

Abstract
Androgens may provide protective effects in the vasculature under pathophysiological conditions. Our past studies have shown that dihydrotestosterone (DHT) decreases expression of cyclooxygenase-2 (COX-2) during cytokine, endotoxin, or hypoxic stimulation in human vascular smooth muscle cells, in an androgen receptor (AR)-independent fashion. Classically DHT is regarded as a pure AR agonist; however, it can be endogenously metabolized to 5α-androstane-3β, 17β-diol (3β-diol), which has recently been shown to be a selective estrogen receptor (ERβ) agonist. Therefore, we hypothesized that DHT’s anti-inflammatory properties following cytokine stimulation are mediated through ERβ. Using primary human brain vascular smooth muscle cells (HBVSMC), we tested whether DHT’s effect on IL-1β induced COX-2 expression was mediated via AR or ERβ. The metabolism of DHT to 3β-diol is a viable pathway in HBVSMC since mRNA for enzymes necessary for the synthesis and metabolism of 3β-diol [3alpha-hydroxysteroid dehydrogenase (HSD), 3β-HSD, 17β-HSD, CYP7B1] was detected. In addition, the expression of AR, ERα, and ERβ mRNA was detected. When applied to HBVSMC, DHT (10 nM; 18 h) attenuated IL-1β-induced increases in COX-2 protein expression. The AR antagonist bicalutamide did not block DHT’s ability to reduce COX-2. Both the non-selective estrogen receptor antagonist ICI 182,780 (1 μM) and the selective ERβ antagonist PHTPP (1 μM) inhibited the effect of DHT, suggesting that DHT actions are ERβ-mediated. In HBVSMC and in rat mesenteric arteries, 3β-diol, similar to DHT, reduced cytokine-induced COX-2 levels. In conclusion, DHT appears to be protective against the progression of vascular inflammation through metabolism to 3β-diol and activation of ERβ.
Keywords: Interleukin-1 beta, Cyclooxygenase-2, Androgen, Vasculature, Vascular smooth muscle
1. Introduction
The cerebral vasculature plays a central role in the pathogenesis of cardiovascular diseases, such as stroke [1], and in the initiation of inflammation after cerebral ischemia, which is a key determinant in stroke outcome [1,2]. Following ischemia, cytokine-induced activation of the transcription factor nuclear factor kappa B (NFκB) leads to an increase in the production of pro-inflammatory mediators, such as cyclooxygenase-2 (COX-2) [3,4]. The induction of pro-inflammatory mediators can be influenced by a number of endocrine factors and we previously reported that the potent androgen receptor (AR) agonist, dihydrotestosterone (DHT), can decrease levels of COX-2 following cytokine administration or hypoxia with glucose deprivation treatment. We further demonstrated that these responses occur independent of AR stimulation [5,6] since they could not be blocked by co-treatment with an AR antagonist.
The mechanism(s) for sex steroid influences on vascular inflammation is complex, given the numerous molecular pathways that these hormones can utilize to alter gene transcription and cellular responses. Both androgens and estrogens have been shown to have anti-inflammatory effects [5,7-9], although androgens have been reported to have some pro-inflammatory effects as well [10,11]. Classically, testosterone can activate the AR directly or indirectly through its conversion by 5 alpha-reductase to the more potent androgen, DHT [12,13]. Alternatively, testosterone can be metabolized to 17β-estradiol (E2) by the aromatase enzyme [14] and subsequently activate estrogen receptor alpha (ERα) or beta (ERβ) [15,16]. A third and less well explored pathway for androgen action is through the conversion of DHT to 5α-androstane-3β, 17β-diol (3β-diol), a known selective ERβ agonist [16-18]. This metabolic pathway occurs by the action of the enzymes 3beta-hydroxysteroid dehydrogenase (3β-HSD), 3alpha-hydroxysteroid dehydrogenase (3α-HSD) or 17beta-hydroxysteroid dehydrogenase (17β-HSD) [18-22]. 3β-Diol is further converted to inactive metabolites by the enzyme CYP7B1 [18,23]. Since blood vessels contain AR, ERα, ERβ, 5α reductase, aromatase, and 3β-HSD [24-30], the expression of these enzymes and receptors allows for any of the potential receptor mediated effects which can influence vascular inflammation.
Similar to testosterone and E2, 3β-diol has recently been shown to reduce expression of inflammatory markers in human umbilical vein endothelial cells [31]. Therefore, we have hypothesized that during cytokine stimulation, DHT decreases expression of the pro-inflammatory mediators via metabolism to 3β-diol and subsequent activation of ERβ in human brain vascular smooth muscle cells (HBVSMC). Additional experiments were performed in rodent peripheral arteries to determine if 3β-diol (ex vivo) was able to attenuate inflammation-induced COX-2 in an intact artery. COX-2 was investigated as a marker for vascular inflammation because of past studies demonstrating that COX-2 inhibition can decrease infarct size in experimental models of stroke [32] and because we have previously shown that in human coronary artery smooth muscle cells, DHT decreases cytokine-induced COX-2 expression via an AR-independent mechanism [5].
2. Experimental
2.1. Primary vascular smooth muscle cell culture and hormone/drug treatment
Primary brain vascular smooth muscle cells (HBVSMC) isolated from two donors were used in this study. HBVSMC from a 20-year-old male donor (Lot #ACBRI 405) were purchased from Cell Systems Corporation (Kirkland, WA) and HBVSMC isolated from a fetal donor (Lot #2733) were purchased from ScienCell Research Laboratories (Carlsbad, CA). Both sets of cells were grown under similar conditions: 5% CO2, room air atmosphere at 37 °C, in Medium 231 (Invitrogen Corporation, Carlsbad, CA) supplemented with smooth muscle growth supplement (ScienCell) and 2% FBS.
2.1.1. Experimental protocol for fetal HBVSMC
In a previous study we demonstrated that DHT reduces hypoxia plus glucose deprivation-induced COX-2 expression via an unidentified AR-independent mechanism in HBVSMC harvested from a fetal donor [6]. In the current study we extended our investigations of the previous study [6] using cryo-preserved cells with the same lot number to determine if DHT’s effect during cytokine stimulation is AR or ER mediated. Hormone/drug treatments were performed on fetal cells at 80–90% confluency and at passage 7 in hormone free media. HBVSMC (passage 7) still expressed the smooth muscle-specific proteins α-actin and smoothelin (data not shown). For the experimental protocol, cells were pre-treated for 1 h with vehicle (0.001% ethanol), the AR antagonist bicalutamide (BIC, 1 μM), or the non-selective ER antagonist ICI 182,780 (ICI, 1 μM; Tocris Bioscience; dissolved in ethanol), followed by 18 h of co-treatment with either vehicle (0.001% ethanol) or DHT (10 nM) then 6 h of IL-1β (5 ng/ml). A 6 h time point for COX-2 induction, a 10 nM DHT dose and a 1 μM antagonist dose were selected for these studies based on dose response curves from our previous studies [5,6].
2.1.2. Experimental protocol for adult HBVSMC
In the experiments involving the adult HBVSMCs the hormone/drug treatments were performed on cells at 80–85% confluency and at passage 9 in hormone free media. HBVSMC at passage 9 still expressed the smooth muscle-specific proteins α-actin and smoothelin (data not shown). Cells were treated with DHT (10 nM), 3β-diol (10 nM) or vehicle (0.001% ethanol) for 18 h followed by interleukin-1beta (IL-1β, 1 ng/ml or 5 ng/ml) for an additional 6 h. In a separate set of experiments, cells were pre-treated for 1 h with vehicle (0.001% ethanol) or the selective ERβ antagonist 4-[2-phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl]phenol (PHTPP, 1 μM; Tocris Bioscience; dissolved in ethanol) followed by 18 h of co-treatment with either vehicle (0.001% ethanol), DHT (10 nM), or 3β-diol (10 nM), then 6 h of IL-1β (5 ng/ml).
2.2. Quantitative real-time RT-PCR
Quantitative real-time reverse transcriptase polymerase chain reaction (qRT-PCR) was used to measure mRNA expression of gonadal steroid hormone receptors and steroid metabolizing enzymes in the adult male HBVSMCs treated for 6 h with hormone-free media, vehicle, or IL-1β (5 ng/ml). RNA was extracted using a standard phenol/chloroform/isoamyl procedure [33] and purity and concentration was confirmed spectrophotometrically using a Nanodrop 2000 (Thermo Scientific, Wilmington, DE). RNA (1 μg) was reverse-transcribed using iSCRIPT (Bio-Rad Laboratories, Hercules, CA) according to the manufacturer’s instructions. The resulting cDNA was quantified using the fluorescent detection reagent Quant-iT OliGreen ssDNA Reagent (Invitrogen, Carlsbad, CA). The quantity of cDNA in each PCR reaction was normalized based on the fluorescent quantification, and real-time RT-PCR was performed on 0.34 pg of template using a LightCycler 480 (Roche Diagnostics Incorporated, Indianapolis, IN). Each reaction included 12 μl of SYBR green master mix (Roche Diagnostics Incorporated), 0.5 μl forward primer, 0.5 μl reverse primer, and 2 μl of template or water (control). Data are reported as relative concentrations of template in comparison to GAPDH. Absolute values in femtogram (fg) of template were calculated based on a standard curve generated by serial diluting the product formed by the respective primers prior to the GAPDH normalization. Primer sequences and product sizes (base pairs) are listed in Table 1. The reaction involved an initial melting step at 95 °C for 10 min followed by 50 cycles of 95 °C (denature) for 10 s, 60 °C (annealing) for 10 s, and 72 °C (elongation) for 6 s. A modification of this protocol with a 65 °C annealing temperature was used for the 17β-HSD and 3α-HSD primer sets and a 55 °C annealing temperature was used for the ERβ primer set. Samples were assayed alongside a cDNA standard curve for each primer to determine the absolute cDNA concentration present. For standard curve generation, cDNA was diluted to a stock concentration of 10 pg/ml then serially diluted from 1 pg/ml to 0.0001 fg/ml in PCR grade sterile water to generate eight working standards for each primer pair. The size of the amplified cDNA was confirmed by 2% agarose gel electrophoresis. Negative controls, where water was used in place of template, were used in all experiments. Levels of mRNA expression for each sample were determined by comparison to the standard curve and reported as the relative concentration of cDNA to GAPDH. Specificity was confirmed via thermal melting curve analysis which showed a single peak at the predicted melting temperature for each primer set.
Table 1.
Primer sequences used for quantitative real time reverse transcriptase polymerase chain reaction.
| Target gene | Forward primer | Reverse primer | Product size |
|---|---|---|---|
| AR | 5′-AGTGGATGGGCTGAAAAA-3′ | 5′-GGGTGTGGAAATAGATGGG-3′ | 305 bp |
| ER α | 5′-AGGGAAAGTAGGGCAGAAA-3’ | 5′-ACGCTGGGAAATGAAGAA-3′ | 172 bp |
| ER β | 5′-AAGAATATCTCTGTGTCAAGGCCATG-3′ | 5′-GGCAATCACCCAAACCAAAG-3′ | 143 bp |
| 3α-HSD | 5′-TGGGGTTGTGGTCCTGGCCA-3′ | 5′-CCACACGCAGGGCCTTCTGG-3′ | 229 bp |
| 3β-HSD | 5′-CAGCCAGGCATGGCCGACTC-3′ | 5′-CTGCTGCCACCTCATGGGCC-3′ | 235 bp |
| 17βHSD | 5′-AGAACCTGCTCTCCGCGCCT-3′ | 5′-TCCATTGGGGCCCCTCCTCC-3′ | 305 bp |
| CYP7B1 | 5′- ATGGCAGCAGTGCGTGACGA-3’ | 5′-CGCACACAGTAGTCCCCGGT-3′ | 221 bp |
| GAPDH | 5′-GCCTTCTCCATGGTGGTGAA-3′ | 5′-GGTCGGAGTCAACGGATTT-3′ | 309 bp |
Primer sequences used for qRT-PCR and product sizes in base pairs are listed for androgen receptor AR), estrogen receptors alpha (ERα) and beta (ERβ), 3β-hydroxysteroid dehydrogenase (HSD), 3β-hydroxysteroid dehydrogenase (3β-HSD), 3β - hydroxysteroid dehydrogenase (3α-HSD) and 17β -hydroxysteroid dehydrogenase (17β-HSD); CYP7B1 (3β-diol inactivation enzyme), and Glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
2.3. Western blot
Levels of COX-2 protein were examined using standard immunoblotting methods, as previously described [5]. Briefly, cells were homogenized in lysis buffer and total protein content of whole cell lysate was determined. Next, samples were diluted in Tris–Glycine SDS sample buffer (Invitrogen) and boiled for 5 min. Two color, fluorescent standard (Li-COR Biosciences, Lincoln, NE) and diluted samples were loaded into 7.5% Smart gels (Li-COR). Proteins were separated via SDS–polyacrylamide gel electrophoresis. Separated proteins were transferred to nitrocellulose membranes and non-specific binding was blocked by incubation at room temperature for 30 min in PBS containing 1% Tween (TPBS) and 3% non-fat dried milk. Membranes were incubated with COX-2 (1:1000) mouse monoclonal antibody (Cayman Chemical, Ann Arbor, MI), AR (N20; epitope mapping at the N-terminus of AR of human origin) (1:500) rabbit polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz CA and β actin (1:5000) mouse monoclonal antibody (Sigma Aldrich Corporation, St. Louis, MO) overnight at 4 °C in TPBS. Following TPBS washes, the membranes were next incubated in Goat Anti-Mouse IR 800 Dye (1:15,000) secondary antibody (Li-COR) for 1 h at room temperature. COX-2 antibody specificity was verified with LPS stimulated Raw-264.7 (mouse macrophage) cell lysate (Santa Cruz Biotechnology, Santa Cruz CA), which is a positive control for COX-2 protein. Vascular smooth muscle phenotype was confirmed (data not shown) in adult male (passage 9) and fetal HBVSMC (passage 7) with anti-alpha actin monoclonal antibody (1:500, Santa Cruz Biotechnology) and anti-smoothelin polyclonal antibody (1:500, Santa Cruz Biotechnology). Following additional TPBS washes, proteins were visualized and quantitated using an Odyssey Infrared Imager and data was analyzed using Odyssey V3.0 software (Li-COR).
2.4. Rat mesenteric artery isolation and hormone/drug treatment (ex vivo)
Animal protocols were approved by the Institutional Animal Care and Use Committee of Arizona State University under subcontract from the University of Arizona. Male Sprague–Dawley rats (250 g) used in this study were purchased from Charles River Laboratories (Portage, Michigan) and maintained in a temperature- and humidity-controlled environment within the Arizona State University Department of Animal Care and Technology (DACT). Animals were kept on 12:12 light dark cycle and food and water were available ad libitum. One week prior to euthanasia rats were transferred to a satellite animal facility at the University of Arizona COM-Phoenix campus in accordance with protocols approved by DACT and housed under the same conditions described above. The University of Arizona COM-Phoenix campus satellite facility is operated under the guidelines of the Arizona State University Institutional Animal Care and Use Committee and accredited by the American Association for Accreditation of Laboratory Animal Care.
2.4.1. Artery isolation
On the day of experiment rats were deeply anesthetized with isoflurane (5%), the abdomen was shaved and skin cleaned with betadine. Under aseptic conditions, a midline incision was made and the thoracic and abdominal cavities were exposed. Heparin (100 U/0.1 mL) was immediately injected into the right ventricle to circumvent clot formation. Following exsanguination via cardiac puncture, the small intestine (i.e. jejunum) and its associated mesentery were removed, rinsed in sterile saline and pinned in a Sylgard®-coated dissection dish (Dow Corning, Midland, MI) containing sterilized ice-cold physiological salt solution (PSS, mM): 129.8 NaCl, 5.4 KCl, 0.83 MgSO4, 0.4 NaH2PO4, 19 NaHCO3, 1.8 CaCl2, and 5.5 glucose aerated with 21% O2, 6% CO2, and balanced N2. Making note not to stretch the vessels, segments of second and third order branches were carefully dissected and immediately transferred to a DMT (Danish Myograph Technology, Aarhus, Denmark) stainless steel tissue bath that was pre-sterilized with ethanol solution and rinsed with sterile PSS. The DMT tissue bath served as an incubation chamber and arteries were gently secured using sterile nylon thread and bathed in fresh PSS bubbled with a 21% O2, 5% CO2, N2 balance gas mixture. Following a 30 min equilibration, arteries were treated with VEH (PSS + 0.001% ethanol), IL-1β (10 ng/ml), or IL-1β (10 ng/ml) + 3β-diol (10 nM) for 3 h. In a separate experiment, artery segment incubations were conducted in the presence of VEH, lipopolysaccharide (LPS; 100 μg/ml) or LPS + 3β-diol for 3 h to investigate the effect of 3β-diol using another inflammatory stimulus. 1.5 h into the incubation the solution containing either the VEH or the treatments was rinsed and replaced with fresh PSS solution plus treatment(s) for an additional 1.5 h incubation. At the end of the 3 h incubations, arteries were removed, placed in an eppendorf tube and flash frozen on dry ice. Arteries were stored at −80 °C until further processed for protein analysis and Western blot.
2.4.2. Artery homogenization and western blot
Arteries were homogenized in tissue lysis buffer containing protease inhibitors and standard Western blot was performed (for details see Western blot protocol in cell section above). Membranes were incubated with a murine polyclonal COX-2 (1:500) antibody raised in rabbit (Cayman Chemical Cat #160126; Ann Arbor, MI) and β actin (1:5000) mouse monoclonal antibody (Sigma Aldrich Corporation, St. Louis, MO) overnight at 4 °C in TPBS. Following TPBS washes, the membranes were incubated in Goat Anti-Mouse IR 800 Dye (1:15,000) secondary antibody (Li-COR) and/or Goat Anti-Rabbit IR 680 Dye (1:15,000) secondary antibody (Li-COR) for 1 h at room temperature. Similar to Western blot for cells, following additional TPBS washes, proteins were visualized and quantitated using an Odyssey Infrared Imager and data were analyzed using Odyssey V3.0 software (Li-COR).
2.5. Reagents and chemicals
All reagents and chemicals were purchased from Sigma Aldrich Corporation (St. Louis, MO) unless otherwise noted.
2.6. Statistical analysis
Samples from each treatment group were run on the same Western blot to allow for direct comparison. For each treatment, individual measures were repeated to achieve an appropriate number for statistical analysis (n ≥ 4). Data from Western blots were expressed as an optical density ratio relative to vehicle and normalized to the optical density values for β actin bands. Multiple vehicles and treatment groups were included on the same gel and normalized to the first vehicle on the gel to account for variance. Normalizing to both a vehicle and a loading control limits variance between blots and allowed pooling of multiple blots from each experiment for comparisons. Each graph represents data obtained from 4 to 8 different membranes. Since the COX-2 bands present as doublets, both bands were analyzed together in a single imaging frame for each protein. Data from qRT-PCR studies are expressed as relative concentration of cDNA normalized to GAPDH and all measures were repeated a sufficient number of times for statistical analysis (n ≥ 7). All values are reported as means ± SEM. Unless otherwise noted, data were compared using one-way analysis of variance (ANOVA) across treatment groups using Prism Software (Irvine, CA), and when indicated, differences were compared post hoc using Student Newman–Keuls test. A level of p < 0.05 was considered significant.
3. Results
3.1. Gonadal steroid receptors and steroid metabolizing enzyme mRNAs are expressed in HBVSMC
Expression of the necessary gonadal steroid receptors and steroid metabolizing enzymes for DHT metabolism/receptor activation was verified using qRT-PCR in adult HBVSMC grown in hormone-free media. The size of the amplified cDNA was confirmed by 2% agarose gel electrophoresis for each primer set. Messenger RNAs for AR, ERα, ERβ, 3α-HSD, 3β-HSD, 17β-HSD, and CYP7B1 were detected in varying amounts in adult HBVSMC (Table 2).
Table 2.
Gonadal steroid receptors and steroid metabolizing enzyme mRNAs are expressed in HBVSMC.
| Target gene | mRNA concentration (fg) |
|---|---|
| AR | 0.7593 ± 0.3215 |
| ER α | 0.0006 ± 0.0001 |
| ER β | 0.0005 ± 0.0000 |
| 3α-HSD | 1.1490 ± 0.1035 |
| 3β-HSD | 0.0006 ± 0.0001 |
| 17β-HSD | 0.1024 ± 0.0131 |
| CYP7B1 | 0.0999 ± 0.0116 |
Messenger RNA expression of hormone receptors androgen receptor (AR), estrogen receptor alpha (ERα), and estrogen receptor beta (ERβ) and steroid metabolism enzymes 3α-hydroxysteroid dehydrogenase (HSD), 3β-HSD, 17β-HSD, and CYP7B1 were assessed in adult male human brain vascular smooth muscle cells (HBVSMC) treated for 6 h with hormone-free media using qRT-PCR. Each data point represents the mean ± SEM of 7–8 determinations.
3.2. AR and ERβ expression is not altered by inflammatory stimuli
To determine if the expression of AR and ERβ mRNA is altered in response to inflammatory stimuli, adult HBVSMC were treated for 6 h with vehicle or IL-1β (5 ng/ml) and mRNA levels were measured by qRT-PCR. Expression of AR and ERβ mRNA was not altered by cytokine treatment (Fig. 1A). Since AR activation has previously been shown to increase the expression of the AR [34], we demonstrated that the AR is fully functional our HBVSM primary cell culture model. Cells were treated with vehicle (VEH) or DHT (10 nM; 6 h) and AR protein levels measured. DHT treatment increased AR protein expression in HBVSMC. Fig. 1B illustrates a representative blot for AR and the loading control β actin. Rat testis lysate served as a positive control for AR. Fig. 1C represents the calculated intensity ratio for AR bands from the representative blot in Fig. 1B (n = 3; p < 0.05).
Fig. 1.
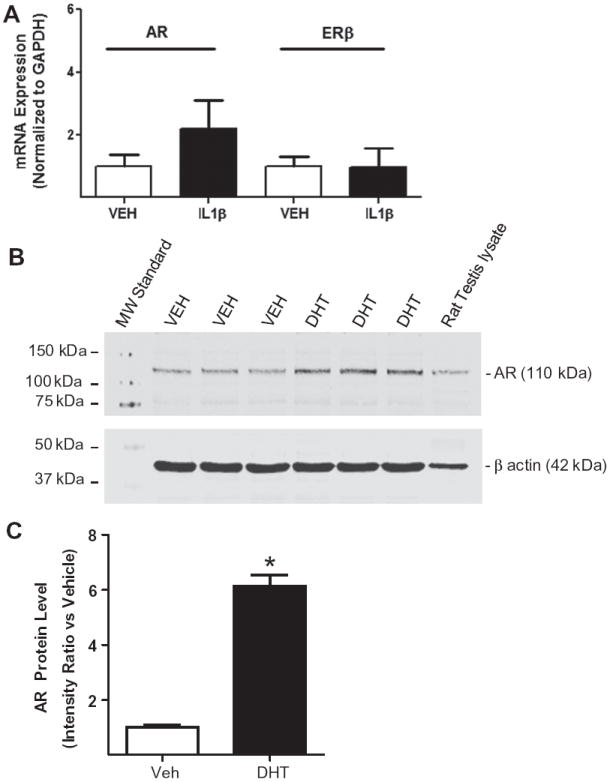
AR and ERβ expression is not altered by inflammatory stimuli. (A) Graph showing AR and ERβ mRNA expression level in adult human brain vascular smooth muscle cells (HBVSMC) treated for 6 h with vehicle (VEH) or interleukin-1 beta (IL-1β; 5 ng/ml) using qRT-PCR. Each bar represents the mean ± SEM for 7–8 determinations. (B) Representative blot of androgen receptor (AR) expression in adult HBVSMC treated with VEH or DHT (10 nM) for 6 h. Rat testis lysate served as a positive control and beta actin verified equal amounts of total protein loaded in each lane. (C) Graph showing AR protein expression in adult HBVSMC expressed as an intensity ratio vs. vehicle. *P < 0.05 vs. VEH (n = 3 per group).
3.3. Effect of DHT on COX-2 during IL-1β-stimulation is mediated via ERβ, not the androgen receptor, in HBVSMC
We have previously shown that DHT decreases cytokine-induced COX-2 expression in human coronary artery vascular smooth muscle cells and in HBVSMC exposed to hypoxia with glucose deprivation. Both effects were found to be AR-independent [5,6]. To test if DHT’s effect to reduce cytokine-induced COX-2 expression in HBVSMC is also AR-independent and to further determine if DHT’s effect is estrogen receptor mediated, fetal HBVSMC were pre-treated for 1 h with vehicle or the AR antagonist BIC (1 μM) or the non-subtype selective ER antagonist ICI 182,780 (ICI, 1 μM), then treated with vehicle or DHT (10 nM; 18 h) followed by vehicle or IL-1β (5 ng/ml; 6 h) in continued presence of hormone/antagonist. One-way ANOVA revealed that IL-1β significantly increased COX-2 protein expression compared to vehicle (p < 0.05; Fig. 2). Furthermore, DHT significantly decreased IL-1β-induced increases in COX-2 protein (p < 0.05 vs. IL-1β; Fig. 2). DHT’s effect was not blocked by the AR antagonist BIC (Fig. 2), but was blocked by the ER antagonist ICI. Antagonist treatments without DHT did not have an effect on COX-2 levels.
Fig. 2.
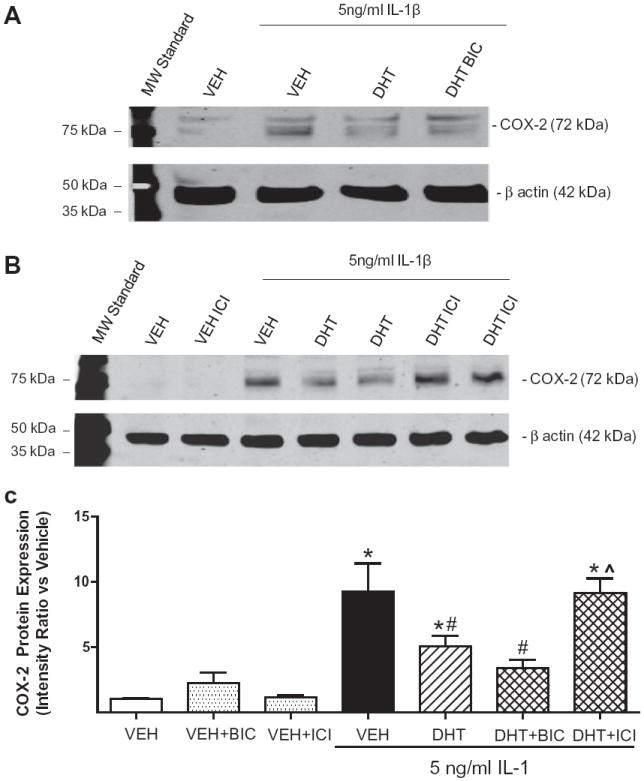
Effect of DHT on COX-2 during IL-1β-stimulation is androgen receptor independent/ER-dependent in HBVSMC. Cyclooxygenase-2 (COX-2) protein expression was assessed in fetal human brain vascular smooth muscle cells (HBVSMC) pre-treated for 1 h with vehicle (VEH), the androgen receptor antagonist bicalutamide (BIC; 1 μM), or the non-selective estrogen receptor antagonist ICI 182,780 (ICI, 1 μM), followed by VEH or dihydrotestosterone (DHT, 10 nM) for 18 h, then exposed to interleukin-1 beta (IL-1β; 5 ng/ml; 6 h) in the continued presence of hormone/antagonist. (A) and (B) Representative blots for COX-2. C: Data analysis for COX-2 levels corrected for loading with beta actin and normalized to vehicle (VEH). *P < 0.01 vs. VEH, #P < 0.05 vs. IL-1β, #P < 0.05 vs. IL-1β, ˆP < 0.05 vs. IL-1β + DHT (n ≥ 4 per group).
3.4. DHT inhibited cytokine-induced increases in COX-2 via an estrogen receptor β-dependent mechanism in HBVSMC
To determine if DHT’s anti-inflammatory actions are ERβ-dependent, adult HBVSMC were pre-treated for 1 h with vehicle or the selective ERβ antagonist PHTPP (1 μM), then treated with vehicle or DHT (10 nM; 18 h) followed by vehicle or IL-1β (5 ng/ml; 6 h) in continued presence of hormone/antagonist. One-way ANOVA revealed that IL-1β significantly increased COX-2 protein expression compared to vehicle (p < 0.05; Fig. 3) in the adult cells, as we observed in the fetal cells. Furthermore, again DHT significantly reduced IL-1β-induced increases in COX-2 protein (p < 0.05 vs. IL-1β; Fig. 3). DHT’s effect was reversed by co-treatment with the selective ERβ antagonist PHTPP (p < 0.01 vs. IL-1β + DHT; Fig. 3).
Fig. 3.
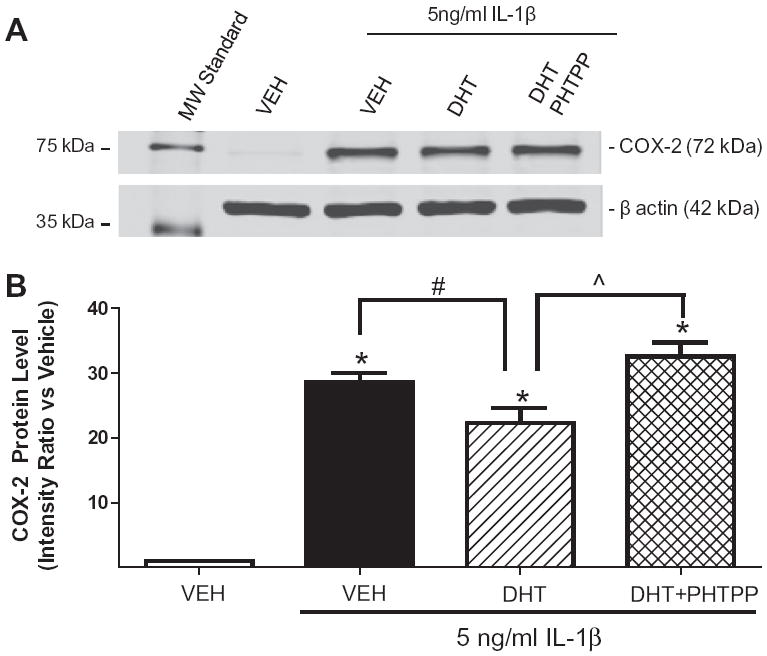
DHT inhibited cytokine-induced increases in COX-2 via an estrogen receptor β-dependent mechanism in HBVSMC. Cyclooxygenase-2 (COX-2) protein expression was assessed in adult human brain vascular smooth muscle cells (HBVSMC) pre-treated for 1 h with vehicle (VEH) or the ERβ antagonist, PHTPP (1 μM), followed by VEH or dihydrotestosterone (DHT; 10 nM) for 18 h, then exposed to interleukin-1 beta (IL-1β; 5 ng/ml; 6 h) in the continued presence of hormone/antagonist. (A) Representative blot for COX-2. B: Graph showing COX-2 levels after treatments. *P < 0.001 vs. VEH, #P < 0.05 vs. IL-1β, ˆP < 0.001 vs. IL-1β + DHT (n ≥ 10 per group).
3.5. DHT and 3β-diol inhibited cytokine-induced increases in COX-2 in HBVSMC
To determine if DHT’s anti-inflammatory actions could be mimicked by 3β-diol, HBVSMC were treated with vehicle, DHT (10 nM), or 3β-diol (10 nM) for 18 h followed by vehicle (VEH) or IL-1β (0.2, 1, or 5 ng/ml; 6 h) in continued presence of hormone. One-way ANOVA showed that both DHT and 3β-diol significantly reduced the effect of the 5 ng/ml or 1 ng/ml doses of IL-1β to increase COX-2 protein (p < 0.05 vs. IL-1β; Fig. 4). The lowest dose of IL-1β (0.2 ng/ml) did not increase COX-2 expression above vehicle levels (data not shown).
Fig. 4.
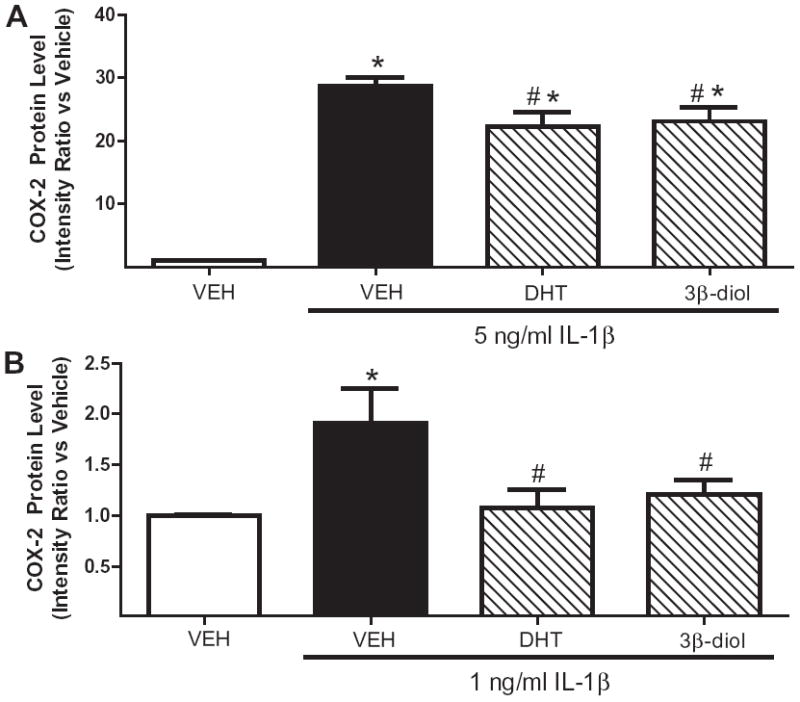
DHT and 3β-diol inhibited cytokine-induced increases in COX-2 in HBVSMC. (A) Cyclooxygenase-2 (COX-2) protein expression was assessed in adult human brain vascular smooth muscle cells (HBVSMC) treated with vehicle (VEH), dihydrotestosterone (DHT; 10 nM), or 3β-diol (10 nM) for 18 h, then exposed to interleukin-1 beta (IL-1β; 5 ng/ml; 6 h) in the continued presence of hormone. *P < 0.001 vs. VEH, #P < 0.05 vs. IL-1β (n ≥ 10 per group). (B) COX-2 protein expression was assessed in adult HBVSMC treated with VEH, DHT (10 nM), or 3β-diol (10 nM) for 18 h, then exposed to a low dose of IL-1β (1 ng/ml; 6 h) in the continued presence of hormone. *P < 0.01 vs. VEH, #P < 0.01 vs. IL-1β (n ≥ 10 per group).
3.6. 3β-Diol inhibited cytokine- and endotoxin-induced increases in COX-2 in rat mesenteric arteries
To assess whether the effects of 3β-diol on COX-2 persist in another experimental vascular model, arteries from rat mesenteric arteries were aseptically isolated and incubated ex vivo for 3 h with either IL-1β (cytokine; n = 1) or LPS (endotoxin; n = 3) in the absence or presence of 3β diol. Fig. 7A illustrates COX-2 protein expression in vehicle (VEH; 0.001% ethanol), IL-1β (10 ng/ml) or IL-1β 10 ng/ml + 3β-diol (10 nM). IL-1β increased COX-2 protein expression and 3β-diol attenuated this effect. Fig. 5B illustrates COX-2 protein expression in arteries incubated with vehicle (PSS), the endotoxin lipopolysaccharide (LPS; 100 μg/ml), or LPS + 3β-diol. Fig. 5C illustrates quantitative data for LPS treatments. One way ANOVA showed LPS increased COX-2 protein levels (p < 0.05 vs. VEH). Increases in endotoxin-induced COX-2 were attenuated in the presence of 3β-diol (#P < 0.05 vs. LPS).
Fig. 5.
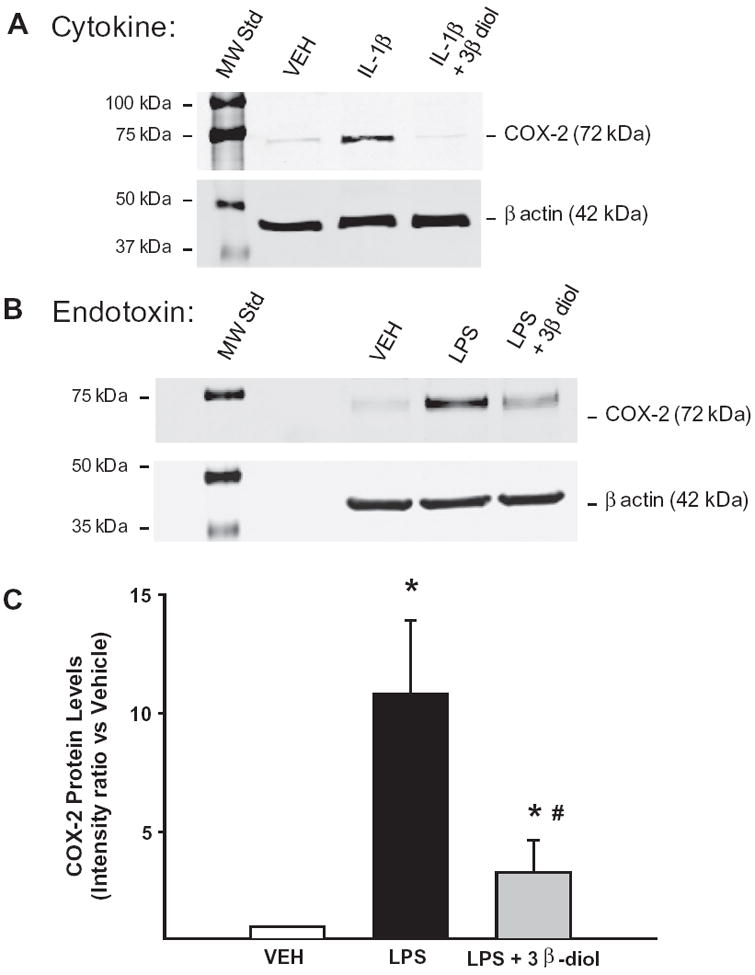
3β-Diol inhibited cytokine- and endotoxin-induced increases in COX-2 in rat mesenteric arteries. Cyclooxygenase-2 (COX-2) protein levels were assessed in isolated mesenteric arteries treated ex vivo with either cytokine or endotoxin in the absence or presence of 3β-diol. Panel A illustrates the Western blot for COX-2 expression for the cytokine experiment: vehicle (VEH), interleukin-1 beta (IL-1β; 10 ng/ml), or IL-1β (10 ng/ml) plus 3β-diol (10 nM). Panel B illustrates the LPS experiment: VEH, the endotoxin lipopolysaccharide (LPS; 100 μg/ml), or LPS + 3β-diol. β Actin served as a loading control. Panel C: Graph showing COX-2 levels after LPS treatment. *P < 0.05 vs. VEH, #P < 0.05 vs. LPS (n = 3).
4. Discussion
The goal of this study was to determine the mechanism underlying DHT’s anti-inflammatory effects in brain vascular smooth muscle cells. Using COX-2 protein expression as a marker for vascular inflammation, we examined the effects of the non-aromatizable androgen, DHT, and its metabolite 3β-diol, on COX-2 protein expression in human brain vascular smooth muscle cells. Furthermore, we examined the effects of 3β-diol ex vivo on cytokine and endotoxin induced COX-2 expression using an isolated rat artery model. The results of these studies support our hypothesis that during cytokine stimulation, DHT decreases COX-2 expression via metabolism to 3β-diol and subsequent activation of ERβ.
Androgens have been shown to have cardioprotective and anti-inflammatory actions. The cardioprotective effects of androgens include inhibition of hypoxic pulmonary hypertension [35] and decreased infarct size after cerebral ischemia in rodents [36-40]. In addition, low testosterone levels predict increased risk of stroke [41] and poorer outcome after stroke in men [42]. Androgens have been shown to reduce inflammation in a variety of cells/tissues including human endothelial [7,43-45] and vascular smooth muscle cells [5]. For instance, we have recently reported that DHT can inhibit both cytokine-induced and hypoxia plus glucose deprivation-induced COX-2 levels in human vascular smooth muscle cells [5,6]. Androgen treatment in vivo has also been shown to decrease inflammatory markers in androgen deficient men [46]. Thus, these data indicate that androgens might protect against vascular inflammation in androgen deficient men already predisposed to cardiovascular events. Since androgen levels decline with age [47], androgen deficiency is not uncommon in aged men. Since our studies and studies by others demonstrate the presence of ERβ in vascular tissues, targeting the 3β-diol/ERβ pathway may be a more direct approach to therapeutically reduce vascular inflammation compared to DHT treatment because activation of the AR receptor pathway results in higher risk for cancer in hormone sensitive target organs, such as prostate tissue.
While the majority of studies have shown anti-inflammatory effects of androgens, a few studies have reported pro-inflammatory effects of androgens in rat cerebral arteries [48,49] and human endothelial cells [10,11,50]. These apparent contradictory findings may be explained by a differential effect of androgens in the absence and presence of inflammation. In our studies, androgens increase vascular inflammation via an AR-dependent mechanism under physiological conditions, but decrease vascular inflammation via an AR-independent mechanism during pathophysiological conditions [5,48]. Since we have found androgens to be protective under pathophysiological conditions [6] but detrimental under healthy physiological conditions [5], many of the studies in healthy young rodents may not yield similar results unless some form of inflammation was induced. Additional differences may be explained by the dose of androgens used. Androgens appear to be protective at low doses, but detrimental at high doses, as has been shown with damage following cerebral ischemia [37] and after induced-inflammation in endothelial cells [7,10,11]. The results obtained in the current studies should be physiologically relevant since low doses of DHT and 3β-diol were used. Future studies will be aimed at determining the nature of the molecular switch(s) that reverses DHT actions from pro- to anti-inflammatory in vascular smooth muscle cells.
The results of our current studies point to ERβ as a novel receptor pathway utilized by DHT during cytokine stimulation. Since DHT can be converted to 3β-diol by the enzymes 3α-HSD, 3β-HSD and 17β-HSD [18-22], we sought to determine if these enzymes are also found in HBVSMC. Our data show that primary HBVSMC express the gonadal steroid receptors AR, ERα, ERβ, as well as enzymes involved in 3β-diol metabolism. This is consistent with previous studies showing that blood vessels contain AR, ERα, ERβ, and 3β-HSD [24-27,29,30]. Additionally, we found that AR and ERβ mRNA expression was not altered by cytokine treatment in our human cell model. Results of our studies also show that both DHT and 3β-diol decrease cytokine-induced COX-2 expression and that DHT’s effects are likely mediated via ERβ. Since 3β-diol can be synthesized from DHT by several enzymes, for which specific inhibitors are not available, we were unable to block 3β-diol synthesis directly. However, we were able to show that an ERβ selective antagonist can block DHT’s ability to inhibit cytokine-induced COX-2 protein expression. These data are supported by a recent study by Norata et al. [31] showing that 3β-diol has anti-inflammatory actions in human umbilical vein endothelial cells and mouse aorta, and that these effects are also mediated by ERβ [31].
Many studies have shown that ERβ activation has a variety of cardioprotective and anti-inflammatory effects. Cardioprotective effects of ERβ activation include protection against cerebral ischemia in rodents [51,52], protection against myocardial ischemia in mice [53-57], and protection against in vitro ischemia in mouse brain endothelial cells [58]. ERβ activation, like androgens, has been shown to have anti-inflammatory effects in a variety of cells/tissues including rat vascular smooth muscle cells [9] and human endothelial cells [31]. A recent study by Saijo et al. identified a mechanism for ERβ-mediated reduction of inflammatory markers in microglia that may help explain the effects we show in vascular cells. They found that 5α-androstene-3β,17β-diol (Adiol), a metabolite of the adrenal androgen, dehydroepiandrosterone, suppresses inflammatory genes by binding to ERβ and recruiting C-terminal binding protein (CtBP), a co-repressor that binds to activator protein-1 dependent promoters, thereby repressing expression of pro-inflammatory genes [59]. Future studies are needed to determine if 3β-diol is similarly capable of directing CtBP recruitment by ERβ or if there are other co-regulatory proteins that are selectively activated by 3β-diol binding to ERβ. Taken together, these studies show that ERβ activation has similar protective effects to androgens, and support the theory that some of the beneficial effects of androgens may occur through activation of ERβ.
Aside from ERβ activation, other AR-independent mechanisms for androgen action have been proposed for DHT’s anti-inflammatory actions. DHT can activate the sex hormone binding globulin receptor to increase cyclic AMP and protein kinase A [60,61]. DHT can also increase intracellular calcium via an unidentified membrane-bound receptor [62,63]. Furthermore, several authors have also hypothesized that a membrane bound AR may exist that is not blocked by classical AR antagonists [62-65]. Whether these alternative pathways for androgen action may also contribute to DHT’s anti-inflammatory effects is yet to be determined and are beyond the scope of the current study.
To investigate the mechanism of DHT’s anti-inflammatory effect we used a unique primary human vascular cell model. Although potentially more clinically relevant compared to rodent studies, we acknowledge that the use of cultured vascular cells does have certain limitations. First, cultured vascular smooth muscle cells may lose their smooth muscle phenotype with later passages [66]. Furthermore, it is also recognized that receptor protein levels for androgens and cytokines may also change with multiple passages in culture. To minimize the risk of changes in culture, all experiments were performed after a small number of passages (7 or 9) and at the time of experiments, HBVSMC still expressed the smooth muscle cell markers smoothelin and alpha actin. A second possible limitation is that each cell type originated from a single donor. However, this is likely not an issue since in our current study (and in other past studies) we utilized vascular smooth muscle cells from various male donors. Additionally, we found that DHT’s anti-inflammatory effect was ER-mediated in both fetal and adult cells. We have also found that 3β-diol reduces IL-1β-induced expression of COX-2 in both smooth muscle cells and freshly isolated rat arteries. Thus, we have provided evidence that the DHT/3β-diol/ERβ pathway may be utilized at different points in development (fetal and adult) and that 3β-diol has anti-inflammatory effects in both the human and rat vasculature. Furthermore, our previous studies in primary human coronary vascular smooth muscle cells, originating from a different donor, also showed that DHT attenuated increases in COX-2 levels following IL-1β stimulation via an AR-independent mechanism [5]. These findings leave open the possibility that this pathway could be active in other vascular beds, an idea which is supported by our data in rat mesenteric arteries and by Norata et al. [31] who showed a similar effect in human umbilical vein endothelial cells [31]. We have also recently reported that DHT decreases hypoxia plus glucose deprivation-induced COX-2 in HBVSMC via an AR-independent mechanism [6]. Whether or not the DHT/3β-diol/ERβ pathway is responsible for this effect is yet to be determined.
Our findings demonstrate that DHT decreases cytokine-induced COX-2 expression via an ERβ-dependent mechanism. These data suggest that this effect of DHT may likely involve the metabolism of DHT to the ERβ agonist 3β-diol (Fig. 6). This is supported by data demonstrating that (1) HBVSMC express ERβ and the necessary enzymes to convert DHT to 3β-diol, (2) 3β-diol mimics the anti-inflammatory effect of DHT, and (3) DHT’s effect to reduce cytokine-induced inflammation is ERβ dependent. Thus, we have identified an ERβ-dependent anti-inflammatory effect for DHT in the human brain vascular cells. A deeper understanding of how androgens modulate inflammation could provide insight into more effective approaches to manage the progression of vascular diseases that eventually lead to devastating end points, such as heart attack or stroke.
Fig. 6.
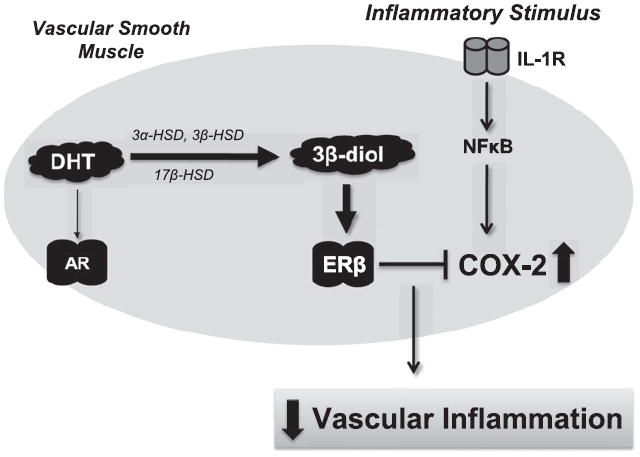
Schematic diagram of proposed mechanism for DHT’s anti-inflammatory actions in vascular tissues. In our proposed model, during interleukin-1 beta (IL-1β) induced inflammation, dihydrotestosterone (DHT) reduces cyclooxygenase-2 (COX-2) expression via metabolism to 3 beta-diol (3β-diol) by the enzymes 3α-hydroxysteroid dehydrogenase (HSD), 3β-HSD, and/or 17β-HSD in vascular smooth muscle cells (thick arrow). 3β-Diol then activates estrogen receptor beta (ERβ) and down regulates COX-2 expression. Although DHT has the ability to bind with AR, it does not appear to down regulate COX-2 expression via this mechanism (thin arrow). AR (androgen receptor), IL-1R (interleukin-1 receptor), NFκB (nuclear factor kappa B).
Acknowledgments
Funding for this project was provided by the American Heart Association Scientific Development Grant (RJG), the American Heart Association Pre-doctoral Fellowship (KLZ), and NIH NS039951 and MH082679 (RJH). Additional support was provided by University of Arizona Sarver Heart Center: Doris Griswold Award for Novel Research in the Area of Cardiovascular Disease and Medicine (KLZ) and a generous gift from the family of Jim and Ann Madson. The authors would like to thank Ms. Rheana Techapinyawat and Mrs. Lakshmi Madhavpeddi for their technical assistance.
Abbreviations
- 3α-HSD
3 alpha hydroxysteroid dehydrogenase
- 3β-diol
5α-androstane-3β,17β-diol
- 3β-HSD
3 beta hydroxysteroid dehydrogenase
- 17β-HSD
17 beta hydroxysteroid dehydrogenase
- Adiol
5α-androstene-3β,17β-diol
- AR
androgen receptor
- COX-2
cyclooxygenase-2
- DHT
dihydrotestosterone
- E2
17β-estradiol
- ERα
estrogen receptor alpha
- ERβ
estrogen receptor beta
- HBVSMC
human brain vascular smooth muscle cells
- ICI
ICI 182,780
- IL-1β
interleukin-1 beta
- LPS
lipopolysaccharide
- NFκB
nuclear factor kappa B
- PHTPP
4-[2-phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl]phenol
- PSS
physiological salt solution
- qRT-PCR
quantitative real-time reverse transcriptase polymerase chain reaction
- VCAM-1
vascular cell adhesion molecule-1
References
- 1.del Zoppo GJ, Mabuchi T. Cerebral microvessel responses to focal ischemia. J Cereb Blood Flow Metab. 2003;23:879–94. doi: 10.1097/01.WCB.0000078322.96027.78. [DOI] [PubMed] [Google Scholar]
- 2.Emsley HCA, Tyrrell PJ. Inflammation and infection in clinical stroke. J Cereb Blood Flow Metab. 2002;22:1399–419. doi: 10.1097/01.WCB.0000037880.62590.28. [DOI] [PubMed] [Google Scholar]
- 3.Brooks A, Menzies-Gow N, Bailey S, Cunningham F, Elliott J. Endotoxin-induced HIF-1α stabilisation in equine endothelial cells: synergistic action with hypoxia. Inflammat Res. 2010;9:689–98. doi: 10.1007/s00011-010-0180-x. [DOI] [PubMed] [Google Scholar]
- 4.Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184:53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Osterlund KL, Handa RJ, Gonzales RJ. Dihydrotestosterone alters cyclooxygenase-2 levels in human coronary artery smooth muscle cells. Am J Physiol Endocrinol Metab. 2010;298:E838–45. doi: 10.1152/ajpendo.00693.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zuloaga KL, Gonzales RJ. Dihydrotestosterone attenuates hypoxia inducible factor-1alpha and cyclooxygenase-2 in cerebral arteries during hypoxia or hypoxia with glucose deprivation. Am J Physiol Heart Circ Physiol. 2011;301:H1882–90. doi: 10.1152/ajpheart.00446.2011. [DOI] [PubMed] [Google Scholar]
- 7.Norata GD, Tibolla G, Seccomandi PM, Poletti A, Catapano AL. Dihydrotestosterone decreases tumor necrosis factor-alpha and lipopolysaccharide-induced inflammatory response in human endothelial cells. J Clin Endocrinol Metab. 2006;91:546–54. doi: 10.1210/jc.2005-1664. [DOI] [PubMed] [Google Scholar]
- 8.Malkin CJ, Pugh PJ, Jones RD, Jones TH, Channer KS. Testosterone as a protective factor against atherosclerosis–immunomodulation and influence upon plaque development and stability. J Endocrinol. 2003;178:373–80. doi: 10.1677/joe.0.1780373. [DOI] [PubMed] [Google Scholar]
- 9.Xing D, Feng W, Miller AP, Weathington NM, Chen Y-F, Novak L, et al. Estrogen modulates TNF-{alpha}-induced inflammatory responses in rat aortic smooth muscle cells through estrogen receptor-beta activation. Am J Physiol Heart Circ Physiol. 2007;292:H2607–12. doi: 10.1152/ajpheart.01107.2006. [DOI] [PubMed] [Google Scholar]
- 10.Death AK, McGrath KCY, Sader MA, Nakhla S, Jessup W, Handelsman DJ, et al. Dihydrotestosterone promotes vascular cell adhesion molecule-1 expression in male human endothelial cells via a nuclear factor-{kappa}B-dependent pathway. Endocrinology. 2004;145:1889–97. doi: 10.1210/en.2003-0789. [DOI] [PubMed] [Google Scholar]
- 11.McCrohon JA, Jessup W, Handelsman DJ, Celermajer DS. Androgen exposure increases human monocyte adhesion to vascular endothelium and endothelial cell expression of vascular cell adhesion molecule-1. Circulation. 1999;99:2317–22. doi: 10.1161/01.cir.99.17.2317. [DOI] [PubMed] [Google Scholar]
- 12.Lephart ED, Lund TD, Horvath TL. Brain androgen and progesterone metabolizing enzymes: biosynthesis, distribution and function. Brain Res Brain Res Rev. 2001;37:25–37. doi: 10.1016/s0165-0173(01)00111-4. [DOI] [PubMed] [Google Scholar]
- 13.Handa RJ, Stadelman HL, Resko JA. Effect of estrogen on androgen receptor dynamics in female rat pituitary. Endocrinology. 1987;121:84–9. doi: 10.1210/endo-121-1-84. [DOI] [PubMed] [Google Scholar]
- 14.Roselli CE, Horton LE, Resko JA. Distribution and regulation of aromatase activity in the rat hypothalamus and limbic system. Endocrinology. 1985;117:2471–7. doi: 10.1210/endo-117-6-2471. [DOI] [PubMed] [Google Scholar]
- 15.Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, et al. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology. 1998;139:4252–63. doi: 10.1210/endo.139.10.6216. [DOI] [PubMed] [Google Scholar]
- 16.Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, et al. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138:863–70. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 17.Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA. 1996;93:5925–30. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weihua Z, Lathe R, Warner M, Gustafsson JA. An endocrine pathway in the prostate, ERbeta, AR, 5alpha-androstane-3beta,17beta-diol, and CYP7B1, regulates prostate growth. Proc Natl Acad Sci USA. 2002;99:13589–94. doi: 10.1073/pnas.162477299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gangloff A, Shi R, Nahoum V, Lin SX. Pseudo-symmetry of C19 steroids, alternative binding orientations, and multispecificity in human estrogenic 17beta-hydroxysteroid dehydrogenase. FASEB J. 2003;17:274–6. doi: 10.1096/fj.02-0397fje. [DOI] [PubMed] [Google Scholar]
- 20.Torn S, Nokelainen P, Kurkela R, Pulkka A, Menjivar M, Ghosh S, et al. Production, purification, and functional analysis of recombinant human and mouse 17beta-hydroxysteroid dehydrogenase type 7. Biochem Biophys Res Commun. 2003;305:37–45. doi: 10.1016/s0006-291x(03)00694-6. [DOI] [PubMed] [Google Scholar]
- 21.Jin Y, Penning TM. Steroid 5alpha-reductases and 3alpha-hydroxysteroid dehydrogenases: key enzymes in androgen metabolism. Best Pract Res Clin Endocrinol Metab. 2001;15:79–94. doi: 10.1053/beem.2001.0120. [DOI] [PubMed] [Google Scholar]
- 22.Steckelbroeck S, Jin Y, Gopishetty S, Oyesanmi B, Penning TM. Human cytosolic 3alpha-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3beta-hydroxysteroid dehydrogenase activity: implications for steroid hormone metabolism and action. J Biol Chem. 2004;279:10784–95. doi: 10.1074/jbc.M313308200. [DOI] [PubMed] [Google Scholar]
- 23.Sundin M, Warner M, Haaparanta T, Gustafsson JA. Isolation and catalytic activity of cytochrome P-450 from ventral prostate of control rats. J Biol Chem. 1987;262:12293–7. [PubMed] [Google Scholar]
- 24.Gonzales RJ, Ansar S, Duckles SP, Krause DN. Androgenic/estrogenic balance in the male rat cerebral circulation: metabolic enzymes and sex steroid receptors. J Cereb Blood Flow Metab. 2007;27:1841–52. doi: 10.1038/sj.jcbfm.9600483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Snyder GD, Krishna UM, Falck JR, Spector AA. Evidence for a membrane site of action for 14,15-EET on expression of aromatase in vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2002;283:H1936–42. doi: 10.1152/ajpheart.00321.2002. [DOI] [PubMed] [Google Scholar]
- 26.Shih H-C, Lin C-L, Wu S-C, Kwan A-L, Hong Y-R, Howng S-L. Upregulation of estrogen receptor alpha and mediation of 17beta-estradiol vasoprotective effects via estrogen receptor alpha in basilar arteries in rats after experimental subarachnoid hemorrhage. J Neurosurg. 2008;109:92–9. doi: 10.3171/JNS/2008/109/7/0092. [DOI] [PubMed] [Google Scholar]
- 27.Nakamura Y, Suzuki T, Inoue T, Tazawa C, Moriya T, Saito H, et al. 3beta-Hydroxysteroid dehydrogenase in human aorta. Endocr J. 2005;52:111–5. doi: 10.1507/endocrj.52.111. [DOI] [PubMed] [Google Scholar]
- 28.Register TC, Adams MR. Coronary artery and cultured aortic smooth muscle cells express mRNA for both the classical estrogen receptor and the newly described estrogen receptor beta. J Steroid Biochem Mol Biol. 1998;64:187–91. doi: 10.1016/s0960-0760(97)00155-6. [DOI] [PubMed] [Google Scholar]
- 29.Ma Y, Qiao X, Falone AE, Reslan OM, Sheppard SJ, Khalil RA. Gender-specific reduction in contraction is associated with increased estrogen receptor expression in single vascular smooth muscle cells of female rat. Cell Physiol Biochem. 2010;26:457–70. doi: 10.1159/000320569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chakrabarti S, Davidge ST. High glucose-induced oxidative stress alters estrogen effects on ERα and ERβ in human endothelial cells: reversal by AMPK activator. J Steroid Biochem Mol Biol. 2009;117:99–106. doi: 10.1016/j.jsbmb.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 31.Norata GD, Cattaneo P, Poletti A, Catapano AL. The androgen derivative 5alpha-androstane-3beta,17beta-diol inhibits tumor necrosis factor alpha and lipopolysaccharide induced inflammatory response in human endothelial cells and in mice aorta. Atherosclerosis. 2010;212:100–6. doi: 10.1016/j.atherosclerosis.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 32.Sugimoto K, Iadecola C. Delayed effect of administration of COX-2 inhibitor in mice with acute cerebral ischemia. Brain Res. 2003;960:273–6. doi: 10.1016/s0006-8993(02)03805-2. [DOI] [PubMed] [Google Scholar]
- 33.Ribaudo R, Gilman M, Kingston RE, Choczynski P, Sacchi N. Preparation of RNA from tissues and cells. Curr Protoc Immunol. 2001;Chapter 10(Unit 10 1) doi: 10.1002/0471142735.im1011s04. [DOI] [PubMed] [Google Scholar]
- 34.González-Cadavid NF, Vernet D, Navarro AF, Rodriguez J, Swerdloff RS, Rajfer J. Up-regulation of the levels of androgen receptor and its mRNA by androgens in smooth-muscle cells from rat penis. Mol Cell Endocrinol. 1993;90:219–29. doi: 10.1016/0303-7207(93)90155-d. [DOI] [PubMed] [Google Scholar]
- 35.Oka M, Karoor V, Homma N, Nagaoka T, Sakao E, Golembeski SM, et al. Dehydroepiandrosterone upregulates soluble guanylate cyclase and inhibits hypoxic pulmonary hypertension. Cardiovascular Res. 2007;74:377–87. doi: 10.1016/j.cardiores.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li ZK, Shen L, Ke H, Li F, Ni LM, Li QH. Effects of androgen on the expression of brain aromatase cytopigment and nerve growth factor in neonatal rats with hypoxic-ischemic brain damage. Zhongguo Dang Dai Er Ke Za Zhi. 2008;10:441–6. [PubMed] [Google Scholar]
- 37.Uchida M, Palmateer JM, Herson PS, Devries AC, Cheng J, Hurn PD. Dose-dependent effects of androgens on outcome after focal cerebral ischemia in adult male mice. J Cereb Blood Flow Metab. 2009;29:1454–62. doi: 10.1038/jcbfm.2009.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pan Y, Zhang H, Acharya AB, Patrick PH, Oliver D, Morley JE. Effect of testosterone on functional recovery in a castrate male rat stroke model. Brain Res. 2005;1043:195–204. doi: 10.1016/j.brainres.2005.02.078. [DOI] [PubMed] [Google Scholar]
- 39.Cheng J, Hu W, Toung TJ, Zhang Z, Parker SM, Roselli CE, et al. Age-dependent effects of testosterone in experimental stroke. J Cereb Blood Flow Metab. 2008;29:486–94. doi: 10.1038/jcbfm.2008.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng J, Uchida M, Zhang W, Grafe MR, Herson PS, Hurn PD. Role of salt-induced kinase 1 in androgen neuroprotection against cerebral ischemia. J Cereb Blood Flow Metab. 2010;31:339–50. doi: 10.1038/jcbfm.2010.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yeap BB, Hyde Z, Almeida OP, Norman PE, Chubb SAP, Jamrozik K, et al. Lower testosterone levels predict incident stroke and transient ischemic attack in older men. J Clin Endocrinol Metab. 2009;94:2353–9. doi: 10.1210/jc.2008-2416. [DOI] [PubMed] [Google Scholar]
- 42.Jeppesen LL, Jorgensen HS, Nakayama H, Raaschou HO, Olsen TS, Winther K. Decreased serum testosterone in men with acute ischemic stroke. Arterioscler Thromb Vasc Biol. 1996;16:749–54. doi: 10.1161/01.atv.16.6.749. [DOI] [PubMed] [Google Scholar]
- 43.Hatakeyama H, Nishizawa M, Nakagawa A, Nakano S, Kigoshi T, Uchida K. Testosterone inhibits tumor necrosis factor-alpha-induced vascular cell adhesion molecule-1 expression in human aortic endothelial cells. FEBS Lett. 2002;530:129–32. doi: 10.1016/s0014-5793(02)03440-3. [DOI] [PubMed] [Google Scholar]
- 44.Jin H, Qiu WB, Mei YF, Wang DM, Li YG, Tan XR. Testosterone alleviates tumor necrosis factor-alpha-mediated tissue factor pathway inhibitor downregulation via suppression of nuclear factor-kappa B in endothelial cells. Asian J Androl. 2009;11:266–71. doi: 10.1038/aja.2008.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mukherjee TK, Dinh H, Chaudhuri G, Nathan L. Testosterone attenuates expression of vascular cell adhesion molecule-1 by conversion to estradiol by aromatase in endothelial cells: Implications in atherosclerosis. Proc Natl Acad Sci USA. 2002;99:4055–60. doi: 10.1073/pnas.052703199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Malkin CJ, Pugh PJ, Jones RD, Kapoor D, Channer KS, Jones TH. The effect of testosterone replacement on endogenous inflammatory cytokines and lipid profiles in hypogonadal men. J Clin Endocrinol Metab. 2004;89:3313–8. doi: 10.1210/jc.2003-031069. [DOI] [PubMed] [Google Scholar]
- 47.Yeap BB. Are declining testosterone levels a major risk factor for ill-health in aging men. Int J Impot Res. 2008;21:24–36. doi: 10.1038/ijir.2008.60. [DOI] [PubMed] [Google Scholar]
- 48.Gonzales RJ, Duckles SP, Krause DN. Dihydrotestosterone stimulates cerebrovascular inflammation through NFkappaB, modulating contractile function. J Cereb Blood Flow Metab. 2009;29:244–53. doi: 10.1038/jcbfm.2008.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Razmara A, Krause DN, Duckles SP. Testosterone augments endotoxin-mediated cerebrovascular inflammation in male rats. Am J Physiol Heart Circ Physiol. 2005;289:H1843–50. doi: 10.1152/ajpheart.00465.2005. [DOI] [PubMed] [Google Scholar]
- 50.Zhang X, Wang LY, Jiang TY, Zhang HP, Dou Y, Zhao JH, et al. Effects of testosterone and 17-beta-estradiol on TNF-alpha-induced E-selectin and VCAM-1 expression in endothelial cells. Analysis of the underlying receptor pathways. Life Sci. 2002;71:15–29. doi: 10.1016/s0024-3205(02)01567-9. [DOI] [PubMed] [Google Scholar]
- 51.Carswell HV, Macrae IM, Gallagher L, Harrop E, Horsburgh KJ. Neuroprotection by a selective estrogen receptor beta agonist in a mouse model of global ischemia. Am J Physiol Heart Circ Physiol. 2004;287:H1501–4. doi: 10.1152/ajpheart.00227.2004. [DOI] [PubMed] [Google Scholar]
- 52.Miller NR, Jover T, Cohen HW, Zukin RS, Etgen AM. Estrogen can act via estrogen receptor alpha and beta to protect hippocampal neurons against global ischemia-induced cell death. Endocrinology. 2005;146:3070–9. doi: 10.1210/en.2004-1515. [DOI] [PubMed] [Google Scholar]
- 53.Pelzer T, Loza PA, Hu K, Bayer B, Dienesch C, Calvillo L, et al. Increased mortality and aggravation of heart failure in estrogen receptor-beta knockout mice after myocardial infarction. Circulation. 2005;111:1492–8. doi: 10.1161/01.CIR.0000159262.18512.46. [DOI] [PubMed] [Google Scholar]
- 54.Nikolic I, Liu D, Bell JA, Collins J, Steenbergen C, Murphy E. Treatment with an estrogen receptor-beta-selective agonist is cardioprotective. J Mol Cell Cardiol. 2007;42:769–80. doi: 10.1016/j.yjmcc.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 55.Wang M, Crisostomo PR, Markel T, Wang Y, Lillemoe KD, Meldrum DR. Estrogen receptor beta mediates acute myocardial protection following ischemia. Surgery. 2008;144:233–8. doi: 10.1016/j.surg.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 56.Wang M, Wang Y, Weil B, Abarbanell A, Herrmann J, Tan J, et al. Estrogen receptor beta mediates increased activation of PI3K/Akt signaling and improved myocardial function in female hearts following acute ischemia. Am J Physiol Regul Integr Comp Physiol. 2009;296:R972–8. doi: 10.1152/ajpregu.00045.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vornehm ND, Wang M, Abarbanell A, Herrmann J, Weil B, Tan J, et al. Acute postischemic treatment with estrogen receptor-alpha agonist or estrogen receptor-beta agonist improves myocardial recovery. Surgery. 2009;146:145–54. doi: 10.1016/j.surg.2009.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo J, Krause DN, Horne J, Weiss JH, Li X, Duckles SP. Estrogen-receptor-mediated protection of cerebral endothelial cell viability and mitochondrial function after ischemic insult in vitro. J Cereb Blood Flow Metab. 2010;30:545–54. doi: 10.1038/jcbfm.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saijo K, Collier Jana G, Li Andrew C, Katzenellenbogen John A, Glass Christopher K. An ADIOL-ER[beta]-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell. 2011;145:584–95. doi: 10.1016/j.cell.2011.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fortunati N, Fissore F, Fazzari A, Becchis M, Comba A, Catalano MG, et al. Sex steroid binding protein exerts a negative control on estradiol action in MCF-7 cells (human breast cancer) through cyclic adenosine 3′,5′-monophosphate and protein kinase A. Endocrinology. 1996;137:686–92. doi: 10.1210/endo.137.2.8593818. [DOI] [PubMed] [Google Scholar]
- 61.Nakhla AM, Rosner W. Stimulation of prostate cancer growth by androgens and estrogens through the intermediacy of sex hormone-binding globulin. Endocrinology. 1996;137:4126–9. doi: 10.1210/endo.137.10.8828467. [DOI] [PubMed] [Google Scholar]
- 62.Benten WP, Lieberherr M, Giese G, Wrehlke C, Stamm O, Sekeris CE, et al. Functional testosterone receptors in plasma membranes of T cells. FASEB J. 1999;13:123–33. doi: 10.1096/fasebj.13.1.123. [DOI] [PubMed] [Google Scholar]
- 63.Benten WP, Lieberherr M, Stamm O, Wrehlke C, Guo Z, Wunderlich F. Testosterone signaling through internalizable surface receptors in androgen receptor-free macrophages. Mol Biol Cell. 1999;10:3113–23. doi: 10.1091/mbc.10.10.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Konoplya EF, Popoff EH. Identification of the classical androgen receptor in male rat liver and prostate cell plasma membranes. Int J Biochem. 1992;24:1979–83. doi: 10.1016/0020-711x(92)90294-b. [DOI] [PubMed] [Google Scholar]
- 65.Heinlein CA, Chang C. The roles of androgen receptors and androgen-binding proteins in nongenomic androgen actions. Mol Endocrinol. 2002;16:2181–7. doi: 10.1210/me.2002-0070. [DOI] [PubMed] [Google Scholar]
- 66.Thyberg J, Roy J, Tran PK, Blomgren K, Dumitrescu A, Hedin U. Expression of caveolae on the surface of rat arterial smooth muscle cells is dependent on the phenotypic state of the cells. Lab Invest. 1997;77:93–101. [PubMed] [Google Scholar]