

Abstract
Acinetobacter sp. strain YAA has five genes (atdA1 to atdA5) involved in aniline oxidation as a part of the aniline degradation gene cluster. From sequence analysis, the five genes were expected to encode a glutamine synthetase (GS)-like protein (AtdA1), a glutamine amidotransferase-like protein (AtdA2), and an aromatic compound dioxygenase (AtdA3, AtdA4, and AtdA5) (M. Takeo, T. Fujii, and Y. Maeda, J. Ferment. Bioeng. 85:17-24, 1998). A recombinant Pseudomonas strain harboring these five genes quantitatively converted aniline into catechol, demonstrating that catechol is the major oxidation product from aniline. To elucidate the function of the GS-like protein AtdA1 in aniline oxidation, we purified it from recombinant Escherichia coli harboring atdA1. The purified AtdA1 protein produced gamma-glutamylanilide (γ-GA) quantitatively from aniline and l-glutamate in the presence of ATP and MgCl2. This reaction was identical to glutamine synthesis by GS, except for the use of aniline instead of ammonia as the substrate. Recombinant Pseudomonas strains harboring the dioxygenase genes (atdA3 to atdA5) were unable to degrade aniline but converted γ-GA into catechol, indicating that γ-GA is an intermediate to catechol and a direct substrate for the dioxygenase. Unexpectedly, a recombinant Pseudomonas strain harboring only atdA2 hydrolyzed γ-GA into aniline, reversing the γ-GA formation by AtdA1. Deletion of atdA2 from atdA1 to atdA5 caused γ-GA accumulation from aniline in recombinant Pseudomonas cells and inhibited the growth of a recombinant Acinetobacter strain on aniline, suggesting that AtdA2 prevents γ-GA accumulation that is harmful to the host cell.
INTRODUCTION
Aniline and its derivatives are very important for the synthesis of chemical products such as dyes, resins, and medicines, but these compounds have toxic, mutagenic, and carcinogenic properties (1–4). Because anilines are widely used in industry and agriculture, they have frequently been detected in aquatic environments, and even in a drinking water treatment plant (5–7). Simple anilines such as aniline and monosubstituted anilines are known to disappear from the environment mainly via biodegradation (8, 9). To understand the mechanisms of aniline biodegradation, many aniline-degrading bacteria have been isolated and characterized (10–25).
Acinetobacter sp. strain YAA has an aniline degradation gene cluster on the plasmid pYA1; the cluster consists of 14 genes (atdA1 to atdA5 and atdRSBCDEFGH) required for the conversion of aniline into tricarboxylic acid (TCA) cycle intermediates (14, 26, 27). The first five genes (atdA1 to atdA5) encode proteins involved in the initial oxidation of aniline to catechol by the release of its amino group, and we tentatively designated them multicomponent aniline dioxygenase (AD) genes (26). The atdA1 gene product shows approximately 30% amino acid (aa) sequence identity with bacterial glutamine synthetases (GSs), while the atdA2 gene product has approximately 30% identity with bacterial glutamine amidotransferases (GATs) (26). The atdA3 to atdA5 gene products share considerable homology with the oxygenase components (large and small subunits) and the reductase component, respectively, of two-component Rieske-type aromatic compound dioxygenases (26, 28, 29). This dioxygenase homolog belongs to group II in the classification of Nam et al. (28), based on large-subunit aa sequence analysis, and to type Iαβ in the later classification of Kweon et al. (29), adding information on the electron transfer chain components to large-subunit sequence analysis. Similar AD gene clusters have been cloned from Pseudomonas putida UCC22 (30), Delftia acidovorans 7N (23), Delftia tsuruhatensis AD9 (18), Frateuria sp. strain ANA-18 (31), Delftia sp. strain AN3 (32), and Comamonas testosteroni I2 (33). However, the mechanism of aniline oxidation has not been defined, as there is little information on the functions of the gene products involved in the process. In particular, the GS-like and GAT-like proteins are quite unique; these proteins have so far not been identified in other aromatic compound dioxygenases, with the only exception being a GS-like protein gene (sadB) in the 4-aminobenzenesulfonate 3,4-dioxygenase gene cluster (sadABD) of Hydrogenophaga sp. strain PBC (34). However, the function of the gene product is still unknown.
de Azevedo Wäsch et al. (35) reported the conversion of isopropylamine into l-alaninol in Pseudomonas sp. strain KIE171. In this process, as shown in Fig. 1A, a GS-like protein (IpuC) catalyzes the formation of γ-glutamylisopropylamide from isopropylamine and l-glutamate, and a GAT-like protein (IpuF) hydrolyzes the amide after the oxidation of the isopropyl group by other enzymes (IpuABDE). These reactions resemble the protection and deprotection of a reactive amino group in chemical synthesis. Similar reactions via γ-glutamylated intermediates occur in the putrescine utilization pathway of Escherichia coli K-12 (36). Considering the functions of IpuC and IpuF and their similarity with AtdA1 (31%) and AtdA2 (26%), we predicted the functions of the Atd proteins in aniline oxidation (Fig. 1B). In this oxidation reaction, a GS-like protein, AtdA1, forms γ-glutamylanilide (γ-GA) from aniline and l-glutamate, and the dioxygenase proteins AtdA3, AtdA4, and AtdA5 oxidize the aromatic-ring of γ-GA. A GAT-like protein, AtdA2, hydrolyzes the oxidized product into l-glutamate and an unstable cyclohexadiene with an amino group, which spontaneously forms catechol and ammonia.
Fig 1.

Isopropylamine oxidation mechanism of Pseudomonas sp. KIE171 (35) (A) and putative aniline oxidation mechanism of Acinetobacter sp. YAA (B).
To confirm this putative pathway, γ-GA was synthesized chemically as an authentic compound, and a Pseudomonas strain that does not degrade catechol was constructed by gene disruption to evaluate the quantitative formation of catechol from aniline or γ-GA. To investigate the first step of the oxidation reaction, AtdA1 was purified from recombinant E. coli harboring atdA1, and aniline conversion to γ-GA was investigated at the enzymatic level. Conversion of γ-GA to catechol was also examined in Pseudomonas strains expressing the remaining dioxygenase genes (atdA3 to atdA5 or atdA2 to atdA5). These experiments revealed that aniline is oxidized via γ-GA into catechol in Acinetobacter sp. YAA and that the γ-GA formation is catalyzed by AtdA1. The putative function of AtdA2 in aniline oxidation is also discussed, based on the results of the gene deletion study.
MATERIALS AND METHODS
Bacterial strains, plasmids, primers, media, and cultivation conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. The primers used in this study are listed in Table S1 in the supplemental material. Escherichia coli strains were cultivated at 37°C, while Pseudomonas and Acinetobacter strains were cultivated at 30°C. Bacterial cultures were maintained on a rotary shaker at 140 rpm. Luria-Bertani (LB) medium was used as a rich medium for growth, and Terrific broth (TB) was used for protein production (37). MSB medium (pH 7.6), consisting of 1 g K2HPO4, 1 g (NH4)2SO4, 0.2 g MgSO4 · 7H2O, 0.02 g FeCl3, 0.1 g NaCl, and 0.1 g CaCl2 (liter−1), was used for the growth test on aniline.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Characteristics | Reference or source |
|---|---|---|
| Strains | ||
| Escherichia coli JM109 | recA1 endA1 gyrA96 thi-1 hsdR17(rK− mK+) e14− (ΔmcrA) supE44 relA1 Δ(lac-proAB) [F′ traD36 proAB+ lacIqZΔM15] | TaKaRa Bio, Kyoto, Japan |
| E. coli S17-1 | RP4-2 (Km::Tn7 Tc::Mu-1) pro-82 recA1 endA1 thiE1 hsdR17 creC510 | 46 |
| E. coli SO58 | K-12 derivative; F− Δggt-2 ΔycjKLC::cat+FRT Δ(aldH-ordL-goaG)::kan+ | 36 |
| Pseudomonas putida KT2440 | mt-2 derivative; hsdR1(r− m+) Cmr | 40 |
| P. putida KT2440-ΔcatA | KT2440 mutant; ΔcatA1 ΔcatA2; catechol degradation negative | This study |
| P. putida KT2440-ΔcatAΔggt | KT2440 mutant; ΔcatA1 ΔcatA2 Δggt-2; catechol and γ-GA degradation negative | This study |
| Acinetobacter baylyi BD413 | Unencapsulated mutant of A. baylyi BD4; ATCC 33305 | 53 |
| Plasmids | ||
| pUC18 | Apr; cloning vector; 2.7 kb; lacOP | TaKaRa Bio, Kyoto, Japan |
| pUC19 | Apr; cloning vector; 2.7 kb; lacOP | TaKaRa Bio, Kyoto, Japan |
| pAS185 | Apr; 18.5-kb SalI fragment of pYA1 in pUC19; atdA1 to atdA5 | 14 |
| pGS18 | Apr; 1.5-kb BamHI fragment containing atdA1 cloned into pUC18 | This study |
| pBBR1MCS-2 | Kmr; broad-host-range vector; 5.1 kb; mob lacOP | 58 |
| pTB01 | 5.3-kb SalI-SmaI fragment containing atdA1 to atdA5 in pBBR1MCS-2 | 48 |
| pK18mobsacB | Kmr; vector for gene disruption; mob sacB lacOP | 45 |
| pKA1 | 1.1-kb fragment containing ΔcatA1 mutation (lacking the 394-bp central part of catA1) in pK18mobsacB | This study |
| pKA2 | 1.2-kb fragment containing ΔcatA2 mutation (lacking the 389-bp central part of catA2) in pK18mobsacB | This study |
| pKGGT-2 | 1.4-kb fragment containing Δggt-2 mutation (lacking the 936-bp central part of ggt-2) in pK18mobsacB | This study |
| pBA2 | 0.7-kb fragment containing atdA2 in pBBR1MCS-2 | This study |
| pTB01-ΔA1 | pTB01 derivative lacking atdA1 | This study |
| pTB01-ΔA2 | pTB01 derivative lacking atdA2 | This study |
| pTB01-ΔA12 | pTB01 derivative lacking atdA1A2 | This study |
Chemical synthesis of γ-GA.
γ-GA was chemically synthesized as shown in Fig. S1 in the supplemental material, and γ-GA and its synthetic intermediates were analyzed by 1H nuclear magnetic resonance (1H-NMR) and Fourier transform infrared spectroscopy (FTIR) as described previously (38, 39). The analytical data are shown in Table S2.
DNA purification, PCRs, electroporation, and other DNA techniques.
Total DNA was extracted from Pseudomonas putida KT2440 (40) or its derivatives by the method of Saito and Miura (41). PCR was performed with a TaKaRa PCR Dice thermal cycler (TaKaRa Bio, Kyoto, Japan). The standard PCR mixture contained 10 ng of template DNA, 10 μmol of each primer, a 2.5 mM concentration of each deoxynucleoside triphosphate (dNTP), 5 μl of 10× Ex Taq buffer, and 0.5 U of Ex Taq polymerase (TaKaRa Bio) in a total volume of 50 μl. The standard PCR conditions were as follows: 94°C for 5 min; 30 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min kb−1; and 72°C for 5 min. For colony PCR, a small amount of bacteria was used instead of the template DNA. DNA fragments were purified from agarose gel by use of TaKaRa Suprec-01 cartridges (TaKaRa Bio), and ligation reactions were performed with a TaKaRa DNA ligation kit (ver.2.1; TaKaRa Bio). P. putida KT2440 was transformed with plasmids (electroporation) as described previously (38), but with a modified voltage (2.5 kV). Plasmid DNA preparation, restriction digestion, and transformation were performed according to standard methods (37).
Construction of recombinant plasmids pGS18 and pBA2.
A 1.5-kb DNA fragment containing atdA1 was amplified from pAS185 (Table 1) by a PCR using primers A1F and A1R and subsequently was digested with BamHI. The fragment was ligated into BamHI-digested pUC18 (Table 1) to form pGS18. Similarly, a 0.7-kb DNA fragment containing atdA2 was amplified from pAS185 by using primers A2F-salI and A2R and then was doubly digested with BamHI and SalI. The fragment was ligated into doubly digested pBBR1MCS-2 (Table 1) to form pBA2. The sequences of the inserts in these plasmids were confirmed by nucleotide sequencing.
Preparation of E. coli cell extract and purification of AtdA1.
E. coli SO58 harboring pGS18 was grown overnight in 300 ml of TB with 0.25 mM isopropyl-β-d-thiogalactoside (IPTG) and 100 mg liter−1 ampicillin. Cells were harvested by centrifugation (4°C, 10,000 × g, 10 min), washed twice with 10 mM sodium phosphate buffer (pH 7.0), and suspended in 15 ml of the same buffer. The bacterial cells were disrupted using a model UD-200 ultrasonic disruptor (Tomy, Tokyo, Japan) (vol. 6, 10 times for 1 min each on ice). After pelleting of the cell debris and unbroken cells by centrifugation (4°C, 15,000 × g, 30 min), the supernatant was used as a cell extract for protein purification.
A Hi-Trap Q Sepharose column (5 ml) (GE Healthcare Japan, Tokyo, Japan) was preequilibrated with phosphate buffer, and the cell extract was loaded onto the column. After washing with phosphate buffer, the target protein was eluted at 2 ml min−1 with an NaCl gradient (total volume, 180 ml) from 0 M to 0.6 M, using a Biologic LP chromatography system (Bio-Rad Japan, Tokyo, Japan), and the last 90 ml of eluate was fractionated into 30 3-ml fractions, which were analyzed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) as described previously (26). Fractions containing the target protein were collected, concentrated with a Vivaspin 6-10K concentrator (GE Healthcare), and desalted using a PD-10 desalting column (GE Healthcare). The desalted solution was concentrated again with the Vivaspin 6-10K concentrator to a volume suitable for the next step. The described anion-exchange chromatography and concentration processes were repeated.
A Hi-Trap butyl FF column (1 ml) (GE Healthcare) was preequilibrated with 10 mM sodium phosphate buffer containing 1 M (NH4)2SO4, and the concentrated protein solution was loaded onto the column. The target protein was eluted at 1 ml min−1 with a gradient of (NH4)2SO4 solution (total volume, 30 ml) from 1 M to 0 M, using the same chromatography system, and was fractionated into 30 1-ml fractions, which were analyzed and purified as described above. The protein concentration was determined by the method of Lowry et al. (42), using bovine serum albumin (Nacalai Tesque, Kyoto, Japan) as a protein standard.
Assay conditions for aniline conversion by AtdA1 and determination of its GS activity.
The standard reaction mixture for the conversion of aniline to γ-GA by AtdA1 contained 0.5 mM aniline, 1 mM l-glutamate, 0.5 mM ATP, 5 mM MgCl2, and 1 μg of AtdA1 in 1 ml of 10 mM sodium phosphate buffer, pH 8.0. The reactions were performed at 40°C. The pH and temperature were optimized prior to fixing these assay conditions (see Fig. S5 in the supplemental material). The GS activity of AtdA1 was measured based on the method of Listrom et al. (43), but employing the above-mentioned reaction mixture and temperature, except for the addition of hydroxylamine instead of aniline to the mixture and the use of an excess amount (10 μg) of AtdA1.
Gel filtration chromatography.
The native molecular mass of AtdA1 was determined at room temperature by use of a Shimadzu Prominent high-performance liquid chromatography (HPLC) system (Shimadzu, Kyoto, Japan) (44) equipped with a TSKgel G3000SWXL column (300-mm length, 7.8-mm internal diameter [ID], 5-μm particles) (Tosoh, Tokyo, Japan) and a refractive index detector (RI-8020; Tosoh). The HPLC conditions were as follows: mobile phase, 0.1 M KH2PO4, 0.1 M Na2SO4, 0.05% NaN3; detection wavelength, 280 nm; flow rate, 1.0 ml min−1; and injection volume, 20 μl.
Gene disruption by homologous recombination.
Gene disruption by homologous recombination in P. putida KT2440 was performed as previously described by Schäfer et al. (45).
(i) Disruption of catechol 1,2-dioxygenase genes (catA1 and catA2).
Two DNA fragments (587 bp and 509 bp), which included the 5′-end region and the 3′-end region of the catechol 1,2-dioxygenase gene of KT2440 (catA2) (912 bp; product accession no. NP_745310), respectively, were amplified from the total KT2440 DNA by PCR with two primer sets: cA2F-F and cA2F-R for the 5′-end region and cA2R-F and cA2R-R for the 3′-end region (see Table S1 in the supplemental material). The PCR conditions were identical to those described above except for the annealing temperature (60°C). The 5′-end-region fragment was digested with EcoRI and BamHI, while the 3′-end-region fragment was digested with BamHI and XbaI. These fragments were cloned together into EcoRI- and XbaI-digested pK18mobsacB (Table 1) to form pKA2, which had an incomplete catA2 gene (ΔcatA2) lacking a 389-bp central region. pKA2 was used to transform E. coli S17-1. pKA2 was then transferred from E. coli S17-1 to P. putida KT2440 by biparental mating (46), and the transconjugants were selected on LB plates containing kanamycin (100 mg liter−1) and chloramphenicol (50 mg liter−1). Kanamycin resistance of the transconjugants originated from pK18mobsacB, whereas chloramphenicol resistance was the original phenotype of P. putida KT2440 (47). This method selected the first-crossover mutants of KT2440, which had the pKA2 sequence inserted into the genome by homologous recombination; pKA2 cannot replicate in KT2440. After colony PCR using primers cA2F-F and cA2R-R to confirm the presence of catA2 and the ΔcatA2 mutation in the transconjugants (see Fig. S2A), one clone was incubated in LB medium at 30°C for 12 h. The culture was spread on LB plates including 10% (wt/vol) sucrose, and the plates were incubated at 30°C for a few days. By this selection, second-crossover mutants or revertants of KT2440 were obtained (see Fig. S2), as the sacB gene in pKA2 is lethal to the host strain grown on sucrose-containing media (45). Colonies containing the ΔcatA2 mutation were checked by colony PCR (see Fig. S2A).
Similarly, two DNA fragments (541 bp and 611 bp) of another catechol 1,2-dioxygenase gene (catA1) (933 bp; product accession no. NP_745846) were amplified from the total KT2440 DNA as described above, using two primer sets: cA1F-F and cA1F-R for the 5′-end region and cA1R-F and cA1R-R for the 3′-end region (see Table S1 in the supplemental material). The resultant fragments were doubly digested with EcoRI and HindIII for the former or with HindIII and XbaI for the latter and were inserted together into EcoRI- and XbaI-digested pK18mobsacB to form plasmid pKA1, which had an incomplete catA1 gene (ΔcatA1) lacking a 394-bp central region. pKA1 was transferred into the ΔcatA2 mutant of KT2440 to make the ΔcatA1 ΔcatA2 double mutant. This catA1 disruption was also confirmed by PCR (see Fig. S2B).
(ii) Disruption of γ-glutamyltranspeptidase gene (ggt-2).
Two DNA fragments (458 bp and 902 bp) of the γ-glutamyltranspeptidase gene (ggt-2) of KT2440 (1,666 bp; product accession no. NP_746768) were amplified from the total DNA by PCR as described above, using two primer sets: ggt-2FF-H and ggt-2FR for the former fragment and ggt-2RF and ggt-2RR for the latter (see Table S1 in the supplemental material). The resultant fragments were doubly digested with XbaI and SalI for the former or with SalI and BamHI for the latter and were inserted together into XbaI- and BamHI-digested pK18mobsacB to form pKGGT-2, which had an incomplete ggt-2 gene (Δggt-2) lacking a 936-bp central region. pKGGT-2 was introduced into the ΔcatA1 ΔcatA2 double mutant as described above to make the ΔcatA1 ΔcatA2 Δggt-2 triple mutant of KT2440.
Construction of pTB01 derivatives lacking atdA1, atdA2, or atdA1 and atdA2 (atdA1A2).
An 8.8-kb DNA fragment was amplified from pTB01 (Table 1) by a PCR using PrimeSTAR Max DNA polymerase (TaKaRa Bio) and primers F-pAS51 and pTB01-A2-5R (see Table S1 in the supplemental material). This primer set was designed to amplify all of the pTB01 sequence except for the atdA1 gene. The fragment was self-ligated to construct pTB01-ΔA1, which was used to transform E. coli JM109, P. putida KT2440, and P. putida derivatives.
Similarly, 9.7-kb and 8.1-kb DNA fragments were independently amplified by PCRs using the same polymerase and two different primer sets (primers A3F and A1R2 and primers A3F and pTB01-A2-5R) (see Table S1 in the supplemental material). The former set was designed to amplify all of the pTB01 sequence except for the atdA2 gene, while the latter was designed to amplify all of the pTB01 sequence except for the atdA1 and atdA2 genes. The amplified fragments were self-ligated to construct pTB01-ΔA2 and pTB01-ΔA12, respectively, and used to transform the host strains. The loss of atdA1, adtA2, or atdA1A2 in these deletion plasmids was confirmed by nucleotide sequencing.
Degradation of aniline and γ-GA in cell suspensions.
Recombinant P. putida KT2440 or its gene-disrupted strains were cultivated in LB medium containing kanamycin (100 mg liter−1) for 24 h, and cells were harvested by centrifugation (4°C, 8,000 × g, 10 min). The cells were washed three times with cold sterile water and suspended in a small amount of 10 mM sodium phosphate buffer (pH 7.0). The suspension was diluted with MSB medium to adjust the optical density at 600 nm (OD600) to 5.0. Aniline or γ-GA was added to the suspension, and degradation experiments were performed at 30°C and 150 rpm on a rotary shaker. Samples were taken at specific intervals, and the substrates and their metabolites were analyzed by HPLC. A Shimadzu Prominent HPLC system (Shimadzu) equipped with a Mightysil RP18 GP Aqua column (150- to 4.6-mm ID by 5 μm) (Kanto Kagaku Kogyo, Tokyo, Japan) (44) was used for the analysis, and the HPLC conditions were as follows: mobile phase, ratio of solution A (5% CH3CN, 95% H2O, 0.1% CH3COOH) to solution B (95% CH3CN, 5% H2O, 0.1% CH3COOH) of 61.9:38.1 or 70:30; detection wavelengths, 254 nm and 277 nm; flow rate, 0.5 ml min−1; and injection volume, 10 μl.
RESULTS
Construction of non-catechol-degrading Pseudomonas putida KT2440.
We previously constructed the recombinant plasmid pTB01 (48), which had a 5.3-kb DNA fragment including the complete AD gene cluster (atdA1 to atdA5), in the broad-host-range plasmid pBBR1MCS-2. An E. coli strain harboring pTB01 showed a strong brown color around the colonies on aniline-containing plates (most likely from catechol formation and auto-oxidation). However, in liquid cultures, a significant decrease in aniline concentration was not detected by HPLC analysis, and only a trace amount of catechol was detected by gas chromatography-mass spectrometry analysis due to the poor enzymatic activity of the gene products expressed in E. coli. In contrast, P. putida KT2440 harboring pTB01 degraded aniline rapidly (48) (the cell suspension at an OD600 of 2 degraded 100 mg liter−1 of aniline within 70 min). Catechol was not detected as the metabolite by HPLC, because this strain could further degrade catechol by use of endogenous catechol 1,2-dioxygenases (encoded by catA1 and catA2 on the genome) (49). To evaluate the degradation of aniline or its intermediates into catechol, it was necessary to quantify the catechol. Therefore, we generated a mutant of KT2440 that could not degrade catechol by disrupting catA1 and catA2.
We constructed two recombinant plasmids, pKA1 and pKA2, which had ΔcatA1 and ΔcatA2 mutations lacking the central segments of catA1 and catA2, respectively, in pK18mobsacB. pKA2 was first introduced into E. coli S17-1 and then transferred into P. putida KT2440 by mating. After the selection of kanamycin- and chloramphenicol-resistant strains (first-crossover mutants) and subsequent sucrose-resistant strains (second-crossover mutants), several ΔcatA2 mutants were obtained, and the deletion in catA2 was confirmed by PCR (see Fig. S2A in the supplemental material). Furthermore, pKA1 was introduced into one of the ΔcatA2 mutants in the same manner. Finally, several ΔcatA1 ΔcatA2 double mutants were obtained (see Fig. S2B). The double mutant was designated P. putida KT2440-ΔcatA. The KT2440-ΔcatA strain accumulated catechol from benzoate stoichiometrically (data not shown), as it had a benzoate degradation pathway via catechol (49). This phenotype strongly supported the disruption of catA1 and catA2.
Conversion of aniline using KT2440-ΔcatA harboring pTB01.
To investigate aniline conversion in KT2440-ΔcatA, pTB01 including the complete AD gene cluster (atdA1 to atdA5) was introduced into this strain, and the resultant strain was used for aniline degradation studies. As shown in Fig. 2, the cell suspension of this strain converted aniline into catechol almost quantitatively, indicating that catechol is a major oxidation product from aniline.
Fig 2.
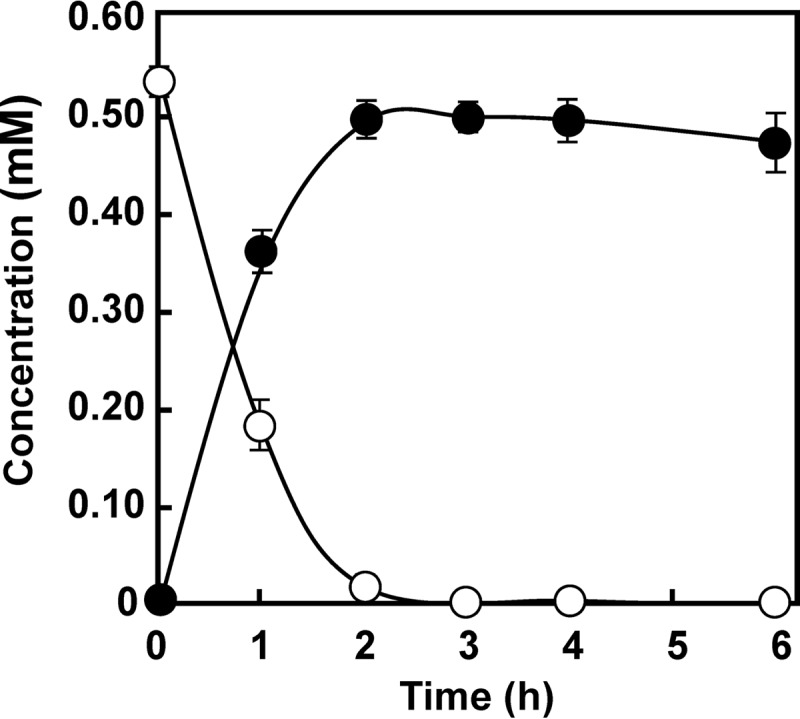
Conversion of aniline into catechol by a cell suspension of P. putida KT2440-ΔcatA harboring pTB01 (atdA1 to atdA5). Symbols: open circles, aniline; closed circles, catechol. The experiment was performed in triplicate, and averages ± standard deviations are shown.
Conversion of aniline by AtdA1.
To examine the function of AtdA1 in aniline oxidation, we planned a similar aniline conversion experiment using cell suspensions of KT2440-ΔcatA harboring only atdA1. However, our preliminary study revealed that the putative product, γ-GA, was easily degraded into aniline and l-glutamate through an unknown endogenous hydrolytic activity of this strain (this problem was essentially solved, as described later). Thus, it was difficult to quantify γ-GA in the degradation tests using this strain. To overcome this limitation, purified AtdA1 was used in an in vitro aniline conversion experiment.
A recombinant E. coli SO58 strain containing atdA1 (in plasmid pGS18) was grown in TB in the presence of IPTG, and its cell extract was prepared. SDS-PAGE of the cell extract showed a 57-kDa protein that was absent in the cell extract of the empty vector control strain and that was very close in size to the predicted molecular mass of AtdA1 (55 kDa). The protein was purified to a single band in SDS-PAGE gels through two cycles of anion-exchange chromatography and hydrophobic chromatography (see Fig. S3 in the supplemental material). The specific aniline conversion activity of the purified protein solution was 22-fold higher than that of the cell extract (see Table S3) (measured under the optimized assay conditions shown below). Gel filtration chromatography showed that its native size was approximately 350 kDa (see Fig. S4), indicating that AtdA1 forms a hexameric structure.
As the aa sequence of AtdA1 showed approximately 30% identity with bacterial GSs (26), aniline conversion by AtdA1 was investigated in a reaction mixture similar to that used for the measurement of GS activity (43, 50), which included l-glutamate, ATP, and MgCl2. Aniline was added to the reaction mixture instead of ammonia. As shown in Fig. 3, aniline (0.5 mM) was reduced by 80% in 8 h, and an almost equal amount of γ-GA was formed. Removal of any of the components (l-glutamate, ATP, or MgCl2) resulted in no conversion (data not shown). Therefore, like GS, AtdA1 requires these components to convert aniline. Maximum activity occurred between pH 8 and pH 10, and the optimum temperature was approximately 40°C (see Fig. S5 in the supplemental material). Under the same conditions, AtdA1 converted the following anilines (with percent conversion listed): aniline (100%), o-chloroaniline (92%), m-chloroaniline (69%), p-chloroaniline (92%), o-methylaniline (40%), m-methylaniline (27%), and p-methylaniline (45%). These results indicate that AtdA1 has broad substrate specificity and prefers o- and p-substituted anilines. AtdA1 showed no GS activity (<0.001 U mg−1), although a positive-control GS from Bacillus stearothermophilus (Unitika, Osaka, Japan) showed considerable GS activity (0.200 U mg−1).
Fig 3.
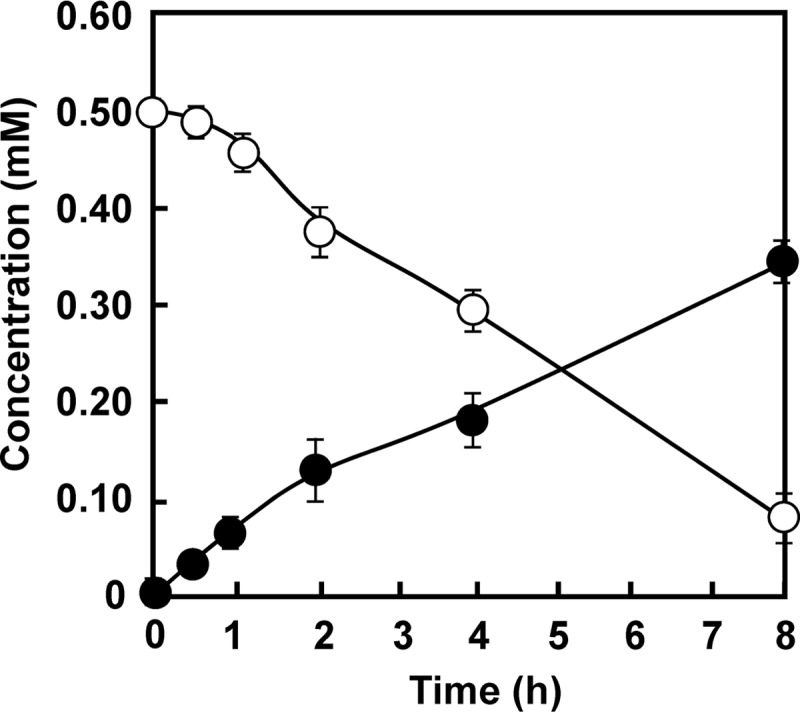
Conversion of aniline into γ-GA by AtdA1. Symbols: open circles, aniline; closed circles, γ-GA. The experiment was performed in triplicate, and averages ± standard deviations are shown.
Construction of ggt-2-disrupted mutant of KT2440-ΔcatA.
As described above, in P. putida KT2440, γ-GA was degraded into aniline by an unknown endogenous hydrolytic activity (see Fig. S6 in the supplemental material). Thus, this strain was not an ideal host for performing γ-GA degradation experiments. Kurihara et al. (36) used E. coli SO58 with a mutation in the γ-glutamyltranspeptidase gene (ggt-2 mutant) to study the putrescine utilization pathway via γ-glutamylated intermediates. γ-Glutamyltranspeptidase catalyzes the hydrolysis of the γ-glutamyl linkage of glutathione (γ-glutamylcysteinylglycine) and uses γ-GA and γ-glutamyl-p-nitroanilide as substrates in colorimetric assays (51, 52). In fact, γ-GA was not degraded in E. coli SO58 (ggt-2) (data not shown). We found two putative γ-glutamyltranspeptidase genes (ggt-1 and ggt-2) and some homologous genes in the registered genome sequence of KT2440 (accession no. NC_002947). These genes were independently disrupted in the same way as that utilized for the catA1 and catA2 disruptions. As expected, when ggt-2 was disrupted in KT2440-ΔcatA, the mutant strain no longer degraded γ-GA (see Fig. S6). Thus, we named this triple mutant KT2440-ΔcatAΔggt and used it for γ-GA degradation studies.
Conversion of γ-GA by KT2440-ΔcatAΔggt harboring pTB01-ΔA1.
We constructed pTB01-ΔA1, which was an atdA1-deleted version of plasmid pTB01. This plasmid was introduced into KT2440-ΔcatAΔggt. From the proposed pathway shown in Fig. 1B, this recombinant strain was expected to convert γ-GA into catechol, because it contained atdA2 to atdA5. Unexpectedly, γ-GA was almost quantitatively converted into aniline (Fig. 4A), although a small amount of catechol was detected (≤0.02 mM). We suspected that AtdA2 catalyzed this unexpected conversion, because AtdA3, AtdA4, and AtdA5 are putative members of the two-component dioxygenase (26, 28, 29). In fact, the cell suspension of KT2440-ΔcatAΔggt harboring only atdA2 (in plasmid pBA2) efficiently degraded γ-GA into aniline (Fig. 4B). To prevent this activity, we constructed an atdA1A2-deleted plasmid, named pTB01-ΔA12, and γ-GA degradation was again carried out, using KT2440-ΔcatAΔggt cells harboring pTB01-ΔA12. As a result, the cell suspension was able to convert γ-GA into catechol (≤0.05 mM), but aniline was not formed as expected (Fig. 4C). This result shows that the two-component dioxygenase (AtdA3, AtdA4, and AtdA5) can convert γ-GA into catechol. The same cell suspension never degraded aniline (data not shown), indicating that aniline is not a direct substrate for the dioxygenase. When the cell suspension of KT2440-ΔcatAΔggt harboring pTB01 (atdA1 to atdA5) was tested for γ-GA degradation, aniline accumulated from γ-GA at the beginning of the reaction, but γ-GA was almost completely converted into catechol (Fig. 5). The presence of both the atdA1 and atdA2 genes significantly improved γ-GA degradation.
Fig 4.

Degradation of γ-GA by cell suspensions of KT2440-ΔcatAΔggt harboring pTB01-ΔA1 (A), pBA2 (B), or pTB01-ΔA12 (C). Symbols: open circles, γ-GA; closed circles, catechol; closed triangles, aniline. The experiments were performed in triplicate, and averages ± standard deviations are shown.
Fig 5.
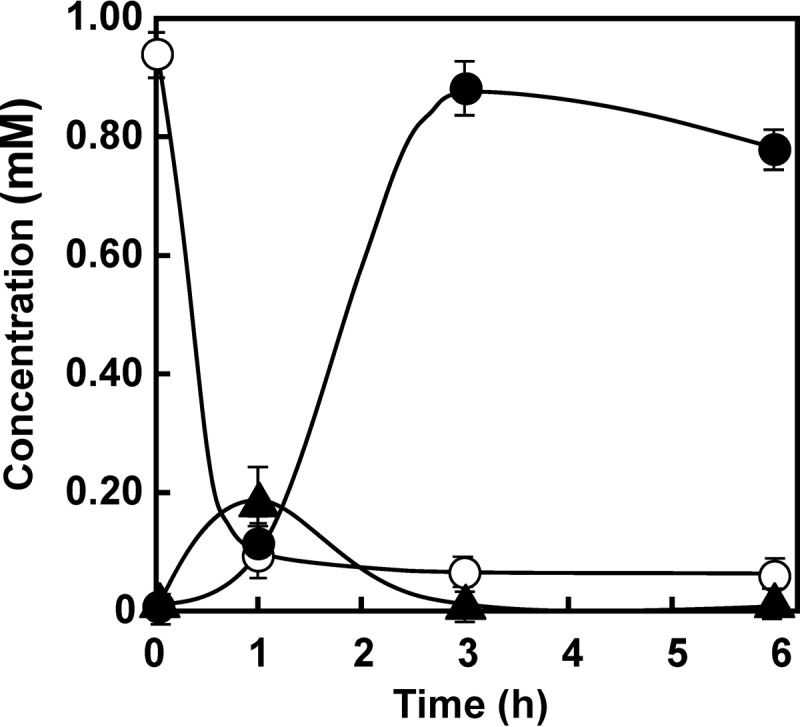
Conversion of γ-GA into catechol by a cell suspension of P. putida KT2440-ΔcatAΔggt harboring pTB01 (atdA1 to atdA5). Symbols: open circles, γ-GA; closed circles, catechol; closed triangles, aniline. The experiment was performed in triplicate, and averages ± standard deviations are shown.
Conversion of aniline by P. putida KT2440-ΔcatAΔggt harboring pTB01-ΔA2.
To understand the function of atdA2 in aniline oxidation, atdA2 was deleted from pTB01 to make pTB01-ΔA2. As shown in Fig. 6A, KT2440-ΔcatAΔggt harboring pTB01 reduced 0.4 mM aniline (from 0.6 mM to 0.2 mM) in 4 h, and a major amount of catechol was detected. A small amount of γ-GA was also detected. In contrast, KT2440-ΔcatAΔggt harboring pTB01-ΔA2 degraded aniline similarly, but a major amount of γ-GA was accumulated (Fig. 6B). This clearly shows that AtdA2 prevents γ-GA accumulation in the cell.
Fig 6.

Conversion of aniline into catechol by cell suspensions of P. putida KT2440-ΔcatAΔggt harboring pTB01 (atdA1 to atdA5) (A) or pTB01-ΔA2 (atdA1 and atdA3 to atdA5) (B). The experiments were performed in triplicate, and averages ± standard deviations are shown. Symbols: open circles, aniline; closed circles, catechol; closed triangles, γ-GA.
Growth of Acinetobacter baylyi BD413 harboring pTB01-ΔA2 on aniline.
To simulate the growth of Acinetobacter sp. YAA lacking atdA2 on aniline, A. baylyi BD413 (formerly termed Acinetobacter sp. or Acinetobacter calcoaceticus) (53) was used as a host strain for pTB01-ΔA2, because in strain YAA, with several cryptic plasmids (14), it was difficult to delete only atdA2 on the aniline degradation plasmid pYA1. The host BD413 strain was able to assimilate catechol but unable to degrade γ-GA (data not shown), indicating that there was no endogenous γ-GA hydrolytic activity. pTB01 and pTB01-ΔA2 were independently introduced into BD413 as described previously (48). The resultant BD413 strains were inoculated into MSB medium containing aniline as the sole carbon source at an OD600 of 0.1 and then cultivated with shaking. BD413 harboring pTB01 completely degraded 0.15 mM aniline within 1 day, as well as additional spikes of aniline administered day by day (Fig. 7A). The growth reached an OD600 of approximately 0.4 after 5 days. During the degradation, γ-GA was not detected at all. In contrast, BD413 harboring pTB01-ΔA2 was unable to degrade 0.15 mM aniline completely even after 2 days of incubation, and a small amount of γ-GA was detected (Fig. 7B). The remaining aniline at day 2 was not reduced during a further 3-day incubation. In a repeated experiment, we added 0.2 mM aniline at day 2, but no further growth was observed (data not shown).
Fig 7.

Growth of Acinetobacter baylyi BD413 harboring pTB01 (atdA1 to atdA5) (A) or pTB01-ΔA2 (atdA1 and atdA3 to atdA5) (B) on aniline. Arrows indicate the addition of aniline. Symbols: open circles, aniline; closed circles, growth (OD600); closed triangles, γ-GA.
DISCUSSION
To date, several AD genes have been cloned and characterized (14, 18, 23, 30–33), but the aniline oxidation mechanisms have not been elucidated, that is, the concrete functions of the products of the AD genes in aniline oxidation are unknown. All the AD gene clusters found so far encode GS-like and GAT-like proteins, which have not been found in other dioxygenases, with one exception (34). In this study, we proposed an aniline oxidation mechanism for Acinetobacter sp. YAA that occurs via γ-GA, referring to the isopropylamide conversion mechanism of Pseudomonas sp. KIE171 (Fig. 1), in which GS-like and GAT-like proteins are involved in the formation and hydrolysis of the γ-glutamyl intermediates (35).
We first proved that catechol is the major oxidation product from aniline by using a ΔcatA1 ΔcatA2 mutant of P. putida KT2440 (Fig. 2). Catechols are known to be the oxidation products from aniline, methylaniline, and chloroaniline (14, 18, 32, 33), but this is the first report to have detected a major amount of catechol from aniline. Next, to prove the presence of γ-glutamyl intermediates in aniline oxidation, we purified the GS-like protein AtdA1 and showed that it can produce γ-GA from aniline and l-glutamate in the presence of ATP and MgCl2 (Fig. 3). γ-GA was previously detected as a water-soluble metabolite in the aniline metabolism of a cattle tick, Boophilus microplus (54), but the formation mechanism was unknown. This enzymatic reaction was almost identical to glutamine synthesis by GS (l-glutamate:ammonia ligase; EC 6.3.1.2) (43, 50), except that aniline was the substrate instead of ammonia. Thus, AtdA1 could be designated an “l-glutamate:aniline ligase,” but here we call it a “γ-GA synthetase” to emphasize the product name γ-GA. Bacterial GS forms a dodecamer consisting of two face-to-face hexameric rings of identical subunits (55). The gel filtration chromatography result suggested that γ-GA synthetase forms a hexameric structure. Therefore, in addition to the sequence similarity with GS, γ-GA synthetase may be structurally similar to GS. Nevertheless, it showed no GS activity. Fukumori and Saint (30) showed that the tdnQ gene of P. putida UCC22 (encoding a GS-like protein) was unable to complement the glutamine requirement of an E. coli glnA (GS gene) mutant. This result is in good agreement with our result.
Without atdA1, aniline was never oxidized, even if all other AD genes were present in KT2440 derivatives (e.g., KT2440-ΔcatAΔggt containing pTB01-ΔA1). This suggests that γ-GA formation is necessary prior to the oxygenation of the aromatic ring of aniline. Fukumori and Saint (30) deleted the tdnQ region from the AD gene cluster of strain UCC22. A recombinant P. putida KT2442 strain harboring tdnTA1A2B no longer showed oxygen uptake for aniline or ammonia-releasing activity. This strain failed to grow on aniline. Murakami et al. (31) also reported that the loss of tdnQ resulted in no oxygen uptake for aniline in recombinant E. coli cells harboring the AD genes from Frateuria sp. ANA-18. Moreover, tdnQ, TdnQ, and their homologs have always been detected in many aniline-degrading bacteria by PCR and proteomic analysis (13, 23, 56). These facts show that GS-like proteins (γ-GA synthetase) are indispensable for aniline oxidation.
The KT2440 derivatives harboring the dioxygenase genes (atdA3 to atdA5) converted γ-GA into catechol (Fig. 4A and C). This is strong evidence demonstrating that γ-GA formation is the starting point of the proposed aniline oxidation mechanism (pathway). Recently, Król et al. (33) reported that recombinant E. coli cells harboring only the dioxygenase genes (dcaA1A2B) from the AD gene cluster (dcaQTA1A2B) of Comamonas testosteroni produced a small amount of 4-chlorocatechol from 3-chloroaniline. Therefore, this dioxygenase may be able to attack anilines directly without GS-like and GAT-like proteins. However, we believe that the major pathway is via γ-glutamylated intermediates, because the gene cluster still keeps dcaQT, encoding the GS-like and GAT-like proteins.
Considering the function of a GAT-like protein, IpuF, in the isopropylamide conversion mechanism of Pseudomonas sp. KIE171 (35) (Fig. 1A), we expected that AtdA2 would catalyze the hydrolysis of the putative oxidized product of γ-GA (Fig. 1B). However, unexpectedly, in the presence of atdA2, γ-GA was easily degraded into aniline (Fig. 4B). This is a reverse reaction against the formation of γ-GA at the expense of ATP by AtdA1 (Fig. 3). Thus, the action of AtdA2 has a confounding effect. One possible explanation for this reverse reaction is control of the cellular γ-GA concentration. In fact, the deletion of atdA2 from atdA1 to atdA5 caused the accumulation of a considerable amount of γ-GA from aniline in recombinant Pseudomonas cells (Fig. 6B). Thus, atdA2 obviously contributes to the prevention of γ-GA accumulation in the host cell. Moreover, A. baylyi BD413 containing the AD gene cluster without atdA2 lost its aniline degradation ability when grown on aniline (Fig. 7B), probably due to γ-GA formation. This host strain has no γ-GA-hydrolyzing activity and thus cannot scavenge γ-GA once it is formed. γ-GA and/or its putative oxidized product must inhibit the dioxygenase activity and be harmful to host cells, because γ-GA degradation always stops at the beginning of the degradation (Fig. 4A and C). The addition of atdA1A2 to the atdA3 to atdA5 genes significantly improved γ-GA degradation (cf. Fig. 4C and 5), as AtdA1 should provide γ-GA for the dioxygenase, while AtdA2 might keep the γ-GA concentration suitable for (or not enough to inactivate) the dioxygenase in the cell.
When aniline conversion was compared between KT2440 strains with and without ggt-2 (cf. Fig. 1 and 6A), the former was more efficient and rapid than the latter under similar conditions. γ-GA was detected only in the latter. This observation suggests that like atdA2, the host ggt-2 gene also prevents γ-GA accumulation and improves aniline degradation. Fukumori and Saint (30) reported that recombinant P. putida KT2442 harboring the AD gene cluster of strain UCC22 without tdnT showed a comparable specific growth rate (μ = 0.16) in mineral salt medium including aniline as the sole carbon source to that of cells with the intact AD gene cluster (μ = 0.17). KT2442 is a spontaneous rifampin-resistant mutant of KT2440 (57) and still has ggt-2 in the genome. Thus, the host ggt-2 gene may have compensated for the lack of tdnT. Hydrogenophaga sp. PBC has the sadABD gene cluster for 4-aminobenzenesulfonate oxidation (34), which encodes a GS-like protein but not a GAT-like protein. However, intriguingly, this strain contains a DNA region encoding a GAT-like protein and a part of an AD large-subunit homolog (whole-genome shotgun sequence contig 46; accession no. AJWL01000044). Thus, this gene product may contribute to the 4-aminobenzenesulfonate oxidation to 4-sulfocatechol by release of the amino group.
At present, two cyclohexadiene diol intermediates (shown in brackets in Fig. 1B) are still hypothetical compounds. Like IpuF, AtdA2 may have activity hydrolyzing one of them to another, with release of l-glutamate, but there is no experimental information on the reaction. To understand the bacterial aniline oxidation mechanism more completely, the involvement of these hypothetical compounds in aniline oxidation and the detailed functions of AtdA2 should be confirmed experimentally.
Supplementary Material
ACKNOWLEDGMENT
We thank H. Suzuki (Kyoto Prefectural University, Kyoto, Japan) for kindly providing E. coli SO58.
Footnotes
Published ahead of print 26 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00397-13.
REFERENCES
- 1.Bhunia F, Saha NC, Kaviraj A. 2003. Effects of aniline—an aromatic amine to some freshwater organisms. Ecotoxicology 12:397–404 [DOI] [PubMed] [Google Scholar]
- 2.Brennan RJ, Schiestl RH. 1997. Aniline and its metabolites generate free radicals in yeast. Mutagenesis 12:215–220 [DOI] [PubMed] [Google Scholar]
- 3.Ferraz ERA, de Oliveira GAR, de Oliveira DP. 2012. The impact of aromatic amines on the environments: risks and damages. Front. Biosci. F4:914–923 [DOI] [PubMed] [Google Scholar]
- 4.Rubino GF, Scansetti G, Piolatto G, Pira E. 1982. The carcinogenic effect of aromatic amines: an epidemiological study on the role of o-toluidine and 4,4′-methylene bis (2-methylaniline) in including bladder cancer in man. Environ. Res. 27:241–245 [DOI] [PubMed] [Google Scholar]
- 5.Akyüz M, Ata S. 2006. Simultaneous determination of aliphatic and aromatic amines in water and sediment samples by ion-pair extraction and gas chromatography-mass spectrometry. J. Chromatogr. A 1129:88–94 [DOI] [PubMed] [Google Scholar]
- 6.Gosetti F, Chiuminatto U, Zampieri D, Mazzucco E, Marengo E, Gennaro MC. 2010. A new on-line solid phase extraction high performance liquid chromatography tandem mass spectrometry method to study the sun light photodegradation of mono-chloroanilines in river water. J. Chromatogr. A 1217:3427–3434 [DOI] [PubMed] [Google Scholar]
- 7.Jurado-Sanchez B, Ballesteros E, Gallego M. 2012. Occurrence of aromatic-amines and N-nitrosoamines in the different steps of a drinking water treatment plant. Water Res. 46:4543–4555 [DOI] [PubMed] [Google Scholar]
- 8.Lyons CD, Katz S, Bartha R. 1984. Mechanisms and pathways of aniline elimination from aquatic environment. Appl. Environ. Microbiol. 48:491–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lyons CD, Katz SE, Bartha R. 1985. Persistence and mutagenic potential of herbicide-derived aniline residues in pond water. Bull. Environ. Contam. Toxicol. 35:696–703 [DOI] [PubMed] [Google Scholar]
- 10.Anson JG, Mackinnon G. 1984. Novel Pseudomonas plasmid involved in aniline degradation. Appl. Environ. Microbiol. 48:868–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aoki K, Ohtsuka K, Shinke R. 1983. Isolation of aniline-assimilating bacteria and physiological characterization of aniline biodegradation in Rhodococcus erythropolis AN-13. Agric. Biol. Chem. 47:2569–2575 [Google Scholar]
- 12.Aoki K, Ohtsuka K, Shinke R, Nishira H. 1984. Rapid biodegradation of aniline by Frateuria species ANA-18. Agric. Biol. Chem. 48:856–872 [Google Scholar]
- 13.Boon NJ, Goris PDV, Verstraete W, Top EM. 2001. Genetic diversity among 3-chloroaniline- and aniline-degrading strains of the Comamonadaceae. Appl. Environ. Microbiol. 67:1107–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujii T, Takeo M, Maeda Y. 1997. Plasmid-encoded genes specifying aniline oxidation from Acinetobacter sp. strain YAA. Microbiology 143:93–99 [DOI] [PubMed] [Google Scholar]
- 15.Fuchs K, Schreiner A, Lingens F. 1991. Degradation of 2-methylaniline and chlorinated isomers of 2-methylaniline by Rhodococcus rhodochrous strain CTM. J. Gen. Microbiol. 137:2033–2039 [DOI] [PubMed] [Google Scholar]
- 16.Kahng HY, Kukor JJ, Oh KH. 2000. Characterization of strain HY99, a novel microorganism capable of aerobic and anaerobic degradation of aniline. FEMS Microbiol. Lett. 190:215–221 [DOI] [PubMed] [Google Scholar]
- 17.Kim YM, Park K, Kim WC, Shin JH, Kim JE, Park HD, Rhee IK. 2007. Cloning and characterization of a catechol-degrading gene cluster from 3,4-dichloroaniline degrading bacterium Pseudomonas sp. KB35B. J. Agric. Food Chem. 55:4722–4727 [DOI] [PubMed] [Google Scholar]
- 18.Liang Q, Takeo M, Chen M, Zhang W, Xu Y, Lin M. 2005. Chromosome-encoded gene cluster for the metabolic pathway that converts aniline to TCA-cycle intermediates in Delftia tsuruhatensis AD9. Microbiology 151:3435–3446 [DOI] [PubMed] [Google Scholar]
- 19.Liu Z, Yang H, Huang Z, Zhou P, Liu SJ. 2002. Degradation of aniline by newly isolated, extremely aniline-tolerant Delftia sp. AN3. Appl. Microbiol. Biotechnol. 58:679–682 [DOI] [PubMed] [Google Scholar]
- 20.Loidl M, Hinteregger C, Ditzelmueller G, Ferschl A, Streichsbier F. 1990. Degradation of aniline and monochlorinated anilines by soil-borne Pseudomonas acidovorans strains. Arch. Microbiol. 155:56–61 [Google Scholar]
- 21.McClure NC, Venable WA. 1986. Adaptation of Pseudomonas putida mt-2 to growth on aromatic amines. J. Gen. Microbiol. 132:2209–2218 [DOI] [PubMed] [Google Scholar]
- 22.Meyers NL. 1992. Molecular cloning and partial characterization of the pathway for aniline degradation in Pseudomonas sp. strain CIT1. Curr. Microbiol. 24:303–310 [Google Scholar]
- 23.Urata M, Uchida E, Nojiri H, Omori T, Obo R, Miyaura N, Ouchiyama N. 2004. Genes involved in aniline degradation by Delftia acidovorans strain 7N and its distribution in the natural environment. Biosci. Biotechnol. Biochem. 68:2457–2465 [DOI] [PubMed] [Google Scholar]
- 24.Vangnai AS, Petchkroh W. 2007. Biodegradation of 4-chloroaniline by bacteria enriched from soil. FEMS Microbiol. Lett. 268:209–216 [DOI] [PubMed] [Google Scholar]
- 25.Yao XF, Khan F, Pandey R, Pandey J, Mourant RG, Jain RK, Guo J, Russell RJ, Oakeshott JG, Pandey G. 2011. Degradation of dichloroaniline isomers by a newly isolated strain, Bacillus megaterium IMT21. Microbiology 157:721–726 [DOI] [PubMed] [Google Scholar]
- 26.Takeo M, Fujii T, Maeda Y. 1998. Sequence analysis of the genes encoding a multicomponent dioxygenase involved in oxidation of aniline and o-toluidine in Acinetobacter sp. strain YAA. J. Ferment. Bioeng. 85:17–24 [Google Scholar]
- 27.Takeo M, Fujii T, Maeda Y. 1998. Cloning and sequencing of a gene cluster for the meta-cleavage pathway of aniline degradation in Acinetobacter sp. strain YAA. J. Ferment. Bioeng. 85:514–517 [Google Scholar]
- 28.Nam JW, Nojiri H, Yoshida T, Habe H, Yamane H, Omori T. 2001. New classification system for oxygenase components involved in ring-hydroxylating oxygenations. Biosci. Biotechnol. Biochem. 65:254–263 [DOI] [PubMed] [Google Scholar]
- 29.Kweon O, Kim S-J, Baek S, Chae J-C, Adjei MD, Baek D-H, Kim Y-C, Cerniglia CE. 2008. A new classification system for bacterial Rieske non-heme iron aromatic ring-hydroxylating oxygenases. BMC Biochem. 9:11. 10.1186/1471-2091-9-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fukumori F, Saint CP. 1997. Nucleotide sequences and regulational analysis involved in conversion of aniline to catechol in Pseudomonas putida UCC22(pTDN1). J. Bacteriol. 179:399–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murakami S, Hayashi T, Maeda T, Takenaka S, Aoki K. 2003. Cloning and functional analysis of aniline dioxygenase gene cluster, from Frateuria species ANA-18, that metabolizes aniline via an ortho-cleavage pathway of catechol. Biosci. Biotechnol. Biochem. 67:2351–2358 [DOI] [PubMed] [Google Scholar]
- 32.Zhang T, Zhang J, Liu S, Liu Z. 2008. A novel and complete gene cluster involved in the degradation of aniline by Delftia sp. AN3. J. Environ. Sci. (China) 20:717–724 [DOI] [PubMed] [Google Scholar]
- 33.Król JE, Penrod JT, McCaslin H, Rogers LM, Yano H, Stancik AD, Dejonghe W, Brown CJ, Parales RE, Wuertz S, Top EM. 2012. Role of IncP-1 β plasmids pWDL7::rfp and pNB8c in chloroaniline catabolism as determined by genomic and functional analyses. Appl. Environ. Microbiol. 78:828–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gan HM, Shahir S, Yahya A. 2012. Cloning and functional analysis of the genes coding for 4-aminobenzenesulfonate 3,4-dioxygenase from Hydrogenophaga sp. PBC. Microbiology 158:1933–1941 [DOI] [PubMed] [Google Scholar]
- 35.de Azevedo Wäsch SI, van der Ploeg JR, Maire T, Lebreton A, Kiener A, Leisinger A. 2002. Transformation of isopropylamine to l-alaninol by Pseudomonas sp. strain KIE171 involves N-glutamylated intermediates. Appl. Environ. Microbiol. 68:2368–2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurihara S, Oda S, Kato K, Kim HG, Koyanagi T, Kumagai H, Suzuki H. 2005. A novel putrescine utilization pathway involves gamma-glutamylated intermediates of Escherichia coli K-12. J. Biol. Chem. 280:4602–4608 [DOI] [PubMed] [Google Scholar]
- 37.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 38.Takeo M, Prabu SK, Kitamura C, Hirai M, Takahashi H, Kato D, Negoro S. 2006. Characterization of alkylphenol degradation gene cluster in Pseudomonas putida MT4 and evidence of oxidation of alkylphenols and alkylcatechols with medium-length alkyl chain. J. Biosci. Bioeng. 102:352–361 [DOI] [PubMed] [Google Scholar]
- 39.Takeo M, Maeda Y, Maeda J, Nishiyama N, Kitamura C, Kato D, Negoro S. 2012. Two identical nonylphenol monooxygenase genes linked to IS6100 and some putative insertion sequence elements in Sphingomonas sp. NP5. Microbiology 158:1796–1807 [DOI] [PubMed] [Google Scholar]
- 40.Nakazawa T. 2002. Travels of a Pseudomonas, from Japan around world. Environ. Microbiol. 4:782–786 [DOI] [PubMed] [Google Scholar]
- 41.Saito H, Miura K. 1963. Preparation of transforming deoxyribonucleic acid by phenol treatment. Biochim. Biophys. Acta 72:619–629 [PubMed] [Google Scholar]
- 42.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265–275 [PubMed] [Google Scholar]
- 43.Listrom CD, Morizono H, Rajagopal BS, McCann MT, Tuchman M, Allewell NM. 1997. Expression, purification, and characterization of recombinant human glutamine synthetase. Biochem. J. 328:159–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takeo M, Murakami M, Niihara S, Yamamoto K, Nishimura N, Kato D, Negoro S. 2008. Mechanism of 4-nitrophenol oxidation in Rhodococcus sp. strain PN1: characterization of the two-component 4-nitrophenol hydroxylase and regulation of its expression. J. Bacteriol. 190:7367–7374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schäfer A, Tauch A, Jager W, Kalinowski J, Thierbach G, Puhler A. 1994. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73 [DOI] [PubMed] [Google Scholar]
- 46.Simon R, Priefer U, Pühler A. 1982. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis of gram negative bacteria. Nat. Biotechnol. 1:784–791 [Google Scholar]
- 47.Fernández M, Conde S, de la Torre J, Molina-Santiago C, Ramos JL, Duque E. 2012. Mechanisms of resistance to chloramphenicol in Pseudomonas putida KT2440. Antimicrob. Agents Chemother. 56:1001–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takeo M, Itoi M, Negoro S. 2000. Expression of the aniline dioxygenase gene cluster from Acinetobacter sp. YAA in Acinetobacter sp. BD413, p 79–82 In Proceedings of the 5th International Symposium on Environmental Biotechnology, Kyoto, Japan [Google Scholar]
- 49.Jiménez JI, Miñambres B, García JL, Díaz E. 2002. Genome analysis of the aromatic catabolic pathways from Pseudomonas putida KT2440. Environ. Microbiol. 4:824–841 [DOI] [PubMed] [Google Scholar]
- 50.Stadtman ER, Ginsburg A. 1974. The glutamine synthetase of Escherichia coli: structure and control, p 755–807 In Boyer PD. (ed), The enzymes, vol. 10. Academic Press, New York, NY [Google Scholar]
- 51.Goldbarg JA, Friedman OM, Pineda EP, Smith EE, Chaterji R, Stein EH, Rutenberg AM. 1960. The colorimetric determination of gamma-glutamyl transpeptidase with a synthetic substrate. Arch. Biochem. Biophys. 91:60–71 [DOI] [PubMed] [Google Scholar]
- 52.Suzuki H, Hashimoto W, Kumagai H. 1993. Escherichia coli K-12 can utilize an exogenous γ-glutamyl peptide as an amino acid source, for which γ-glutamyltranspeptidase is essential. J. Bacteriol. 175:6038–6040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Juni E, Janik A. 1969. Transformation of Acinetobacter calco-aceticus (Bacterium anitratum). J. Bacteriol. 98:281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Willox S, Weatherby RP, Holder GM. 1976. Aniline metabolism in two species of Arachniad: water-soluble metabolites. Xenobiotica 6:565–573 [DOI] [PubMed] [Google Scholar]
- 55.Yanchunas J, Jr, Dabrowski MJ, Schurke P, Atkins WM. 1994. Supramolecular self-assembly of Escherichia coli glutamine synthetase: characterization of dodecamer stacking and high order association. Biochemistry 33:14949–14956 [DOI] [PubMed] [Google Scholar]
- 56.Breugelmans P, Leroy B, Bers K, Dejonghe W, Wattiez R, De Mot R, Springael D. 2010. Proteomic study of linuron and 3,4-dichloroaniline degradation by Variovorax sp. WDL1: evidence for the involvement of an aniline dioxygenase-related multicomponent protein. Res. Microbiol. 161:208–218 [DOI] [PubMed] [Google Scholar]
- 57.Franklin FCH, Bagdasarian M, Bagdasarian MM, Timmis KN. 1981. Molecular and functional analysis of the TOL plasmid pWW0 from Pseudomonas putida and cloning of the genes for the entire regulated aromatic ring-cleavage pathway. Proc. Natl. Acad. Sci. U. S. A. 78:7458–7462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.