collection contained mutations upstream of secY, we sequenced approximately 1,000 bp upstream of secY in the 13 additional extragenic suppressor strains. One additional suppressor, identified as 24S (strain NR151), contained a single C → G polymorphism located 134 bp upstream of the translational start site for secY (Fig. 2A). The close proximity of the 4S and 24S mutations suggests that they act in a similar manner to suppress the secA2 K129R allele.
4S and 24S suppressor mutations are located within the secY promoter.
Because the mutations identified in suppressors 4S and 24S are located upstream of the secY coding sequence, we hypothesized that they would map to the secY promoter and suppress the secA2 K129R phenotypes through an effect on secY transcription. Mycobacterial promoters do not always resemble the classical promoter structure defined for E. coli and other bacteria, and it can be difficult to predict their location. However, manual inspection of the sequence upstream of secY revealed two potential M. smegmatis −10 regions, each beginning with the highly conserved nucleotides TA (27) and matching at least 3/6 nucleotides of the −10 region consensus sequence. These two −10 regions, positioned at nucleotides −132 and −140 upstream of the secY start codon, are the closest potential −10 regions to secY and are located in the same region as the 4S and 24S mutations (Fig. 2A). No obvious −35 region was found, but this was not surprising, given that mycobacteria have low homology at this region (27) and may not even require the −35 region for promoter function (28).
The location of the potential promoter −10 regions suggests that the 4S and 24S mutations could directly impact transcription. The 4S mutation is an insertion that improves the −10 region at nucleotide −140 from a 3/6 nucleotide match to the −10 region consensus to a 4/6 nucleotide match. The 24S mutation is a C → G polymorphism upstream of the −10 region at nucleotide −132. Interestingly, this mutation creates a TGn motif upstream of the −10 region. Addition of a TGn motif to a −10 region can result in a 3- to 5-fold increase in transcription (29).
To determine the location of the secY transcriptional start site in relation to the 4S and 24S mutations, we performed 5′-RLM-RACE on M. smegmatis RNA (using primer R1) (Fig. 2B). Following the RACE procedure, we PCR amplified the 5′ ends of cDNA created from secY transcripts by using a nested PCR strategy. The first round of PCR (using primer R2) amplified out from the secY coding sequence and yielded multiple products. A second round of PCR with a primer upstream of secY (primer R3) narrowed in on the longest secY transcripts. This nested PCR amplification produced two products (Fig. 2B) that we cloned and sequenced to determine the 5′ ends of each species. The longer of the two species represented secY transcripts with a 5′ end located 127 to 129 bp upstream of the secY translational start site, and the shorter of the two species represented secY transcripts with a 5′ end located 75 bp upstream of secY. The transcripts beginning 127 to 129 bp upstream of secY were the longest species detected, making it likely that they represent true transcriptional start sites. This start site region corresponds perfectly with the potential −10 regions we identified upstream of secY and supports the conclusion that the 4S and 24S suppressor mutations are located within the secY promoter. The transcripts beginning 75 bp upstream of secY could result from an alternate transcriptional start site, although there are no obvious promoter sequences located adjacent to this site. Alternatively, these shorter transcripts could be a result of transcript processing/degradation at this location.
4S and 24S suppressor mutations cause increased secY transcript levels.
Given the location of the 4S and 24S mutations, we next tested whether the mutations affect secY expression by measuring secY transcript levels in the complemented secA2, secA2 deletion mutant, secA2 K129R mutant, and 4S and 24S suppressor strains. Total RNA was isolated from each strain and was analyzed by quantitative RT-PCR (Fig. 3A). There was no significant difference in secY transcript levels between the complemented secA2, secA2 deletion mutant, and secA2 K129R mutant strains. However, relative to the secA2 K129R parent strain, both suppressors 4S and 24S exhibited a reproducible increase in secY transcript levels, although this result was statistically significant only for suppressor 24S.
Fig 3.
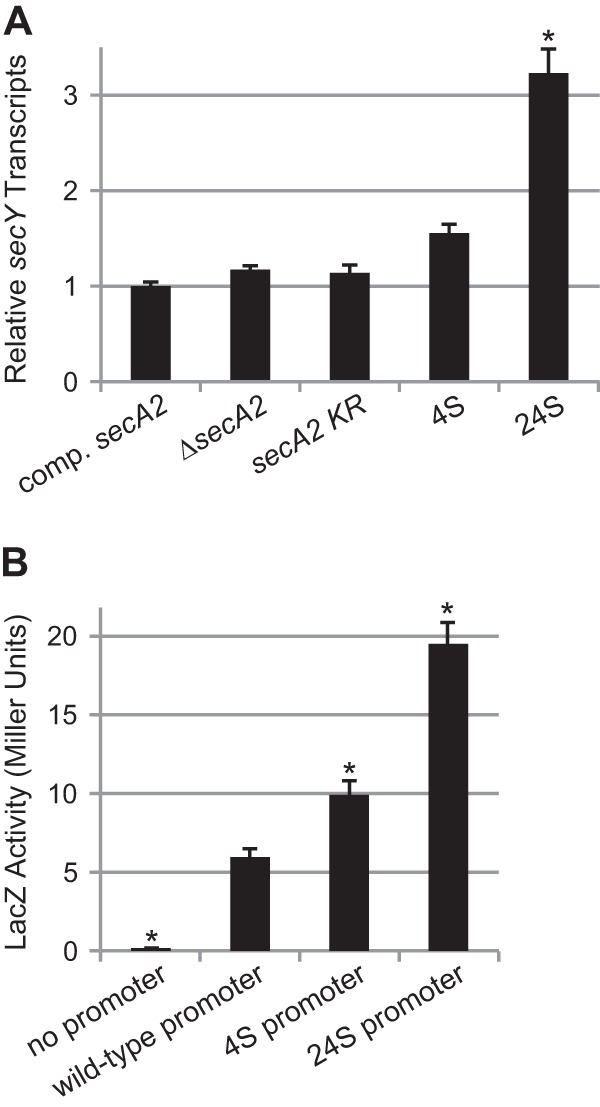
Suppressor mutations cause increased secY expression. (A) secY transcript levels measured by quantitative RT-PCR, relative to rpoB transcript levels. The secY transcript level in the complemented secA2 strain was set to 1. Data represent the means of three biological replicates, and error bars represent standard errors. *, statistically different from the SecA2 K129R strain (P < 0.001) based on one-way analysis of variance (ANOVA) with the Student-Newman-Keuls test. Strains tested were the following: complemented secA2 (NR172), ΔsecA2 + empty plasmid (NR160), secA2 K129R (NR178), 4S (NR236), and 24S (NR151). (B) LacZ-fusion activity assay results. The secY promoter regions plus 34 bp of the secY coding sequence from wild-type and suppressor strains were translationally fused to lacZ and tested in wild-type M. smegmatis mc2155. Data represent the means of six biological replicates, and error bars represent standard errors. *, statistically different from the wild-type promoter strain (P < 0.05) by one-way ANOVA on ranks with the Student-Newman-Keuls test. LacZ fusions tested were the following: no promoter (pCV125), wild-type secY promoter (pLL11), 4S secY promoter (pLL8), and 24S secY promoter (pLL15).
We also tested the effects of the 4S and 24S mutations in the absence of the secA2 K129R allele (i.e., in a wild-type background) by using secY′-′lacZ fusions. To do this, we constructed plasmids containing 379 bp upstream of secY plus 34 bp of secY coding sequence translationally fused to lacZ, introduced these plasmids into wild-type M. smegmatis mc2155, and measured LacZ (β-galactosidase) activity as a measure of secY expression (Fig. 3B). Relative to the wild-type secY promoter, constructs containing the 4S or 24S secY promoters produced significantly higher levels of LacZ activity. These results reinforced the conclusion that the 4S and 24S mutations increase secY expression.
SecY protein is undetectable in the secA2 K129R strain but was recovered in 4S and 24S suppressor strains.
We next tested for an increase in SecY protein levels in the 4S and 24S suppressors. To measure SecY protein, we generated an antibody against a peptide matching the C terminus of M. smegmatis SecY and performed anti-SecY Western blot assays on WCL and on subcellular fractions containing the cell envelope (ENV) or the cytoplasm (SOL). The purity of the subcellular fractions was confirmed by Western blot analysis with antibodies to the cell envelope protein MspA and the cytoplasmic protein GroEL1 (data not shown). Anti-SecY Western blotting on the WCL of the complemented secA2 strain detected a prominent protein species at 37 kDa (Fig. 4). Although the predicted size of M. smegmatis SecY is 48 kDa, full-length SecY in E. coli (30) and Synechococcus sp. PCC7942 (31) is also observed to migrate aberrantly at about 37 kDa. Additionally, lower-molecular-weight species were sometimes apparent on the anti-SecY Western blots, which were likely degradation products, similar to those detected for E. coli SecY (8, 32). In support of the 37-kDa species being full-length SecY, strains expressing higher levels of SecY exhibited an increase in this product (discussed below). In addition, this full-length M. smegmatis SecY species was detected almost exclusively in the cell envelope fraction, as expected for an integral membrane protein.
Fig 4.
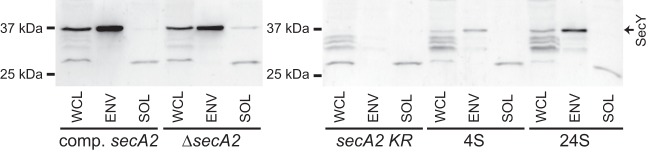
Suppressor mutations increase SecY protein levels relative to the secA2 K129R strain. Whole-cell lysates and subcellular fractions were separated by SDS-PAGE, and SecY protein was detected by Western blotting. All samples were equally loaded. Strains tested were the following: complemented secA2 (NR172), ΔsecA2 + empty plasmid (NR160), secA2 K129R (NR178), 4S (NR236), and 24S (NR151).
Next, we performed anti-SecY Western blot assays on complemented secA2, secA2 deletion mutant, secA2 K129R mutant, and 4S and 24S suppressor strains (Fig. 4). Both complemented secA2 and secA2 deletion mutant strains exhibited strong signals for full-length SecY protein. In stark contrast, we observed a complete loss of detectable full-length SecY protein in the secA2 K129R mutant, revealing a link between SecA2 K129R and SecY. As there was no transcriptional effect on secY observed in the secA2 K129R strain (Fig. 3A), the change in SecY protein levels in the secA2 K129R mutant does not appear to be caused by a difference in secY expression. An alternate possibility is that stability of the SecY protein is reduced by SecA2 K129R. This explanation is supported by an increase in the SecY degradation products observed in the secA2 K129R strain. In addition, experiments in E. coli have shown that jamming export through the SecYEG channel results in degradation of SecY (33). Therefore, the most likely explanation for the lack of detectable SecY protein in the secA2 K129R strain is that SecA2 K129R causes stress on the housekeeping SecY export channel, resulting in drastic SecY degradation.
Relative to the secA2 K129R strain, suppressors 4S and 24S exhibited increased levels of full-length SecY protein. While the SecY levels in 4S and 24S were still lower than that in the complemented secA2 or secA2 deletion mutant strains, the increase relative to the secA2 K129R strain was consistent with the increased transcription of secY (Fig. 3A). This increase in secY expression would not be expected to prevent the SecY degradation caused by SecA2 K129R. In fact, the presumed SecY degradation products remained high in both suppressor strains. Thus, it appears that the 4S and 24S mutations are able to suppress secA2 K129R by altering the balance between secY expression and SecY degradation, shifting the steady-state SecY protein level high enough to allow the recovered growth seen in these strains.
Increased secY expression is sufficient to suppress secA2 K129R.
The above results demonstrated that the 4S and 24S mutations cause increased expression of secY. However, the question remained whether this relatively small increase in secY expression alone was sufficient to suppress the secA2 K129R phenotypes. To address this question, we created integrating plasmids carrying the secY gene under the control of its native promoter, either with no mutation (wild type) or carrying the 4S mutation upstream of secY. These plasmids were then introduced into the secA2 K129R strain, creating merodiploid strains that contained the endogenous secY gene at its chromosomal location and a second copy of secY integrated in the chromosome at the Tweety mycobacteriophage attachment site. Addition of an extra copy of secY, whether driven by the 4S promoter or the native promoter, was indeed sufficient to suppress secA2 K129R phenotypes, and both versions suppressed secA2 K129R equally as well as the original 4S suppressor (Fig. 5A). In support of this result, Western blotting revealed that when a second copy of secY was introduced into the secA2 K129R strain, the level of full-length SecY protein increased to the same level as seen in the 4S suppressor strain (Fig. 5B). These results indicated that the increased secY expression observed in the 4S and 24S suppressor strains can indeed explain their abilities to suppress secA2 K129R phenotypes.
Fig 5.
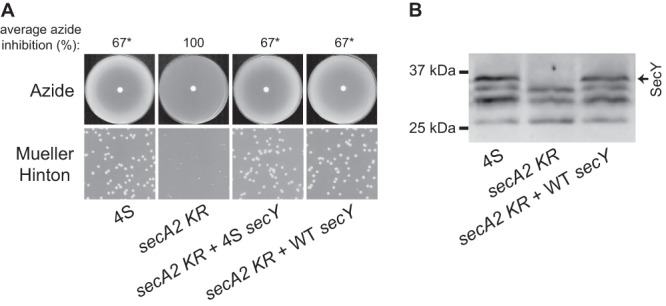
Increased SecY levels are sufficient to suppress secA2 K129R. Integrating plasmids containing secY under the control of the wild-type promoter or the 4S promoter were added to the secA2 K129R strain and tested for azide sensitivity, colony size on Mueller-Hinton agar, and SecY protein level. Strains tested were the following: 4S suppressor + empty plasmid (NR236 + pLL2), secA2 K129R + empty plasmid (NR178 + pLL2), secA2 K129R + 4S secY (NR178 + pLL23), secA2 K129R + wild-type (WT) secY (NR178 + pLL21). (A) Azide sensitivity and Mueller-Hinton agar growth phenotypes. Average azide inhibition was calculated by measuring the diameter of the zone of azide inhibition as a percentage of the plate diameter and is the mean of three biological replicates. *, statistically different from the secA2 K129R strain (P < 0.001) by one-way analysis of variance with the Student-Newman-Keuls test. (B) Whole-cell lysates separated by SDS-PAGE and SecY protein detected by Western blotting. All samples were equally loaded.
Additional extragenic suppressors are also associated with increased SecY levels.
Given the above results, we selected six additional extragenic suppressors and tested for an effect on SecY. These six suppressors expressed normal levels of SecA2 K129R protein and did not carry mutations in the secY gene or promoter (data not shown). Relative to the secA2 K129R mutant, each of these suppressors exhibited improved azide resistance and growth on Mueller-Hinton agar (Fig. 6A), with the degree of suppression varying from strain to strain. Western blotting for SecY revealed that all six additional suppressors had an increased steady-state level of SecY protein relative to the parent secA2 K129R strain (Fig. 6B). However, unlike the case with the 4S and 24S suppressors, none of these six additional suppressors had altered secY transcription, as assessed by quantitative RT-PCR (Fig. 6C). Therefore, these suppressors must affect SecY levels in a completely different manner.
Fig 6.
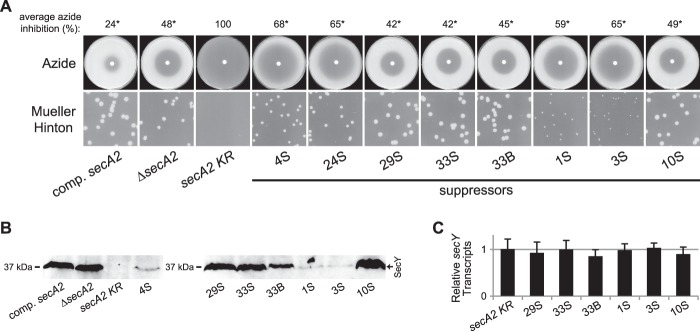
Additional extragenic suppressors increase SecY protein levels but not secY transcript levels. Strains tested were the following: complemented secA2 (NR172), ΔsecA2 + empty plasmid (NR160), secA2 K129R (NR178), 4S (NR236), 24S (NR151), 29S (NR154), 33S (NR155), 33B (NR156), 1S (NR230), 3S (NR234), and 10S (NR248). (A) Azide sensitivity and Mueller-Hinton agar growth phenotypes. Average azide inhibition was calculated by measuring the diameters of the zones of azide inhibition as a percentage of the plate diameter and is the mean of two to four biological replicates. *, statistically different from the secA2 K129R strain (P < 0.001) by one-way analysis of variance (ANOVA) with the Student-Newman-Keuls test. (B) Cell envelope fractions separated by SDS-PAGE and SecY protein detected by Western blotting. All samples were equally loaded. (C) secY transcript levels measured by quantitative RT-PCR, relative to rpoB transcript levels. The secY transcript level for the secA2 K129R strain was set to 1. Data represent the means of six biological replicates, and error bars represent standard errors. No statistical differences were detected (by one-way ANOVA).
The finding that multiple extragenic suppressors affect the SecY protein level, despite differences in their genotypes and phenotypes, reinforces the conclusion that SecA2 K129R has a detrimental effect on SecY. Furthermore, the level of SecY protein observed correlates with the phenotype of the respective strain. The secA2 K129R mutant had no detectable SecY protein, little resistance to azide, and very poor growth on Mueller-Hinton agar. Intermediate suppressors, such as 4S, 24S, 1S, and 3S, had low but detectable SecY protein levels and moderate resistance to azide and growth on Mueller-Hinton agar. Strong suppressors, such as 29S, 33S, 33B, and 10S, had near-wild-type levels of SecY protein and the strongest azide resistance and growth on Mueller-Hinton agar observed, comparable to the phenotypes of the secA2 deletion mutant. Based on this pattern, we concluded that a critical problem caused by the presence of SecA2 K129R is a reduced SecY protein level and that extragenic suppressor mutations can correct this problem by increasing the amount of SecY protein to a functional level.
DISCUSSION
While the conserved Sec export system of bacteria is well understood, even the most basic steps of SecA2-dependent export remain to be characterized. For the mycobacterial SecA2-dependent export system, there is no obvious SecY2 available to serve as a dedicated export channel. This type of SecA2 pathway is therefore termed a “SecA2-only” system (14). One of the many basic questions to be answered about the SecA2 pathway of mycobacteria is whether it utilizes the SecYEG translocase or an unknown apparatus to export its specific set of proteins. Here, we showed that a SecA2 K129R dominant negative protein leads to reduced levels of the sole SecY of mycobacteria and that increased SecY levels can suppress the severe phenotypes of a secA2 K129R mutant. These data are significant in revealing a link between SecA2 and the housekeeping SecY, which provides strong support for the idea that SecA2 works with the canonical SecYEG translocase to export proteins.
The effect of SecA2 K129R on SecY levels indicates a relationship between SecA2-dependent export and the canonical Sec pathway. However, this result also raises the question of why SecA2 K129R leads to lower SecY levels. There is precedent for SecY degradation occurring in response to stress at the Sec translocase. In E. coli, when the SecYEG channel complex is artificially “jammed” by attempted export of a folded protein, the SecY protein is degraded by FtsH protease, which serves to remove the nonfunctional “jammed” translocon (33). Our results are consistent with there being a similar stress on the SecYEG channel in the presence of the ATP-binding-defective SecA2 K129R in M. smegmatis. There is also a homolog of ftsH in M. smegmatis that could potentially be responsible for SecY degradation, as in E. coli.
The housekeeping SecA protein drives export of an individual protein in a stepwise fashion through successive cycles of ATP binding and hydrolysis, during which SecA repeatedly releases and reassociates with the translocon. Furthermore, ATP hydrolysis is specifically necessary for SecA to dissociate from the translocase during this process (10). Therefore, a SecA protein that cannot bind and/or hydrolyze ATP (such as a Walker box KR variant) will become trapped at the membrane translocon and will fail to complete protein export. A Walker box substitution in E. coli SecA (SecA K108R) that is defective in ATP binding and hydrolysis (34) shifts SecA localization from the soluble fraction toward the cell envelope fraction (35), as does the corresponding SecA2 K129R variant of M. smegmatis (17). Thus, the most likely explanation for the effect of SecA2 K129R on SecY is that SecA2 K129R is locked in nonfunctional complexes with the housekeeping Sec export pathway, which results in SecY degradation in an effort by the cell to eliminate SecA2 K129R-jammed translocons. By analogy to the housekeeping Sec pathway (36), SecA2 could physically “dock” with SecYEG during protein export. However, we were unable to detect any physical interaction between SecA2 and SecY by chemical cross-linking in M. smegmatis (data not shown). Therefore, the link we detected between SecA2 export and the SecY channel may reflect either a transient physical interaction or an indirect interaction involving another component of the export machinery.
Because SecY is critical for the export of many essential proteins, the reduced SecY levels observed in the presence of SecA2 K129R would be extremely detrimental to the cell. This helps explain the severe growth inhibition caused by SecA2 K129R. However, it is unlikely that there is no functional SecY protein in the secA2 K129R mutant, even though there was no full-length SecY detectable by Western blotting for this strain (Fig. 4). SecY is thought to be essential for growth in all bacteria, including M. tuberculosis (37); therefore, a complete loss of SecY would be lethal. Most likely, the amount of SecY in the secA2 K129R strain was below the level of detection with our anti-SecY antibody.
Given the reduced SecY levels in the secA2 K129R strain, it is logical that extragenic suppressor mutations might act to restore SecY abundance to a functional level. In suppressors 4S and 24S, this is accomplished by increasing transcription of secY, which must allow sufficient SecY production to replace the SecY channels lost to degradation. Interestingly, while suppressor 4S consistently displayed a less robust effect on secY expression than suppressor 24S (Fig. 3), these two suppressors were phenotypically identical (Fig. 1B). This indicates that the relatively weak effects of the 4S mutation are sufficient to reach a threshold SecY level necessary to restore functional export through the SecYEG translocon.
Six additional extragenic suppressors also restored SecY protein levels to various degrees, though not by affecting secY transcription. These additional suppressor mutations could act in an alternate way to increase SecY production, for example, by increasing the efficiency of secY translation. Another possibility is that these additional suppressor mutations serve to avoid the detrimental interaction between SecA2 K129R and the Sec pathway, thereby preventing SecY degradation in the first place. The latter possibility is especially interesting, as it may point to additional components of the machinery required for SecA2-dependent export and help elucidate the interaction points between the SecA2 pathway and housekeeping Sec export. While it is formally possible that these suppressors might completely bypass the need for SecA2 in protein export, this possibility seems unlikely, because even the strongest suppressors only restored phenotypes to the level of a secA2 deletion mutant (i.e., none of the suppressors restored phenotypes to the level seen with wild-type secA2). In any case, the correlation between the suppressor phenotype and restoration of detectable SecY protein levels makes a strong case for reduced SecY levels being responsible for the secA2 K129R mutant phenotype.
The results of the current study build upon our previous results showing that the canonical SecA of mycobacteria (SecA1) is important for SecA2-dependent protein export, and they suggest that the SecA2 export pathway is fully integrated into housekeeping Sec export. It remains possible, however, that there exist additional specialty components that are important for SecA2-dependent protein export. We recently showed that features of a protein's mature domain (i.e., not the signal peptide) determine the requirement for SecA2. One interesting possibility is that the defining feature of the mature domain of SecA2-exported proteins is a propensity to fold in the cytoplasm prior to export (18). As an integrated component of the Sec pathway, SecA2 could assist the canonical Sec pathway to greater or lesser degrees with the export of proteins that are difficult to export due to folding or other features.
A protein export pathway in which SecA2 works with SecYEG may be a common feature of “SecA2-only” type systems. In fact, recent data from the Clostridium difficile “SecA2-only” system (7) showed that the corresponding Walker box substitution in the SecA2 of this system (SecA2 K106R) was dominant negative and caused severe growth inhibition when overexpressed, suggesting that it similarly interferes with an essential pathway. These and other data are consistent with the idea that SecA1 and SecA2 may also share use of the SecYEG channel in C. difficile. It would be interesting to see whether the detrimental effects of C. difficile SecA2 K106R also involve SecY degradation.
In conclusion, we uncovered a connection between SecA2 and the housekeeping SecY of mycobacteria by using a classical genetic approach. Our results indicate that the “SecA2-only” system of mycobacteria utilizes the housekeeping SecYEG channel to export its select subset of proteins, addressing a key question about the mechanism of SecA2-dependent export. This pathway for SecA2-dependent export is distinct from that employed by SecA2-SecY2 systems. Because our study indicated that the mycobacterial SecA2 export system is actually an adaptation of the housekeeping Sec system, continued study of this system will not only increase our understanding of SecA2-dependent export but may also shed light on the conserved Sec pathway utilized by all bacteria.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant AI054540 awarded to M.B.
We thank Adrie Steyn for providing plasmid pCV125 and William Jacobs for providing plasmid pYUB2063. We also thank Anthony Richardson for technical assistance with 5′-RLM-RACE and quantitative RT-PCR and members of the Braunstein laboratory for critical reading of the manuscript.
Footnotes
Published ahead of print 2 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00630-13.
REFERENCES
- 1.Braunstein M, Brown AM, Kurtz S, Jacobs WR., Jr 2001. Two nonredundant SecA homologues function in mycobacteria. J. Bacteriol. 183:6979–6990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lenz LL, Portnoy DA. 2002. Identification of a second Listeria secA gene associated with protein secretion and the rough phenotype. Mol. Microbiol. 45:1043–1056 [DOI] [PubMed] [Google Scholar]
- 3.Bensing BA, Sullam PM. 2002. An accessory sec locus of Streptococcus gordonii is required for export of the surface protein GspB and for normal levels of binding to human platelets. Mol. Microbiol. 44:1081–1094 [DOI] [PubMed] [Google Scholar]
- 4.Siboo IR, Chaffin DO, Rubens CE, Sullam PM. 2008. Characterization of the accessory Sec system of Staphylococcus aureus. J. Bacteriol. 190:6188–6196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nguyen-Mau SM, Oh SY, Kern VJ, Missiakas DM, Schneewind O. 2012. Secretion genes as determinants of Bacillus anthracis chain length. J. Bacteriol. 194:3841–3850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caspers M, Freudl R. 2008. Corynebacterium glutamicum possesses two secA homologous genes that are essential for viability. Arch. Microbiol. 189:605–610 [DOI] [PubMed] [Google Scholar]
- 7.Fagan RP, Fairweather NF. 2011. Clostridium difficile has two parallel and essential Sec secretion systems. J. Biol. Chem. 286:27483–27493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brundage L, Hendrick JP, Schiebel E, Driessen AJ, Wickner W. 1990. The purified E. coli integral membrane protein SecY/E is sufficient for reconstitution of SecA-dependent precursor protein translocation. Cell 62:649–657 [DOI] [PubMed] [Google Scholar]
- 9.Schiebel E, Driessen AJ, Hartl FU, Wickner W. 1991. Delta mu H+ and ATP function at different steps of the catalytic cycle of preprotein translocase. Cell 64:927–939 [DOI] [PubMed] [Google Scholar]
- 10.van der Wolk JP, de Wit JG, Driessen AJ. 1997. The catalytic cycle of the Escherichia coli SecA ATPase comprises two distinct preprotein translocation events. EMBO J. 16:7297–7304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shiba K, Ito K, Yura T, Cerretti DP. 1984. A defined mutation in the protein export gene within the spc ribosomal protein operon of Escherichia coli: isolation and characterization of a new temperature-sensitive secY mutant. EMBO J. 3:631–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hou JM, D'Lima NG, Rigel NW, Gibbons HS, McCann JR, Braunstein M, Teschke CM. 2008. ATPase activity of Mycobacterium tuberculosis SecA1 and SecA2 proteins and its importance for SecA2 function in macrophages. J. Bacteriol. 190:4880–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bensing BA, Sullam PM. 2009. Characterization of Streptococcus gordonii SecA2 as a paralogue of SecA. J. Bacteriol. 191:3482–3491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rigel NW, Braunstein M. 2008. A new twist on an old pathway: accessory Sec systems. Mol. Microbiol. 69:291–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibbons HS, Wolschendorf F, Abshire M, Niederweis M, Braunstein M. 2007. Identification of two Mycobacterium smegmatis lipoproteins exported by a SecA2-dependent pathway. J. Bacteriol. 189:5090–5100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Braunstein M, Espinosa BJ, Chan J, Belisle JT, Jacobs WR., Jr 2003. SecA2 functions in the secretion of superoxide dismutase A and in the virulence of Mycobacterium tuberculosis. Mol. Microbiol. 48:453–464 [DOI] [PubMed] [Google Scholar]
- 17.Rigel NW, Gibbons HS, McCann JR, McDonough JA, Kurtz S, Braunstein M. 2009. The accessory SecA2 system of mycobacteria requires ATP binding and the canonical SecA1. J. Biol. Chem. 284:9927–9936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feltcher ME, Gibbons HS, Ligon LS, Braunstein M. 2013. Protein export by the mycobacterial SecA2 system is determined by the preprotein mature domain. J. Bacteriol. 195:672–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Snapper SB, Lugosi L, Jekkel A, Melton RE, Kieser T, Bloom BR, Jacobs WR., Jr 1988. Lysogeny and transformation in mycobacteria: stable expression of foreign genes. Proc. Natl. Acad. Sci. U. S. A. 85:6987–6991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pham TT, Jacobs-Sera D, Pedulla ML, Hendrix RW, Hatfull GF. 2007. Comparative genomic analysis of mycobacteriophage Tweety: evolutionary insights and construction of compatible site-specific integration vectors for mycobacteria. Microbiology 153:2711–2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Larsen MH, Biermann K, Tandberg S, Hsu T, Jacobs WR., Jr. 2007. Genetic manipulation of Mycobacterium tuberculosis. Curr. Protoc. Microbiol. Chapter 10:Unit 10A.12 [DOI] [PubMed] [Google Scholar]
- 22.Li R, Li Y, Kristiansen K, Wang J. 2008. SOAP: short oligonucleotide alignment program. Bioinformatics 24:713–714 [DOI] [PubMed] [Google Scholar]
- 23.Kent WJ. 2002. BLAT: the BLAST-like alignment tool. Genome Res. 12:656–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Badejo AC, Badejo AO, Shin KH, Chai YG. 2013. A gene expression study of the activities of aromatic ring-cleavage dioxygenases in Mycobacterium gilvum PYR-GCK to changes in salinity and pH during pyrene degradation. PLoS One 8(2):e58066. 10.1371/journal.pone.0058066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alland D, Steyn AJ, Weisbrod T, Aldrich K, Jacobs WR., Jr 2000. Characterization of the Mycobacterium tuberculosis iniBAC promoter, a promoter that responds to cell wall biosynthesis inhibition. J. Bacteriol. 182:1802–1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oliver DB, Cabelli RJ, Dolan KM, Jarosik GP. 1990. Azide-resistant mutants of Escherichia coli alter the SecA protein, an azide-sensitive component of the protein export machinery. Proc. Natl. Acad. Sci. U. S. A. 87:8227–8231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Newton-Foot M, Gey van Pittius NC. 2013. The complex architecture of mycobacterial promoters. Tuberculosis (Edinb.) 93:60–74 [DOI] [PubMed] [Google Scholar]
- 28.Kenney TJ, Churchward G. 1996. Genetic analysis of the Mycobacterium smegmatis rpsL promoter. J. Bacteriol. 178:3564–3571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bashyam MD, Tyagi AK. 1998. Identification and analysis of “extended −10” promoters from mycobacteria. J. Bacteriol. 180:2568–2573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ito K. 1984. Identification of the secY (prlA) gene product involved in protein export in Escherichia coli. Mol. Gen. Genet. 197:204–208 [DOI] [PubMed] [Google Scholar]
- 31.Nakai M, Sugita D, Omata T, Endo T. 1993. Sec-Y protein is localized in both the cytoplasmic and thylakoid membranes in the cyanobacterium Synechococcus PCC7942. Biochem. Biophys. Res. Commun. 193:228–234 [DOI] [PubMed] [Google Scholar]
- 32.Akiyama Y, Ito K. 1990. SecY protein, a membrane-embedded secretion factor of E. coli, is cleaved by the ompT protease in vitro. Biochem. Biophys. Res. Commun. 167:711–715 [DOI] [PubMed] [Google Scholar]
- 33.van Stelten J, Silva F, Belin D, Silhavy TJ. 2009. Effects of antibiotics and a proto-oncogene homolog on destruction of protein translocator SecY. Science 325:753–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitchell C, Oliver D. 1993. Two distinct ATP-binding domains are needed to promote protein export by Escherichia coli SecA ATPase. Mol. Microbiol. 10:483–497 [DOI] [PubMed] [Google Scholar]
- 35.Jilaveanu LB, Zito CR, Oliver D. 2005. Dimeric SecA is essential for protein translocation. Proc. Natl. Acad. Sci. U. S. A. 102:7511–7516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zimmer J, Nam Y, Rapoport TA. 2008. Structure of a complex of the ATPase SecA and the protein-translocation channel. Nature 455:936–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sassetti CM, Boyd DH, Rubin EJ. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48:77–84 [DOI] [PubMed] [Google Scholar]
- 38.Snapper SB, Melton RE, Mustafa S, Kieser T, Jacobs WR., Jr 1990. Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol. Microbiol. 4:1911–1919 [DOI] [PubMed] [Google Scholar]
- 39.Stover CK, de la Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, Bansal GP, Young JF, Lee MH, Hatfull GF, Snapper SB, Barletta RG, Jacobs WR, Jr, Bloom BR. 1991. New use of BCG for recombinant vaccines. Nature 351:456–460 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.