

Abstract
Aims: Infusion of purified haptoglobin (Hp) functions as an effective hemoglobin (Hb) scavenging therapeutic in animal models of hemolysis to prevent cardiovascular and renal injury. Epidemiologic studies demonstrate the phenotype heterogeneity of human Hp proteins and suggest differing vascular protective potential imparted by the dimeric Hp1-1 and the polymeric Hp2-2. Results: In vitro experiments and in vivo studies in guinea pigs were performed to evaluate phenotype-specific differences in Hp therapeutics. We found no differences between the two phenotypes in Hb binding and intravascular compartmentalization of Hb in vivo. Both Hp1-1 and Hp2-2 attenuate Hb-induced blood pressure response and renal iron deposition. These findings were consistent with equal prevention of Hb endothelial translocation. The modulation of oxidative Hb reactions by the two Hp phenotypes was not found to be different. Both phenotypes stabilize the ferryl (Fe4+) Hb transition state, provide heme retention within the complex, and prevent Hb-driven low-density lipoprotein (LDL) peroxidation. Hb-mediated peroxidation of LDL resulted in endothelial toxicity, which was equally blocked by the addition of Hp1-1 and Hp2-2. Innovation and Conclusion: The present data do not provide support for the concept that phenotype-specific Hp therapeutics offer differential efficacy in mitigating acute Hb toxicity. Antioxid. Redox Signal. 19, 1619–1633.
Introduction
Hemoglobin (Hb) is released from red blood cells during multiple disease states, including hereditary hemolytic anemias (e.g., sickle cell disease), malaria, and extracorporeal circulation, as well as during massive blood transfusion. In these conditions, extracellular Hb is both a disease marker and a toxin (11). Ligand binding of extracellular Hb can alter nitric oxide (NO) and redox homeostasis, which can lead to acute pulmonary and systemic hypertensive responses, endothelial dysfunction, and oxidative tissue damage (38). Moreover, the pathophysiology caused by acute and chronic extracellular Hb exposure may contribute to tissue-specific sequelae associated with hemolytic conditions (38).
The physiologic detoxification systems for free heme and Hb comprise a group of plasma scavenger proteins and their respective cellular clearance receptors (38). In the case of Hb, haptoglobin (Hp) binds Hb dimers in an irreversible large protein complex, which is cleared by Hb:Hp scavenger receptors (1,11). In humans, this receptor has been identified as the Hb scavenger receptor CD163, which enables endocytosis of the complex by macrophages and peripheral blood monocytes (20,37,39). Binding of Hb to Hp appears to fundamentally alter its potential to induce pathophysiology, as the Hb:Hp complex protects against Hb toxicity by three primary mechanisms (38): (i) Hp binds Hb dimers and sequesters the Hb:Hp complex within the intravascular compartment, thereby limiting tissue exposure; (ii) Hp stabilizes the redox chemistry and the structure of bound Hb; and (iii) Hp prevents release of heme when oxidized to its ferric (Fe3+) transition state. The potential therapeutic efficacy of Hp has been suggested in preclinical models of hemolysis and transfusion of significant quantities of stored blood, where infusion of a plasma-derived Hp concentrate attenuates Hb-driven vascular dysfunction, hemoglobinuria, and acute kidney injury (4). Specifically, by providing intravascular sequestration of Hb within the circulation, Hp prevented accumulation of free Hb in the kidneys of transfused animals. Renal tubulus function and clearance capacity remained unchanged in Hp-treated guinea pigs (4). The use of Hp as a treatment modality is also supported by clinical experience with a human plasma-derived Hp product approved for use in Japan (38). Based on this evidence, several development projects have recently commenced in the United States and Europe, with the aim to explore the potential therapeutic benefit of plasma-derived Hb scavenger proteins.
Innovation.
Haptoglobin (Hp) therapeutics are in the early stages of development, when critical strategic approaches to production process, proof-of-concept, nonclinical safety, and early phase clinical study design define the future success of novel pharmaceuticals. Hp is a multiphenotype plasma protein suggested to have endogenous genotype-specific vascular protective effects. Hp1-1 and 2-2 differences in pathophysiologic outcomes are largely based on transgenic mouse models and epidemiological analyses that find Hp1-1 to more effectively attenuate disease processes. The present studies demonstrate that within the context of treating acute hemolytic events, these two major phenotypes of Hp (1-1 and 2-2) both display comparable efficacy in vitro and in an established guinea pig model. These data provide early proof of concept that is critical to Hp therapeutics development optimization.
In humans, Hp has a genetic polymorphism that leads to expression of three primary Hp phenotypes that share a common Hb-binding β-chain, yet differ in their α-chain composition (34,42). Hp phenotype 1-1 expresses a smaller α-chain (α1), which can form only one disulfide bond to one other α-β subunit. The resulting Hp is a dimeric (α1)2β2 Hp molecule with a molecular weight of ∼85 kDa. In contrast, Hp2-2 has a longer α chain (α2) that can form two disulfide bonds with other Hp α-chains. The resulting Hp is a large heterogeneous polymeric molecule composed of multiple α and β subunits. Hp2-1 has a mixed α1 and α2 chain composition (21). Epidemiological studies have linked Hp2-2 genotype/phenotype carrier status to a higher risk of cardiovascular complications in patients with diabetes (13,21,22,26,33). Findings of these and similar studies suggested that a “good” and a “bad” endogenous Hp phenotype may differentially impact vascular disease states with underlying hemolytic components. However, it remains unclear whether this evidence should imply that phenotype-specific Hp therapeutics must be developed in order to exert maximum therapeutic efficacy in hemolysis. During routine Cohn fractionation of pooled human plasma, Hp1-1 and Hp2-2 enrich in different fractions. Therefore, production strategies to develop phenotype-specific Hp therapeutics at large scale are feasible and currently employed by some developers (15). For example, the Japanese Hp therapeutic produced by Benesis contains only Hp2-2, while an investigational product developed by the British plasma fractionation company Bio Products Laboratory (BPL) contains predominantly Hp1-1.
The purpose of the present study is to understand the differences between Hp1-1 and Hp2-2 interactions with Hb in a series of in vitro and in vivo experiments and evaluate the efficacy of the individual phenotype Hp preparations. The third major phenotype, Hp2-1, was not examined in this study because industrial isolation of this phenotype from pooled plasma appears not to be less feasible. Data from this series of experiments suggest that both Hp1-1 and Hp2-2 demonstrate therapeutic efficacy in hemolytic conditions.
Results
Molecular characterization of two phenotype-specified Hp therapeutics
Characterization of the composition and Hb binding of two human plasma-derived and phenotype-specific Hp therapeutics has been performed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and mass spectrometry. SDS-PAGE indicates that both products are predominantly composed of the respective Hp phenotype (Fig. 1A). The phenotype specificity of the two products was also confirmed by liquid chromatography–mass spectrometry (LC-MS/MS) analysis (Supplementary Fig. S1). One of the Hp products is approved as a therapeutic plasma protein in Japan and comprises polymeric Hp phenotype 2-2 (characterized by the larger α-2 chain in a reducing SDS-PAGE). The second product is an Hp1-1-specified investigational therapeutic, characterized by the smaller α-1 Hp chain. Impurities in the two human plasma-derived products were identified by LC-MS/MS and appear to be comparable (Fig. 1B). In both preparations >90% of the protein was identified as Hp. Subsequent quantitative mass spectrometry analysis confirmed that the Hb α/β chain contamination in both products is <2%, relative to the amount of Hp (data not shown). Total Hb binding capacity (Fig. 1C and Supplementary Fig. S2) and Hb association (Fig. 1D) of the two Hp phenotype-specific products was determined by mass spectrometry and surface plasmon resonance (SPR), respectively, and no significant differences were noted. This data demonstrates that the Hp phenotype-specific therapeutics investigated in these studies do not demonstrate quantitative differences in purity or Hb binding.
FIG. 1.

Molecular characterization of phenotype-specified Hp therapeutics. (A) The phenotype-specified Hp preparations that were used throughout these studies were analyzed by reducing (left) and nonreducing (right) SDS-PAGE electrophoresis. Hp2-2 is comprised of a longer α2 chain and displays (in the nonreduced state) a heterogeneous multimeric state compared to Hp1-1. (B) The major contaminants of plasma-derived Hp preparations have been determined by LC-MS/MS of trypsin-digested Hp preparations. (C) Binding capacities of Hp phenotypes have been measured by quantitative mass spectrometry. The input of the chromatography purified Hb:Hp complexes into the mTRAQ mass spectrometry analysis was quantified by UV-VIS spectrophotometry (=equal heme, left panel). The output provides relative quantification of Hp and Hb peptides in the two complexes. The right panel displays ion current chromatograms of representative Hb (Hb727) and Hp (Hp560) peptides, respectively. In these chromatograms, peptides derived from Hb:Hp1-1 (red) and Hb:Hp2-2 (blue) were discriminated by their specific mass shift that was introduced by the mTRAQ labeling. For details of MS spectra see Supplementary Figure S1. (D) Surface plasmon resonance recordings of Hb binding to Hp2-2 (left) and Hp1-1 (right) immobilized on a GLM Proteon Sensor chip. Hp, haptoglobin; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; LC-MS/MS, liquid chromatography–mass spectrometry; Hb, hemoglobin; mTRAQ, mass differential Tags for Relative and Absolute Quantification; UV-VIS, UV-VIS spectrophotometry. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Attenuation of Hb-mediated hypertension is not Hp phenotype dependent
One of the most extensively studied adverse effects of extracellular Hb is an acute hypertensive response (11). In earlier studies, we have demonstrated that increased Hp expression, Hb:Hp complex infusion, and supra-physiologic co-infusion of Hp attenuates the hypertensive response induced by Hb exposure in dog and guinea pig (4,9). In the present study we explored whether Hp therapeutics attenuate hypertension in guinea pigs when extracellular Hb is present in the circulation and hypertension is established. Such a scenario has not yet been studied, but is more comparable to the actual therapeutic situation where Hp may be administered. Both Hp phenotype-specific therapeutics significantly reduced blood pressure compared to the (non-Hp-treated) control animals when infused 10 min after exposure to human oxy-Hb (at a stable maximum blood pressure response) (Fig. 2A). The mean±standard error (SE) maximum blood pressure reduction was −67.4%±14.6% and −76.1%±9.1% following Hp1-1 and Hp2-2 infusion, respectively (Fig. 2B). Hb infusion alone maintained a stable blood pressure for ∼80 min. The mean area under the curve (AUC)±SE mean arterial blood pressure (MAP) elevation from time 0 to 80 min was 1360±84.9 in the Hb-only-treated animals. AUCs were significantly lower following Hp1-1 (600±158) and Hp2-2 (532±228) infusion, respectively (Fig. 2C). These values did not differ for Hp1-1 and Hp2-2 at Hb equivalent doses of infused Hp protein. Supplementary Figure S3 shows separate experiments where the two Hp products were infused into control animals that were not pretreated with Hb. This study confirms that Hp per se has no hypotensive effect. NO consumption of purified Hb and the two Hb:Hp complexes (Fig. 3A), as well as of plasma before and immediately after Hp infusion, did not differ (Fig. 3B). These findings support our previous assumption that the attenuation of free Hb-mediated hypertension by Hp is not caused by changes in global NO scavenging within the intra-vascular space. Therefore, the (local) NO sparing effect is more likely due to prevention of Hp-bound Hb from accessing the subendothelial space, where NO consumption is most deleterious.
FIG. 2.

Equal efficacy of Hp phenotype-specified therapeutics against Hb-induced hypertension. (A) % change (Δ) in MAP from baseline values to 180 mg Hb is shown as control (gray circles), Hp1-1 (open circles), and Hp2-2 (dark circles) groups (n=5/group). In the Hp1-1 and Hp2-2 groups, each Hp phenotype was administered in gram equivalent dose 10 min after the Hb infusion. Animals were monitored continuously for 80 min. (B) % change (Δ) maximum MAP was determined as the change from maximum Hb-induced MAP response to MAP after the Hp infusion (15 min). (C) Area under the MAP curve (AUC) was determined by the linear trapezoidal rule for determining AUC in the interval 0–80 min using individual data plotted in (A). All data are representative of mean values±SE. Statistical significance was set at p<0.05. Systolic and diastolic blood pressures were measured using a Gould pressure transducer with data acquisition record using a BioPak data acquisition system and AcqKnowledge software for data analysis and MAP calculation. MAP, mean arterial blood pressure; SE, standard error.
FIG. 3.

Hp1-1 and Hp2-2 do not change ex vivo NO consumption by extracellular Hb. NO consumption of purified Hb, Hb:Hp1-1, and Hb:Hp2-2 (A) or plasma samples from Hb infused animals (B) has been measured with an NO-sensitive electrode and serial 15 μl (of 62.5 μM Hb/Hb:Hp complex solution) and 7.5 μl (of plasma) injections, respectively, into a closed equilibrated NO-producing system. The data represent means of triplicate recordings (n=3±SD), with three serial injections. In (B), the three samples represent plasma collected after Hb infusion at the time of peak blood pressure response (Hb, black trace) and after therapeutic Hp application at the time of maximum Hp-mediated blood pressure attenuation (Hp2-2, blue; or Hp1-1, red) (heme concentration=542±59 μM [Hb alone group], 556±27 μM [Hp1-1 group], 550±49 μM [Hp2-2 group]). NO, nitric oxide. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Hp1-1 and Hp2-2 restrict trans-endothelial Hb redistribution
Previous studies suggest that the vascular protective effects of Hp could be related to the ability of the large Hb:Hp complexes to restrict passive diffusion and/or active transport of free Hb across the endothelium into the subendothelial space. This biological function of Hp may explain epidemiological differences in vascular protection by (the smaller) Hp1-1 and (the larger) Hp2-2 phenotypes. To test this hypothesis, we investigated redistribution of Hb and phenotype-specific Hb:Hp complexes across confluent monolayers of human umbilical vein endothelial cells (HUVECs) (Fig. 4). In the absence of Hp, we observed significant redistribution of Hb across the endothelium, with 53.5%±3.6% of total Hb appearing in the abluminal compartment after 6 h (Fig. 4A, B). During the redistribution process Hb did undergo autoxidation such that the final composition in both compartments was ∼50% ferrous (Fe2+), 45% ferric (Fe3+), and 5% hemichrome. However, Hb remained predominantly intact, as indicated by size-exclusion chromatography (SEC) of the abluminal protein demonstrating an identical chromatographic pattern to Hb recovered from the luminal compartment (Fig. 4C). In addition, the quantity of free heme, as determined by a dialysis assay, remained below detection limit in all conditions (data not shown). This suggests that in this system all heme remains bound to some protein and/or lipid components. When Hp was present in the luminal medium, 97.6%±0.3% of the Hp1-1 and 97.7%±0.2% of the Hp2-2-bound Hb was sequestered, significantly limiting transendothelial redistribution of both Hb:Hp complexes. The end study composition of Hp-bound Hb was ∼55% ferrous (Fe2+) and 45% ferric (Fe3+) with no measurable hemichrome. The potential free Hb redistribution mechanisms are passive diffusion or active transport of the protein across the intact endothelial monolayer. An alternative explanation for this phenomenon could be that free Hb or Hb oxidation/degradation products damage the endothelium in our experiments. In this case, the high concentration of abluminal Hb would be a marker of endothelial damage and subsequent “leakiness.” In an attempt to evaluate these scenarios, we have performed identical experiments as described above in an electric cell impedance substrate measurement (ECIS) instrument, as this enables measurement of transendothelial electrical resistance in real time to determine monolayer integrity during Hb and Hb:Hp exposure. In these experiments, we did not observe any changes in transendothelial resistance with either free Hb or Hb:Hp complex incubation (Fig. 4D). Accordingly, at the end of the experiments, endothelial cells exposed to the different conditions displayed equal adherence junction morphology (Fig. 4E). Further, neither Hb nor any of the Hb:Hp complexes impaired HUVEC mitochondrial function as evidenced by unaltered cellular ATP concentrations at the end of the 6-h incubation period (Fig. 4F). We can thus conclude that redistribution of free Hb across the endothelium is a significant pathophysiological component of free Hb toxicity that is effectively prevented by Hp therapeutics of either phenotype.
FIG. 4.

Hp1-1 and Hp2-2 equally restrict trans-endothelial diffusion of extracellular Hb. Confluent HUVEC monolayers grown on a permeable trans-well cell culture insert were incubated on the “luminal” side with 300 μM Hb, Hb:Hp1-1, or Hb:Hp2-2. After 6 h of incubation, total heme concentrations on the “luminal” (A) and “abluminal” (B) side of the endothelial monolayer were quantified by UV-VIS spectrophotometry and by SEC (C), respectively. The results indicate that the large molecular size of the Hb:Hp complexes fully restrict trans-endothelial diffusion of Hb. (D) The integrity of the endothelial monolayer during Hb and Hb:Hp incubation was confirmed by identical experiments performed in an ECIS instrument in order to measure trans-endothelial resistance over time. In absence or presence of Hp of either phenotype, Hb treatment does not affect monolayer electrical resistance. (E) Adherence junction morphology was examined on the ECIS electrodes at the end of the experiments shown in (D), where green indicates β-catenin, blue shows 4′,6-diamidino-2-phenylindole stained nuclei. (F) Cellular ATP was quantified after 6 h of incubation under the same condition as in the translocation assay with a luminescent assay. There was no significant difference among treatments. Data are shown as mean±SE (n=8). The negative control samples (negative) were complete assay reaction mix without cells. HUVECs, human umbilical vein endothelial cells; SEC, size-exclusion chromatography; ECIS, electric cell impedance substrate measurement. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Hp1-1 and Hp2-2 prevent free Hb redistribution in guinea pigs
To validate the intravascular sequestration hypothesis in vivo, we evaluated free and Hp-bound Hb plasma concentrations in guinea pigs after infusion of Hb (60 and 180 mg) alone and following treatment with the phenotype-specific Hp products (Fig. 5). In the absence of Hp, Hb is rapidly cleared from plasma. This short circulation time is primarily the result of glomerular filtration and Hb distribution to the kidneys. In contrast, following Hp treatment, the complexed Hb remained in circulation over an extended period of time, consistent with previous studies in this and several other species (9,30,31). In the present study two maximum plasma concentrations of Hb (expressed as heme) were evaluated following intravenous infusion of 60 mg (Cmax=224±5.5 μM plasma heme) and 180 mg (Cmax=556±60 μM plasma heme) (Fig. 5A–D). Milligram equivalent doses of Hp1-1 and Hp2-2 were infused 10 min after Hb exposure. In the 60 mg Hb group Hb:Hp was cleared from plasma by ∼30 h post-Hp administration. Pharmacokinetic parameters of Hb (expressed as heme) bound to Hp1-1 and Hp2-2 were derived from plasma concentration versus time curves (Fig. 5A, B). Comparison of the primary exposure parameters (AUC0–30h and plasma clearance) and the terminal half-life demonstrated no difference between Hb bound to Hp1-1 or Hp2-2 (Fig. 5E–G). The mean±SE for exposure parameters were as follows: AUC0–30h 1526±72 h·μg/ml (Hb:Hp1-1) and 1322±55 h·μg/ml (Hb:Hp2-2); plasma Cl, 157±8.0 ml/h (Hb:Hp1-1) and 180±8.0 ml/h (Hb:Hp2-2). The terminal half-life was 8.2±0.5 h for Hb:Hp1-1 and 8.4±2.3 h for Hb:Hp2-2. The Cmax for Hb alone was 220±12 μM plasma heme, AUC0–4h was 231±30 h·μg/ml, plasma Cl, 1004±127 ml/h and the terminal half-life was determined to be 1.0 h. In the 180 mg Hb exposure group plasma concentrations of Hb:Hp leveled off at ∼200 μM heme. This observation suggests saturable kinetics within the system and did not allow for meaningful pharmacokinetic analysis. However, the long circulation time of the Hb:Hp complex at the 180 mg exposure may be considered reflective of the intravascular Hp sequestration of Hb. Similar to the previous in vitro trans-endothelial redistribution findings, we did not observe significant differences in the redistribution of Hb to the kidneys following sequestration within Hp1-1 or Hp2-2 complex. Renal iron deposition determined from the 180 mg dosing group was increased in Hb-infused animals (Fig. 6A); however, both Hp phenotypes appeared to limit iron deposition. Percent area analysis of kidney tissue stained positive for iron was 55.7%±8.6%, as measured 24 h after Hb infusion. Following Hp1-1 and Hp2-2 infusion, the percentage of areas staining positive for iron were 6.93%±1.2% and 11.49%±0.9%, respectively (Fig. 6B). These values were lower than those corresponding to Hb alone. However, they were not different from each other, suggesting that both Hp1-1 and 2-2 prevent renal iron deposition. To quantitatively compare the intravascular Hb sequestration by the two Hp products in vivo, we have quantified complex formation and Hb binding in post-infusion plasmas of Hp-treated guinea pigs over a range of Hb concentrations. These data are shown in Figure 6C and in Supplementary Figure S4. Also in these studies, we did not detect a significant difference in the total Hb binding capacities of the two Hp phenotypes in vivo.
FIG. 5.

Extravascular decompartmentalization of Hb, Hp1-1, and Hp2-2. Plasma concentration versus time curves of plasma Hb (expressed as heme concentration). (A) Hemoglobin infusion (60 mg) followed by Hp1-1 (60 mg) infused at 10 min post-Hb infusion. (B) Hemoglobin infusion (60 mg) followed by Hp2-2 (60 mg) infused at 10 min post-Hb infusion. (C) Hemoglobin infusion (180 mg) followed by Hp1-1 (180 mg) infused at 10 min post-Hb infusion. (D) Hemoglobin infusion (180 mg) followed by Hp2-2 (180 mg) infused at 10 min post-Hb infusion. In both panels, black and white open circles and dark squares represent bound and free Hb, respectively, whereas open circles represent the plasma concentration–time curve of Hb alone. Comparison of primary pharmacokinetic data from the 60 mg Hb exposure group (E) AUC0–4h Hb, AUC0–30h Hb:Hp1-1 and Hb:Hp2-2. (F) Plasma clearance (dose/AUC) based on Hb AUC0–4h, and Hb:Hp AUC0–30h. (G) Terminal elimination half-life based on the negative value of the terminal slope (k) of the log-linear plasma concentration–time curve for Hb, Hb:Hp1-1, and Hb:Hp2-2. Parameters were all significantly different (p<0.001) between Hb and Hb:Hp complexes, but not between Hb:Hp1-1 and Hb:Hp2-2 based on a nonparametric analyses (Kruskal–Wallis test followed by a Dunns post-test for group comparisons). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 6.

Equal prevention of renal iron deposition by Hp1-1 and Hp2-2. (A) Representative Perls iron stained kidney tissue with 3,3′-diaminobenzidine intensification showing nontreated (upper left), Hb (upper right), Hb+Hp1-1 (lower left), and Hb+Hp2-2 (lower right). (B) Image analysis was performed using ImageJ to determine the ratio of iron-stained area to the total area of 20 images (10×) of n=5 individual animals. Percentage areas are represented as mean±SE. (C) Ex vivo Hb binding of guinea pig plasmas was measured by SEC after infusion with either Hp1-1 or Hp2-2. The figure shows chromatogram areas (AU*min) of Hb-bound Hp components (=Hb:Hp complex) plotted against Hb concentration. Details are provided in Supplementary Figure S4. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Ferryl Hb stabilization, heme release, and attenuation of oxidative toxicity are not related to Hp phenotypes
Stabilization of the Hb structure and its redox chemistry have been considered essential components of Hp protection against Hb toxicity (8,10,14,32,41). In the present study, Hb oxidation/reduction reactions were monitored by UV-VIS spectrometry in a system with glucose oxidase (GOX) as a source of continuous low-level hydrogen peroxide (H2O2). The different reaction products were calculated from the raw data by spectral deconvolution (Fig. 7). In the absence and in the presence of Hp, we observed a comparable rapid transition of oxy-Hb(Fe2+) to ferryl-Hb(Fe4+), although, in the presence of Hp, the decay of Hb(Fe4+) was slower than in the absence of Hp (Fig. 7A). The modeled ferryl kinetics revealed slower decay rates k2 (min−1) for Hb:Hp than for free Hb over a range of glucose oxidase (GOX) concentrations. The rates for Hb:Hp1-1 and Hb:Hp2-2 were not statistically different (Fig. 7B). A structurally nondefined porphyrin degradation product appeared delayed when Hp of either phenotype was present in the reaction. To further confirm the stability of ferryl-Hb(Fe4+) in the Hb:Hp complex, we performed additional experiments in which an excess of catalase was added—in order to terminate Hb(Fe3+) to Hb(Fe4+) transition—at a time when all Hb was oxidized to Hb(Fe4+) (∼12 min after the reaction commenced) and followed the decay of Hb(Fe4+) over time (Fig. 7C). Compared to Hb alone, the stability of Hb(Fe4+) was markedly enhanced in the presence of Hp1-1 and 2-2. Again, no apparent differences between the two Hp phenotypes were observed.
FIG. 7.

Hb:Hp1-1 and Hb:Hp2-2 stabilize the Hb ferryl state. (A) Hb-Fe2+ (12.5 μM) or the respective Hp complexes were incubated with 40 mU of glucose-oxidase (in phosphate buffered saline 20 mM) as a source of continuous hydrogen peroxide at 37°C. The absorbance over the wavelength range of 300–700 nm was measured during 5 h. The amounts of different Hb reaction products were extracted by spectral deconvolution. While Hp1-1 and Hp2-2 complexes of Hb indicate identical reaction patterns over time, free Hb shows a more rapid decline of ferryl (Fe4+) concentrations (red line) with a concurrent, more rapid increase of a structurally nondefined porphyrin degradation product (black line). (B) Ferryl decay rates (k2) over a range of GOX concentrations. Experimental design and readout as shown in (A). Red: free Hb; blue: Hb:Hp1-1; gray: Hb:Hp2-2. The plotted line represents a linear regression±95% confidence interval. N=3 experiments. (C) Identical experiments were repeated with 200 nM of bovine catalase added after the first 12 min. The recordings demonstrate that, in the presence of both Hp phenotypes, the ferryl (Fe4+) state of Hb is much more stabile compared to free Hb. GOX, glucose oxidase. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Heme release from ferric (Fe3+) Hb was measured over time in an Hb-hemopexin (Hpx) heme transfer assay (Fig. 8). In a reaction mixture containing ferric (Fe3+) Hb and Hpx, we noted a gradual disappearance of the characteristic HbFe3+ UV-VIS spectrum (red trace) and an increase of the spectrum that is specific for the heme-Hpx complex (blue trace) (Fig. 8A). This spectral transition reflects the transfer of heme from Hb to Hpx. The concentrations over time of met-Hb and heme-Hpx were calculated by spectral deconvolution and the decay rate of met-Hb calculated by fitting the data to a double exponential model, which might be reflective of the different heme release kinetics of Hb α- and β- chains, respectively (18) (Fig. 8B and Table 1). Compared to free Hb, there was no significant heme transfer detected with the Hb:Hp complexes of either phenotype (Fig. 8C, D). One potential mechanism of Hb toxicity is related to the transfer of heme from HbFe3+ to lipoproteins, particularly low-density lipoprotein (LDL), with subsequent oxidation of the LDL. We measured LDL oxidation by HbFe3+ and the HbFe3+:Hp complexes, respectively, and found that Hp of both phenotypes prevented LDL oxidation equivalently (Fig. 8E). In vivo, endothelial damage by heme-oxidized LDL is a critical toxicity associated with Hb following hemolysis. We have therefore established an in vitro model of endothelial toxicity that is driven by in situ oxidation of LDL. As shown in Figure 8F, Hp of both phenotypes significantly delays the breakdown of endothelial barrier function that occurs when the endothelium is exposed to LDL and Hb in the presence of low (nontoxic, 5–10 μM) H2O2 concentrations.
FIG. 8.

Hp1-1 and Hp2-2 restrict heme release and protect against LDL peroxidation-driven endothelial damage. (A–D) Heme release from free met-Hb (Fe3+) or from the respective complexes with Hp1-1 and Hp2-2 was probed using an Hb-Hpx heme transfer assay. Serial UV-VIS spectra were recorded over time (15 h at 37°C) with reaction mixtures containing 12.5 μM HbFe3+(A), HbFe3+:Hp1-1 (C), and HbFe3+:Hp2-2 (D) and an excess of Hpx. The first spectrum (t0) is highlighted in red and the last spectrum (t15h) in blue. The relative quantities of met-Hb (solid lines) and heme-Hpx (dashed lines) were calculated by spectral deconvolution. (E) Hb-driven LDL peroxidation. About 5 μM heme equivalent of free Hb, Hb:Hp1-1, or Hb:Hp2-2 was injected into a 0.1 mg/ml LDL solution in a closed system. Oxygen was monitored as a surrogate parameter of LDL peroxidation in the system using an oxygen sensor. Data are represented as mean±SE of triplicate experiments. (F) LDL peroxidation-driven endothelial damage. Confluent monolayers of HUVECs were grown in an ECIS instrument and treated with 0.1 mg/ml LDL and 2.5 mU GOX as a source of continuous low-level hydrogen peroxide exposure. In the absence of Hb, no change in electrical resistance was observed. Endothelial integrity was rapidly damaged when free Hb was added to the reaction (decrease of endothelial resistance). This endothelial damage was attenuated/delayed in the presence of either Hp1-1 or Hp2-2. LDL, low-density lipoprotein; Hpx, hemopexin. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Table 1.
Rates of met-Hemoglobin Decay Resulting from Heme Transfer to Hemopexin
| k1(s−1) | k2(s−1) | R2 | |
|---|---|---|---|
| met–Hb | 3.51 | 0.60 | 5.35 |
| met-Hb:Hp1-1 | 0.05 | −0.03 | 3.51 |
| met-Hb:Hp2-2 | 0.07 | −0.03 | 14.79 |
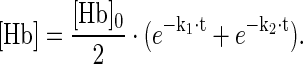 |
Hb, hemoglobin; Hp, haptoglobin.
Discussion
Epidemiological gene association studies suggest that expression of the Hp2-2 genotype might be a risk factor for morbidity and mortality in a variety of vascular diseases associated with local or systemic hemolysis (13,21). The mechanisms that support this hypothesis are based on the observations of decreased clearance of Hp2-2-bound Hb and increased potential for oxidative stress mediated by heme and iron release (2,3,26,33). Human plasma-derived Hp is approved and marketed in Japan for a wide range of hemolytic conditions (38). Recently, certain U.S. and European plasma fractionators have initiated development programs to isolate Hb-binding proteins and are currently evaluating potential disease-state indications (15). Given these important developments in this field and the extensive knowledge of a potential Hp phenotype-specific predictor of poor disease outcome, the experiments described here were designed to detect differences in Hp1-1 and Hp2-2 interactions with Hb when considered as a therapeutic agent. The experimental results were able to address the relevance of phenotype for Hp therapeutics in acute hemolytic conditions and serve as a proof of concept for future drug development initiatives.
Our studies demonstrate that the protein composition of the evaluated commercial Hp products are predominantly dimeric (Hp1-1) and multimeric (Hp2-2), respectively. Protein impurity content was found to be nearly identical in both preparations, based on quantitative mass spectrometry. Certain studies suggest that the overall binding capacity of Hp1-1 is greater than that of Hp2-2 (3,23), supporting the concept that Hp2-2 may be a less effective Hb binding protein in vivo. In the present study, SPR analysis demonstrated similar Hb association kinetics for each phenotype, whereas quantitative mass spectrometry analysis found no differences in the binding capacity for Hb within Hp1-1 and Hp2-2. These findings suggest that in vivo sequestration of Hb should be similar for the two phenotypes. This concept is supported by data presented in Figure 2, where infusion of Hb, followed by either Hp1-1 or Hp2-2, demonstrated similar blood pressure attenuating capability. Additionally, curves depicted in Figure 5 show similar temporal changes in plasma heme concentration for both Hp1-1- and Hp2-2-bound and free Hb. This data correlated with renal iron deposition, suggesting that the Hb was effectively compartmentalized within the vascular space by both Hp phenotypes. The present data was derived from dosing on a milligram Hb to milligram Hp basis. However, it is critical to note that clinical dosing will likely differ significantly. Given the dynamic range of Hb concentrations that can occur, relevant dosing approaches in complex clinical conditions may require titration and or dose escalation within the range allowed by defined safety margins.
Although definitive mechanism for the MAP attenuating effects of Hp remains difficult to resolve, it has been suggested that Hp can prevent Hb extravasation from the circulation into the peri-vascular space (4,19,28). Data presented in Figure 3 demonstrate that blood/plasma sampled after Hb infusion consumes NO ex vivo to the same extent as blood/plasma sampled after Hp infusion (when Hb was predominantly Hp bound). This is consistent with previously published data using the same assay to confirm NO consumption in hemolysing sickle cell patient blood/plasma (35) and in previous studies showing equivalent NO consumption with the Hb:Hp complex (4). Collectively, these studies establish no differential NO binding between Hb and the Hb:Hp complex of either phenotype. Data shown in Figure 4 suggest that Hb transport across the vascular endothelium could be equally inhibited over time by the addition of Hp1-1 and Hp2-2. Almost 100% of Hp-bound Hb was prevented from crossing cultured endothelial monolayers, while Hb was able to extravasate to a maximum of 45% over a 6-h period. Hb translocation was not associated with endothelial resistance changes or significant adherence junction disruption, potentially indicating that an active transport mechanism for Hb perivascular translocation exists. This transport mechanism may be not accessible for Hb:Hp complexes, either due to its large molecular size or altered surface charge state.
The capacity for clearance of the Hb:Hp complex appears to differ among mammalian species. Studies in mice, guinea pigs, and dogs suggest a slow circulatory clearance of Hb:Hp complex that becomes saturated, particularly at supra-physiological Hb:Hp concentrations (4,9). In mice, this may be associated with lower Hb:Hp binding affinity to CD163 (17). In humans, no specific studies have been performed to address supra-physiologic circulatory exposure to Hb:Hp. The pharmacokinetic capacity of humans to effectively clear Hb:Hp by the CD163-monocyte/macrophage system remains a critical unanswered question. However, within the physiological range of Hp levels, Hb appears to be effectively cleared from human circulation (29). The capacity of specific clearance of human Hb:Hp by the guinea pig macrophage system is not known. Therefore, the comparable rates of delayed complex disappearance of Hb:Hp1-1 and Hb:Hp2-2 in our studies should be primarily interpreted as indicative of the equal capacity of the two phenotypes to sequester Hb within the circulation (e.g., prevention of renal filtration and prevention of trans-endothelial translocation of Hb into the extravascular space). Earlier in vitro studies have suggested that Hb:Hp1-1 might be cleared faster by human CD163 than Hb:Hp2-2. However, when examined in our previously reported endocytosis model, which is based on a human CD163-transduced HEK-293 cell line (12,36,39), we found a slightly increased complex uptake and enhanced heme-oxygenase (HO-1) expression with Hb:Hp2-2 (Supplementary Fig. S5). These findings appear to be better compatible with the higher affinity of Hb:Hp2-2 that was reported in the initial description of the Hb scavenger receptor (20). Collectively, these conflicting results together with the known species differences in the Hb scavenger system underpin the limitation of studies focusing on receptor-mediated Hb clearance in cell-based assays and in experimental animal models. Therefore, separate studies in human could most effectively address the potential for phenotype-dependent Hb:Hp clearance during early clinical trials.
The antioxidant function of Hp has previously been suggested to attenuate the following sequence: heme iron oxidation to the Hb(Fe4+) transition state, peroxide-driven redox cycling of Hb(Fe4+) to Hb(Fe3+), free radical generation and heme release, radical and/or heme transfer to LDL, and accumulation of oxidized LDL (oxLDL) (24,25). Free heme and oxLDL are potential mediators of tissue injury and understanding the contribution of Hp1-1 and Hp2-2 phenotypes to slowing down the generation of these intermediates may provide insight into the potential therapeutic benefits specific to either Hp1-1 or Hp2-2 (5–7,27). In vitro data presented here suggest that Hb redox cycles between Hb(Fe4+) and Hb(Fe3+) in vitro and that both Hp phenotypes stabilize the Hb(Fe4+) transition. Moreover, the two phenotypes prevent heme release from Hb and subsequent peroxidation of LDL. Functionally, these observations appear to be associated with protection against endothelial damage that is mediated by Hb-driven LDL peroxidation.
Collectively, our findings suggest that Hp1-1 and Hp2-2 would likely provide equivalent therapeutic efficacy in the treatment of acute hemolysis. Equal Hb protection by the two Hp phenotypes is also suggested by the recently resolved structure of the Hb:Hp complex, which confirmed that Hb binding is exclusively provided by the conserved Hb β-chain, without any involvement of the heterogeneous α-chain (1). Our findings remain a separate physiologic concept from known epidemiologic differences in disease susceptibility to Hp genotype. However, it is likely that disease associations may not be explained by differences in the primary Hb detoxification functions of Hp phenotypes, but by other properties of the molecule. The different molecular conformations of Hp phenotypes may affect post-translational processing and secretion, which may lead to altered plasma and/or tissue concentrations of endogenous Hp. Lower total plasma concentrations and correlated Hb binding capacities have indeed been described in some populations with the Hp2-2 phenotype (16,43). Alternatively, other functions of Hp such as its immune-modulatory effects might be phenotype dependent (40).
In conclusion, our studies support Hp's in vitro and in vivo therapeutic efficacy against Hb toxicity for a wide range of biochemical and physiological processes. Due to inherent experimental and statistical limitations, proving equal activity of two compounds is difficult, and we cannot exclude that some minor difference do exists in the Hb protective activity of Hp1-1 and Hp2-2, or eventually of the third phenotype, Hp2-1, which was not examined in these studies. However, it remains unlikely that such minor differences will measurably affect the Hb protective therapeutic efficacy of an Hp product. The presented data represent an initial proof of concept that both Hp1-1 and Hp2-2 protect against Hb-driven toxicity. These observations may help therapeutic development decisions to increase production yield and decrease production cost of Hp therapeutics while maintaining therapeutic efficacy.
Materials and Methods
Haptoglobin therapeutics and Hb
Human Hb was purified to remove catalase by ion exchange chromatography on a DEAE Sephadex column (GE-Healthcare). Hb endotoxin levels were <0.05 endotoxin units/ml (Pyrogent Plus; Lonza). A Hp1-1 phenotype-specified investigational therapeutic was from BPL, whereas the Hp2-2-specified product was provided by Benesis Corporation. Phenotype-specified Hb:Hp complexes were separated from excessive (nonbound) Hb by gel chromatography on HiLoad 26/60 Superdex 200 chromatography column (GE Healthcare Lifesciences). Throughout the study quantities of Hb and Hb:Hp complexes are given as heme molarity measured by spectrophotometry. For the in vivo experiments where (Hb free) Hp therapeutics are administered, Hp quantities are indicated by protein mass (mg). The reason for this is that the Hb binding capacity of Hp is proportional to the protein mass but not to the molar quantity of the heterogeneous Hp2-2 molecules.
SPR measurement
SPR measurements were performed on a Proteon XPR36 Instrument (BioRad). Hp1-1/Hp2-2 was immobilized on a ProteOn Sensor Chip GLM using the amino coupling reagents (BioRad). To deactivate the remaining surface groups in the activated channels, 1 M ethanolamine-HCl (pH 8.5; BioRad) was injected. The running buffer used for the association studies with Hb was 150 mM sodium chloride (NaCl), 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic, 3 mM ethylenediaminetetraacetic acid, and 0.005% Tween-20 (pH 7.4).
In vivo studies in guinea pigs
Male Hartley guinea pigs (Charles Rivers Laboratories) were acclimated for 1 week upon arrival to the Food and Drug Administration (FDA)/Center for Biologics Evaluation and Research (CBER) animal care facility. All animals weighed 400–450 g before surgery. Animal protocols were approved by the FDA/CBER Institutional Animal Care and Use Committee with all experimental procedures performed in adherence to the National Institutes of Health guidelines on the use of experimental animals. Surgical preparation was performed as previously described (8). Following a 24-h recovery period, basal blood pressure was collected over 30 min via a right carotid artery catheter connected to Gould P23 XL pressure transducer (Gould Instrument Systems Inc.). Arterial blood pressure was recorded continuously at 100 Hz using an MP100A-CE data acquisition system (Biopac Systems, Inc.). Four groups were evaluated at the 180 mg dose: (I) nontreated, (II) Hb alone (180 mg), (III) Hb (180 mg)+Hp1-1 (180 mg), and (IV) Hb (180 mg)+Hp2-2 (180) (group size n=5). For Group I animals received 3 ml 0.9% NaCl (3 ml/min). Group II animals received 180 mg Hb infused over 3 min. For groups III and IV, Hb (180 mg) was infused over 3 min (1 ml/min) to establish a blood pressure response. The Hb dose was tailored to reach a peak plasma concentration of 500–600 μM. Our previous experience showed that this treatment evokes strong and reproducible blood pressure responses and iron deposition in the kidneys (9). Ten minutes after the start of Hb administration, 180 mg of Hp1-1 or 2-2 was infused over 3 min (1 ml/min). Blood pressure parameters were analyzed off-line using AcqKnowledge software (Biopac Systems, Inc.). At 30 h after the initial infusion of Hb or 0.9% NaCl, animals were humanely euthanized with a 100 mg/kg solution of Euthasol administered intra-peritoneal. Animals were immediately perfused of blood via the arterial catheter with 60 ml cold saline, kidneys were excised and fixed in 10% formalin or frozen at −80°C.
Plasma Hb measurements
Concentrations of ferrous (Fe2+) Hb (oxy/deoxy), ferric (Fe3+) Hb, and hemichromes were determined using a multicomponent analysis based on the extinction coefficients for each Hb species. Hp-bound and unbound fractions of Hb were determined by SEC–high-performance liquid chromatography (SEC-HPLC) using a Waters 600 controller, a Waters 600 pump, and a Waters 2499 photodiode array detector (Waters, Corp.). Plasma samples were separated on BioSep-SEC-S3000 (600×7.5 mm) column (Phenomenex) with 0.1 M potassium phosphate as the mobile phase. All samples were monitored at λ=280 nm and λ=405 nm in dual-wavelength mode. Hb-bound and unbound in plasma was determined by dividing the Hb peak area and the Hb:Hp peak area by additive areas under the Hb:Hb chromatographic peak (15 min elution Hp2-2:Hb and 17 min elution Hp1-1:Hb) and the Hb chromatographic peak (20 min elution) measured at λ=405 nm.
Pharmacokinetic analysis
Four groups were evaluated at 60 mg Hb exposure: (I) nontreated, (II) Hb alone (60 mg), (III) Hb (60 mg)+Hp1-1 (60 mg), and (IV) Hb (60 mg)+Hp2-2 (60) (group size n=3–5). Another four groups were evaluated at 180 mg Hb exposure: (I) nontreated, (II) Hb alone (180 mg), (III) Hb (180 mg)+Hp1-1 (180 mg), and (IV) Hb (180 mg)+Hp2-2 (180) (group size n=5). Plasma concentration versus time data were evaluated for bound and unbound Hb parameters determined using noncompartmental pharmacokinetic methods. WinNonlin version 5.2.1 (Pharsight Corp.) were used to calculate PK parameter estimates. The area under the plasma concentration–time curve (AUC0-INF) was estimated using the linear trapezoidal rule to the last measurable concentration (AUC0-INF last) where Clast is the last measurable plasma concentration (30 hours). Extrapolation to infinity (AUCC last-INF) was calculated by dividing Clast by the negative value of the terminal slope (k) of the log-linear plasma concentration–time curve. Thus AUC0-INF is equal to the sum of AUC0-C last and AUCC last-INF. Additional parameters were calculated as follows: the plasma clearance (CL) was calculated as the dose (μg heme) divided by AUC0-INF, the mean residence time (MRT) was calculated as k−1, the apparent volume of distribution (Vdss) was calculated as the product of CL and MRT, and half-life (t1/2) was calculated as ln(2) divided by k. Bound Hb following the 60 mg exposure declined to approximately zero by 30 h post-Hb and Hp1-1 and Hp2-2 exposure. This dose allowed for a pharmacokinetic comparison between the two Hp variants. In 180 mg Hb exposure group Hb:Hp persisted in the plasma at similar concentrations well beyond 30 h (∼72 h). Saturated plasma concentrations prohibited pharmacokinetic assessment in this group. Iron histopathology was performed as described (9). Pharmacokinetic parameter comparisons were performed based a nonparametric analyses (Kruskal–Wallis test followed by a Dunns post-test for group comparisons) using GraphPad Prism 5 software.
Endothelial permeability assay
HUVECs (Lonza) were grown to confluence on 0.4 μm polyester membrane (Costar No. 3460) transwell plates. HUVECs were cultured in phenol red free endothelial growth medium (EBM) (Lonza, Clonetics). After 24 h of incubation new media (1.5 ml) was added to each transwell with 300 μM catalase free Hb, purified Hb:Hp1-1 complex or purified Hb:Hp2-2 complex. After 6 h of incubation, the medium was collected from the top and bottom of the transwell and centrifuged for 10 min at 15,000 rpm. The supernatant from top well (lumen) and the bottom well (ablumen) were analyzed for Hb concentration and bound and free Hb by SEC-HPLC as described in the blood collection and Hb measurement section of the Materials and Methods.
In vitro NO consumption assay
Constant nitric oxide (NO) levels were generated in a closed stirred 2 ml glass chamber by decay of the long half-life NO donor DETA NONOate (Enzo Life Sciences). The reaction was performed with 5 mM DETA NONOate in argon purged, essentially anaerobic phosphate buffered saline (PBS) (pH 7.4; w/o Ca/Mg) at 23°C. NO was monitored at a 1/s recording rate with a Clark-type NO microsensor (Unisense). Hb or Hb:Hp complexes were injected into the system at a concentration of 62.5 μM (heme equivalent) with a gas-tight Hamilton syringe as three consecutive identical boluses at 30 s intervals. Alternatively, pre- or post-infusion plasma samples were injected at a fixed volume of 7.5 μl. Data were recorded using SensorTrace basic software (Unisense). Statistical analysis of replicate measurements was performed with GraphPad Prism Software Version 5.01.
Spectral analyses of Hb H2O2 reactions
A solution containing PBS (pH 7.2; Gibco) enriched with glucose (20 mM) and a total concentration of 12.5 μM heme equivalent of either Hb, Hb:Hp1-1, or Hb:Hp2-2 was prepared in 1 ml spectrophotometer cuvettes and pre-warmed at 37°C. Basic ferrous Hb (Fe2+) spectra were measured using a Cary 60 UV-Vis spectrophotometer (Agilent Technologies) across 300–700 nm wavelength range, in steps of 2 nm. GOX was subsequently added to a final concentration of 40 mU/L and full spectra were recorded at every minute over the period of 5 h. In an equivalent parallel setting, bovine catalase was added to the reaction mixture after 12 min. During the entire measurement process, the temperature was maintained at 37°C using a PCB 1500 Water Peltier System (Varian). Reference spectra of ferrous oxy-Hb (Fe2+), ferric-Hb (Fe3+), and ferryl-Hb (Fe4+) were recorded at 37°C in PBS (pH 7.2). For each time point, the concentration of oxy-Hb, ferric-Hb, and ferryl-Hb in the reaction mixtures were deconvoluted out of the full recorded spectrum by applying Lawson–Hanson's Non Negative Least Squares algorithm of SciPy (www.scipy.org; see Supplementary Fig. S6 for details of the deconvolution procedure). The decay (k2) of ferryl (shown in Fig. 7A, B) was modeled according to the following reaction:
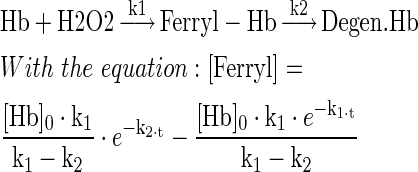 |
Hb-Hpx heme transfer
met-Hb (Fe3+) (10 μM in PBS [pH 7.4]) was incubated with a twofold molar excess of Hpx (Athens Research and Technology, Inc). Serial UV-VIS spectra were recorded using a Cary 60 UV-VIS Spectrophotometer (Agilent Technologies) in order to follow the transition of met-Hb to heme-Hpx over time. For each time-point, the concentrations of met-Hb and heme-Hpx in the reaction mixtures were deconvoluted out of the full spectrum by applying Lawson–Hanson's Non Negative Least Squares algorithm of SciPy (www.scipy.org). Rates of heme loss from met-Hb were calculated by fitting the following model to the data by nonlinear regression using R (www.r-project.org):
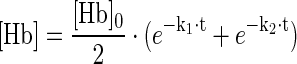 |
The two rates (fast and slow) may represent the differing stability of a-globin heme and b-globin heme, respectively (13).
LDL peroxidation
Human plasma-derived LDL was donated by Dr. L. Rohrer, Institute of Clinical Chemistry, University Hospital of Zurich. LDL (0.1 g/L) oxidation was measured in an airtight micro-respiration chamber (Unisense) in PBS (pH 7.45) at 37°C. The LDL solution was referenced as 100% of available oxygen. After a period of signal stabilization, Hb or Hb:Hp complexes (100% ferric) were injected at a final concentration of 5 μM. Oxygen consumption during Hb-driven LDL peroxidation was measured by an oxygen sensor (OX-MR; Unisense) connected to a picoampere-meter PA2000 and an OXY-Meter ADC216USB (Unisense). Data were recorded using the Unisense SensorTrace BASIC Software version 3.0.2. Before each experiment, the oxygen sensor was calibrated in an anoxic solution containing ascorbic acid and sodium hydroxide, both to a final concentration of 0.1 M.
Endothelial ECIS and viability measurement
Human umbilical vein endothelial cells (Lonza) were cultured in phenol red free EBM (Lonza, Clonetics) and seeded on collagen-coated ECIS Cultureware Disposable Electrode Arrays 8W10E+72 h before commencing the experiments. Electrical impedance was recorded in the multiple frequency mode in a ECIS ZTheta System (Applied BioPhysics) and analyzed with ECIS software (data given in the figures correspond to the 4000 Hz recordings). Relative endothelial ATP concentrations were measured with a luminescent cell viability assay (CellTiter-Glo® Luminescent Cell Viability Assay; Promega).
Mass spectrometry measurements of relative Hb binding capacity
Phenotype-specified Hb-saturated and HPLC purified Hb:Hp complexes were quantified by UV-VIS spectrophotometry and equal amounts of heme-equivalent complex were used for the isotope labeling procedure. Mass differential Tags for Relative and Absolute Quantification (mTRAQ)-4 and mTRAQ-0 label was applied to Hb:Hp1-1 and Hb:Hp2-2 complex, respectively. Next, 100 μg heme equivalent of each complex was digested with sequencing grade trypsin (Promega) and labeled with the respective tag according to the mTRAQ reagents protocol (Applied Biosystems). Samples were desalted with ZipTip (Millipore). Mass spectrometry analysis was performed on an LTQ-Orbitrap-Velos (Thermo) coupled to an Eksigent nanoHPLC. Peptides were identified by information-dependent acquisition of fragmentation spectra of multiple-charged peptides. Up to 20 data-dependent MS/MS spectra were acquired in the linear ion trap for each full scan spectrum acquired in the Orbitrap. Collision-induced dissociation with collision energy 35 was chosen for fragmentation. Proteins were identified and quantified using MaxQuant Software (www.maxquant.org) Version 1.3.0.5 and Hb and Hp peptide quantification was performed by integrating the MS1 ion signal intensity over time using XCalibur Software (Thermo).
Supplementary Material
Abbreviations Used
- AUC
area under the curve
- CBER
Center for Biologics Evaluation and Research
- Cl
plasma clearance
- Clast
last measurable plasma concentration
- EBM
endothelial growth medium
- ECIS
electric cell impedance substrate
- FDA
Food and Drug Administration
- GOX
glucose oxidase
- H2O2
hydrogen peroxide
- Hb
hemoglobin
- HO-1
heme-oxygenase 1
- Hp
haptoglobin
- HPLC
high-performance liquid chromatography
- Hpx
hemopexin
- HUVECs
human umbilical vein endothelial cells
- k
terminal slope (k) of the log-linear plasma concentration-time curve
- LC-MS/MS
liquid chromatography mass spectrometry
- LDL
low-density lipoprotein
- MAP
mean arterial blood pressure
- MRT
mean residence time
- NaCl
sodium chloride
- NO
nitric oxide
- oxLDL
oxidized LDL
- PBS
phosphate buffered saline
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SEC
size-exclusion chromatography
- SPR
surface plasmon resonance
- t1/2
terminal elimination half-life
- UV-VIS
UV-VIS spectrophotometry
- Vdss
apparent volume of distribution
Acknowledgments
This work was supported by the Swiss National Science Foundation (grants 310030/120658 and 31003A/138500), the University of Zurich Research Priority Program “Integrative Human Physiology,” the Swiss Federal Commission for Technology and Innovation (CTI), and FDA Internal Funding. The findings and conclusions in this article have not been formally disseminated by the FDA and should not be construed to represent any agency determination or policy.
Author Disclosure Statement
The authors disclose no conflict of interest. This study is not financially supported by or part of an industry-sponsored drug development project.
References
- 1.Andersen CB. Torvund-Jensen M. Nielsen MJ. de Oliveira CL. Hersleth HP. Andersen NH. Pedersen JS. Andersen GR. Moestrup SK. Structure of the haptoglobin-haemoglobin complex. Nature. 2012;489:456–459. doi: 10.1038/nature11369. [DOI] [PubMed] [Google Scholar]
- 2.Asleh R. Guetta J. Kalet-Litman S. Miller-Lotan R. Levy AP. Haptoglobin genotype- and diabetes-dependent differences in iron-mediated oxidative stress in vitro and in vivo. Circ Res. 2005;96:435–441. doi: 10.1161/01.RES.0000156653.05853.b9. [DOI] [PubMed] [Google Scholar]
- 3.Asleh R. Marsh S. Shilkrut M. Binah O. Guetta J. Lejbkowicz F. Enav B. Shehadeh N. Kanter Y. Lache O. Cohen O. Levy NS. Levy AP. Genetically determined heterogeneity in hemoglobin scavenging and susceptibility to diabetic cardiovascular disease. Circ Res. 2003;92:1193–1200. doi: 10.1161/01.RES.0000076889.23082.F1. [DOI] [PubMed] [Google Scholar]
- 4.Baek JH. D'Agnillo F. Vallelian F. Pereira CP. Williams MC. Jia Y. Schaer DJ. Buehler PW. Hemoglobin-driven pathophysiology is an in vivo consequence of the red blood cell storage lesion that can be attenuated in guinea pigs by haptoglobin therapy. J Clin Invest. 2012;122:1444–1458. doi: 10.1172/JCI59770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balla G. Jacob HS. Eaton JW. Belcher JD. Vercellotti GM. Hemin: a possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler Thromb. 1991;11:1700–1711. doi: 10.1161/01.atv.11.6.1700. [DOI] [PubMed] [Google Scholar]
- 6.Balla J. Jacob HS. Balla G. Nath K. Eaton JW. Vercellotti GM. Endothelial-cell heme uptake from heme proteins: induction of sensitization and desensitization to oxidant damage. Proc Natl Acad Sci U S A. 1993;90:9285–9289. doi: 10.1073/pnas.90.20.9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balla J. Vercellotti GM. Jeney V. Yachie A. Varga Z. Jacob HS. Eaton JW. Balla G. Heme, heme oxygenase, and ferritin: how the vascular endothelium survives (and dies) in an iron-rich environment. Antioxid Redox Signal. 2007;9:2119–2137. doi: 10.1089/ars.2007.1787. [DOI] [PubMed] [Google Scholar]
- 8.Banerjee S. Jia Y. Siburt CJ. Abraham B. Wood F. Bonaventura C. Henkens R. Crumbliss AL. Alayash AI. Haptoglobin alters oxygenation and oxidation of hemoglobin and decreases propagation of peroxide-induced oxidative reactions. Free Radic Biol Med. 2012;53:1317–1326. doi: 10.1016/j.freeradbiomed.2012.07.023. [DOI] [PubMed] [Google Scholar]
- 9.Boretti FS. Buehler PW. D'Agnillo F. Kluge K. Glaus T. Butt OI. Jia Y. Goede J. Pereira CP. Maggiorini M. Schoedon G. Alayash AI. Schaer DJ. Sequestration of extracellular hemoglobin within a haptoglobin complex decreases its hypertensive and oxidative effects in dogs and guinea pigs. J Clin Invest. 2009;119:2271–2280. doi: 10.1172/JCI39115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buehler PW. Abraham B. Vallelian F. Linnemayr C. Pereira CP. Cipollo JF. Jia Y. Mikolajczyk M. Boretti FS. Schoedon G. Alayash AI. Schaer DJ. Haptoglobin preserves the CD163 hemoglobin scavenger pathway by shielding hemoglobin from peroxidative modification. Blood. 2009;113:2578–2586. doi: 10.1182/blood-2008-08-174466. [DOI] [PubMed] [Google Scholar]
- 11.Buehler PW. D'Agnillo F. Schaer DJ. Hemoglobin-based oxygen carriers: from mechanisms of toxicity and clearance to rational drug design. Trends Mol Med. 2010;16:447–457. doi: 10.1016/j.molmed.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 12.Buehler PW. Vallelian F. Mikolajczyk MG. Schoedon G. Schweizer T. Alayash AI. Schaer DJ. Structural stabilization in tetrameric or polymeric hemoglobin determines its interaction with endogenous antioxidant scavenger pathways. Antioxid Redox Signal. 2008;10:1449–1462. doi: 10.1089/ars.2008.2028. [DOI] [PubMed] [Google Scholar]
- 13.Cahill LE. Levy AP. Chiuve SE. Jensen MK. Wang H. Shara NM. Blum S. Howard BV. Pai JK. Mukamal KJ. Rexrode KM. Rimm EB. Haptoglobin genotype is a consistent marker of coronary heart disease risk among individuals with elevated glycosylated hemoglobin. J Am Coll Cardiol. 2013;61:728–737. doi: 10.1016/j.jacc.2012.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cooper CE. Schaer DJ. Buehler PW. Wilson MT. Reeder BJ. Silkstone G. Svistunenko DA. Bulow L. Alayash AI. Haptoglobin binding stabilizes hemoglobin ferryl iron and the globin radical on tyrosine beta145. Antioxid Redox Signal. 2013;18:2264–2273. doi: 10.1089/ars.2012.4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dalton J. Podmore A. Enriched haptoglobin polymers for the treatment of disease. US Patent Application 20110021418. 2011 [Google Scholar]
- 16.Delanghe J. Allcock K. Langlois M. Claeys L. De Buyzere M. Fast determination of haptoglobin phenotype and calculation of hemoglobin binding capacity using high pressure gel permeation chromatography. Clin Chim Acta. 2000;291:43–51. doi: 10.1016/s0009-8981(99)00194-1. [DOI] [PubMed] [Google Scholar]
- 17.Etzerodt A. Kjolby M. Nielsen MJ. Maniecki M. Svendsen P. Moestrup SK. Plasma clearance of hemoglobin and haptoglobin in mice and effect of CD163 gene targeting disruption. Antioxid Redox Signal. 2013;18:2254–2263. doi: 10.1089/ars.2012.4605. [DOI] [PubMed] [Google Scholar]
- 18.Gattoni M. Boffi A. Sarti P. Chiancone E. Stability of the heme-globin linkage in alphabeta dimers and isolated chains of human hemoglobin. A study of the heme transfer reaction from the immobilized proteins to albumin. J Biol Chem. 1996;271:10130–10136. doi: 10.1074/jbc.271.17.10130. [DOI] [PubMed] [Google Scholar]
- 19.Gladwin MT. Kanias T. Kim-Shapiro DB. Hemolysis and cell-free hemoglobin drive an intrinsic mechanism for human disease. J Clin Invest. 2012;122:1205–1208. doi: 10.1172/JCI62972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kristiansen M. Graversen JH. Jacobsen C. Sonne O. Hoffman HJ. Law SK. Moestrup SK. Identification of the haemoglobin scavenger receptor. Nature. 2001;409:198–201. doi: 10.1038/35051594. [DOI] [PubMed] [Google Scholar]
- 21.Levy AP. Asleh R. Blum S. Levy NS. Miller-Lotan R. Kalet-Litman S. Anbinder Y. Lache O. Nakhoul FM. Asaf R. Farbstein D. Pollak M. Soloveichik YZ. Strauss M. Alshiek J. Livshits A. Schwartz A. Awad H. Jad K. Goldenstein H. Haptoglobin: basic and clinical aspects. Antioxid Redox Signal. 2010;12:293–304. doi: 10.1089/ars.2009.2793. [DOI] [PubMed] [Google Scholar]
- 22.Levy AP. Levy JE. Kalet-Litman S. Miller-Lotan R. Levy NS. Asaf R. Guetta J. Yang C. Purushothaman KR. Fuster V. Moreno PR. Haptoglobin genotype is a determinant of iron, lipid peroxidation, and macrophage accumulation in the atherosclerotic plaque. Arterioscler Thromb Vasc Biol. 2007;27:134–140. doi: 10.1161/01.ATV.0000251020.24399.a2. [DOI] [PubMed] [Google Scholar]
- 23.Levy AP. Purushothaman KR. Levy NS. Purushothaman M. Strauss M. Asleh R. Marsh S. Cohen O. Moestrup SK. Moller HJ. Zias EA. Benhayon D. Fuster V. Moreno PR. Downregulation of the hemoglobin scavenger receptor in individuals with diabetes and the Hp 2–2 genotype: implications for the response to intraplaque hemorrhage and plaque vulnerability. Circ Res. 2007;101:106–110. doi: 10.1161/CIRCRESAHA.107.149435. [DOI] [PubMed] [Google Scholar]
- 24.Miller YI. Altamentova SM. Shaklai N. Oxidation of low-density lipoprotein by hemoglobin stems from a heme-initiated globin radical: antioxidant role of haptoglobin. Biochemistry. 1997;36:12189–12198. doi: 10.1021/bi970258a. [DOI] [PubMed] [Google Scholar]
- 25.Miller YI. Smith A. Morgan WT. Shaklai N. Role of hemopexin in protection of low-density lipoprotein against hemoglobin-induced oxidation. Biochemistry. 1996;35:13112–13117. doi: 10.1021/bi960737u. [DOI] [PubMed] [Google Scholar]
- 26.Moreno PR. Purushothaman KR. Purushothaman M. Muntner P. Levy NS. Fuster V. Fallon JT. Lento PA. Winterstern A. Levy AP. Haptoglobin genotype is a major determinant of the amount of iron in the human atherosclerotic plaque. J Am Coll Cardiol. 2008;52:1049–1051. doi: 10.1016/j.jacc.2008.06.029. [DOI] [PubMed] [Google Scholar]
- 27.Nagy E. Eaton JW. Jeney V. Soares MP. Varga Z. Galajda Z. Szentmiklosi J. Mehes G. Csonka T. Smith A. Vercellotti GM. Balla G. Balla J. Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler Thromb Vasc Biol. 2010;30:1347–1353. doi: 10.1161/ATVBAHA.110.206433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakai K. Sakuma I. Ohta T. Ando J. Kitabatake A. Nakazato Y. Takahashi TA. Permeability characteristics of hemoglobin derivatives across cultured endothelial cell monolayers. J Lab Clin Med. 1998;132:313–319. doi: 10.1016/s0022-2143(98)90045-2. [DOI] [PubMed] [Google Scholar]
- 29.Noyes WD. Garby L. Rate of haptoglobin in synthesis in normal man. Determinations by the return to normal levels following hemoglobin infusion. Scand J Clin Lab Invest. 1967;20:33–38. [PubMed] [Google Scholar]
- 30.Osada J. Elimination from rat circulation of goat and sheep haptoglobin and their complexes with rat haemoglobin. Acta Biochim Pol. 1988;35:169–175. [PubMed] [Google Scholar]
- 31.Osada J. Nowacki W. Elimination of goat haemoglobin and its complexes with goat haptoglobin from goat and rat circulation. Acta Biochim Pol. 1989;36:365–369. [PubMed] [Google Scholar]
- 32.Pimenova T. Pereira CP. Gehrig P. Buehler PW. Schaer DJ. Zenobi R. Quantitative mass spectrometry defines an oxidative hotspot in hemoglobin that is specifically protected by haptoglobin. J Proteome Res. 2010;9:4061–4070. doi: 10.1021/pr100252e. [DOI] [PubMed] [Google Scholar]
- 33.Purushothaman KR. Purushothaman M. Levy AP. Lento PA. Evrard S. Kovacic JC. Briley-Saebo KC. Tsimikas S. Witztum JL. Krishnan P. Kini A. Fayad ZA. Fuster V. Sharma SK. Moreno PR. Increased expression of oxidation-specific epitopes and apoptosis are associated with haptoglobin genotype: possible implications for plaque progression in human atherosclerosis. J Am Coll Cardiol. 2012;60:112–119. doi: 10.1016/j.jacc.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 34.Ratanasopa K. Chakane S. Ilyas M. Nantasenamat C. Bulow L. Trapping of human hemoglobin by haptoglobin: molecular mechanisms and clinical applications. Antioxid Redox Signal. 2013;18:2364–2374. doi: 10.1089/ars.2012.4878. [DOI] [PubMed] [Google Scholar]
- 35.Reiter CD. Wang X. Tanus-Santos JE. Hogg N. Cannon RO., 3rd Schechter AN. Gladwin MT. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8:1383–1389. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- 36.Schaer CA. Schoedon G. Imhof A. Kurrer MO. Schaer DJ. Constitutive endocytosis of CD163 mediates hemoglobin-heme uptake and determines the noninflammatory and protective transcriptional response of macrophages to hemoglobin. Circ Res. 2006;99:943–950. doi: 10.1161/01.RES.0000247067.34173.1b. [DOI] [PubMed] [Google Scholar]
- 37.Schaer CA. Vallelian F. Imhof A. Schoedon G. Schaer DJ. CD163-expressing monocytes constitute an endotoxin-sensitive Hb clearance compartment within the vascular system. J Leukoc Biol. 2007;82:106–110. doi: 10.1189/jlb.0706453. [DOI] [PubMed] [Google Scholar]
- 38.Schaer DJ. Buehler PW. Alayash AI. Belcher JD. Vercellotti GM. Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood. 2013;121:1276–1284. doi: 10.1182/blood-2012-11-451229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schaer DJ. Schaer CA. Buehler PW. Boykins RA. Schoedon G. Alayash AI. Schaffner A. CD163 is the macrophage scavenger receptor for native and chemically modified hemoglobins in the absence of haptoglobin. Blood. 2006;107:373–380. doi: 10.1182/blood-2005-03-1014. [DOI] [PubMed] [Google Scholar]
- 40.Shen H. Song Y. Colangelo CM. Wu T. Bruce C. Scabia G. Galan A. Maffei M. Goldstein DR. Haptoglobin activates innate immunity to enhance acute transplant rejection in mice. J Clin Invest. 2012;122:383–387. doi: 10.1172/JCI58344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vallelian F. Pimenova T. Pereira CP. Abraham B. Mikolajczyk MG. Schoedon G. Zenobi R. Alayash AI. Buehler PW. Schaer DJ. The reaction of hydrogen peroxide with hemoglobin induces extensive alpha-globin crosslinking and impairs the interaction of hemoglobin with endogenous scavenger pathways. Free Radic Biol Med. 2008;45:1150–1158. doi: 10.1016/j.freeradbiomed.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 42.Wicher KB. Fries E. Evolutionary aspects of hemoglobin scavengers. Antioxid Redox Signal. 2010;12:249–259. doi: 10.1089/ars.2009.2760. [DOI] [PubMed] [Google Scholar]
- 43.Wuyts B. Delanghe JR. Kasvosve I. Langlois MR. De Buyzere ML. Janssens J. A new method for fast haptoglobin phenotyping and hemoglobin binding capacity calculation based on capillary zone electrophoresis. Clin Chem Lab Med. 2000;38:715–720. doi: 10.1515/CCLM.2000.101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.