

Abstract
The small pH stress-sensing chaperone HdeA helps pathogenic enteric E. coli survive passage through the severely acidic environment of the mammalian stomach. Under stress conditions, HdeA transitions from an inactive folded dimer to a chaperone-active unfolded monomer to prevent the acid-induced aggregation of periplasmic proteins. Here we use a topology-based Gō-like model to delineate the relationship between dimer interface formation and monomer folding and to better understand the structural details of the chaperone activation mechanism. Free energy surfaces show that dimer interface formation and monomer folding proceed concurrently through an on-pathway dimeric intermediate in which one monomer is partially unfolded. The absence of a preexisting fully folded monomer in the proposed binding mechanism is in agreement with HdeA’s rapid chaperone response. Binding between unfolded monomers exhibits an enhancement of molecular recognition reminiscent of the fly-casting mechanism. Overall, our simulations further highlight the efficient nature of HdeA’s chaperone response and we anticipate that knowledge of a dimeric intermediate will facilitate the interpretation of experimental studies.
Keywords: HdeA, chaperone stress response, conditional disorder, protein dimerization, Gō model
Introduction
Pathogenic enteric bacteria have developed mechanisms to survive the harshly acidic environment of the mammalian stomach and colonize the intestine. While the cytoplasm is relatively insulated and maintained near neutral pH by several decarboxylase systems and the ability of bacteria to reverse their inner membrane potential, the periplasmic space is more vulnerable to changes in environmental pH due to the permeable nature of the outer membrane.1 In the periplasm, the small, ATP-independent chaperone HdeA promotes bacterial survival by rapidly binding to other periplasmic proteins and preventing their acid-induced aggregation.1–4
Under non-stress conditions (neutral pH), HdeA exists as a well-folded, inactive homodimer (Fig. 1). Upon exposure to low pH (< 3), the dimer rapidly disassembles and partially unfolds into a chaperone-active monomer.3,4 Thus, HdeA belongs to a class of newly discovered “conditionally disordered” proteins that lose structure in order to gain function.5–7 As demonstrated by Förster resonance energy transfer (FRET) measurements, the highly flexible nature of the chaperone-active monomer allows HdeA to adopt different conformations depending on the particular substrate.4 This feature helps to explain why HdeA binds to a broad range of much larger client proteins,8 even though it is one of the smallest known proteinaceous chaperones (9.7 kDa per subunit). Moreover, the hydrophobic dimer interface of HdeA doubles as the substrate recognition region, inherently regulating chaperone activity as the neighboring chains in the inactive dimer conceal the hydrophobic residues that play a role in binding unfolded substrate during acid stress.4 Upon return to neutral pH, HdeA slowly releases substrate in order to keep the local concentration of acid-denatured proteins low, thereby reducing the propensity of substrate aggregation and thus facilitating the refolding process.9
Figure 1.
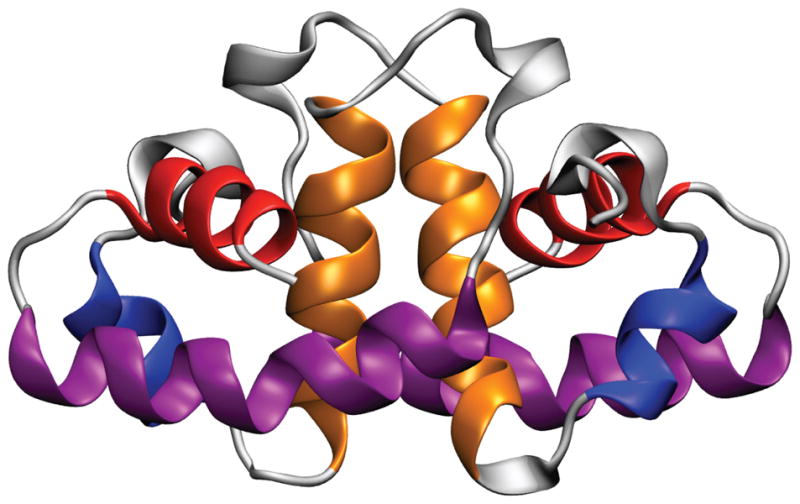
Crystal structure of the HdeA homodimer10 with helices A (blue), B (orange), C (purple), and D (red) indicated. Helices A and D are toward the N- and C-termini, respectively, and helices B and C as well as the loop between them participate in the dimer interface.
Aside from a novel structure prediction approach in which HdeA served as a test case,11 only one other computational study has been performed for this system.12 In the latter, the mechanism of HdeA dimer dissociation was investigated through a combination of constant pH molecular dynamics simulation and umbrella sampling.12 pKa calculations from this study identified several residues that contribute significantly to dimer interface stability and motivated the experimental design of several mutants to alter pH-sensing in HdeA.13 Remarkably, one designed mutant exhibited constitutive chaperone activity. Near neutral pH, the mutant exists as a partially unfolded, chaperone-active monomer, while under the same conditions wild type HdeA is a well-folded, inactive dimer. This finding provides a fundamentally important link between protein disorder and function.13 However, the atomic-level structural and dynamic information available for HdeA is limited, and the relationship between monomer disordering and dimer dissociation during chaperone activation remains unknown. Knowledge of how these two events transpire with respect to one another would provide key mechanistic insight into the chaperone response of the protein.
The binding and folding of homodimers typically proceeds through a two- or three-state mechanism, depending on whether an intermediate is involved in the reaction pathway.14 In the case of HdeA, a two-state mechanism between the folded complex and unfolded monomers would provide a direct route to the active-state ensemble, thereby facilitating a quick chaperone response. However, considering that complete unfolding is not necessary to elicit chaperone activity,13 a stable intermediate along the binding pathway may exhibit structural features advantageous for chaperone function. In either scenario, the binding mechanism of HdeA (e.g., “folding upon binding” of two largely disordered chains or “conformational selection” arising from the association between well structured intermediates) could very well translate to the manner in which the chaperone interacts with substrate.
In this study, we focus on understanding the relationship between HdeA dimer interface formation and folding by using a native topology-based Gō model.15–17 Gō models are centered on the principle of minimal frustration,18,19 which reasons that protein sequences have evolved to diminish the occurrence of non-native interactions during folding so as to efficiently arrive at a robust native state. The view that native-state topology governs protein folding is consistent with a smooth, funneled energy landscape.20 Akin to folding, association between components of biological complexes also proceeds through a conformational search on a funneled energy surface.21,22 Strong evidence supporting the notion that native-state topology drives protein binding on a minimally frustrated landscape stems from the success of Gō models in recapitulating the experimental binding mechanisms for several homodimers.23–25 Since binding events are relatively rare, we employ replica exchange enhanced sampling26 in combination with a Gō-like model that accounts for sequence effects.27 The details of the binding mechanism revealed by our simulations yield new insight into the efficient chaperone response of HdeA.
Computational Methods
Gō-like model
We apply the sequence-flavored Gō-like potential developed by Karanicolas and Brooks27 to study HdeA dimerization and folding. In this model, each residue is represented as a single bead located at the position of the Cα atom and with the mass of its corresponding amino acid. Virtual bonds along the protein main-chain connect the beads to one another. The potential energy of the system is pairwise additive, comprising both bonded and non-bonded terms. For the bonded interactions, virtual bonds and angles are described by a harmonic potential with the minimum placed at their value in the experimental structure (“native state”). Virtual dihedral angle probability distributions for each of the 400 possible ordered residue pairs are used to construct potentials reflecting the secondary structure propensity of consecutive amino acid pairs due to chirality and differences in side-chain size and geometry.
For the non-bonded interactions, residue pairs participating in backbone hydrogen bonding or with side-chain heavy atoms within 4.5 Å in the native structure are used to define the set of native contacts. Contacting residues in the native state interact favorably through a 12-10-6, Lennard-Jones-type potential. Compared to a standard 12-6 Lennard-Jones potential, this potential exhibits a narrower minimum and a slight energy barrier, which reflects the desolvation penalty experienced by two residues before forming a contact. Sequence effects are also taken into account through this term, as the strength of the interaction is scaled in proportion to the statistical contact energies for specific residue pairs reported by Miyazawa and Jernigan.28 All residue pairs not in contact in the native structure experience an unfavorable interaction energy that takes the form of a standard 12-6 Lennard-Jones potential. A more comprehensive description of this Gō-like model is presented in references 27 and 29. This model successfully reproduced the folding features of a large test set of experimentally characterized proteins30 and has been applied to uncover key characteristics of antiparallel β-sheet formation,31 subdomain competition during folding,32 and the folding and binding of intrinsically disordered proteins.29
System set-up and simulation
The starting configuration of the HdeA dimer for simulation is taken from a crystal structure (PDB ID: 1BG8).10 The asymmetric unit comprises three chains (A, B, and C) and the biological dimer formed between chains C and an image of C are chosen for simulation such that both subunits are initially identical, both in sequence and in structure. To delineate the two monomers in the text, they are simply referred to as chains A and B. All observable residues from the crystal structure (10–85 out of 89 total per subunit) are included such that the simulated dimer comprises 152 total Cα atoms to be considered in defining the native-state topology. One disulfide bond is present in each subunit between residues 18 and 66 and is modeled with a harmonic restraint with a spring constant of a carbon-carbon bond and an equilibrium distance of 4.8 Å (the value in the crystal structure).
We perform several simulations of the Gō-like model over a broad range of temperatures to determine the folding/unfolding and binding/dissociation transition temperature of HdeA. Each run consists of 1.5 × 108 dynamics steps and the heat capacity (Cv) is used to monitor the transitions. Cv is calculated as , where is the fluctuation in energy at temperature T and kB is the Boltzmann constant. Based on the profile of Cv as a function of T/Tm, where Tm (304 K) is the melting temperature (Fig. 2A), the Gō-like model is simulated in combination with the replica exchange (REX) molecular dynamics algorithm26 with 16 total replicas spanning temperature windows exponentially distributed between 0.95Tm and 1.11Tm. (Calculations of Cv from the constant temperature runs and “Gō-REX” simulations yield similar results, and the profile constructed from the latter is shown in Figure 2A.) Recent work demonstrated the suitability of using increased temperatures to expedite conformational sampling in Gō models.29 The “Gō-REX” simulations are implemented as in the Multiscale Modeling Tools for Structural Biology (MMTSB) tool set.33 After equilibration at each temperature state for 105 time steps (0.02 ps each), exchanges are attempted every 2.5 × 103 time steps until a total of 105 exchanges (2.5 × 108 total steps). Coordinates are saved at each exchange for all temperature states. A Langevin thermostat with a friction coefficient of 1.36 ps−1 and periodic boundary conditions with a box size of 90 Å3 are used for both the Gō-REX and constant temperature simulations. Periodic boundaries ensure a high local concentration of protein such that sufficient binding events can be observed within a practical simulation time. This procedure is repeated six times with similar results and the data are combined for the analyses presented in the text.
Figure 2.

(A) Specific heat as a function of temperature, scaled by Tm. (B) Free energy as a function of the fraction of Qtotal at and near Tm. Folded complexes and unfolded chains have values closer to one and zero, respectively. We note that the unfolded ensemble maintains roughly a third of the native contacts.
Analysis
Throughout the simulations, native contacts are counted as being formed if they are within 1 Å of their distance in the crystal structure. Coordinates from the last half of simulation are used to construct the free energy surfaces by the temperature-weighted histogram analysis method (T-WHAM).27,34 Based on the free energy surface in the plane of the total number of inter- and intramolecular native contacts (Qinter and Qintra, respectively), the system is divided into four regions: the dimer state (N2), a dimeric intermediate (I2), a transition state (TS) and the unfolded state (U). These are defined as follows: N2 has Qinter > 20, Qintra > 270; I2 has Qinter > 20, Qintra ≤ 270; TS has Qinter ≤ 20, Qintra > 180; and U has Qinter ≤ 20, Qintra ≤ 180. Representative structures of the four states are computed using an ensemble of at least 284 randomly chosen structures in each region, and determining a median structure by measuring the all-to-all root-mean-square deviation (RMSD) of atomic coordinates within each ensemble. The median for each region is then determined as the structure that has the lowest RMSD to the other ensemble members.
Results
The specific heat curve for HdeA exhibits one dominant peak centered at Tm (Fig. 2A). The absence of two separate peaks indicates the coupling of monomer folding/unfolding and dimer association/dissociation. Coupled folding and binding is a characteristic of homodimers that bind through a two-state mechanism (between unfolded monomers and a folded dimer).14 The mechanism of dimerization (i.e., two- or three-state) can be also be inferred by comparing the number of inter- and intra-monomeric contacts per residue as well as by the size and hydrophobicity of the interface.23,35 While the binding mode for HdeA is unclear from the ratio between the number of interfacial (51) and intra-monomer contacts (162), the dimer interface buries over 20% of the total surface area of two unbound folded monomers (2158 Å2 versus 10,150 Å2) and is relatively hydrophobic (average hydrophobicity per residue of 0.63).36 Homodimers with more extensive and hydrophobic interfaces typically undergo a two-state binding mechanism.35,37 Yet, in the case of HdeA, the free energy profile constructed along the fraction of native contacts (Qtotal) at Tm displays two larger minima and a smaller one appended to the native basin (Fig. 2B). The additional minimum is maintained at temperatures above and below Tm and indicates the presence of an intermediate state.
To further investigate the mechanism of HdeA dimer interface formation and its relationship to monomer folding, we construct free energy surfaces at Tm (Fig. 3). Figure 3A shows the surface constructed in the plane of the distance between the centers of mass of the two subunits (dCM) and Qtotal. The landscape displays a narrow free energy minimum at the native folded dimer (N2; high Qtotal, low dCM) and a less stable minimum (I2) located adjacent to the native basin. A higher energy transition region separates the native-like dimers from a broad basin representing unfolded monomers (U; low Qtotal, high dCM). The absence of a minimum at intermediate values of Qtotal and larger distances indicates that fully folded monomers are only present when the two subunits are relatively close together (dCM ~ 18–25 Å).
Figure 3.
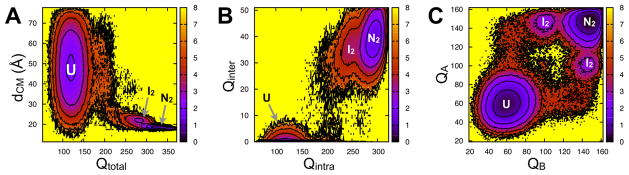
Free energy surfaces reporting on the relationship between dimer interface formation and monomer folding. Surfaces are constructed in the plane of several reaction coordinates at Tm: (A) dCM and Qtotal, (B) Qinter and Qintra, and (C) QA and QB. The color bars indicate the free energy in units of kcal/mol.
Next, Qtotal is divided into subsets of intermolecular (Qinter) and intramolecular (Qintra) contacts, which are analyzed against one another in the free energy surface presented in Figure 3B. The native basin N2 is located at high Qinter and high Qintra. Similar to Figure 3A, adjoined to N2 is the “intermediate” minimum I2. This minimum is located in a region with well over half of the intermolecular contacts formed (Qinter > 25) while many (~50) intramolecular contacts are lost, indicating that it represents an intermediate species that is still largely associated but partially unfolded. Between the native-like and unfolded basins, both Qinter and Qintra decrease across the transition region, indicating that the loss of contacts across the dimer interface proceeds concurrently with unfolding. The observation that only I2 and not N2 largely connects to the unfolded ensemble emphasizes that I2 is an “on-pathway,” or obligatory, intermediate, as has been previously observed in experiment for the Trp repressor protein.38 To analyze the behavior of the individual subunits during binding, we construct a free energy surface in the plane of the intra-monomer native contacts (QA and QB, Fig. 3C). In addition to the fully folded dimer (N2, high QA and high QB), intermediate states (I2) in which one monomer is partially unfolded (QA or QB ~ 100) are observed. The transition region proceeds from the intermediate basin directly to the unfolded ensemble (U). The absence of a minimum at high QA and low QB (and vice versa) emphasizes that dimerization/dissociation bypasses a preexisting fully folded monomer in simulation. Overall, the free energy surfaces support the notion that HdeA interface formation and folding proceed concurrently with one another and through an on-pathway dimeric intermediate.
To garner further structural insight from the simulated ensemble, contact maps for native intra- and intermolecular residue-residue contacts are constructed for the subpopulations corresponding to N2, I2, the transition state (TS), and U from the free energy surface in Figure 3B (Fig. 4). Representative structures for each region are displayed next to the maps. Folded dimers similar to N2 exhibit a pattern in which both intra- and intermolecular native contacts are satisfied throughout simulation (Fig. 4, N2). Compared to N2, structures from the I2 basin also maintain contacts about the dimer interface toward the N-terminus, while for one of the monomers many intramolecular contacts involving helix D at the C-terminus are largely unformed (Fig. 4, I2). The breaking of intramolecular contacts extends to the N-terminus and the interfacial contacts almost completely disappear in the TS ensemble (Fig. 4, TS). In the unfolded ensemble, the dimer interface and essentially all non-local intramolecular contacts are absent (Fig. 4, U). The small number of non-local contacts between the regions of helices A and C in the unfolded ensemble are due to the presence of the intramolecular disulfide bond. Local intramolecular contacts within the helices are still present to a notable degree in the unfolded ensemble. The continual loss of both intra- and intermolecular contacts from N2 to U further emphasizes that unfolding and dissociation occur simultaneously.
Figure 4.

Contact maps for each of the four states, with representative structures. Each contact map is computed at Tm, and shows the probability that each native contact is satisfied (red indicates a contact is always satisfied, and blue indicates that it is never satisfied). Since monomers A and B are indistinguishable, the intramolecular contact maps show the average over the two monomers. Similarly, the intermolecular contact maps should be perfectly symmetric about the diagonal in the limit of infinite sampling. The non-local contact in the unfolded ensemble with a probability of 1 (red, located between the regions of helices A and C) corresponds to the intramolecular disulfide bond. Representative structures for each region are shown in the rightmost column, and are determined as described in the methods section.
We also analyze the free energy as a function of the separation distance between the monomer centers of mass at Tm (Fig. 5). A separation distance of zero corresponds to the center-of-mass distance between the two monomers in the crystal structure. The computed free energy from simulation is normalized by the volume of a spherical shell with radius dCM. The modest decrease in free energy at larger distances reflects the gain in conformational entropy once the monomers are fully dissociated.39,40 At separation distances less than ~7 Å, the free energy decreases by ~1 kcal/mol into a shallow minimum before dropping more significantly by ~3 kcal/mol to the global minimum at the native separation distance. This strong attraction between the two monomers during binding is reminiscent of the fly-casting speedup for protein association.41
Figure 5.
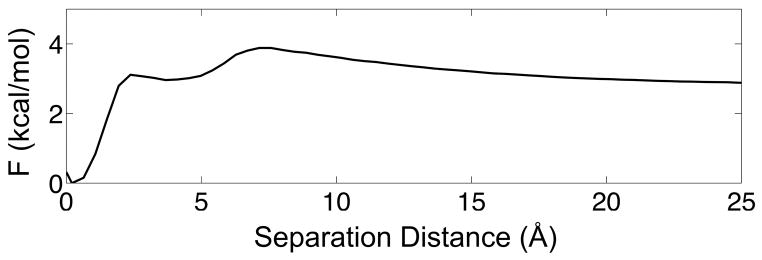
Free energy along the separation distance between the two monomers at Tm. The calculated free energy is normalized by the volume of a spherical shell with radius dCM. The separation distance is defined as dCM – dCM0,24 where dCM0 is the distance between the subunits in the crystal structure (18.1 Å).
Discussion
HdeA is an intriguingly efficient chaperone. To achieve its chaperone-active disordered state, HdeA takes advantage of the same acid stress conditions that promote the unfolding and inactivation of other periplasmic proteins. After dissociation, the exposed hydrophobic dimer interface region doubles as the substrate interaction site. The conformational lability of the active state ensemble and exposure of a large hydrophobic patch appears to permit binding to a host of much larger client proteins.4 Moreover, HdeA functions without the need for ATP or other energy cofactors, a key attribute in the ATP-deficient periplasm. Taken collectively, the details of the coupled binding and folding of HdeA observed in our topology-based simulations further highlight an efficient chaperone response.
In our proposed mechanism, monomer unfolding and dimer dissociation occur in concert with one another. Although binding and folding are coupled, these events do not strictly follow a two-state mechanism and instead proceed through an on-pathway dimeric intermediate in which one monomer is partially unfolded. Recent experiments showed that only partial unfolding is necessary for HdeA to prevent acid-induced substrate aggregation.13 Given that we observe a partially unfolded dimeric intermediate in our simulations, HdeA would be poised to interact with unfolded substrate immediately upon dissociation, without the requirement of additional large conformational change. Monomer unfolding commences in the C-terminus while the dimer interface is still formed at the N-terminus, suggesting that hydrophobic residues elsewhere in the protein become exposed and may contribute along with the interfacial region to provide a relatively large surface area for substrate interaction. Although such a picture is in agreement with the rapid response of HdeA in inhibiting substrate aggregation,4 the chaperone-active monomers released from the dimer interface would still have to diffuse toward the substrate, limiting the onset of the chaperone response. Nevertheless, acidic conditions significantly induce expression of the hdeA gene,42 which would increase the local concentration of HdeA in the periplasm and thus increase the likelihood that a dissociation event would occur in close proximity to an unfolding substrate.
Our simulations also offer insight into the deactivation of HdeA after return to neutral pH, where the chaperone slowly releases substrate before re-forming the dimer interface.9 We observe a strong attraction between HdeA monomers as the separation distance between them decreases. Previous all-atom molecular dynamics simulations of HdeA dimer dissociation revealed a similar trend for a potential of mean force constructed along the center-of-mass distance between the two monomers.12 However, the all-atom simulations used folded models of the HdeA monomer for the purpose of pKa calculations and thus did not identify a partially unfolded intermediate species. Our Gō-like model simulations account for both binding and folding and as a result further delineate the behavior of HdeA subunit association by revealing a partially folded dimeric intermediate along the pathway. The attraction between monomers we observe in simulation initiates between unfolded chains and becomes stronger when a partially structured subunit folds upon interface formation. This scenario supports the notion that dimerization, and thus chaperone deactivation, is facilitated by a more structured monomer serving as a template for folding of the neighboring chain, and is indicative of the enhancement in molecular recognition outlined by the fly-casting mechanism.41 It is tempting to speculate that such a “folding-upon-binding” mechanism is also at play in HdeA-substrate interaction, as it would permit high-specificity/low-affinity binding to a broad range of partner proteins.43,44
Previous experiments characterized pH-induced conformational change and dimer dissociation in HdeA separately using intrinsic tryptophan fluorescence and FRET measurements.4 Our Gō-like model simulations yield additional insight into experiment as we can monitor both processes simultaneously. While the coupled binding and folding observed in the simulations is in agreement with the similar rates for conformational change and monomerization measured by fluorescence, an intermediate species was not reported in experiment. However, the presence of a dimeric intermediate would have likely gone undetected by the FRET measurements. The donor and acceptor fluorophores used to track dissociation were located at two sites (residue 27 of both subunits) that oppose each other about the dimer interface. Since these two residues have a similar average Cα-Cα distance between them in the ensemble of structures represented by the native (N2) and intermediate (I2) dimers during simulation (9.3 and 9.9 Å, respectively), the formation of the dimeric intermediate would not have significantly altered the FRET signal in experiment. We anticipate that knowledge of a dimeric intermediate in the activation mechanism of HdeA will aid in the interpretation of ongoing experimental efforts aiming to characterize the conformational behavior of the HdeA monomer and its interaction with substrate.
Conclusions
Under acidic conditions, HdeA dissociates from an inactive, well-folded dimer to a chaperone-active, partially unfolded monomer. Understanding how dimer dissociation and monomer unfolding proceed with respect to one another is key for building a complete understanding of HdeA’s chaperone function. We addressed this question by employing a native topology based Gō-like model to elucidate the mechanism of HdeA dimerization. Our simulations support a picture in which dimerization and folding occur simultaneously, albeit through an on-pathway dimeric intermediate comprising a partially unfolded monomer. The presence of partially unfolded, and thus chaperone-active, monomers immediately upon dimer dissociation would contribute to the speedy chaperone response of HdeA during acid stress. The relatively strong attraction between unfolded chains during binding would facilitate shutting off chaperone activity upon return to non-stress conditions. Overall, the mechanism of coupled folding and dimerization observed in simulation highlights the role of conformational flexibility in regulating the chaperone function of HdeA. The current study provides a foundation for an all-atom and pH-dependent description of the disordered monomeric active state ensemble and its interaction with substrate.
Acknowledgments
LSA thanks the Brooks’ group members Karunesh Arora, Sean Law, and Afra Panahi for methodological input as well as Linda Foit, Loic Salmon, James Bardwell, and Hashim M. Al-Hashimi for helpful discussions about HdeA. We are grateful for support from the Center for Theoretical Biological Physics (CTBP) funded by the NSF (PHY0216576), and the Center for Multiscale Modeling Tools for Structural Biology (MMTSB), funded by the NIH (RR012255).
References
- 1.Hong W, Wu YE, Fu X, Chang Z. Chaperone-Dependent Mechanisms for Acid Resistance in Enteric Bacteria. Trends Microbiol. 2012;20:328–335. doi: 10.1016/j.tim.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Gajiwala KS, Burley SK. Hdea, a Periplasmic Protein That Supports Acid Resistance in Pathogenic Enteric Bacteria. J Mol Biol. 2000;295:605–612. doi: 10.1006/jmbi.1999.3347. [DOI] [PubMed] [Google Scholar]
- 3.Hong W, Jiao W, Hu J, Zhang J, Liu C, Fu X, Shen D, Xia B, Chang Z. Periplasmic Protein Hdea Exhibits Chaperone-Like Activity Exclusively within Stomach Ph Range by Transforming into Disordered Conformation. J Biol Chem. 2005;280:27029–27034. doi: 10.1074/jbc.M503934200. [DOI] [PubMed] [Google Scholar]
- 4.Tapley TL, Korner JL, Barge MT, Hupfeld J, Schauerte JA, Gafni A, Jakob U, Bardwell JC. Structural Plasticity of an Acid-Activated Chaperone Allows Promiscuous Substrate Binding. Proc Natl Acad Sci U S A. 2009;106:5557–5562. doi: 10.1073/pnas.0811811106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bardwell JC, Jakob U. Conditional Disorder in Chaperone Action. Trends Biochem Sci. 2012;37:517–525. doi: 10.1016/j.tibs.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaya N, Garcia V, Vierling E. Substrate Binding Site Flexibility of the Small Heat Shock Protein Molecular Chaperones. Proc Natl Acad Sci U S A. 2009;106:15604–15609. doi: 10.1073/pnas.0902177106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reichmann D, Xu Y, Cremers CM, Ilbert M, Mittelman R, Fitzgerald MC, Jakob U. Order out of Disorder: Working Cycle of an Intrinsically Unfolded Chaperone. Cell. 2012;148:947–957. doi: 10.1016/j.cell.2012.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang M, Lin S, Song X, Liu J, Fu Y, Ge X, Fu X, Chang Z, Chen PR. A Genetically Incorporated Crosslinker Reveals Chaperone Cooperation in Acid Resistance. Nat Chem Biol. 2011;7:671–677. doi: 10.1038/nchembio.644. [DOI] [PubMed] [Google Scholar]
- 9.Tapley TL, Franzmann TM, Chakraborty S, Jakob U, Bardwell JC. Protein Refolding by Ph-Triggered Chaperone Binding and Release. Proc Natl Acad Sci U S A. 2010;107:1071–1076. doi: 10.1073/pnas.0911610107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang F, Gustafson KR, Boyd MR, Wlodawer A. Crystal Structure of Escherichia Coli Hdea. Nat Struct Biol. 1998;5:763–764. doi: 10.1038/1796. [DOI] [PubMed] [Google Scholar]
- 11.Papoian GA, Ulander J, Eastwood MP, Luthey-Schulten Z, Wolynes PG. Water in Protein Structure Prediction. Proc Natl Acad Sci U S A. 2004;101:3352–3357. doi: 10.1073/pnas.0307851100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang BW, Brunetti L, Brooks CL., III Probing pH-Dependent Dissociation of HdeA Dimers. J Am Chem Soc. 2011;133:19393–19398. doi: 10.1021/ja2060066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foit L, George JS, Zhang BW, Brooks CL, III, Bardwell JC. Chaperone Activation by Unfolding. Proc Natl Acad Sci U S A. 2013;110:E1254–1262. doi: 10.1073/pnas.1222458110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rumfeldt JAO, Galvagnion C, Vassall KA, Meiering EM. Conformational Stability and Folding Mechanisms of Dimeric Proteins. Prog Biophys Mol Biol. 2008;98:61–84. doi: 10.1016/j.pbiomolbio.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 15.Clementi C, Jennings PA, Onuchic JN. How Native-State Topology Affects the Folding of Dihydrofolate Reductase and Interleukin-1beta. Proc Natl Acad Sci U S A. 2000;97:5871–5876. doi: 10.1073/pnas.100547897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shea JE, Onuchic JN, Brooks CL., III Exploring the Origins of Topological Frustration: Design of a Minimally Frustrated Model of Fragment B of Protein A. Proc Natl Acad Sci U S A. 1999;96:12512–12517. doi: 10.1073/pnas.96.22.12512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taketomi H, Ueda Y, Gō N. Studies on Protein Folding, Unfolding and Fluctuations by Computer Simulation. I. The Effect of Specific Amino Acid Sequence Represented by Specific Inter-Unit Interactions. Int J Pept Protein Res. 1975;7:445–459. [PubMed] [Google Scholar]
- 18.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Funnels, Pathways, and the Energy Landscape of Protein Folding: A Synthesis. Proteins. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 19.Gō N. Theoretical Studies of Protein Folding. Annu Rev Biophys Bioeng. 1983;12:183–210. doi: 10.1146/annurev.bb.12.060183.001151. [DOI] [PubMed] [Google Scholar]
- 20.Wolynes PG. Recent Successes of the Energy Landscape Theory of Protein Folding and Function. Q Rev Biophys. 2005;38:405–410. doi: 10.1017/S0033583505004075. [DOI] [PubMed] [Google Scholar]
- 21.Tsai CJ, Kumar S, Ma B, Nussinov R. Folding Funnels, Binding Funnels, and Protein Function. Protein Sci. 1999;8:1181–1190. doi: 10.1110/ps.8.6.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsai CJ, Ma B, Nussinov R. Folding and Binding Cascades: Shifts in Energy Landscapes. Proc Natl Acad Sci U S A. 1999;96:9970–9972. doi: 10.1073/pnas.96.18.9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levy Y, Cho SS, Onuchic JN, Wolynes PG. A Survey of Flexible Protein Binding Mechanisms and Their Transition States Using Native Topology Based Energy Landscapes. J Mol Biol. 2005;346:1121–1145. doi: 10.1016/j.jmb.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 24.Levy Y, Wolynes PG, Onuchic JN. Protein Topology Determines Binding Mechanism. Proc Natl Acad Sci U S A. 2004;101:511–516. doi: 10.1073/pnas.2534828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levy Y, Onuchic JN. Mechanisms of Protein Assembly: Lessons from Minimalist Models. Acc Chem Res. 2006;39:135–142. doi: 10.1021/ar040204a. [DOI] [PubMed] [Google Scholar]
- 26.Sugita Y, Okamoto Y. Replica-Exchange Molecular Dynamics Method for Protein Folding. Chem Phys Lett. 1999;314:141–151. [Google Scholar]
- 27.Karanicolas J, Brooks CL., III The Origins of Asymmetry in the Folding Transition States of Protein L Protein G. Protein Sci. 2002;11:2351–2361. doi: 10.1110/ps.0205402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyazawa S, Jernigan RL. Residue-Residue Potentials with a Favorable Contact Pair Term and an Unfavorable High Packing Density Term, for Simulation and Threading. J Mol Biol. 1996;256:623–644. doi: 10.1006/jmbi.1996.0114. [DOI] [PubMed] [Google Scholar]
- 29.Ganguly D, Chen JH. Topology-Based Modeling of Intrinsically Disordered Proteins: Balancing Intrinsic Folding and Intermolecular Interactions. Proteins. 2011;79:1251–1266. doi: 10.1002/prot.22960. [DOI] [PubMed] [Google Scholar]
- 30.Karanicolas J, Brooks CL., III Improved Go-Like Models Demonstrate the Robustness of Protein Folding Mechanisms Towards Non-Native Interactions. J Mol Biol. 2003;334:309–325. doi: 10.1016/j.jmb.2003.09.047. [DOI] [PubMed] [Google Scholar]
- 31.Karanicolas J, Brooks CL., III The Structural Basis for Biphasic Kinetics in the Folding of the Ww Domain from a Formin-Binding Protein: Lessons for Protein Design? Proc Natl Acad Sci U S A. 2003;100:3954–3959. doi: 10.1073/pnas.0731771100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hills RD, Brooks CL., III Subdomain Competition Cooperativity and Topological Frustration in the Folding of Chey. J Mol Biol. 2008;382:485–495. doi: 10.1016/j.jmb.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feig M, Karanicolas J, Brooks CL., III Mmtsb Tool Set: Enhanced Sampling and Multiscale Modeling Methods for Applications in Structural Biology. J Mol Graphics Modell. 2004;22:377–395. doi: 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 34.Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman PA. Multidimensional Free-Energy Calculations Using the Weighted Histogram Analysis Method. J Comput Chem. 1995;16:1339–1350. [Google Scholar]
- 35.Xu D, Tsai CJ, Nussinov R. Mechanism and Evolution of Protein Dimerization. Protein Sci. 1998;7:533–544. doi: 10.1002/pro.5560070301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kyte J, Doolittle RF. A Simple Method for Displaying the Hydropathic Character of a Protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 37.Tsai CJ, Xu D, Nussinov R. Structural Motifs at Protein-Protein Interfaces: Protein Cores Versus Two-State and Three-State Model Complexes. Protein Sci. 1997;6:1793–1805. doi: 10.1002/pro.5560060901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gloss LM, Matthews CR. Mechanism of Folding of the Dimeric Core Domain of Escherichia Coli Trp Repressor: A Nearly Diffusion-Limited Reaction Leads to the Formation of an On-Pathway Dimeric Intermediate. Biochemistry. 1998;37:15990–15999. doi: 10.1021/bi981511p. [DOI] [PubMed] [Google Scholar]
- 39.Tidor B, Karplus M. The Contribution of Cross-Links to Protein Stability - a Normal Mode Analysis of the Configurational Entropy of the Native-State. Proteins. 1993;15:71–79. doi: 10.1002/prot.340150109. [DOI] [PubMed] [Google Scholar]
- 40.Woo HJ, Roux B. Calculation of Absolute Protein-Ligand Binding Free Energy from Computer Simulations. Proc Natl Acad Sci U S A. 2005;102:6825–6830. doi: 10.1073/pnas.0409005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shoemaker BA, Portman JJ, Wolynes PG. Speeding Molecular Recognition by Using the Folding Funnel: The Fly-Casting Mechanism. Proc Natl Acad Sci U S A. 2000;97:8868–8873. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tucker DL, Tucker N, Conway T. Gene Expression Profiling of the pH Response in Escherichia Coli. J Bacteriol. 2002;184:6551–6558. doi: 10.1128/JB.184.23.6551-6558.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spolar RS, Record MT., Jr Coupling of Local Folding to Site-Specific Binding of Proteins to DNA. Science. 1994;263:777–784. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
- 44.Uversky VN, Oldfield CJ, Dunker AK. Intrinsically Disordered Proteins in Human Diseases: Introducing the D2 Concept. Annu Rev Biophys. 2008;37:215–246. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]