

Abstract
Reduced NMDA-receptor (NMDAR) function has been implicated in the pathophysiology of neuropsychiatric disease, most strongly in schizophrenia but also recently in autism spectrum disorders (ASD). To determine the direct contribution of NMDAR dysfunction to disease phenotypes, a mouse model with constitutively reduced expression of the obligatory NR1 subunit has been developed and extensively investigated. Adult NR1neo−/− mice show multiple abnormal behaviors, including reduced social interactions, locomotor hyperactivity, self-injury, deficits in prepulse inhibition, and sensory hypersensitivity, among others. Whereas such phenotypes have largely been interpreted in the context of schizophrenia, these behavioral abnormalities are rather non-specific and are frequently present across models of diseases characterized by negative symptom domains. This study investigated auditory electrophysiological and behavioral paradigms relevant to autism, to determine whether NMDAR hypofunction may be more consistent with adult ASD-like phenotypes. Indeed, transgenic mice demonstrated behavioral deficits relevant to all core ASD symptoms, including decreased social interactions, altered ultrasonic vocalizations, and increased repetitive behaviors. NMDAR disruption recapitulated clinical endophenotypes including reduced prepulse inhibition, auditory-evoked response N1 latency delay, and reduced gamma synchrony. Auditory electrophysiological abnormalities more closely resembled those seen in clinical studies of autism than schizophrenia. These results suggest that NMDA-receptor hypofunction may be associated with a continuum of neuropsychiatric diseases, including schizophrenia and autism. Neural synchrony abnormalities suggest an imbalance of glutamatergic and GABAergic coupling and may provide a target, along with behavioral phenotypes, for preclinical screening of novel therapeutics.
Keywords: autism, mouse model, NMDA-receptor, behavior, endophenotype
Introduction
Reduced NMDA-receptor mediated glutamatergic signaling has been strongly linked to the pathophysiology of schizophrenia (Tsai & Coyle, 2002). A number of animal models have been created to investigate this connection, including acute administration of NMDAR antagonists (Jentsch & Roth, 1999) and NR1neo−/− mice, which were generated to have constitutive ~85% downregulation of the obligate NMDA-NR1 subunit (Mohn et al., 1999). NR1neo−/− mice have extensive behavioral abnormalities, including reduced social interactions and mating, locomotor hyperactivity, deficits in sensorimotor gating, self-injury, and cognitive inflexibility (Duncan et al., 2006, Duncan et al., 2004, Dzirasa et al., 2009, Mohn et al., 1999, Moy et al., 2008). Although these results were interpreted in the context of schizophrenia, such behavioral deficits are largely non-specific and the specificity of these findings for schizophrenia has been recently questioned (Barkus et al., 2012). To more specifically examine endophenotypic deficits in this model, we previously investigated auditory and visual event-related potentials (ERPs), which show a complex pattern of disruption in schizophrenia (Amann et al., 2010, Bodarky et al., 2009, Halene et al., 2009a). However, NMDA-NR1 deficiency caused significant sensory disinhibition, a pattern not closely resembling that seen in the clinical population. Such findings suggested that NR1neo−/− mice might be a more robust model for disorders characterized by negative symptoms and sensory hypersensitivity, such as autism (Tannan et al., 2008).
Emerging evidence has indicated that disrupted NMDA-receptor signaling may contribute to the pathogenesis of idiopathic autism (Carlson, 2011, Carlsson, 1998, Gai et al., 2011, Moskal et al., 2011, Myers et al., 2011, O’roak et al., 2011) and related clinical syndromes, including Rett and Fragile X Syndrome (Krueger et al., 2011, Maliszewska-Cyna et al., 2010, Yun & Trommer, 2011). Autism is a highly disabling neurodevelopmental disorder defined by a triad of core symptoms, including impairments in reciprocal social interactions, deficits in expressive and receptive language function, and repetitive or restricted interests and behaviors. In addition, autism is associated with a number of comorbid symptoms and disorders, including cognitive disability, attentional problems, disrupted sensory processing, obsessive-compulsive disorder (OCD), self-injury, epilepsy, and sleep dysfunction (Levy et al., 2009). Auditory processing is disrupted in autism, with a delayed profile and reduced gamma synchrony (Gandal et al., 2010) – a pattern distinct from schizophrenia.
This study investigated the relationship between NMDA-receptor downregulation and ASD-like phenotypes in adult mice, to more rigorously establish a connection between NMDAR-dysfunction and disease pathophysiology. Using established behavioral paradigms, we demonstrate that NR1neo−/− mice have deficits in all core domains of autism, including sociability, communication, and repetitive behaviors (Silverman et al., 2010). Likewise, these mice recapitulate electrophysiological endophenotypes seen in autism more closely than in schizophrenia. Finally, these findings suggest that NMDA-receptor dysregulation could contribute to core ASD symptoms and clinical biomarkers.
Materials and Methods
Animals
The NR1 transgenic mice were bred and genotyped in house, as previously published (Bodarky et al., 2009, Halene et al., 2009a, Mohn et al., 1999). Mice were maintained on a first-generation (F1) C57BL/6J x 129S6/SvEvTac hybrid background. The line was maintained on the two separate backgrounds (B6 and 129) and was crossed to generate F1 test subjects. Only F1 subjects were used in this study (not backcrossed). Only adult male NR1 transgenic mice and WT littermates were used in this study. Three separate sets of mice were tested. The first group (age 5.4 +/− 1.3 months) was assessed for male-female social interactions & vocalizations, then tail suspension, and finally 5-day locomotor activity. The second group (age 4.1 +/−0.1 months) was measured for auditory startle followed by auditory electrophysiology. The final group (age 10.1 +/− 0.3 months) was assessed for male-male social interactions followed by T-maze performance. Tests in the same mice were separated by at least six days. Animals were housed 4–5 per cage on a 12-hour light/dark cycle in a temperature-controlled facility with food and water available ad libitum. After electrode implantation surgery, mice were housed individually. All testing, with the exception of 5-day locomotor activity, was performed exclusively in the light-cycle. All protocols were approved by the University of Pennsylvania Institutional Animal Care and Use Committees and were conducted in accordance with National Institutes of Health guidelines. Throughout the study, all possible effort was taken to minimize pain and discomfort in experimental subjects.
Social Approach/Avoidance Paradigm
Sociability was measured in NR1 (n=7) and WT mice (n=8) using a standard social choice task as previously published (Gandal et al., 2010, Halene et al., 2009a). Briefly, test mice were placed in a large arena (50×35×40cm) covered with 2 cm of bedding in a dimly lit room. In phase 1 of the experiment, two identical Plexiglas cylinders were placed at opposite ends of the chamber. The test mouse was allowed to freely explore the arena and cylinders for 10 minutes, tracked by an overhead camera interfaced with TopScan software (Cleversys, Inc, VA). In phase 2, an unfamiliar wildtype A/J stimulus mouse was placed under one of the two cylinders. The test mouse was reintroduced to the arena for an additional 10 minutes. The cylinders had multiple holes (1 cm in diameter) to allow for auditory, visual, and olfactory investigation between mice. Equipment was thoroughly cleaned between tests, and the “social cylinder” was counterbalanced among mice. NR1neo−/− and WT mice were assessed for the total time spent sniffing social and non-social chambers. Sniffing time was measured offline by a single blinded experimenter using two stopwatches and was defined as the time a mouse spent with its nose oriented toward the cylinder at a distance of less than 1 cm. Significance was assessed with a group x condition repeated-measures ANOVA, followed by Bonferroni posttests where appropriate.
Male-Female Social Interaction and Vocalizations
Mice were well handled and acclimated to the testing room for 1 hour in low light. A single male mouse (WT or NR1neo−/− in alternating order, n=8/group) was then removed from its home cage and placed into a large, clean testing chamber (46×24×20 cm) with no bedding. An ultrasonic microphone (Pettersson Elektronik AB, Sweden; Model D940) was placed approximately 10 cm above the setup. The microphone is sensitive to frequencies 10–200 kHz and was interfaced with Spike2 software through Power1401 hardware at a 250 kHz sampling rate (CED; Cambridge, UK). Male mice were allowed to acclimate to the testing chamber for 2 minutes, during which time no ultrasonic vocalizations (USVs) were detected. Next, an unfamiliar receptive WT female mouse was placed in the cage. Social interactions initiated by the male were timed with a silent stopwatch and USVs emitted by the male were recorded during a 5 minute testing session. At no point was any mounting behavior observed. Mice were then placed in a holding cage until all males from the same homecage had been tested. The testing chamber was cleaned with 70% ethanol and allowed to dry for at least 5 minutes between sessions. Sonograms were created in Spike2 using the Fast Fourier Transform (FFT; length 1024 points, Hanning window). Vocalizations were hand counted by a single, blinded experimenter offline. Peak frequency was measured by FFT (2048 point, 0.1 kHz resolution) for 20 randomly sampled calls per animal. Significance was assessed by unpaired t-tests.
Modified Tail Suspension Test
A modified tail suspension task (TST) was performed on adult mice (n=10–11/group). Mice were held upside down 20 cm from a clean table surface. Mice were videotaped for 2 minutes and measured for repetitive, forepaw handwringing behavior. Handwringing was defined as repeated clasping of forepaws while shaking them up and down in front of the face (see Supplemental Videos). Wildtype mice generally spread their forepaws during tail suspension. Again, a single blinded experimenter rated the videos as to whether handwringing behavior was present at any time during the 2 minute testing session. Signifiance was assessed using a Chi-square test.
Auditory Electrophysiology
Animals (n=8/group) were anesthetized with isoflurane and underwent stereotaxic implantation of tripolar electrode assemblies (PlasticsOne, Roanoke, VA, USA) for non-anesthetized recording of auditory event-related potentials (ERPs), according to previously published methods (Ehrlichman et al., 2009a, Gandal et al., 2008, Halene et al., 2009a, Lazarewicz et al., 2010). A low-impedance stainless steel macroelectrode (<5 kΩ, 1000 Hz) was placed in the right CA3 hippocampal region (1.8mm posterior, 2.65mm right lateral, and 2.75mm deep) referenced to the ipsilateral frontal sinus. The pedestal was secured to the skull with cyanoacrylate and dental cement. This low-impedance, differential recording configuration captures both early and late components of the auditory evoked potential, including the acoustic brainstem response, mid-latency P1 (e.g., human P50/M50) and N1 (e.g., human N100/M100), as well as the late P2 and P3a peaks, as published (Connolly et al., 2004, Connolly et al., 2003, Siegel et al., 2003) with strong analogy to human scalp EEG (Gandal et al., 2010, Gandal et al., 2008).
Recording of auditory ERPs was performed at least 1 week after electrode implantation in a home-cage environment, as previously described (Ehrlichman et al., 2009a, Gandal et al., 2008, Halene et al., 2009a, Lazarewicz et al., 2010). Cages were placed in a sound attenuated recording chamber inside a Faraday electrical isolation cage. Electrode cables were connected to a high-impedance differential AC amplifier (A–M Systems; Seattle, WA). Stimuli were generated by Micro1401 hardware with Spike2 software (CED; Cambridge, UK) and were delivered through speakers attached to the cage top. The sequencer file consisted of a series of 1000 white-noise bursts presented at 80 dB SPL (10 ms in duration) with a 1-s interstimulus (ISI)interval. Recording sessions were preceded by a 15 minute acclimation phase. Raw EEG was filtered between 1 and 500 Hz. Individual sweeps were rejected for movementartifact based on a criterion of 2 times the root mean squaredamplitude per mouse.
Grand average waveforms were created for each mouse and N1 latency was measured as the local minimum with zero slope between 20 and 60 ms. Waveforms that did not meet these criteria were not included (1 mouse). Time-frequency decomposition of EEG signal was performed with the EEGLab toolbox in Matlab (Delorme & Makeig, 2004). Single trial epochs between −0.3 and 0.8 sec relative were extracted from the continuous data sampled at 1667 Hz. Phase-locking (e.g., intertrial coherence) was calculated using Morlet wavelets in 100 linearly spaced frequency bins between 5–100 Hz, with wavelet cycles linearly increasing from 3 to 6, as published (Gandal et al., 2010). For each subject, phase-locking was averaged over the peak response from 0–100 ms post-stimulus in the gamma frequency range (30–80 Hz).
Delayed Alternating T-maze Paradigm
Spatial cognition was measured using a discrete-trial, spontaneous alternation T-maze task with a 5-second delay, according to published protocols (Deacon & Rawlins, 2006). The maze consisted of three identical arms (30 cm long, 10 cm wide, 12 cm tall), with a central partition and guillotine doors for each arm. Wild-type mice have a natural tendency to alternate when placed in such a chamber, achieving spontaneous alternation rates up to 90% (Deacon & Rawlins, 2006). Mice were well handled and acclimated to the room and apparatus prior to testing, which occurred in low-lux red light to minimize anxiety. To begin the testing session, mice were placed in the central arm of the partition with the guillotine door closed. The door was then opened and mice were allowed to choose one of two side arms. Once a mouse had entered the side arm, the guillotine door was closed and mice were confined to that arm for 30 seconds. Mice were then returned to the central partition, which was closed off for 5-seconds (delay period), the guillotine door was opened and mice were allowed to make a second choice of goal arms. Mice (n=7–9/group) were tested for a total of 10 trials.
Prepulse Inhibition (PPI)
Prepulse inhibition of acoustic startle was assessed in mice (n=8/group) using a four-chamber system (San Diego Instruments, San Diego; CA) as published (Gandal et al., 2010). Briefly, 10 trials of startle responses were measured to 40-ms pulses at 0 and 90–120 dB sound pressure (5 dB increments), randomly presented five times each with an inter-stimulus interval from 10–20s. 5 total pre-pulse trials consisted of a 20-ms pulse at 69, 73, or 81-dB followed by a 40-ms pulse of 120 dB 100 ms later. PPI was calculated as the percent difference in startle units following the pre-pulse/startle pair as compared to the startle tone alone. Significance was assessed with a group x intensity repeated-measures ANOVA.
Locomotor Activity (LMA)
The effect of constitutive NMDAR-hypofunction was assessed on locomotor activity, as previously published (Ehrlichman et al., 2009b). Mice (n=4–5/group) were allowed to acclimate to the testing room for 30+ minutes on two consecutive days prior to testing. After acclimation, mice were placed in a new home cage environment with clean bedding inside an automated locomotor activity photobeam frame (30×24×8 cm) with sensors arranged in an 8-beam array strip with 3.175 cm spacing (MED Associates, VT). For 120 hours, locomotor activity was recorded in 120-minute bins by a computer using MED PC software set to detect the number of times that horizontal light beams were broken. The testing room was set to continue the standard 12-hour light/dark cycle. Data was analyzed with a group x time-period repeated-measures ANOVA.
Statistical Analysis
Significance was assessed using the statistical tests as described above (T-test, Chi-square, or ANOVA) with GraphPad Prism software (v5, La Jolla, CA). Normality was assessed with a Shapiro-Wilkes test and non-normally distributed data was log transformed prior to analysis. Significance was set at P<0.05 for all measures.
Results
Behavioral Phenotypes Relevant to the Core Symptoms of Autism
Social Deficits
Social function was assessed in two separate paradigms. In a mating paradigm, transgenic male mice and wildtype littermates were paired with an unfamiliar, wildtype female. In this task, NR1neo−/− mice initiated significantly less social contact, as measured by total interaction time (Fig 1; t14=6.03, P<0.0001). In addition, sociability to a same-sex, unfamiliar wildtype mouse was measured with a standard 3-chambered social approach/avoidance paradigm. In this task, NR1neo−/− mice again showed significantly reduced sociability (Fig 1; group x condition interaction: F3,68=4.667, P=0.004; Bonferroni posttest P<0.001). These results are consistent with previously published studies demonstrating social deficits in mice with reduced NMDA-receptor signaling (Duncan et al., 2004, Halene et al., 2009a, Mohn et al., 1999).
Figure 1.

Constitutive NMDA-receptor downregulation impairs sociability in mice. (A) Adult male WT or NR1neo−/− mice were paired with a receptive, WT female for 5 minutes. Mating vocalizations, emitted by the male mouse, were recorded with an ultrasonic-range microphone placed above the testing chamber. (B) Transgenic mice demonstrated significantly reduced interest in the female mouse, as measured by the total time of male initiated interactions. (C) Sociability to a gonadectomized, same-sex wildtype stimulus mouse was measured using a 3-chambered approach/avoidance paradigm. Adult NR1-transgenic mice showed significantly reduced social interest in this task as well. Plots show mean +/− S.E.M. (***P<0.001).
Communication Impairments
Mice emit ultrasonic vocalizations (USVs) in several contexts that have communicative significance (Panksepp et al., 2007, Scattoni et al., 2009). When paired with a receptive female, male mice will reliably emit 70 kHz “pre-mating” vocalizations prior to mount followed by 40 kHz “mating” calls, thought to facilitate copulation (Ricceri et al., 2007, White et al., 1998). Such pre-mating USVs were recorded during the male-female social interaction described above. Over the 5-minute testing session, NR1neo−/− mice emitted significantly fewer vocalizations compared with wildtype littermates (Fig 2; t14=2.271, P<0.0001). Interestingly, the number of calls emitted by WT males was highly correlated with the amount of social interaction time, indicating the social significance of USVs (Fig 2C; R2=0.8, P<0.003). This association did not hold for NR1-transgenic mice, indicating a lack of appropriate social communication function (R2=0.003 P=0.9). In addition, the peak frequency of calls emitted by NR1neo−/− mice was significantly lower than that of the WT group (NR1: 40.0 ± 5.4 kHz, WT: 73.5 ± 5.5 kHz, t9=4.06, P=0.003). These findings indicate that transgenic mice fail to emit vocalizations in socially appropriate contexts.
Figure 2.

GRIN1 mutant mice demonstrate abnormalities in social communication, as measured by ultrasonic vocalizations (USVs). (A) Male mice emit 70 kHz “pre-mating” vocalizations and 40 kHz “mating” USVs when paired with a receptive female. This sonogram demonstrates four pre-mating calls, recorded from a WT mouse. (B) NR1neo−/− emit significantly fewer ultrasonic pre-mating calls than do WT littermates. (C) In WT mice, there was a significant, linear association (on a log-log plot) between the number of pre-mating vocalizations emitted and male initiated social interactions, highlighting the relevance of such ultrasonic calls for murine social communication. This relationship was absent for NR1neo−/− mice. Plots show mean +/− S.E.M. (**P<0.01, ***P<0.001).
Repetitive, Restricted Interests and Behaviors
The third core symptom of autism is characterized by motor stereotypies, repetitive or restricted interests, self-injury, and resistance to change. Stereotyped hand movements (e.g., hand-flapping, ringing, or rocking) are frequently observed in patients with autism and Rett Syndrome (RTT), and related phenotypes of abnormal forepaw clasping have been reported in mouse models of RTT (Carter et al., 2010, Chao et al., 2010, Pierce & Courchesne, 2001). During tail suspension, NR1-transgenic mice exhibited a similar forepaw clasping/wringing behavior at a significantly greater rate than wildtype littermates (Fig 3A; X2=17.33, P<0.0001). Likewise, NR1neo−/− mice were found to have significantly more skin lesions caused by self-injurious, over-grooming behavior (Fig 3B; X2=5.193, P<0.02). Along similar lines, previous studies have identified cognitive inflexibility in NR1neo−/− mice, as measured by repetitive nose poke responses in a novel hole board paradigm (Moy et al., 2008) and perseverative errors in an eight-arm radial maze task (Dzirasa et al., 2009).
Figure 3.

NMDA-receptor hypofunction leads to increased repetitive behaviors and self-injury. (A) During a tail-suspension test, 90% of NR1 mutant mice demonstrate a repetitive, hand-wringing stereotypy that was only observed in 10% of WT littermates. See supplemental information for a video. (B) Approximately 25% of NR1neo−/− mice developed skin lesions due to self-injurious over-grooming behavior. Such lesions were not observed in any WT littermates. Plots show mean +/− S.E.M. (*P<0.05, ***P<0.001).
Behavioral Phenotypes Relevant to ASD Comorbidities
Cognitive Deficits
Intellectual disability is a common comorbidity associated with ASD (Levy et al., 2009). We assessed cognitive function using a discrete T-maze paradigm with a 5 second delay, which measures spatial working memory (Deacon & Rawlins, 2006). NR1-transgenic mice showed significantly reduced alternation, consistent with a deficit in cognitive function (WT: 85±6%, n=9, NR1:64±5%, n=7; t14=2.36, P<0.05). Similar results were observed in an eight-arm radial maze task (Dzirasa et al., 2009).
Abnormal Activity Patterns
Clinical studies have reported various activity level disturbances in subjects with autism using actigraphs and polysomnography (Miano et al., 2007, Souders et al., 2009). In our study, we monitored the activity patterns in NR1neo−/− mice across the light-dark cycle over a 120-hour period. A main effect of genotype (Fig 4; F1,7=6.52, P<0.04) indicated increased locomotor activity in NR1-transgenic mice, an effect that was most apparent during the light (inactive) cycle. Elevated locomotor activity has been reported in other mouse models of autism (Penagarikano et al., 2011) and is frequently observed in patients with autism, along with other symptoms of attention-deficit hyperactivity disorder (Lee & Ousley, 2006).
Figure 4.
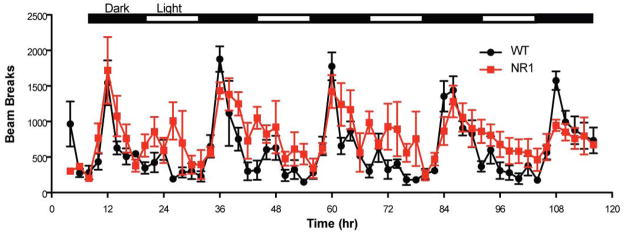
NR1 mutant mice show increased locomotor activity during light cycle. Group averages of activity are shown over 120-hour testing session.
Endophenotypes Relevant to Autism
Auditory Startle
Autism is characterized by hypersensitivity to auditory stimuli coupled with reduced prepulse inhibition (PPI) of the acoustic startle response, an endophenotype which has been correlated with core symptoms of the disorder (Levy et al., 2009, Mcalonan et al., 2002, Perry et al., 2007, Yuhas et al., 2011). NR1neo−/− mice showed enhanced startle amplitudes, indicative of increased sensory-motor reactivity (Fig 5A; main effect of group, F1, 14=5.2, P<0.04). Conversely, transgenic mice exhibited a significant reduction in prepulse inhibition (Fig 5B; F1, 14=8.1; P<0.02), recapitulating clinical observations.
Figure 5.

NMDA-receptor disruption recapitulates several auditory endophenotypes observed in autism. (A) Transgenic mice show elevated acoustic startle responses and (B) reduced prepulse inhibition (PPI). (C) Developmental NMDA-receptor downregulation leads to delayed latency of the N1 component of the auditory-evoked response, as observed in children with autism. (D) Grand-average auditory-evoked response waveforms demonstrating P1 and N1 peaks. (E, F) Transient auditory-evoked phase-locking (PLF; e.g., intertrial coherence) was assessed in the gamma frequency range (30–80 Hz). NR1neo−/− mice showed significantly reduced gamma synchrony, mirroring results from the clinical population. Plots show mean +/− S.E.M. (*P<0.05).
Auditory Evoked-Responses
Clinical studies measuring auditory event-related potentials (ERPs) have demonstrated that early auditory encoding processes are abnormal in autism (Jeste & Nelson, 2009, Roberts et al., 2008). Using pure-tone or click paradigms, recent work has reported delayed profiles of auditory processing in subjects with autism (Bruneau et al., 1999, Gage et al., 2003, Oram Cardy et al., 2005b, Roberts et al., 2010, Sokhadze et al., 2009) as well as in unaffected relatives to a lesser degree (Maziade et al., 2000). Such findings have been shown to be predictive of communication deficits in ASD (Oram Cardy et al., 2005a, Oram Cardy et al., 2008, Roberts et al., 2011). NR1neo−/− mice were evaluated in an analogous auditory-evoked response paradigm. Auditory-evoked responses are highly analogous between mice and humans, with corresponding P1 (mouse P20, human P50) and N1 (mouse N40, human N100) peaks (Amann et al., 2010). Transgenic mice showed a significant, 10–15% delay in the latency of the N1 component of the auditory evoked-response (Fig 5C; t14=2.2, P<0.05), identical to that observed in clinical studies of autistic children (Gandal et al., 2010, Roberts et al., 2010). The amplitudes of P1 and N1 peaks did not differ between groups, consistent with the clinical phenotype in ASD (Lincoln et al., 1995, Roberts et al., 2010).
Gamma Synchrony
Recent work has demonstrated abnormal neural synchrony in autism, which may contribute to core phenotypes (Uhlhaas & Singer, 2006, Uhlhaas & Singer, 2007, Welsh et al., 2005). In particular, high-frequency, gamma (30–80 Hz) oscillations have been shown to mediate a host of cognitive and sensory functions, including sensory encoding, selective attention, and memory (Herrmann et al., 2010). Several studies have identified reduced transient auditory-evoked gamma-band responses in autism, thought to reflect abnormal cognitive and perceptual functioning (Braeutigam et al., 2008, Gandal et al., 2010, Rojas et al., 2008, Wilson et al., 2007). Likewise, the phase-locked early auditory gamma-band response was significantly reduced in NR1neo−/− mice (Fig 5; t14=2.28, P<0.05), recapitulating the clinical endophenotype. Such gamma frequency abnormalities are in accordance with previous reports of disrupted selective attention and gamma-theta phase coherence following NMDAR disruption (Bickel et al., 2008, Dzirasa et al., 2009).
Discussion
Model Validity
This study has demonstrated that constitutive NMDA-receptor hypofunction in mice is associated with deficits in social and communicative functioning as well as elevated repetitive and self-injurious behaviors. This pattern of behavioral dysfunction is consistent with the negative symptoms of schizophrenia as well as autism-like phenotypes. Results in the social interaction paradigms complement and replicate previously published findings (Duncan et al., 2004, Halene et al., 2009a, Mohn et al., 1999). The strong reduction of mating vocalizations fits with reports of sexual dysfunction in NR1neo−/− mice (Mohn et al., 1999) and levels of self-injury were highly consistent with previous work (Moy et al., 2008). In addition, transgenic mice demonstrated several comorbid symptoms and endophenotypes relevant to autism, including deficits in prepulse inhibition, which have been widely reported in the clinical population (Mcalonan et al., 2002, Perry et al., 2007, Yuhas et al., 2011). As such, adult NR1neo−/− mice show strong face validity for autism-like phenotypes (Crawley, 2007). The emergence of clinical and preclinical evidence implicating reduced NMDA-receptor signaling as a pathophysiological deficit in autism bolsters the construct validity of this model for the disease. Predictive validity is harder to assess, given the lack of viable treatments currently available for autism. The antipsychotic risperidone has demonstrated efficacy for some symptoms of autism, including the repetitive, restricted interests and behaviors (Mcdougle et al., 2005). Similarly, antipsychotics have been shown to reduce motor stereotypies in NR1-hypomorphic mice as well as reverse PPI deficits (Duncan et al., 2006, Mohn et al., 1999).
As with any preclinical model of a neuropsychiatric disease, it is also important to establish the specificity of phenotypic deficits (Silverman et al., 2010). Global neurologic or functional impairment could confound observed results and would look less like autism, which is characterized by deficits in specific domains. Here, we demonstrate that auditory function, exploratory ability, and the capacity to vocalize are all intact. Likewise, we and others have previously reported that visual and olfactory function is unaltered in NR1neo−/− mice, using behavioral and electrophysiological measures (Duncan et al., 2004, Halene et al., 2009b). Pain sensation was shown to be present, and perhaps even enhanced, in a previous study (Bickel et al., 2007). Finally, no deficit in motor coordination was found in either rotarod or swim tests (Duncan et al., 2004, Fradley et al., 2005). These results indicate that developmental NMDAR-disruption does not cause global neurological impairment. While these findings suggest that gross sensory and motor function is intact, future studies employing more complex cognitive and sensory testing are necessary to demonstrate true disease specificity.
Model & Disease Specificity
NR1neo−/− mice were originally developed as a model for the glutamate hypothesis of schizophrenia (Mohn et al., 1999) and have been extensively investigated as such (Duncan et al., 2006, Duncan et al., 2004, Halene et al., 2009a, Moy et al., 2006). Our results indicate that these mice recapitulate autism-like phenotypic deficits in core and several associated symptom domains. Indeed, the most severe phenotypes were profound reductions in social interactions and social vocalizations, deficits more pronounced in autism than schizophrenia (Solomon et al., 2011). This highlights the challenge modeling a complex human behavioral disorder in rodents using behavioral phenotypes alone. To overcome this difficulty, we also investigated auditory evoked response endophenotypes, which have distinct profiles in each disorder. For example, delayed latencies in the early components of the auditory-evoked response (as observed here) have been reported in ASD patients and their first-degree unaffected relatives and have been associated with language impairment in autism (Maziade et al., 2000, Oram Cardy et al., 2008, Roberts et al., 2010). In contrast, latencies of these peaks are not different in schizophrenia (Hanlon et al., 2005). Reduced peak amplitudes of the auditory event-related potential (not observed here) are well replicated in schizophrenia, with a strong genetic contribution (Turetsky et al., 2008), and have been observed in multiple mouse models of the disorder (Carlson et al., 2011, Lazarewicz et al., 2010). Subjects with ASD, however, do not show reduced amplitudes of these ERP peaks (Roberts et al., 2010). Indeed, we previously demonstrated increased ERP amplitudes in NR1neo−/− mice, likely elicited by longer interstimulus (ISI) intervals. The current study employed a shorter ISI to more closely match parameters used in clinical studies (Lincoln et al., 1995, Roberts et al., 2010). Similar deficits in auditory-evoked gamma synchrony have been reported across both disorders and may reflect shared neural abnormalities, as recently suggested (Gandal et al., 2010, Gandal et al., 2011). The continued translation of such endophenotypes to preclinical settings will provide additional measures with which to investigate disease-specific pathophysiology.
The clinical overlap between autism and schizophrenia further complicates the ability to discern the specificity of a given preclinical model. Although autism and schizophrenia are clearly distinct disorders, they share a significant number of common clinical characteristics, including behavioral phenotypes, comorbid symptoms, epidemiology, neuroimaging findings, and genetics (Cheung et al., 2010, King & Lord, 2011). Both disorders disproportionately affect males, have an estimated population prevalence of 1–2%, are highly heritable but with complex non-Mendelian genetics, and are neurodevelopmental in origin (Levy et al., 2009, Van Os & Kapur, 2009). Many genes originally linked to autism have been recently associated with schizophrenia, including NRXN1 and SHANK3 (Gauthier et al., 2010, Levinson et al., 2011). The converse has been observed as well, for genes like DISC1 (Kilpinen et al., 2008). In addition, common copy number variants (e.g., 22q11 and 16p11.2) have been linked to both disorders with strong penetrance (Levinson et al., 2011, Sahoo et al., 2011). Common environmental risk factors include prenatal infection, maternal stress, and perinatal hypoxia. Behaviorally, both disorders show impairments in social interactions, communicative function, and stereotyped behaviors. Of note, recent studies that directly compared social cognition in subjects with ASD and schizophrenia have reported highly similar deficits and underlying neural substrates (Couture et al., 2010, Pinkham et al., 2008). Other shared phenotypes include sleep disruption, epilepsy, ADHD, and cognitive abnormalities (Levy et al., 2009, Qin et al., 2005, Siegel & Ralph, 2011). Finally, recent evidence suggests that common phenotypic deficits may stem from shared circuit insults, such as interneuron dysfunction or disrupted excitatory/inhibitory balance, both of which can be caused by reduced NMDA-receptor signaling (Levitt et al., 2004, Lewis et al., 2005, Rubenstein & Merzenich, 2003, Yizhar et al., 2011). Despite the aforementioned commonalities, a number of characteristics clearly distinguish these disorders, such as age of onset, psychosis, and the degree of language impairment. As such, future work is needed to determine the biological mechanisms that dictate the pleiotropy of identified risk-genes and ensuing phenotypic heterogeneity.
Conclusions and Future Directions
In contrast to the premise of this study, there has been some suggestion that autism is characterized by glutamatergic hyperfunction (Fatemi, 2008). This hypothesis is based on elevated blood levels of glutamate in high-functioning autism coupled with some mouse models showing enhanced long-term potentiation and/or elevated NMDA-receptor levels (Etherton et al., 2011, Rinaldi et al., 2007, Shimmura et al., 2011). Overexpression of NMDA-receptors has been shown to enhance some forms of learning and memory in mice and could account for the savant-like abilities reported in some individuals with autism (Tang et al., 1999). Like many neural pathways, it is likely that optimal NMDA-receptor signaling follows an “inverted U” profile, where deviations in either direction from a homeostatic set point would cause deteriorated behavioral function. This could also account for the significant degree of phenotypic heterogeneity observed in individuals with autism, some with severe intellectual disability and others with normal, or even elevated, cognitive function. To more rigorously test this hypothesis, future work should investigate the effect of NMDA-receptor overexpression of target phenotypes, such as sociability and EEG biomarkers. A second potential limitation focuses on the age of the mice employed in this study. Only adults were used despite the fact that the age of disease onset is an important characteristic that separates autism from schizophrenia. Future work will focus on juvenile NR1neo−/− and WT mice to determine the onset of phenotypic and endophenotypic deficits.
In conclusion, this study demonstrates that genetic disruption of NMDA-receptor signaling leads to deficits in social and communicative function as well as increased repetitive and self-injurious behaviors the core symptoms of autism. In addition, NMDAR disruption recapitulates auditory-evoked response endophenotypes characteristic of the ASD population, including a delayed profile of auditory processing and reduced gamma-band synchrony. Such electrophysiological biomarkers help to better characterize preclinical models of neuropsychiatric diseases and provide insight into circuit-level mechanisms that may underlie phenotypic deficits. Gamma synchrony is known to require tight coupling of excitatory and inhibitory balance as well as the appropriate function of fast-spiking, parvalbumin-expressing (PV+) interneurons (Sohal et al., 2009). As such, results implicate disrupted E/I balance and/or PV cell function as a potential mechanism underlying the behavioral deficits following NMDAR-hypofunction and suggest that restoration of such balance with glutamatergic or GABAergic agents would be a rational approach for pharmacologic intervention in neuropsychiatric diseases associated with impaired NMDAR signaling.
Supplementary Material
Acknowledgments
The authors work like to thank Tony Thieu, Mili Mehta, Gerald Jonak, and Yuling Liang for assistance with the experiments presented in this manuscript. This study was funded by the National Institutes of Health Grants R01-DA023210 (SJS), F30-MH087071 (MJG), and R01-DC008871 (TR). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Dr. Siegel reports having received grant support from Eli Lilly, AstraZeneca, NuPathe, and Pfizer that is unrelated to the content of this paper and consulting payments from NuPathe, Merck, Sanofi, and Wyeth that are unrelated to this work. Dr. Roberts would like to thank the Oberkircher family for the Oberkircher Family Chair in Pediatric Radiology at the Children’s Hospital of Philadelphia.
Footnotes
Dr. Roberts also discloses a financial medical advisory board relationship with Prism Clinical Imaging, which had no overlap with the present study. All other authors report no biomedical financial interests or potential conflicts of interest.
References
- Amann LC, Gandal MJ, Halene TB, Ehrlichman RS, White SL, McCarren HS, Siegel SJ. Mouse behavioral endophenotypes for schizophrenia. Brain Res Bull. 2010;83:147–161. doi: 10.1016/j.brainresbull.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Barkus C, Dawson LA, Sharp T, Bannerman DM. GluN1 hypomorph mice exhibit wide-ranging behavioral alterations. Genes Brain Behav. 2012;11:342–351. doi: 10.1111/j.1601-183X.2012.00767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickel S, Lipp HP, Umbricht D. Impaired attentional modulation of auditory evoked potentials in N-methyl-D-aspartate NR1 hypomorphic mice. Genes Brain Behav. 2007;6:558–568. doi: 10.1111/j.1601-183X.2006.00283.x. [DOI] [PubMed] [Google Scholar]
- Bickel S, Lipp HP, Umbricht D. Early auditory sensory processing deficits in mouse mutants with reduced NMDA receptor function. Neuropsychopharmacology. 2008;33:1680–1689. doi: 10.1038/sj.npp.1301536. [DOI] [PubMed] [Google Scholar]
- Bodarky CL, Halene TB, Ehrlichman RS, Banerjee A, Ray R, Hahn CG, Jonak G, Siegel SJ. Novel environment and GABA agonists alter event-related potentials in N-methyl-D-aspartate NR1 hypomorphic and wild-type mice. J Pharmacol ExpTher. 2009;331:308–318. doi: 10.1124/jpet.109.150938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braeutigam S, Swithenby SJ, Bailey AJ. Contextual integration the unusual way: a magnetoencephalographic study of responses to semantic violation in individuals with autism spectrum disorders. Eur J Neurosci. 2008;27:1026–1036. doi: 10.1111/j.1460-9568.2008.06064.x. [DOI] [PubMed] [Google Scholar]
- Bruneau N, Roux S, Adrien JL, Barthelemy C. Auditory associative cortex dysfunction in children with autism: evidence from late auditory evoked potentials (N1 wave-T complex) Clin Neurophysiol. 1999;110:1927–1934. doi: 10.1016/s1388-2457(99)00149-2. [DOI] [PubMed] [Google Scholar]
- Carlson GC. Glutamate receptor dysfunction and drug targets across models of autism spectrum disorders. Pharmacol Biochem Behav. 2011 doi: 10.1016/j.pbb.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson GC, Talbot K, Halene TB, Gandal MJ, Kazi HA, Schlosser L, Phung QH, Gur RE, Arnold SE, Siegel SJ. Dysbindin-1 mutant mice implicate reduced fast-phasic inhibition as a final common disease mechanism in schizophrenia. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1109625108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson ML. Hypothesis: is infantile autism a hypoglutamatergic disorder? Relevance of glutamate -serotonin interactions for pharmacotherapy. J Neural Transm. 1998;105:525–535. doi: 10.1007/s007020050076. [DOI] [PubMed] [Google Scholar]
- Carter P, Downs J, Bebbington A, Williams S, Jacoby P, Kaufmann WE, Leonard H. Stereotypical hand movements in 144 subjects with Rett syndrome from the population-based Australian database. Mov Disord. 2010;25:282–288. doi: 10.1002/mds.22851. [DOI] [PubMed] [Google Scholar]
- Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, Ekker M, Rubenstein JL, Noebels JL, Rosenmund C, Zoghbi HY. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung C, Yu K, Fung G, Leung M, Wong C, Li Q, Sham P, Chua S, McAlonan G. Autistic disorders and schizophrenia: related or remote? An anatomical likelihood estimation. PLoS One. 2010;5:e12233. doi: 10.1371/journal.pone.0012233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly PM, Maxwell C, Liang Y, Kahn JB, Kanes SJ, Abel T, Gur RE, Turetsky BI, Siegel SJ. The effects of ketamine vary among inbred mouse strains and mimic schizophrenia for the P80, but not P20 or N40 auditory ERP components. Neurochem Res. 2004;29:1179–1188. doi: 10.1023/b:nere.0000023605.68408.fb. [DOI] [PubMed] [Google Scholar]
- Connolly PM, Maxwell CR, Kanes SJ, Abel T, Liang Y, Tokarczyk J, Bilker WB, Turetsky BI, Gur RE, Siegel SJ. Inhibition of auditory evoked potentials and prepulse inhibition of startle in DBA/2J and DBA/2Hsd inbred mouse substrains. Brain Res. 2003;992:85–95. doi: 10.1016/j.brainres.2003.08.035. [DOI] [PubMed] [Google Scholar]
- Couture SM, Penn DL, Losh M, Adolphs R, Hurley R, Piven J. Comparison of social cognitive functioning inschizophrenia and high functioning autism: more convergence than divergence. Psychol Med. 2010;40:569–579. doi: 10.1017/S003329170999078X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley JN. Mouse behavioral assays relevant to the symptoms of autism. Brain Pathol. 2007;17:448–459. doi: 10.1111/j.1750-3639.2007.00096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon RM, Rawlins JN. T-maze alternation in the rodent. Nat Protoc. 2006;1:7–12. doi: 10.1038/nprot.2006.2. [DOI] [PubMed] [Google Scholar]
- Delorme A, Makeig S. EEGLAB: an open source toolbox for analysis of single-trial EEG dynamics including independent component analysis. J Neurosci Methods. 2004;134:9–21. doi: 10.1016/j.jneumeth.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Duncan GE, Moy SS, Lieberman JA, Koller BH. Effects of haloperidol, clozapine, and quetiapine on sensorimotor gating in a genetic model of reduced NMDA receptor function. Psychopharmacology (Berl) 2006;184:190–200. doi: 10.1007/s00213-005-0214-1. [DOI] [PubMed] [Google Scholar]
- Duncan GE, Moy SS, Perez A, Eddy DM, Zinzow WM, Lieberman JA, Snouwaert JN, Koller BH. Deficits in sensorimotor gating and tests of social behavior in a genetic model of reduced NMDA receptor function. Behav Brain Res. 2004;153:507–519. doi: 10.1016/j.bbr.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Dzirasa K, Ramsey AJ, Takahashi DY, Stapleton J, Potes JM, Williams JK, Gainetdinov RR, Sameshima K, Caron MG, Nicolelis MA. Hyperdopaminergia and NMDA receptor hypofunction disrupt neural phase signaling. J Neurosci. 2009;29:8215–8224. doi: 10.1523/JNEUROSCI.1773-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlichman RS, Gandal MJ, Maxwell CR, Lazarewicz MT, Finkel LH, Contreras D, Turetsky BI, Siegel SJ. N-methyl-d-aspartic acid receptor antagonist-induced frequency oscillations in mice recreate pattern of electrophysiological deficits in schizophrenia. Neuroscience. 2009a;158:705–712. doi: 10.1016/j.neuroscience.2008.10.031. [DOI] [PubMed] [Google Scholar]
- Ehrlichman RS, Luminais SN, White SL, Rudnick ND, Ma N, Dow HC, Kreibich AS, Abel T, Brodkin ES, Hahn CG, Siegel SJ. Neuregulin 1 transgenic mice display reduced mismatch negativity, contextual fear conditioning and social interactions. Brain Res. 2009b;1294:116–127. doi: 10.1016/j.brainres.2009.07.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etherton M, Foldy C, Sharma M, Tabuchi K, Liu X, Shamloo M, Malenka RC, Sudhof TC. Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc Natl Acad Sci U S A. 2011;108:13764–13769. doi: 10.1073/pnas.1111093108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH. The hyperglutamatergic hypothesis of autism. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:911. doi: 10.1016/j.pnpbp.2007.11.004. author reply 912–913. [DOI] [PubMed] [Google Scholar]
- Fradley RL, O’Meara GF, Newman RJ, Andrieux A, Job D, Reynolds DS. STOP knockout and NMDA NR1 hypomorphic mice exhibit deficits in sensorimotor gating. Behav Brain Res. 2005;163:257–264. doi: 10.1016/j.bbr.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Gage NM, Siegel B, Callen M, Roberts TP. Cortical sound processing in children with autism disorder: an MEG investigation. Neuroreport. 2003;14:2047–2051. doi: 10.1097/00001756-200311140-00008. [DOI] [PubMed] [Google Scholar]
- Gai X, Xie HM, Perin JC, Takahashi N, Murphy K, Wenocur AS, D’Arcy M, O’Hara RJ, Goldmuntz E, Grice DE, Shaikh TH, Hakonarson H, Buxbaum JD, Elia J, White PS. Rare structural variation of synapse and neurotransmission genes in autism. Mol Psychiatry. 2011 doi: 10.1038/mp.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Edgar JC, Ehrlichman RS, Mehta M, Roberts TP, Siegel SJ. Validating gamma oscillations and delayed auditory responses as translational biomarkers of autism. Biol Psychiatry. 2010;68:1100–1106. doi: 10.1016/j.biopsych.2010.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Edgar JC, Klook K, Siegel SJ. Gamma synchrony: Towards a translational biomarker for the treatment-resistant symptoms of schizophrenia. Neuropharmacology. 2011 doi: 10.1016/j.neuropharm.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Ehrlichman RS, Rudnick ND, Siegel SJ. A novel electrophysiological model of chemotherapy-induced cognitive impairments in mice. Neuroscience. 2008;157:95–104. doi: 10.1016/j.neuroscience.2008.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier J, Champagne N, Lafreniere RG, Xiong L, Spiegelman D, Brustein E, Lapointe M, Peng H, Cote M, Noreau A, Hamdan FF, Addington AM, Rapoport JL, Delisi LE, Krebs MO, Joober R, Fathalli F, Mouaffak F, Haghighi AP, Neri C, Dube MP, Samuels ME, Marineau C, Stone EA, Awadalla P, Barker PA, Carbonetto S, Drapeau P, Rouleau GA, Team SD. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc Natl Acad Sci U S A. 2010;107:7863–7868. doi: 10.1073/pnas.0906232107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halene TB, Ehrlichman RS, Liang Y, Christian EP, Jonak GJ, Gur TL, Blendy JA, Dow HC, Brodkin ES, Schneider F, Gur RC, Siegel SJ. Assessment of NMDA receptor NR1 subunit hypofunction in mice as a model for schizophrenia. Genes Brain Behav. 2009a;8:661–675. doi: 10.1111/j.1601-183X.2009.00504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halene TB, Talmud J, Jonak GJ, Schneider F, Siegel SJ. Predator odor modulates auditory event-related potentials in mice. Neuroreport. 2009b;20:1260–1264. doi: 10.1097/WNR.0b013e3283300cde. [DOI] [PubMed] [Google Scholar]
- Hanlon FM, Miller GA, Thoma RJ, Irwin J, Jones A, Moses SN, Huang M, Weisend MP, Paulson KM, Edgar JC, Adler LE, Canive JM. Distinct M50 and M100 auditory gating deficits in schizophrenia. Psychophysiology. 2005;42:417–427. doi: 10.1111/j.1469-8986.2005.00299.x. [DOI] [PubMed] [Google Scholar]
- Herrmann CS, Frund I, Lenz D. Human gamma-band activity: a review on cognitive and behavioral correlates and network models. Neurosci Biobehav Rev. 2010;34:981–992. doi: 10.1016/j.neubiorev.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1999;20:201–225. doi: 10.1016/S0893-133X(98)00060-8. [DOI] [PubMed] [Google Scholar]
- Jeste SS, Nelson CA., 3rd Event related potentials in the understanding of autism spectrum disorders: an analytical review. J Autism Dev Disord. 2009;39:495–510. doi: 10.1007/s10803-008-0652-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpinen H, Ylisaukko-Oja T, Hennah W, Palo OM, Varilo T, Vanhala R, Nieminen-von Wendt T, von Wendt L, Paunio T, Peltonen L. Association of DISC1 with autism and Asperger syndrome. Mol Psychiatry. 2008;13:187–196. doi: 10.1038/sj.mp.4002031. [DOI] [PubMed] [Google Scholar]
- King BH, Lord C. Is schizophrenia on the autism spectrum? Brain Res. 2011;1380:34–41. doi: 10.1016/j.brainres.2010.11.031. [DOI] [PubMed] [Google Scholar]
- Lazarewicz MT, Ehrlichman RS, Maxwell CR, Gandal MJ, Finkel LH, Siegel SJ. Ketamine modulates theta and gamma oscillations. J Cogn Neurosci. 2010;22:1452–1464. doi: 10.1162/jocn.2009.21305. [DOI] [PubMed] [Google Scholar]
- Lee DO, Ousley OY. Attention-deficit hyperactivity disorder symptoms in a clinic sample of children and adolescents with pervasive developmental disorders. J Child Adolesc Psychopharmacol. 2006;16:737–746. doi: 10.1089/cap.2006.16.737. [DOI] [PubMed] [Google Scholar]
- Levinson DF, Duan J, Oh S, Wang K, Sanders AR, Shi J, Zhang N, Mowry BJ, Olincy A, Amin F, Cloninger CR, Silverman JM, Buccola NG, Byerley WF, Black DW, Kendler KS, Freedman R, Dudbridge F, Pe’er I, Hakonarson H, Bergen SE, Fanous AH, Holmans PA, Gejman PV. Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am J Psychiatry. 2011;168:302–316. doi: 10.1176/appi.ajp.2010.10060876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt P, Eagleson KL, Powell EM. Regulation of neocortical interneuron development and the implications for neurodevelopmental disorders. Trends Neurosci. 2004;27:400–406. doi: 10.1016/j.tins.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Levy SE, Mandell DS, Schultz RT. Autism. Lancet. 2009;374:1627–1638. doi: 10.1016/S0140-6736(09)61376-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Lincoln AJ, Courchesne E, Harms L, Allen M. Sensory modulation of auditory stimuli in children with autism and receptive developmental language disorder: event-related brain potential evidence. J Autism Dev Disord. 1995;25:521–539. doi: 10.1007/BF02178298. [DOI] [PubMed] [Google Scholar]
- Maziade M, Merette C, Cayer M, Roy MA, Szatmari P, Cote R, Thivierge J. Prolongation of brainstem auditory-evoked responses in autistic probands and their unaffected relatives. Arch Gen Psychiatry. 2000;57:1077–1083. doi: 10.1001/archpsyc.57.11.1077. [DOI] [PubMed] [Google Scholar]
- McAlonan GM, Daly E, Kumari V, Critchley HD, van Amelsvoort T, Suckling J, Simmons A, Sigmundsson T, Greenwood K, Russell A, Schmitz N, Happe F, Howlin P, Murphy DG. Brain anatomy and sensorimotor gating in Asperger’s syndrome. Brain. 2002;125:1594–1606. doi: 10.1093/brain/awf150. [DOI] [PubMed] [Google Scholar]
- McDougle CJ, Scahill L, Aman MG, McCracken JT, Tierney E, Davies M, Arnold LE, Posey DJ, Martin A, Ghuman JK, Shah B, Chuang SZ, Swiezy NB, Gonzalez NM, Hollway J, Koenig K, McGough JJ, Ritz L, Vitiello B. Risperidone for the core symptom domains of autism: results from the study by the autism network of the research units on pediatric psychopharmacology. Am J Psychiatry. 2005;162:1142–1148. doi: 10.1176/appi.ajp.162.6.1142. [DOI] [PubMed] [Google Scholar]
- Miano S, Bruni O, Elia M, Trovato A, Smerieri A, Verrillo E, Roccella M, Terzano MG, Ferri R. Sleep in children with autistic spectrum disorder: a questionnaire and polysomnographic study. Sleep Med. 2007;9:64–70. doi: 10.1016/j.sleep.2007.01.014. [DOI] [PubMed] [Google Scholar]
- Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–436. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- Moskal JR, Burgdorf J, Kroes RA, Brudzynski SM, Panksepp J. A novel NMDA receptor glycine-site partial agonist, GLYX-13, has therapeutic potential for the treatment of autism. Neurosci Biobehav Rev. 2011;35:1982–1988. doi: 10.1016/j.neubiorev.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Poe MD, Nonneman RJ, Young NB, Koller BH, Crawley JN, Duncan GE, Bodfish JW. Development of a mouse test for repetitive, restricted behaviors: relevance to autism. Behav Brain Res. 2008;188:178–194. doi: 10.1016/j.bbr.2007.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy SS, Perez A, Koller BH, Duncan GE. Amphetamine-induced disruption of prepulse inhibition in mice with reduced NMDA receptor function. Brain Res. 2006;1089:186–194. doi: 10.1016/j.brainres.2006.03.073. [DOI] [PubMed] [Google Scholar]
- Myers RA, Casals F, Gauthier J, Hamdan FF, Keebler J, Boyko AR, Bustamante CD, Piton AM, Spiegelman D, Henrion E, Zilversmit M, Hussin J, Quinlan J, Yang Y, Lafreniere RG, Griffing AR, Stone EA, Rouleau GA, Awadalla P. A population genetic approach to mapping neurological disorder genes using deep resequencing. PLoS Genet. 2011;7:e1001318. doi: 10.1371/journal.pgen.1001318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, Rieder MJ, Nickerson DA, Bernier R, Fisher SE, Shendure J, Eichler EE. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oram Cardy JE, Flagg EJ, Roberts W, Brian J, Roberts TP. Magnetoencephalography identifies rapid temporal processing deficit in autism and language impairment. Neuroreport. 2005a;16:329–332. doi: 10.1097/00001756-200503150-00005. [DOI] [PubMed] [Google Scholar]
- Oram Cardy JE, Flagg EJ, Roberts W, Roberts TP. Delayed mismatch field for speech and non-speech sounds in children with autism. Neuroreport. 2005b;16:521–525. doi: 10.1097/00001756-200504040-00021. [DOI] [PubMed] [Google Scholar]
- Oram Cardy JE, Flagg EJ, Roberts W, Roberts TP. Auditory evoked fields predict language ability and impairment in children. Int J Psychophysiol. 2008;68:170–175. doi: 10.1016/j.ijpsycho.2007.10.015. [DOI] [PubMed] [Google Scholar]
- Panksepp JB, Jochman KA, Kim JU, Koy JJ, Wilson ED, Chen Q, Wilson CR, Lahvis GP. Affiliative behavior, ultrasonic communication and social reward are influenced by genetic variation in adolescent mice. PLoS One. 2007;2:e351. doi: 10.1371/journal.pone.0000351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, Sonnenblick LI, Gruver R, Almajano J, Bragin A, Golshani P, Trachtenberg JT, Peles E, Geschwind DH. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011;147:235–246. doi: 10.1016/j.cell.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry W, Minassian A, Lopez B, Maron L, Lincoln A. Sensorimotor gating deficits in adults with autism. Biol Psychiatry. 2007;61:482–486. doi: 10.1016/j.biopsych.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Pierce K, Courchesne E. Evidence for a cerebellar role in reduced exploration and stereotyped behavior in autism. Biol Psychiatry. 2001;49:655–664. doi: 10.1016/s0006-3223(00)01008-8. [DOI] [PubMed] [Google Scholar]
- Pinkham AE, Hopfinger JB, Pelphrey KA, Piven J, Penn DL. Neural bases for impaired social cognition in schizophrenia and autism spectrum disorders. Schizophr Res. 2008;99:164–175. doi: 10.1016/j.schres.2007.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin P, Xu H, Laursen TM, Vestergaard M, Mortensen PB. Risk for schizophrenia and schizophrenia-like psychosis among patients with epilepsy: population based cohort study. BMJ. 2005;331:23. doi: 10.1136/bmj.38488.462037.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricceri L, Moles A, Crawley J. Behavioral phenotyping of mouse models of neurodevelopmental disorders: relevant social behavior patterns across the life span. Behav Brain Res. 2007;176:40–52. doi: 10.1016/j.bbr.2006.08.024. [DOI] [PubMed] [Google Scholar]
- Rinaldi T, Kulangara K, Antoniello K, Markram H. Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc Natl Acad Sci U S A. 2007;104:13501–13506. doi: 10.1073/pnas.0704391104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts TP, Cannon KM, Tavabi K, Blaskey L, Khan SY, Monroe JF, Qasmieh S, Levy SE, Edgar JC. Auditory magnetic mismatch field latency: a biomarker for language impairment in autism. Biol Psychiatry. 2011;70:263–269. doi: 10.1016/j.biopsych.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts TP, Khan SY, Rey M, Monroe JF, Cannon K, Blaskey L, Woldoff S, Qasmieh S, Gandal M, Schmidt GL, Zarnow DM, Levy SE, Edgar JC. MEG detection of delayed auditory evoked responses in autism spectrum disorders: towards an imaging biomarker for autism. Autism Res. 2010;3:8–18. doi: 10.1002/aur.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts TP, Schmidt GL, Egeth M, Blaskey L, Rey MM, Edgar JC, Levy SE. Electrophysiological signatures: magnetoencephalographic studies of the neural correlates of language impairment in autism spectrum disorders. Int J Psychophysiol. 2008;68:149–160. doi: 10.1016/j.ijpsycho.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas DC, Maharajh K, Teale P, Rogers SJ. Reduced neural synchronization of gamma-band MEG oscillations in first-degree relatives of children with autism. BMC Psychiatry. 2008;8:66. doi: 10.1186/1471-244X-8-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo T, Theisen A, Rosenfeld JA, Lamb AN, Ravnan JB, Schultz RA, Torchia BS, Neill N, Casci I, Bejjani BA, Shaffer LG. Copy number variants of schizophrenia susceptibility loci are associated with a spectrum of speech and developmental delays and behavior problems. Genet Med. 2011;13:868–880. doi: 10.1097/GIM.0b013e3182217a06. [DOI] [PubMed] [Google Scholar]
- Scattoni ML, Crawley J, Ricceri L. Ultrasonic vocalizations: a tool for behavioural phenotyping of mouse models of neurodevelopmental disorders. Neurosci Biobehav Rev. 2009;33:508–515. doi: 10.1016/j.neubiorev.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimmura C, Suda S, Tsuchiya KJ, Hashimoto K, Ohno K, Matsuzaki H, Iwata K, Matsumoto K, Wakuda T, Kameno Y, Suzuki K, Tsujii M, Nakamura K, Takei N, Mori N. Alteration of plasma glutamate and glutamine levels in children with high-functioning autism. PLoS One. 2011;6:e25340. doi: 10.1371/journal.pone.0025340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel SJ, Connolly P, Liang Y, Lenox RH, Gur RE, Bilker WB, Kanes SJ, Turetsky BI. Effects of strain, novelty, and NMDA blockade on auditory-evoked potentials in mice. Neuropsychopharmacology. 2003;28:675–682. doi: 10.1038/sj.npp.1300087. [DOI] [PubMed] [Google Scholar]
- Siegel SJ, Ralph L. Demystifying schizophrenia for the general practitioner. Jones and Bartlett Publishers; Sudbury, Mass: 2011. [Google Scholar]
- Silverman JL, Yang M, Lord C, Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci. 2010;11:490–502. doi: 10.1038/nrn2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokhadze E, Baruth J, Tasman A, Sears L, Mathai G, El-Baz A, Casanova MF. Event-related potential study of novelty processing abnormalities in autism. Appl Psychophysiol Biofeedback. 2009;34:37–51. doi: 10.1007/s10484-009-9074-5. [DOI] [PubMed] [Google Scholar]
- Solomon M, Olsen E, Niendam T, Ragland JD, Yoon J, Minzenberg M, Carter CS. From lumping to splitting and back again: atypical social and language development in individuals with clinical-high-risk for psychosis, first episode schizophrenia, and autism spectrum disorders. Schizophr Res. 2011;131:146–151. doi: 10.1016/j.schres.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souders MC, Mason TB, Valladares O, Bucan M, Levy SE, Mandell DS, Weaver TE, Pinto-Martin J. Sleep behaviors and sleep quality in children with autism spectrum disorders. Sleep. 2009;32:1566–1578. doi: 10.1093/sleep/32.12.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Tannan V, Holden JK, Zhang Z, Baranek GT, Tommerdahl MA. Perceptual metrics of individuals with autism provide evidence for disinhibition. Autism Res. 2008;1:223–230. doi: 10.1002/aur.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai G, Coyle JT. Glutamatergic mechanisms in schizophrenia. Annu Rev Pharmacol Toxicol. 2002;42:165–179. doi: 10.1146/annurev.pharmtox.42.082701.160735. [DOI] [PubMed] [Google Scholar]
- Turetsky BI, Greenwood TA, Olincy A, Radant AD, Braff DL, Cadenhead KS, Dobie DJ, Freedman R, Green MF, Gur RE, Gur RC, Light GA, Mintz J, Nuechterlein KH, Schork NJ, Seidman LJ, Siever LJ, Silverman JM, Stone WS, Swerdlow NR, Tsuang DW, Tsuang MT, Calkins ME. Abnormal auditory N100 amplitude: a heritable endophenotype in first-degree relatives of schizophrenia probands. Biol Psychiatry. 2008;64:1051–1059. doi: 10.1016/j.biopsych.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. Neural synchrony in brain disorders: relevance for cognitive dysfunctions and pathophysiology. Neuron. 2006;52:155–168. doi: 10.1016/j.neuron.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. What do disturbances in neural synchrony tell us about autism? Biol Psychiatry. 2007;62:190–191. doi: 10.1016/j.biopsych.2007.05.023. [DOI] [PubMed] [Google Scholar]
- van Os J, Kapur S. Schizophrenia. Lancet. 2009;374:635–645. doi: 10.1016/S0140-6736(09)60995-8. [DOI] [PubMed] [Google Scholar]
- Welsh JP, Ahn ES, Placantonakis DG. Is autism due to brain desynchronization? Int J Dev Neurosci. 2005;23:253–263. doi: 10.1016/j.ijdevneu.2004.09.002. [DOI] [PubMed] [Google Scholar]
- White NR, Prasad M, Barfield RJ, Nyby JG. 40-and 70-kHz vocalizations of mice (Mus musculus) during copulation. Physiol Behav. 1998;63:467–473. doi: 10.1016/s0031-9384(97)00484-8. [DOI] [PubMed] [Google Scholar]
- Wilson TW, Rojas DC, Reite ML, Teale PD, Rogers SJ. Children and adolescents with autism exhibit reduced MEG steady-state gamma responses. Biol Psychiatry. 2007;62:192–197. doi: 10.1016/j.biopsych.2006.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, Huguenard JR, Hegemann P, Deisseroth K. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuhas J, Cordeiro L, Tassone F, Ballinger E, Schneider A, Long JM, Ornitz EM, Hessl D. Brief report: Sensorimotor gating in idiopathic autism and autism associated with fragile X syndrome. J Autism Dev Disord. 2011;41:248–253. doi: 10.1007/s10803-010-1040-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.