

Abstract
This review discusses the multiple roles of the CagA protein encoded by the cag pathogenicity island of Helicobacter pylori and highlights the CagA degradation activities on p53. By subverting the p53 tumor suppressor pathway CagA induces a strong antiapoptotic effect. Helicobacter pylori infection has been always associated with an increased risk of gastric cancer. The pro-oncogenic functions of CagA also target the tumor suppressor ASPP2. In the absence of tumor suppressor genes, cells survive and proliferate at times and in places where their survival and proliferation are inappropriate.
Helicobacter pylori infection is associated with an increased risk of gastric cancer. Its genome encodes CagA, which targets the p53 and ASPP2 tumor suppressors.
Helicobacter pylori (H. pylori) is a Gram-negative, helix-shaped, and microaerophilic bacterium that colonizes the mucosa of the stomach lifelong and has succeeded in living in a hostile niche where most other microorganisms fail to thrive, and in which competition is limited or absent. It is one of the most common human infections, affecting half of the world population and it is the causative agent of peptic ulcer disease (PUD), chronic gastritis, and is associated with an increased risk of gastric adenocarcinoma and MALT lymphoma. More than 80% of individuals are asymptomatic. During their lives, 10%–20% are at risk of PUD, 1%–3% will develop a gastric adenocarcinoma, and 0.1% MALT lymphoma. Infections are more frequent in the developing world than in Western Europe, North America, and Australasia. The age of acquisition is critical because it is associated with more severe outcomes. Transmission is by fecal–oral or oral–oral routes; it occurs within families in developed nations. In developing countries it is community acquired.
H. pylori was acquitted by a host jump from an unknown source in Africa, where two discrete superlineages differentiated. One of the superlineages was predominantly associated until very recently with San (hpAfrica2) and large felines (Hac), whereas the second is widespread throughout Africa (hpAfrica1, hpNEAfrica) and accompanied anatomically modern humans during their first “out of Africa” migration. Subsequent migration resulted in the Asian and Oceanic lineages hpAsia2, hpAsia, and hpSahul. After new migratory waves ancestors of the African hpNEAfrica and/or the Asian hpAsia2 populations resulted in the admixed hpEurope population, which then became the predominant population of extant H. pylori in Europe, the middle East, and western Asia (Moodley et al. 2012). The distribution of human languages is quite a sensitive indicator of the dispersal of modern human beings around the globe. When cluster analysis was applied to a set of randomly selected gene samples, six H. pylori strains isolated from East Asia clustered with a strain from Peru. It turned out that the Peruvian isolate “was from an ethnic Japanese living in Peru” (Achtman et al. 1999). DNA fingerprinting analysis in Maori and Pacific Islanders by O’Toole et al. suggested that Polynesians are infected by a group of H. pylori strains that are distinct from European New Zealanders (Campbell et al. 1997). Likewise, isolates from Australian Aborigines are distinct from European origin Australians and indicated that the bacterial lineages first arrived in Australia with the earliest human migrations. New results lend support for two distinct waves of migrations into the Pacific. First are the early migrations to New Guinea and Australia accompanied by hpSahul and, second, a much later dispersal of hspMaori from Taiwan through the Pacific by the Malayo-Polynesian-speaking Lapita culture. Each sampling area yielded either hpSahul or hspMaori haplotypes, but not both (Moodley et al. 2009).
The major determinant of virulence is the cag pathogenicity island (PAI), a chromosomal segment of 40 kb containing 30 genes (Blum et al. 1994; Covacci et al. 1999). In comparison with partially deleted cag PAI strains, those with a functional intact cag PAI would increase the risk of gastric carcinoma 10-fold in infected subjects (Censini et al. 1996; Akopyants et al. 1998; Nguyen et al. 2008). Encoded in the cag PAI are the CagA antigen (Covacci et al. 1993; Tummuru et al. 1993) and the cag type IV secretion system (TFSS) (Covacci and Rappuoli 2000). The TFSS can be seen as a surface organelle, membrane-sheeted, forming a conduit for translocation of the substrate, the CagA protein (Rohde et al. 2003). Recent data suggest that α5β1 integrin acts a receptor for CagA translocation (Kaplan-Türköz et al. 2012).
THE CagA PROTEIN
We are updating our knowledge on both the type IV secretion system of H. pylori and on a specific role of CagA in the p53 pathway. Together with essential notions on the microorganism we will provide an insight into CagA-host interactions. Furthermore, new data on the TFSS of a conjugative plasmid will help to explain the first phase of CagA secretion. Great progress has been made on the study of coevolution of H. pylori with its human host and the use of H. pylori as a marker for the study of human migrations. We are approaching a phase in which genome-wide association studies will merge with data about migration of humans infected by H. pylori, providing a scaffold to understand language dispersal and anthropology as well as transmission of the infectious agents within families, persistent infection, and tumor progression. The last part will concentrate on the cellular mechanisms exploited by CagA in epithelial damage (read lesions) and the earliest phase of tumorigenic progression in concert with the tumor suppressor ASPP2 and p53 degradation.
Scanning electron microscopy images of human gastric mucosa revealed the presence of large amounts of H. pylori cells at the level of junctions (Steer 1984; Hazell et al. 1986). In addition, during coculture of bacterial and epithelial cells of animal or human origin massive elongations of the host cells were observed and named “hummingbird phenotype.” CagA molecules, once translocated via the cag TFSS into the host cells, are tyrosine-phosphorylated within the repetitive sequence motif EPIYA and this activation has a central role in inducing changes in the host-cell morphology (Stein et al. 2000).
CagA tyrosine phosphorylation initiates host-cell signaling events via interaction with a SH2 or SH3 domain. This leads to the induction of a signaling cascade that mimics growth factor-like responses. In addition, inhibitors specific for the Src kinase family abolish CagA tyrosine phosphorylation in vitro and in tissue culture infection experiments, and two members of the Src kinase family, c-Src and Lyn, are the major CagA kinases (Stein et al. 2002). Src family kinases are strongly implicated in the development, growth, progression, and metastasis of a number of human cancers. They belong to the family of nonreceptor kinases and are posttranslationally modified through covalent attachment of a 14-carbon fatty acid moiety, myristate. Together with six basic amino acid residues the myristylation acts as the amino-terminal signal for membrane anchorage of the Src kinases. The main target is the plasma membrane, but perinuclear and endosomal membranes are also targeted. In the plasma membrane, Src kinases are found associated with focal adhesions, the actin cytoskeleton and adherens junctions. Src responses include mitogenic signaling, through association with growth factor receptors, cell adhesion and spreading, cell migration, and regulation of cell–cell contact formation (Segal et al. 1999; Asahi et al. 2000; Backert et al. 2000; Odenbreit et al. 2000).
CagA and Host–Cell Interactions
H. pylori adheres to cells in the proximity of the apical–junctional complex. The epithelial apical–junctional complex forms a network of transmembrane, scaffolding, and signaling proteins, and serves as a barrier, adhesion site, and pathways network to control cell polarity, proliferation, and differentiation processes. Simultaneous visualization of CagA, ZO-1, and H. pylori showed that injected CagA colocalized with ZO-1 at sites of bacterial attachment, as well as at cell–cell contacts. In Madin-Darby canine kidney (MDCK) monolayers infected with wild-type H. pylori, individual junctions showed barrier dysfunction with leakage of ruthenium red into the basolateral space. MDCK cells elongated and extended processes between adjacent cells, resulting in dysplastic cell shapes. Intraepithelial dysplasia depended on the injection of CagA and was reversible if monolayers were treated with antibiotics. CagA interferes with the assembly of functional tight junctions and alters the composition of junctional protein complexes (Amieva et al. 2003). The number of copies of the repeated region is strain specific; size variability of CagA ranges from 128 kDa (absence of 102 bp repeat) to 144 kDa (four 102 bp repeats in tandem) (Fig. 1) (Xiang et al. 1995). CagA molecules lacking the EPIYA motifs are translocated, but not phosphorylated. Unlike the parental strain the mutants failed to induce signaling events that govern cellular elongation and suggest that tyrosine phosphorylation is critical for CagA activation (Hayashi et al. 2012).
Figure 1.

Repeats present in CagA are a site for tyrosine phosphorylation. Within the cagA gene, duplication of sequence motifs in the 5′-end region assembled into a repeat at nucleotide position 3406. The anatomy of a repeat is based on three domains: D1, D2, and D3. Repeats are 102 bp causing an increment in size of the protein of 4 kDa. The EPIYA motifs are shown.
THE TYPE IV SECRETION SYSTEM MACHINERY: FROM THE INNER CORE OF CONJUGATION SYSTEMS TO THE INNER CORE OF cag TFSS
Bordetella pertussis, Legionella pneumophila, and Bartonella henselae are members of a large bacterial family that share versions of the TFSS. New structural data were not specific for the H. pylori TFSS so that the latter might be different. However, there are good reasons to examine this large structure. It spans the periplasmic space and forms a chamber where other components are assembled. This is the minimal set of VirB subunits that could be useful for most, if not all, TFSSs. Our understanding of the conjugation system was developed with studies of the F plasmid of Escherichia coli. Conjugation, a contact-dependent process, bears a strong mechanistic resemblance to the type III secretion systems (TTSSs) and TFSSs. Pathogenic bacteria of humans and plants have coopted conjugation systems to export virulence factors to eukaryotic host cells. Thanks to the efforts of Christie et al. (Christie 2009; Sarkar et al. 2013) and Waksman et al. (Chandran et al. 2009; Fronzes et al. 2009a,b; Rivera-Calzada et al. 2013) we are now capable of describing the inner mechanics of the core complex (CC) and the effector delivery subsystem.
CC
The VirB7, VirB9, and VirB10 proteins assemble into a 1.07 megadalton (MDa) CC spanning the inner and outer membranes. This core consists of 14 copies of each of the proteins around which all other components assemble. The main body structure has two polar rings or “restrictions” that intercept the outer membrane (cap) and the inner membrane (base), respectively. The wall of the inner layer consisted of the amino-terminal domain of VirB10 and VirB9 homologs and is anchored in the inner membrane, resembling a cup that is opened at the base by a 55 Å diameter hole. The wall of the outer layer revealed a double-walled structure composed by the carboxy-terminal domain of the VirB10 homolog partially overlapping with the first half of the inner wall of the cap made by the VirB7 and VirB9 homologs. VirB7 and VirB9 contain signal peptides that target them to the periplasm. VirB7 is a small lipoprotein that interacts with and stabilizes VirB9 (Fig. 2).
Figure 2.
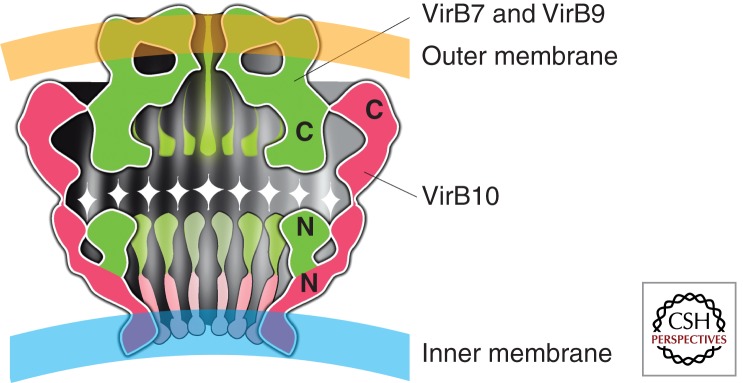
An artistic impression of the core complex as reported by Waksman et al. is shown. It spans the periplasmic space and is composed of a semispherical cup formed by monomers of the VirB10 protein and by a cap of assembled VirB7 and VirB9 elements. The dark areas are the inner capsular spaces.
The Energizers
The cytoplasmic ATPases VirD4, VirB11, and VirB4 are essential for secretion and for powering the secretory engine. VirD4 belong to the substrate receptor because they recruit substrates for secretion through the translocation channel. VirD4 are tethered to the inner membrane by an amino-terminal membrane anchor sequence that is required for assembly into hexameric structures. VirB11 is a typical traffic ATPase in a dynamic and regulated equilibrium between the cytoplasm and the membrane localization. Electron microscopy visualization of VirB11 homologs showed that they organize into hexameric rings. VirB4 forms homodimers, but the dimer might represent an intermediate state for the assembly of higher-order complexes. All of them are likely to form very large complexes of ATPases involved in energy supply for the transport of substrates along the secretory engine.
VirB7, VirB9, and VirB10 form a stable CC that spans the cell envelope and aggregate spontaneously. VirB6 and VirB8 stabilize the ATPase complex—VirB4, VirB11, and VirD4—to dock the translocation channel at the inner membrane. The VirB2 and VirB5 pilus subunits are involved in the latter stage of pilus biogenesis.
CagA AND HOST-CELL DIFFERENTIATION
CagA Induction of Abnormal Apical and Basolateral Markers: Loss of Polarity
The reconstruction of the evolutionary path of a bacterial species should allow us to infer its population structure, the diversification of the virulence determinants that are responsible for discontinuity in clinicopathological manifestations. During lifelong infection CagA persistence increases the risk of long-term complications for the host, such as the development of peptic ulcers and gastric adenocarcinoma (Blaser and Atherton 2004; Cover and Blaser 2009; Correa and Piazuelo 2012). CagA induced abnormal localization of the gp135 apical marker to the basolateral membrane in MDCK cells. This loss of polarity occurred rapidly, because gp135 associated with the basolateral membrane is detected before changes in cell morphology are usually seen. At later stages, CagA-expressing cells elongated and freely migrated underneath neighboring cells, and gp135 localized throughout the basolateral membrane and concentrated at the trailing pole. The localization of basolateral membrane proteins is also altered in CagA-expressing cells. E-cadherin is usually absent from the apical surface of epithelia and exposed if cell polarity is lost or if the fence function of the junctions, which controls the diffusion of integral membrane proteins, fails. Antibodies to the extracellular domain of E-cadherin added to the apical side of nonpermeabilized epithelial monolayers do not stain the apical surface and cannot reach basolateral epitopes (Gumbiner and Simons 1987; Shore and Nelson 1991). However, these antibodies stained the apical surface of CagA-expressing cells but not neighboring control cells. The paracellular barrier of the tight junctions is disrupted in CagA-expressing cells, because these antibodies applied to the apex of nonpermeable cells penetrated the paracellular space to stain basolateral E-cadherin (Bagnoli et al. 2005).
CagA-Dependent Loss of Cell Adhesion
CagA-expressing cells extend pseudopodial processes between adjacent cells and also move between and under neighboring cells in a manner consistent with the loss of cell–cell adhesion. By tracking the movement of individual CagA-expressing cells and surrounding control cells CagA-expressing cells translocated almost four times further from their points of origin indicating a loss of cell adhesion and movement independent of their neighbors. The acquisition of migratory behavior in the context of loss of apicobasal polarity and the severing of intercellular junctions is reminiscent of a differentiation process known as epithelial-to-mesenchymal transition (EMT) (Birchmeier et al. 1996). This morphogenetic developmental program is observed at specialized sites during embryogenesis but is also thought to play an important part in the loss of epithelial characteristics that occurs during tumorigenic progression (Thiery 2002). Epithelial cells that undergo this mesenchymal transition are able to invade the extracellular matrix by expressing matrix metalloproteinases (MMPs) and degrading their underlying basement membrane (Yin et al. 2010).
CagA and Matrix Invasion
MDCK monolayers on filters coated with a basement membrane-like substrate containing type IV collagen and laminin indicated that CagA-expressing cells generated basal pseudopodia that invaded the matrix and penetrated into the underlying filter pores 40 times more than control cells. When GM6001, a synthetic peptide inhibitor of MMPs (Boghaert et al. 1994) is added to the matrix a four-times reduction was observed, suggesting that the process is MMPs dependent and supporting the notion that CagA induces invasive behavior in epithelial cells (Kitadai et al. 2003; El-Etr et al. 2004). Different domains of the CagA protein were expressed in polarized MDCK monolayers. The expression of the carboxy-terminal domain containing the EPIYA motifs were found tyrosine phosphorylated; cells elongated, extending basal pseudopodia that advance and retract between and below adjacent cells. The carboxyl terminus of CagA was sufficient, however, to induce MMP activity because its expression induced the formation of pseudopodia that were able to penetrate basement membranes. The amino terminus of the CagA protein, lacking the EPIYA motifs, was found sufficient to target CagA to the junctions and the plasma membrane but did not cause detectable redistribution of junctional proteins, nor did it disrupt cell polarity. Furthermore, it did not induce morphological changes, cell migration, or invasive behavior. Together, these findings show that the signaling triggered by the EPIYA motifs of CagA is necessary to cause cellular elongation and pseudopodial activity but additional conformational motifs are necessary.
In summary, CagA, a cancer-associated effector protein of H. pylori, is sufficient to disrupt the mechanisms that maintain normal epithelial differentiation, including cell adhesion, cell polarity, and the inhibition of migration.
CagA AND CELL INITIATION
CagA Associates with the ASPP2 Tumor Suppressor
CagA is one of the most important factors that link infection with H. pylori to the development of gastric cancer. Mongolian gerbils challenged with CagA+ H. pylori strains develop gastric dysplasia and adenocarcinoma 12 wk postinfection (Franco et al. 2008). CagA has been suggested to interact with the RTK c-Met (Churin et al. 2003), the tyrosine phosphatase SHP-2 (Higashi et al. 2002), and the adaptor protein Grb2 (Mimuro et al. 2002). CagA has no clear amino acid homology with eukaryotic proteins involved in signaling pathways, but has structural similarities to proteins that can act on regulators of differentiation. For example, CagA share some of the functional properties of Gab proteins, docking molecules that serve as scaffolds to mediate the activation of multiple signals downstream from RTK (Hatakeyama 2003; Higashi et al. 2004). CagA and Gab proteins have been implicated in the activation of the Ras/MAPK (Meyer-ter-Vehn et al. 2000) and PI3-kinase pathways (Churin et al. 2003), both of which have been linked to EMT (Bakin et al. 2000; Grände et al. 2002). Earlier reports have linked injection or exogenous expression of CagA to the activation of several oncogenic pathways. For example, destabilization of the E-cadherin/β-catenin complex by CagA induces abnormal activation of the wingless/int (WNT)/β-catenin pathway (Franco et al. 2005; Botham et al. 2008; Neal et al. 2013). The role of the individual domains of CagA was investigated using an in vivo biotinylation approach combined with affinity purification to identify interacting proteins. LC-MS/MS analysis of a 150 kDa polypeptide unambiguously identified ASPP2, which interacted with full-length CagA and its amino terminus but not with its carboxyl terminus (Buti et al. 2011). The interaction between ASPP2 and p53 results in proteasomal degradation of p53 and consequently, inhibition of the apoptotic response of the host cell. CagA thus hijacks ASPP2 and alters its activity in a manner that promotes cell survival and favors transformation.
CagA Delocalizes ASPP2
ASPP2 is a member of the ASPP family, which includes the proapoptotic ASPP1 and the antiapoptotic iASPP (Bergamaschi et al. 2003). Both ASPP1 and ASPP2 are known activators of the tumor suppressor p53 in response to DNA damage or oncogenic stimuli (Samuels-Lev et al. 2001). The carboxyl terminus of ASPP2 transiently interacts with the DNA-binding domain of p53, and after this interaction, p53 induces the expression of genes involved in apoptosis. ASPP2 also binds other factors with roles in apoptosis (Bcl-2 and Yes-associated protein) (Naumovski and Cleary 1994; Yang et al. 1999; Espanel and Sudol 2001) and control of cell growth (p65/RelA, a subunit of NF-κB, APC tumor suppressor, and protein phosphatase-1) (Helps et al. 1995; Nakagawa et al. 2000). AGS cells infected with Hp and lysed in 0.5% Triton X-100 indicated that ASPP2 was delocalized to a Triton-resistant fraction, which contains cell remnants, nuclei, Triton-resistant membranes, and bacteria. Coexpression of Cherry-ASPP2 with GFP-CagA shows strong colocalization, and CagA recruits ASPP2 to a region close to the plasma membrane. Although there is no clear consensus as to the exact localization of ASPP2 (Sun et al. 2008; Uhlmann-Schiffler et al. 2009), a recent study described the targeting of ASPP2 to tight junctions (Sottocornola et al. 2010). Because full-length CagA is also found close to the plasma membrane and junctional complexes, CagA likely recruits ASPP2 to form a complex near the plasma membrane.
ASPP2 and p53
Injection of CagA facilitates the interaction between p53 and ASPP2. Treatment of cells with Doxorubicin (Dox), a DNA damaging agent that activates p53, induced the association of ASPP2 with p53 as well as apoptosis. Association between CagA and ASPP2 was detected already 90 min postinfection, increasing as time progressed. In contrast, the association between ASPP2 and cytoplasmic p53 was observed not earlier than 3 h and reached a peak at 7 h. This suggested that ASPP2 recruits the cytoplasmic pool of p53 after ASPP2 engaged by CagA (Buti et al. 2011).
Proteosomal Degradation of p53
p53 is a transcription factor that regulates expression on a variety of target genes. It is rapidly degraded by the proteasome but strongly stabilized after DNA damage or cellular stress. Because ASPP2 recruits cytoplasmic p53 during H. pylori infection, the transcriptional activity of p53 might be altered during translocation of CagA. Several lines of research suggested that CagA-mediated inhibition of p53 expression is ASPP2 dependent and CagA-induced degradation of p53 is mediated by the proteasome. Under normal conditions, the tumor suppressor function of the ASPP2-p53 pathway is exerted mainly through induction of the apoptotic response. However, in H. pylori infected cells, CagA promotes the interaction between p53 and ASPP2, leading to an enhanced degradation of p53, and therefore inhibition of its transcriptional activity. CagA inhibits apoptosis by binding the tumor suppressor ASPP2 and through this interaction p53 is degraded and its apoptotic function inhibited (Mimuro et al. 2007; Buti et al. 2011).
CagA Antiapoptotic Effect
Several viral oncogenes (e.g., T-antigen E1B-55Kd and HPV E6 and E7) inactivate the tumor suppressor activity of the host cell by targeting and degrading p53 when the infection is established (Sarnow et al. 1982; Schutzbank et al. 1982; Werness et al. 1990). In addition, H. pylori infection is enhanced when the apoptotic response of gastric epithelial cells is impaired. CagA injection induces the up-regulation of the antiapoptotic protein MCL-1, and consequently, the infected cells become more resistant to normal cell turnover in the stomach. CagA-mediated degradation of p53 results in resistance to the apoptotic response in an ASPP2-dependent manner. Of note, RUNX3 is also sent to the proteasome after CagA interaction, suggesting a link between CagA and the proteasomal machinery of the infected cell (Tsang et al. 2010).
ASPP2 Interactions
ASPP2 is best known for its role as tumor suppressor, and as such, it enhances the apoptotic response of the cell. The amino terminus of ASPP2 is required to enhance the transcriptional activity of p53 (Gorina and Pavletich 1996). Cytosolic DDA3 binds ASPP2 and prevents activation of p53 without affecting the ASPP2-p53 interaction (Sun et al. 2008). CagA may dislocate p53 through ASPP2, thereby altering its function. Indeed, cytoplasmic ASPP1 functions as oncogene, whereas nuclear ASPP1 acts as tumor suppressor (Vigneron et al. 2010). The function of ASPP2 depends on its localization, and CagA may interfere with its localization. The function of ASPP2 is not restricted to activation of p53 but includes regulation of cell–cell adhesion and polarity. Drosophila dASPP localizes at adherens junctions and regulates the activity of carboxy-terminal kinase (dCsk). Loss of function of dASPP increases cell spreading and apoptosis. In mammalian cells, ASPP2 associates with Par-3 (Sottocornola et al. 2010), a complex crucial for the formation and localization of the apical-junctional complex. ASPP2-depleted cells are defective in the formation of tight junctions and acquire a migratory phenotype (Langton et al. 2007, 2009; Cong et al. 2010). Thus, ASPP2 acts as a tumor suppressor by enhancing the transcriptional activity of p53 (Bunz et al. 1998; Wei et al. 2010); then it may act as a regulator of cell–cell adhesion in a p53-independent manner. Interactors of CagA identified by LC-MS/MS not only reveal the presence of ASPP2 but also Par-3 (Fig. 3) (Buti et al. 2011). Several articles have also identified abnormal p53 after exposure to H. pylori, including truncated forms that are promoting increased survival of the host cells (Wei et al. 2010, 2012). MALT lymphomas can be distinguished from other lymphomas because they occur in various extranodal locations, primarily the stomach, where they are preceded by H. pylori infection (Vicente-Dueñas et al. 2012). Malt lymphomas are associated with chromosomal translocations producing the oncogene MALT-1. Mouse animal model of the disease has been generated and progression to activated B-cell diffuse large-cell lymphoma (ADC-DLBCL) is associated with loss of p53, mimicking the human form of the disease (Vicente-Dueñas et al. 2012). Loss of p53 is a crucial factor for inducing a totipotent phenotype and generating pluripotent stem cells (Marión et al. 2009; Kawamura et al. 2009). The association between H. pylori and diseases might hinge on the effect that controls the epigenetic status of the cells. Not only does CagA cause the functional ablation of the ASPP2/p53 tumor suppressor pathways and the altered terminal differentiation program (EMT), but may predispose to initiation/promotion before the further stage of tumorigenic progression.
Figure 3.

Schematic model of the effects induced by the CagA-ASPP2 interaction. CagA binds ASPP2, and this complex is responsible for proteasome-dependent p53 degradation. Cells harboring CagA are unable to cause apoptosis. In addition, they acquire the ability to migrate, invade, and they lack a terminal differentiation program (EMT).
CONCLUDING REMARKS
The acquisition of the cag PAI has targeted the mechanisms of virulence of H. pylori at the intracellular level.
CagA, the major effector of virulence is conveyed within the host cell by a dedicated TFSS. The contact-dependent secretion system relies on a large complex of ATPases able to translocate CagA molecules at a sustained peak rate.
Tyrosine-phosphorylated CagA is responsible for acting at the level of cellular junctions. The clinical lesions (ulceration) associated with H. pylori infection should be seen as resulting from loss of contact among cells.
H. pylori has infected man before the human migration and has established a status of quasispecies. Through a deep-sequencing approach migrations can be traced at an unprecedented level and factors affecting persistence, tissue specificity, and disease outcomes can be identified in the host.
At the epithelial level the CagA protein causes altered terminal differentiation, preneoplastic lesions like ETM, and abnormal cell migration.
Degradation of p53 is a consequence of the recruitment and misregulation of ASPP2 by CagA. We suggest that altered localization and/or posttranslational modification of ASPP2 might be responsible for decay in tumor suppressor function resulting in aberrant complex with p53.
ACKNOWLEDGMENTS
We thank C. Montecucco, Fabio Bagnoli, Manuel Amieva, Hidde Ploegh, Stanley Falkow, Jörg Hacker, Derya Unutmaz, and Rino Rappuoli for helpful discussions and critical reading of the manuscript and Riccardo Beltrami, Claudio Donati, and Duccio Medini for granting access to an unpublished set of data. We acknowledge the National Center for Biotechnology Information, EBI, EMBL, and The Broad Institute at MIT (Cambridge, MA) for granting access to their genomic software, and Dr. N. Figura (University of Siena, Italy) for H. pylori clinical isolates. We gratefully acknowledge G. Corsi for the artwork and Barbara Acosta (Cold Spring Harbor Laboratory Press) for excellent editorial assistance. A.C. thanks the “external reviewer” for previous comments on the manuscript.
Footnotes
Editors: Pascale Cossart and Stanley Maloy
Additional Perspectives on Bacterial Pathogenesis available at www.perspectivesinmedicine.org
REFERENCES
- Achtman M, Azuma T, Berg DE, Ito Y, Morelli G, Pan ZJ, Suerbaum S, Thompson SA, Van der Ende A, Van Doorn LJ 1999. Recombination and clonal groupings within Helicobacter pylori from different geographical regions. Mol Microbiol 32: 459–470 [DOI] [PubMed] [Google Scholar]
- Akopyants NS, Clifton SW, Kersulyte D, Crabtree JE, Youree BE, Reece CA, Bukanov NO, Drazek ES, Roe BA, Berg DE 1998. Analyses of the cag pathogenicity island of Helicobacter pylori. Mol Microbiol 28: 37–53 [DOI] [PubMed] [Google Scholar]
- Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S 2003. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 300: 1430–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahi M, Azuma T, Ito S, Ito Y, Suto H, Nagai Y, Tsubokawa M, Tohyama Y, Maeda S, Omata M, et al. 2000. Helicobacter pylori Caga protein can be tyrosine phosphorylated in gastric epithelial cells. J Exp Med 191: 593–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backert S, Ziska E, Brinkmann V, Zimny-Arndt U, Fauconnier A, Jungblut PR, Naumann M, Meyer TF 2000. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell Microbiol 2: 155–164 [DOI] [PubMed] [Google Scholar]
- Bagnoli F, Buti L, Tompkins L, Covacci A, Amieva MR 2005. Helicobacter pylori CagA induces a transition from polarized to invasive phenotypes in MDCK cells. Proc Natl Acad Sci 102: 16339–16344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL 2000. Phosphatidylinositol 3-kinase function is required for transforming growth factor β-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem 275: 36803–36810 [DOI] [PubMed] [Google Scholar]
- Bergamaschi D, Samuels Y, O’Neil NJ, Trigiante G, Crook T, Hsieh J-K, O’Connor DJ, Zhong S, Campargue I, Tomlinson ML, et al. 2003. iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat Genet 33: 162–167 [DOI] [PubMed] [Google Scholar]
- Birchmeier C, Birchmeier W, Brand-Saberi B 1996. Epithelial-mesenchymal transitions in cancer progression. Acta Anatomica 156: 217–226 [DOI] [PubMed] [Google Scholar]
- Blaser MJ, Atherton JC 2004. Helicobacter pylori persistence: Biology and disease. J Clin Invest 113: 321–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum G, Ott M, Lischewski A, Ritter A, Imrich H, Tschäpe H, Hacker J 1994. Excision of large DNA regions termed pathogenicity islands from tRNA-specific loci in the chromosome of an Escherichia coli wild-type pathogen. Infect Immun 62: 606–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boghaert ER, Chan SK, Zimmer C, Grobelny D, Galardy RE, Vanaman TC, Zimmer SG 1994. Inhibition of collagenolytic activity relates to quantitative reduction of invasion in vitro in a c-Ha-ras transfected glial cell line. J Neurooncol 21: 141–150 [DOI] [PubMed] [Google Scholar]
- Botham CM, Wandler AM, Guillemin K 2008. A transgenic Drosophila model demonstrates that the Helicobacter pylori CagA protein functions as a eukaryotic Gab adaptor. PLoS Pathog 4: e1000064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B 1998. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282: 1497–1501 [DOI] [PubMed] [Google Scholar]
- Buti L, Spooner E, Van der Veen AG, Rappuoli R, Covacci A, Ploegh HL 2011. Helicobacter pylori cytotoxin-associated gene A (CagA) subverts the apoptosis-stimulating protein of p53 (ASPP2) tumor suppressor pathway of the host. Proc Natl Acad Sci 108: 9238–9243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell S, Fraser A, Holliss B, Schmid J, O’Toole PW 1997. Evidence for ethnic tropism of Helicobacter pylori. Infect Immun 65: 3708–3712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, Rappuoli R, Covacci A 1996. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci 93: 14648–14653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran V, Fronzes R, Duquerroy S, Cronin N, Navaza J, Waksman G 2009. Structure of the outer membrane complex of a type IV secretion system. Nature 462: 1011–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie PJ 2009. Structural biology: Translocation chamber’s secrets. Nature 462: 992–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churin Y, Al-Ghoul L, Kepp O, Meyer TF, Birchmeier W, Naumann M 2003. Helicobacter pylori CagA protein targets the c-Met receptor and enhances the motogenic response. J Cell Biol 161: 249–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong W, Hirose T, Harita Y, Yamashita A, Mizuno K, Hirano H, Ohno S 2010. ASPP2 regulates epithelial cell polarity through the PAR complex. Curr Biol 20: 1408–1414 [DOI] [PubMed] [Google Scholar]
- Correa P, Piazuelo MB 2012. The gastric precancerous cascade. J Digest Dis 13: 2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covacci A, Rappuoli R 2000. Tyrosine-phosphorylated bacterial proteins: Trojan horses for the host cell. J Exp Med 191: 587–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covacci A, Censini S, Bugnoli M, Petracca R, Burroni D, Macchia G, Massone A, Papini E, Xiang Z, Figura N 1993. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc Natl Acad Sci 90: 5791–5795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covacci A, Telford JL, Del Giudice G, Parsonnet J, Rappuoli R 1999. Helicobacter pylori virulence and genetic geography. Science 284: 1328–1333 [DOI] [PubMed] [Google Scholar]
- Cover TL, Blaser MJ 2009. Helicobacter pylori in health and disease. Gastroenterology 136: 1863–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Etr SH, Mueller A, Tompkins LS, Falkow S, Merrell DS 2004. Phosphorylation-independent effects of CagA during interaction between Helicobacter pylori and T84 polarized monolayers. J Infect Dis 190: 1516–1523 [DOI] [PubMed] [Google Scholar]
- Espanel X, Sudol M 2001. Yes-associated protein and p53-binding protein-2 interact through their WW and SH3 domains. J Biol Chem 276: 14514–14523 [DOI] [PubMed] [Google Scholar]
- Franco AT, Israel DA, Washington MK, Krishna U, Fox JG, Rogers AB, Neish AS, Collier-Hyams L, Perez-Perez GI, Hatakeyama M, et al. 2005. Activation of β-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci 102: 10646–10651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco AT, Johnston E, Krishna U, Yamaoka Y, Israel DA, Nagy TA, Wroblewski LE, Piazuelo MB, Correa P, Peek RM 2008. Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res 68: 379–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fronzes R, Christie PJ, Waksman G 2009a. The structural biology of type IV secretion systems. Nat Rev Microbiol 7: 703–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fronzes R, Schäfer E, Wang L, Saibil HR, Orlova EV, Waksman G 2009b. Structure of a type IV secretion system core complex. Science 323: 266–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorina S, Pavletich NP 1996. Structure of the p53 tumor suppressor bound to the Ankyrin and SH3 domains of 53BP2. Science 274: 1001–1005 [DOI] [PubMed] [Google Scholar]
- Grände M, Franzen Å, Karlsson J-O, Ericson LE, Heldin N-E, Nilsson M 2002. Transforming growth factor-β and epidermal growth factor synergistically stimulate epithelial to mesenchymal transition (EMT) through a MEK-dependent mechanism in primary cultured pig thyrocytes. J Cell Sci 115 : 4227–4236 [DOI] [PubMed] [Google Scholar]
- Gumbiner B, Simons K 1987. The role of uvomorulin in the formation of epithelial occluding junctions. Ciba Found Symp 125: 168–186 [DOI] [PubMed] [Google Scholar]
- Hatakeyama M 2003. Helicobacter pylori CagA—A potential bacterial oncoprotein that functionally mimics the mammalian Gab family of adaptor proteins. Microb Infect 5: 143–150 [DOI] [PubMed] [Google Scholar]
- Hayashi T, Senda M, Morohashi H, Higashi H, Horio M, Kashiba Y, Nagase L, Sasaya D, Shimizu T, Venugopalan N, et al. 2012. Tertiary structure-function analysis reveals the pathogenic signaling potentiation mechanism of Helicobacter pylori oncogenic effector CagA. Cell Host Microbe 12: 20–33 [DOI] [PubMed] [Google Scholar]
- Hazell SL, Lee A, Brady L, Hennessy W 1986. Campylobacter pyloridis and gastritis: Association with intercellular spaces and adaptation to an environment of mucus as important factors in colonization of the gastric epithelium. J Infect Dis 153: 658–663 [DOI] [PubMed] [Google Scholar]
- Helps NR, Barker HM, Elledge SJ, Cohen PT 1995. Protein phosphatase 1 interacts with p53BP2, a protein which binds to the tumour suppressor p53. FEBS Lett 377: 295–300 [DOI] [PubMed] [Google Scholar]
- Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, Hatakeyama M 2002. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 295: 683–686 [DOI] [PubMed] [Google Scholar]
- Higashi H, Nakaya A, Tsutsumi R, Yokoyama K, Fujii Y, Ishikawa S, Higuchi M, Takahashi A, Kurashima Y, Teishikata Y, et al. 2004. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J Biol Chem 279: 17205–17216 [DOI] [PubMed] [Google Scholar]
- Kaplan-Türköz B, Jiménez-Soto LF, Dian C, Ertl C, Remaut H, Louche A, Tosi T, Haas R, Terradot L 2012. Structural insights into Helicobacter pylori oncoprotein CagA interaction with β1 integrin. Proc Natl Acad Sci 109: 14640–14645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, Wahl GM, Izpisúa Belmonte JC 2009. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 460: 1140–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitadai Y, Sasaki A, Ito M, Tanaka S, Oue N, Yasui W, Aihara M, Imagawa K, Haruma K, Chayama K 2003. Helicobacter pylori infection influences expression of genes related to angiogenesis and invasion in human gastric carcinoma cells. Biochem Biophys Res Commun 311: 809–814 [DOI] [PubMed] [Google Scholar]
- Langton PF, Colombani J, Aerne BL, Tapon N 2007. Drosophila ASPP regulates C-terminal Src kinase activity. Dev Cell 13: 773–782 [DOI] [PubMed] [Google Scholar]
- Langton PF, Colombani J, Chan EHY, Wepf A, Gstaiger M, Tapon N 2009. The dASPP-dRASSF8 complex regulates cell-cell adhesion during Drosophila retinal morphogenesis. Curr Biol 19: 1969–1978 [DOI] [PubMed] [Google Scholar]
- Marión RM, Strati K, Li H, Murga M, Blanco R, Ortega S, Fernandez-Capetillo O, Serrano M, Blasco MA 2009. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 460: 1149–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-ter-Vehn T, Covacci A, Kist M, Pahl HL 2000. Helicobacter pylori activates mitogen-activated protein kinase cascades and induces expression of the proto-oncogenes c-fos and c-jun. J Biol Chem 275: 16064–16072 [DOI] [PubMed] [Google Scholar]
- Mimuro H, Suzuki T, Tanaka J, Asahi M, Haas R, Sasakawa C 2002. Grb2 is a key mediator of Helicobacter pylori CagA protein activities. Mol Cell 10: 745–755 [DOI] [PubMed] [Google Scholar]
- Mimuro H, Suzuki T, Nagai S, Rieder G, Suzuki M, Nagai T, Fujita Y, Nagamatsu K, Ishijima N, Koyasu S, et al. 2007. Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe 2: 250–263 [DOI] [PubMed] [Google Scholar]
- Moodley Y, Linz B, Yamaoka Y, Windsor HM, Breurec S, Wu J-Y, Maady A, Bernhöft S, Thiberge J-M, Phuanukoonnon S, et al. 2009. The peopling of the Pacific from a bacterial perspective. Science 323: 527–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moodley Y, Linz B, Bond RP, Nieuwoudt M, Soodyall H, Schlebusch CM, Bernhöft S, Hale J, Suerbaum S, Mugisha L, et al. 2012. Age of the association between Helicobacter pylori and man. PLoS Patho 8: e1002693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Koyama K, Murata Y, Morito M, Akiyama T, Nakamura Y 2000. APCL, a central nervous system-specific homologue of adenomatous polyposis coli tumor suppressor, binds to p53-binding protein 2 and translocates it to the perinucleus. Cancer Res 60: 101–105 [PubMed] [Google Scholar]
- Naumovski L, Cleary M 1994. Bcl2 inhibits apoptosis associated with terminal differentiation of HL-60 myeloid leukemia cells. Blood 83: 2261–2267 [PubMed] [Google Scholar]
- Neal JT, Peterson TS, Kent ML, Guillemin K 2013. H. pylori virulence factor CagA increases intestinal cell proliferation by Wnt pathway activation in a transgenic zebrafish model. Disease Model Mech 6: 802–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LT, Uchida T, Murakami K, Fujioka T, Moriyama M 2008. Helicobacter pylori virulence and the diversity of gastric cancer in Asia. J Med Microbiol 57: 1445–1453 [DOI] [PubMed] [Google Scholar]
- Odenbreit S, Püls J, Sedlmaier B, Gerland E, Fischer W, Haas R 2000. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287: 1497–1500 [DOI] [PubMed] [Google Scholar]
- Rivera-Calzada A, Fronzes R, Savva CG, Chandran V, Lian PW, Laeremans T, Pardon E, Steyaert J, Remaut H, Waksman G, et al. 2013. Structure of a bacterial type IV secretion core complex at subnanometre resolution. EMBO J 32: 1195–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde M, Püls J, Buhrdorf R, Fischer W, Haas R 2003. A novel sheathed surface organelle of the Helicobacter pylori cag type IV secretion system. Mol Microbiol 49: 219–234 [DOI] [PubMed] [Google Scholar]
- Samuels-Lev Y, O’Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S, Campargue I, Naumovski L, Crook T, Lu X 2001. ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell 8: 781–794 [DOI] [PubMed] [Google Scholar]
- Sarkar MK, Husnain SI, Jakubowski SJ, Christie PJ 2013. Isolation of bacterial type IV machine subassemblies. Method Mol Biol 966: 187–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarnow P, Ho YS, Williams J, Levine AJ 1982. Adenovirus E1b-58kd tumor antigen and SV40 large tumor antigen are physically associated with the same 54 kd cellular protein in transformed cells. Cell 28: 387–394 [DOI] [PubMed] [Google Scholar]
- Schutzbank T, Robinson R, Oren M, Levine AJ 1982. SV40 large tumor antigen can regulate some cellular transcripts in a positive fashion. Cell 30: 481–490 [DOI] [PubMed] [Google Scholar]
- Segal ED, Cha J, Lo J, Falkow S, Tompkins LS 1999. Altered states: Involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc Natl Acad Sci 96: 14559–14564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore EM, Nelson WJ 1991. Biosynthesis of the cell adhesion molecule uvomorulin (E-cadherin) in Madin-Darby canine kidney epithelial cells. J Biol Chem 266: 19672–19680 [PubMed] [Google Scholar]
- Sottocornola R, Royer C, Vives V, Tordella L, Zhong S, Wang Y, Ratnayaka I, Shipman M, Cheung A, Gaston-Massuet C, et al. 2010. ASPP2 binds Par-3 and controls the polarity and proliferation of neural progenitors during CNS development. Dev Cell 19: 126–137 [DOI] [PubMed] [Google Scholar]
- Steer HW 1984. Surface morphology of the gastroduodenal mucosa in duodenal ulceration. Gut 25: 1203–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein M, Rappuoli R, Covacci A 2000. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc Natl Acad Sci 97: 1263–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein M, Bagnoli F, Halenbeck R, Rappuoli R, Fantl WJ, Covacci A 2002. c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol Microbiol 43: 971–980 [DOI] [PubMed] [Google Scholar]
- Sun W-T, Hsieh P-C, Chiang M-L, Wang M-C, Wang F-F 2008. p53 target DDA3 binds ASPP2 and inhibits its stimulation on p53-mediated BAX activation. Biochem Biophys Res Commun 376: 395–398 [DOI] [PubMed] [Google Scholar]
- Thiery JP 2002. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2: 442–454 [DOI] [PubMed] [Google Scholar]
- Tsang YH, Lamb A, Romero-Gallo J, Huang B, Ito K, Peek RM, Ito Y, Chen LF 2010. Helicobacter pylori CagA targets gastric tumor suppressor RUNX3 for proteasome-mediated degradation. Oncogene 29: 5643–5650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tummuru MK, Cover TL, Blaser MJ 1993. Cloning and expression of a high-molecular-mass major antigen of Helicobacter pylori: Evidence of linkage to cytotoxin production. Infect Immun 61: 1799–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann-Schiffler H, Kiermayer S, Stahl H 2009. The DEAD box protein Ddx42p modulates the function of ASPP2, a stimulator of apoptosis. Oncogene 28: 2065–2073 [DOI] [PubMed] [Google Scholar]
- Vicente-Dueñas C, Fontán L, Gonzalez-Herrero I, Romero-Camarero I, Segura V, Aznar MA, Alonso-Escudero E, Campos-Sanchez E, Ruiz-Roca L, Barajas-Diego M, et al. 2012. Expression of MALT1 oncogene in hematopoietic stem/progenitor cells recapitulates the pathogenesis of human lymphoma in mice. Proc Natl Acad Sci 109: 10534–10539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneron AM, Ludwig RL, Vousden KH 2010. Cytoplasmic ASPP1 inhibits apoptosis through the control of YAP. Genes Dev 24: 2430–2439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Nagy TA, Vilgelm A, Zaika E, Ogden SR, Romero-Gallo J, Piazuelo MB, Correa P, Washington MK, El-Rifai W, et al. 2010. Regulation of p53 tumor suppressor by Helicobacter pylori in gastric epithelial cells. Gastroenterology 139: 1333–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Noto J, Zaika E, Romero-Gallo J, Correa P, El-Rifai W, Peek RM, Zaika A 2012. Pathogenic bacterium Helicobacter pylori alters the expression profile of p53 protein isoforms and p53 response to cellular stresses. Proc Natl Acad Sci 109: E2543–E2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werness BA, Levine AJ, Howley PM 1990. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 248: 76–79 [DOI] [PubMed] [Google Scholar]
- Xiang Z, Censini S, Bayeli P, Telford J, Figura N, Rappuoli R, Covacci A 1995. Analysis of expression of CagA and VacA virulence factors in 43 strains of Helicobacter pylori reveals that clinical isolates can be divided into two major types and that CagA is not necessary for expression of the vacuolating cytotoxin. Infect Immun 63: 94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JP, Hori M, Takahashi N, Kawabe T, Kato H, Okamoto T 1999. NF-κB subunit p65 binds to 53BP2 and inhibits cell death induced by 53BP2. Oncogene 18: 5177–5186 [DOI] [PubMed] [Google Scholar]
- Yin Y, Grabowska AM, Clarke PA, Whelband E, Robinson K, Argent RH, Tobias A, Kumari R, Atherton JC, Watson SA 2010. Helicobacter pylori potentiates epithelial:mesenchymal transition in gastric cancer: Links to soluble HB-EGF, gastrin and matrix metalloproteinase-7. Gut 59: 1037–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]