

ABSTRACT
The purpose of this study was to evaluate the composition and richness of bacterial communities associated with low-birthweight (LBW) infants in relation to host body site, individual, and age. Bacterial 16S rRNA genes from saliva samples, skin swabs, and stool samples collected on postnatal days 8, 10, 12, 15, 18, and 21 from six LBW (five premature) infants were amplified, pyrosequenced, and analyzed within a comparative framework that included analogous data from normal-birthweight (NBW) infants and healthy adults. We found that body site was the primary determinant of bacterial community composition in the LBW infants. However, site specificity depended on postnatal age: saliva and stool compositions diverged over time but were not significantly different until the babies were 15 days old. This divergence was primarily driven by progressive temporal turnover in the distal gut, which proceeded at a rate similar to that of age-matched NBW infants. Neonatal skin was the most adult-like in microbiota composition, while saliva and stool remained the least so. Compositional variation among infants was marked and depended on body site and age. Only the smallest, most premature infant received antibiotics during the study period; this heralded a coexpansion of Pseudomonas aeruginosa and a novel Mycoplasma sp. in the oral cavity of this vaginally delivered, intubated patient. We conclude that concurrent molecular surveillance of multiple body sites in LBW neonates reveals a delayed compositional differentiation of the oral cavity and distal gut microbiota and, in the case of one infant, an abundant, uncultivated oral Mycoplasma sp., recently detected in human vaginal samples.
IMPORTANCE
Complications of premature birth are the most common cause of neonatal mortality. Colonization by the indigenous microbiota, which begins at delivery, may predispose some high-risk newborns to invasive infection or necrotizing enterocolitis (NEC), and protect others, yet neonatal microbiome dynamics are poorly understood. Here, we present the first cultivation-independent time series tracking microbiota assembly across multiple body sites in a synchronous cohort of hospitalized low-birthweight (LBW) neonates. We take advantage of archived samples and publically available sequence data and compare our LBW infant findings to those from normal-birthweight (NBW) infants and healthy adults. Our results suggest potential windows of opportunity for the dispersal of microbes within and between hosts and support recent findings of substantial baseline spatiotemporal variation in microbiota composition among high-risk newborns.
INTRODUCTION
The composition of the human microbiota is body site specific in healthy adults (1–3), yet this is not the case in newborns shortly after delivery (4). While the postnatal assembly of an adult-like distal gut microbiota has been studied in healthy infants (5–7), relatively little is known about the development of the microbiota at extraintestinal sites (8, 9) or about the compositional differentiation of the microbiota across multiple sites during the neonatal period. Knowledge of these spatiotemporal dynamics is particularly lacking for low-birthweight (LBW) infants, who are at high risk of invasive infection and other serious perinatal complications, including necrotizing enterocolitis (NEC), a disease linked in part to microbial colonization (10, 11). LBW infants are often premature, and often receive antibiotics, experience delays in the initiation of enteral feedings, and/or require prolonged hospital stays—all of which can influence, and be influenced by, interactions with microbes. Certain complications, such as sepsis and NEC, are characterized by onset timing (12, 13); for example, the postnatal age at the onset of NEC is inversely correlated with the gestational age at delivery (14). These patterns underscore a need to understand better the temporal dynamics of microbiome development in high-risk neonates.
Postnatal microbial colonization prompts the terminal maturation of host intestinal structures, mediates the development of the immune system, and induces resistance to invasion by would-be pathogens (15–17). Furthermore, early life colonization deficiencies have been associated with alterations in host metabolism and immune function (18, 19). In the neonatal intensive care unit (NICU), however, the promotion of potentially beneficial host-microbe interactions must be carefully balanced against the control of pathogen spread among a highly vulnerable patient population (20, 21). This is distinctively challenging with regard to the prevention and treatment of NEC, a disease in which the interrelated roles of antibiotic exposure, enteral feedings, and changes in the intestinal microbiota are imprecisely defined (10, 11). Recent studies of the fecal microbiota of premature infants using cultivation-independent approaches have revealed a low level of diversity, high interindividual variability, and a capacity for abrupt temporal shifts in species- and strain-level composition (22–32). However, most of these studies have been limited to a relatively small number of samples and to a single body site, the distal gut.
In the present study, we simultaneously tracked the distal gut, oral cavity, and skin surface microbiota of six hospitalized LBW infants, including 2 sets of twins, over the 2nd and 3rd weeks of life. Our analysis focused on factors underpinning compositional variation during this critical time span. For the distal gut microbiota, we also made comparisons to age-matched normal-birthweight (NBW) infants using archived samples from a prior study (5); and for all sites, we made comparisons to adults using publically available sequence data (1, 2). Although the infants sampled here were unaffected by sepsis or NEC, their age range represents an important window of vulnerability for both of these conditions.
RESULTS
LBW infant cohort characteristics.
Five of the six infants (all but baby 6) were premature; these five had completed <32 weeks of gestation at the time of delivery. Among the premature infants, three were born weighing <1.5 kg, placing them in the category of “very LBW” (VLBW) and at highest risk for complications of preterm birth. These three infants were born at Comer Children’s Hospital, whereas the others were born at outside hospitals and then transferred to Comer’s NICU prior to enrollment. The cohort included two sets of premature twins, both delivered via Cesarean section. All infants received antibiotics in the first week of life (Table 1). None of their mothers received antepartum antibiotics.
TABLE 1 .
LBW infant characteristics and clinical information
| Babya | Sexb | Delivery modec | Birth wt (kg) | Gestational age at delivery | Birth locatione | Postdelivery antibiotics and length of treatmentf | Medical conditionsg | Complication(s) during pregnancy and/or deliveryh |
|---|---|---|---|---|---|---|---|---|
| 1 | F | Cs | 1.82 | 31d | Not at a UC hospital | Ap + Gm 48 hi | Premature, respiratory distress, hyperbilirubinemia | Loss of fetal heart tones |
| 2 | M | Cs | 1.74 | 31 | Not at a UC hospital | Ap + Gm 48 hi | Premature, respiratory distress, hyperbilirubinemia | Loss of fetal heart tones |
| 3 | M | V | 0.75 | 24 | UC hospital | Ap + Gm 7 days | Premature, respiratory distress, hyperbilirubinemia | PPROM |
| 4 | M | Cs | 1.38 | 30 | UC hospital | Ap + Gm 48 h | Premature, hyperbilirubinemia, GE reflux, AOP | Preeclampsia, preterm labor |
| 5 | M | Cs | 1.05 | 30 | UC hospital | Ap + Gm 48 h | Premature, respiratory distress, hyperbilirubinemia, GE reflux, AOP | Preeclampsia, preterm labor |
| 6 | M | V | 1.72 | 38 | Not at a UC hospital | Ap + Gm +Cx 48-72 hj | IUGR, chromosome 4p deletion syndromek | None noted |
Babies 1 and 2 are dizygotic (DZ) twins; babies 4 and 5 are monozygotic (MZ) twins (monochorionic diamniotic).
F, female; M, male.
Cs, Cesarean section; V, vaginal.
31, 31 weeks and 1 day.
UC, University of Chicago.
Ap, ampicillin; Cx, cefotaxime; Gm, gentamicin.
GE, gastroesophageal; AOP, anemia of prematurity; IUGR, intrauterine growth restriction.
PPROM, preterm premature rupture of membranes.
A third antibiotic may have been given (non-UC chart unclear/unavailable).
The most likely treatment duration (non-UC chart unclear/unavailable).
Including various syndrome-associated medical problems.
Baby 3, the smallest, most premature infant in the study, was intubated and mechanically ventilated throughout the sampling period, whereas the others either had no history of endotracheal intubation (babies 4 and 5) or had been extubated by the time sampling commenced (babies 1, 2, and 6). Baby 3 was also treated with antibiotics on days 13 to 19 for a suspected case of sepsis (Table 2), but all cultures (blood, urine, and cerebrospinal fluid [CSF] samples) were negative; no respiratory tract samples were cultured. Baby 3 was the only subject to receive antibiotics during the sampling period. Finally, in some cases, modifications to the infants’ feeding regimens and/or hospital locations were made during the sampling period (Table 2). Most feedings were delivered via nasogastric or orogastric tube.
TABLE 2 .
LBW infant age-related events and informationa
| Baby | Feature | Postnatal age in daysb |
|||||
|---|---|---|---|---|---|---|---|
| 8 | 10 | 12 | 15 | 18 | 21 | ||
| 1 | Feeding | BMtr | BMfull | BMfull | BMtr, Ftr | BMfull | BMfull |
| Wt (kg) | 1.72 | 1.79 | 1.725 | 1.8 | 1.91 | 1.974 | |
| Antibiotic(s) | None noted | None noted | None noted | None noted | None noted | None noted | |
| Location | NICU | NICU | NICU step-down | NICU step-down | NICU step-down | NICU step-down | |
| 2 | Feeding | BMtr | BMtr | BMtr | BMtr, Ftr | BMtr | Ffull |
| Wt (kg) | 1.71 | 1.805 | 1.785 | 1.86 | 1.86 | 1.96 | |
| Antibiotic(s) | None noted | None noted | None noted | None noted | None noted | None noted | |
| Location | NICU | NICU | NICU step-down | NICU step-down | NICU step-down | NICU step-down | |
| 3 | Feeding | BMtr | BMtr, Ftr | BMtr, Ftr | Ftr | Ftr | Ffull |
| Wt (kg) | 0.84 | 0.86 | 0.92 | 0.94 | 0.9 | 0.97 | |
| Antibiotic(s) | None noted | None noted | None noted | Vm + Gm + Cxc | Gm + Cxc | None noted | |
| Location | NICU | NICU | NICU | NICU | NICU | NICU | |
| 4 | Feeding | Ftr | BMtr | BMtr | Ffull | Ffull | Ffull |
| Wt (kg) | 1.3 | 1.35 | 1.53 | 1.62 | 1.6 | 1.67 | |
| Antibiotic(s) | None noted | None noted | None noted | None noted | None noted | None noted | |
| Location | NICU | NICU | NICU | NICU | NICU | NICU | |
| 5 | Feeding | Ftr | BMtr | BMtr | Ffull | Ffull | Ffull |
| Wt (kg) | 1.05 | 1.115 | 1.205 | 1.245 | 1.234 | 1.26 | |
| Antibiotic(s) | None noted | None noted | None noted | None noted | None noted | None noted | |
| Location | NICU | NICU | NICU | NICU | NICU | NICU | |
| 6 | Feeding | Ffull | Ffull | Ffull | Ffull | Ffull | Ffull |
| Wt (kg) | 1.725 | 1.65 | 1.675 | 1.735 | 1.77 | 1.765 | |
| Antibiotic(s) | None noted | None noted | None noted | None noted | None noted | None noted | |
| Location | NICU | NICU | NICU | Transitional floor | Transitional floor | Transitional floor | |
Respiratory support, baby 3 was intubated from day of life (DOL) ~1 to 44. Babies 1, 2, and 6 were intubated DOL 1 and 2, 1 and 2, and 4 to 6, respectively. Babies 4 and 5 were not intubated. Most babies received oxygen via nasal cannulae throughout the study. Feeding support, most feedings were delivered via a naso- or orogastric tube.
BM, pumped or stored maternal breast milk; F, formula; tr, trophic (i.e., minimal); Vm, vancomycin; Gm, gentamicin; Cx, cefotaxime.
Sepsis ruleout. Vm from DOL 13 to 15, Gm from DOL 13 to 19, and Cx from DOL 14 to 19.
Baby 3 received antibiotics for a suspected case of NEC around day of life (DOL) 40, but his clinical signs resolved quickly without further intervention. To our knowledge, none of the other infants went on to have invasive infections or NEC after DOL 21.
Overview of bacterial taxonomic representation.
Of the 108 samples collected for the study, 106 yielded sufficient quantities of 16S rRNA gene sequences to warrant subsequent analysis (range, 219 to 1,914 sequences/sample; median, 1,066 sequences/sample). Due to low sequencing yield, two samples were dropped.
Overall, nine bacterial phyla were represented (Fig. 1). On average, the most abundant were the Firmicutes (71.6%), Proteobacteria (21.4%), Bacteroidetes (5.4%), Tenericutes (1.0%), and Actinobacteria (0.5%). Rare phyla (those with average abundances of ≤0.01%) included the Cyanobacteria, Deinococcus-Thermus, Chloroflexi, and Fusobacteria. In total, 119 bacterial genera were detected, the most abundant of which are displayed in Fig. 1. Dominant genera were as follows: from the phylum Firmicutes, Staphylococcus, Streptococcus, Enterococcus, and Gemella; from the class Gammaproteobacteria, Klebsiella/Enterobacter (genera indistinguishable using the available gene fragment), Haemophilus, Citrobacter, Proteus, and Pseudomonas; and from the phylum Bacteroidetes, the genus Bacteroides.
FIG 1 .

Stacked bar plots depicting the relative abundances of the 30 most abundant genus-level taxa in the LBW infants. Taxa were ranked according to their mean abundance across all samples (percentages at right). Ten taxa had mean abundances of >1.00% (percentages in bold type within parentheses). Cs, Cesarean section delivery; V, vaginal delivery.
The identities of the abundant taxa found here are generally consistent with those observed in prior studies of LBW infants (22, 24–26, 28, 33), including studies of premature infants recruited from the same NICU as that which served as the setting for the current study (23, 27, 29).
Microbiota composition is primarily shaped by body site.
Patterns of bacterial community-wide compositional variation were evaluated using the unweighted UniFrac metric. Pairs of samples containing similar (i.e., closely related) lineages have relatively small UniFrac distances, whereas those containing divergent (i.e., distantly related) lineages have relatively large ones (34). The unweighted UniFrac metric is incidence based (i.e., presence/absence based); thus, branch lengths associated with high- and low-abundance taxa count equally.
Exploratory analysis using UniFrac-based principal coordinate analysis (PCoA) revealed that, as in healthy adults (1, 2), body site—i.e., whether the community was from a saliva, skin, or stool sample—was the primary determinant of bacterial community composition in the LBW infants (Fig. 2). Indeed, microbiota composition differed significantly across the three sites (permutational multivariate analysis of variance [PERMANOVA] main test, P < 0.001). This factor (“body site”) remained significant when hierarchically nested within “individuals” (i.e., when examining within-infant distances only; PERMANOVA main test, P < 0.001); however, in pairwise a posteriori tests, baby 4’s saliva and stool communities were undifferentiated overall (P = 0.323).
FIG 2 .
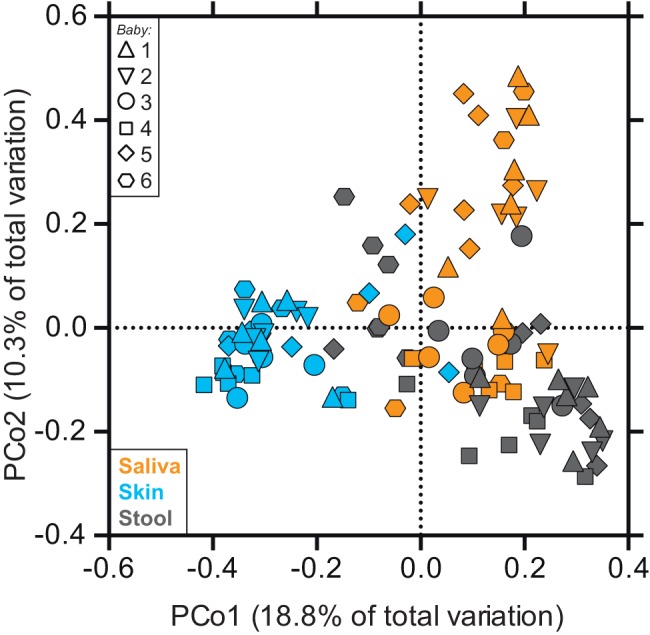
Unweighted UniFrac-based principal coordinate analysis (PCoA) of LBW infant-associated bacterial communities. Each symbol represents the value for a sample, with the shape of the symbol indicating the infant (infants 1 to 6) and the color indicating the body site. The percentages of variation explained by the plotted principal coordinates (PCo1 and PCo2) are indicated on the axes.
The relative abundance of seven genera differed significantly across the three body sites (ANOVA adjusted for Bonferroni’s correction, P < 0.001). Among those with an average abundance of >1.0%, Klebsiella/Enterobacter (genera indistinguishable using the available gene fragment), Enterococcus, and Citrobacter were particularly abundant in stool, as was Staphylococcus (largely Staphylococcus epidermidis) on skin, and Streptococcus in saliva (Fig. 1). Controlling for sequencing effort, the number of operational taxonomic units (OTUs) on skin was significantly higher than the number in saliva or stool (see Fig. S2 in the supplemental material).
Notably, Staphylococcus and Streptococcus, which are characteristically found on skin and in saliva, respectively, were surprisingly abundant at other sites in the LBW infants (Fig. 1), and the level of body site-driven compositional differentiation in the LBW infants (as shown in Fig. 2) seemed lower than that reported for healthy adults (1–3). Indeed, when we compared these groups directly (see Fig. S1 in the supplemental material), we found that the effect of “body site” was smaller in LBW infants (PERMANOVA η2 = 0.21) than in healthy adults (η2 = 0.34). This direct comparison also revealed that, among the three sites examined, LBW infant skin was the most adult-like in terms of microbiota composition (Fig. 3).
FIG 3 .
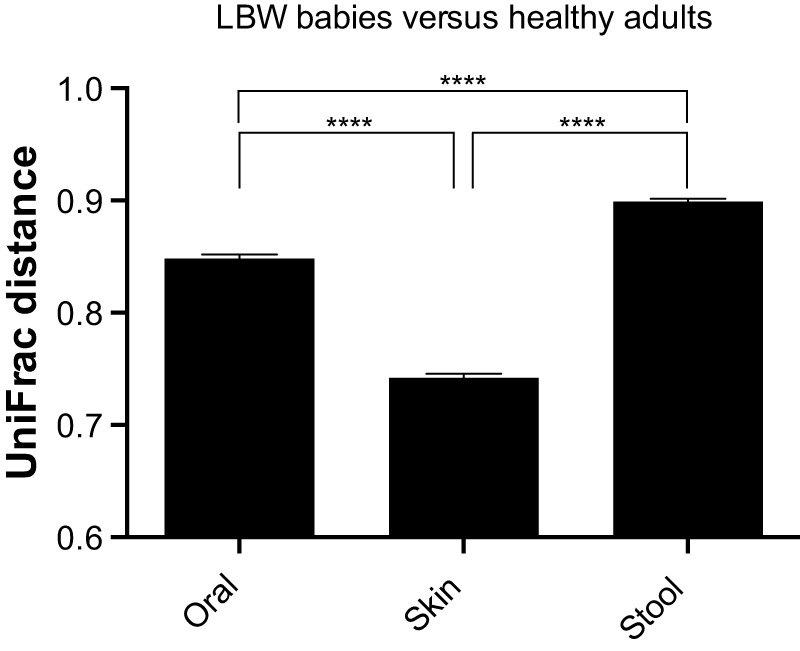
Average unweighted UniFrac distances between LBW infants (present study) and healthy adults (references 1, 2, and 5; see Materials and Methods) for oral, skin surface, and stool microbiota (250 sequences per sample). Values that are significantly different by Tukey’s posthoc tests are indicated by bars and 4 asterisks (P < 0.0001). Error bars represent 95% confidence intervals.
Neonatal personalization of microbiota composition.
Compositional variation existed among the LBW infants (PERMANOVA main test, P < 0.001), but the effect of “individual” (PERMANOVA η2 = 0.13) was smaller than the effect of “body site” (η2 = 0.21) (Fig. 2). It was also not the case that every baby harbored a highly personalized microbiota: in pairwise a posteriori tests, the microbiota of babies 1 and 2 (the dizygotic [DZ] twins) were compositionally similar to each other and to the microbiota of baby 5 (P values of >0.05). By day 21, the genus-level profiles for the fecal bacterial communities of co-twins were remarkably similar (Fig. 1c); as follows, overall interindividual variability for the distal gut decreased modestly as the cohort grew older (see Fig. S3 in the supplemental material). Throughout their hospitalization, co-twins were generally colocated; however, specific aspects of their care may have varied. For example, on DOL 21, babies 1 and 2 (the DZ twins) received different diets (Table 2).
The relative abundance of three genera differed significantly among the six infants (ANOVA adjusted for Bonferroni’s correction, P < 0.001). Bacteroides (B. caccae) was particularly abundant in baby 6’s stool samples (at all ages), as was Proteus (P. mirabilis) in baby 3’s saliva and stool samples (early ages), and Haemophilus (H. parainfluenzae) in baby 4’s saliva and stool samples (early ages; also present at low abundance in monozygotic [MZ] co-twin) (Fig. 1). Baby 6 was the only term infant in the study; he was also delivered vaginally. Numerous studies link vaginal delivery to early colonization by Bacteroides (35, 36).
A high degree of interindividual variation in fecal microbiota composition has been observed in preterm (23, 24, 28) and term (5, 7) infants. Our data suggest that this pattern extends to the neonatal skin and oral microbiota. The ultimate cause of interindividual variation may be difficult to ascertain—e.g., despite receiving remarkably similar medical treatment (Tables 1 and 2), clear differences existed between the microbiomes of the MZ co-twins (Fig. 1).
Delayed compositional divergence of gut and oral communities.
As a categorical predictor, infant age was not associated with differences in bacterial community composition (PERMANOVA main test, P = 0.935), and the relative abundance of only one genus (Staphylococcus) changed consistently as the cohort grew older (modest decline in stool and saliva; linear correlation, r = −0.485 and −0.387, respectively; adjusted for Bonferroni’s correction, P = 0.011 and 0.064, respectively). Thus, microbiota composition was more stable over time (here, DOL 8, 10, 12, 15, 18, and 21) than across body sites and host individuals.
Next, we addressed whether the degree of body site-associated compositional differentiation depended on the age of the infant. We found that at all ages, microbiota composition on skin was significantly different from that in saliva and stool; however, we also found that the microbiota compositions of saliva and stool were not significantly different from each other until the babies were at least 15 days old (Table 3). Indeed, saliva and stool compositions grew progressively more distinct as the infants grew older (Table 3). Furthermore, on average within infants, the compositional difference between saliva and stool samples increased significantly with infant age (linear regression, R2 = 0.7075, P = 0.0359). We asked whether this divergence was driven by compositional turnover in the distal gut, oral cavity, or both. Pairwise a posteriori tests mainly implicated the distal gut, where the amount of variation explained by time was positively correlated with the size of the time step (see Table S1 in the supplemental material)—a pattern that was not as apparent in saliva or on skin (Table S2). These results were well supported by correlation tests, which further emphasize that the temporal pattern of neonatal microbiome assembly depends on the observed body site (Table 4).
TABLE 3 .
Effect of body site on the composition of LBW infant-associated bacterial communities over timea
| Test | Age | Body sites (age)b | F/t | P valuec | No. of permutations |
|---|---|---|---|---|---|
| Main test | All | 2.9245 | 0.001 | 996 | |
| Pairwise tests | DOL 8 | Saliva, skin | 1.9426 | 0.002 | 402 |
| Saliva, stool | 0.90264 | 0.696 | 407 | ||
| Skin, stool | 2.0107 | 0.005 | 405 | ||
| DOL 10 | Saliva, skin | 2.1699 | 0.001 | 402 | |
| Saliva, stool | 0.96292 | 0.544 | 401 | ||
| Skin, stool | 2.0022 | 0.001 | 407 | ||
| DOL 12 | Saliva, skin | 1.4472 | 0.013 | 414 | |
| Saliva, stool | 1.0734 | 0.314 | 405 | ||
| Skin, stool | 1.9793 | 0.004 | 407 | ||
| DOL 15 | Saliva, skin | 1.8748 | 0.001 | 409 | |
| Saliva, stool | 1.416 | 0.025 | 411 | ||
| Skin, stool | 1.9982 | 0.004 | 416 | ||
| DOL 18 | Saliva, skin | 1.5016 | 0.014 | 399 | |
| Saliva, stool | 1.7443 | 0.010 | 416 | ||
| Skin, stool | 2.1177 | 0.006 | 411 | ||
| DOL 21 | Saliva, skin | 1.5612 | 0.004 | 395 | |
| Saliva, stool | 1.6077 | 0.006 | 407 | ||
| Skin, stool | 2.2291 | 0.001 | 402 |
Results of main and pairwise a posteriori using unweighted UniFrac-based permutational multivariate ANOVA and the t statistic. DOL, day of life.
The model is “body site” nested within levels of “age.”
The P values in bold type highlight the divergence of saliva and stool sample values over time.
TABLE 4 .
Correlation between the compositional dissimilarity of LBW infant-associated bacterial communities and elapsed timea
| Parameter | Body site |
||
|---|---|---|---|
| Saliva | Skin | Stool | |
| No. of pairs | 85 | 85 | 90 |
| Spearman’s r | 0.2919 | 0.0351 | 0.4225 |
| 95% confidence interval | 0.0776 to 0.4805 | −0.1856 to 0.2524 | 0.2301 to 0.5831 |
| P value (one-tailed) | 0.0034 | 0.7500 | <0.0001 |
Spearman’s rank order correlation measuring the dependence of community distance (unweighted UniFrac metric) on temporal distance (number of days) within subjects for each body site.
Stool microbiota development in LBW and NBW infants.
We compared stool microbiota dynamics in LBW and age-matched (i.e., time series spanning 8 to 21 days in age) NBW infants. To do this, we pyrosequenced bacterial 16S rRNA genes amplified from archived fecal DNA samples from NBW infants who were enrolled in a prior study (5) (see Materials and Methods). In both cohorts, compositional variation depended positively on elapsed time (Fig. 4a, P values of <0.05). We also found that there was no significant difference between the cohorts with respect to the rate of compositional turnover (Fig. 4a, P = 0.7911). On average, the stool microbiotas of LBW infants were slightly enriched in the observed number of OTUs (controlling for sequencing effort; P = 0.01) and significantly enriched in OTUs assigned to Enterobacter, the Enterobacteriaceae, Enterococcus, and Staphylococcus (adjusted for Bonferroni’s correction, P values of <0.001). Escherichia was abundant in the NBW infants and virtually absent from the LBW infants (P < 0.001). Despite these differences, over time, the community-wide composition of LBW infant stool grew more similar to that of 21-day-old NBW infant stool (i.e., to that of a healthy reference group; Fig. 4b). These results suggest that while gestational age at delivery, delivery mode, or other factors may affect gut microbiota makeup, its rate of development may depend more on intrinsic community-level factors, e.g., the amount of time the site has been available to colonists, microbe-microbe interactions, microbe-host interactions (that are independent of host gestational age), or increasing hypoxia/anaerobiosis.
FIG 4 .
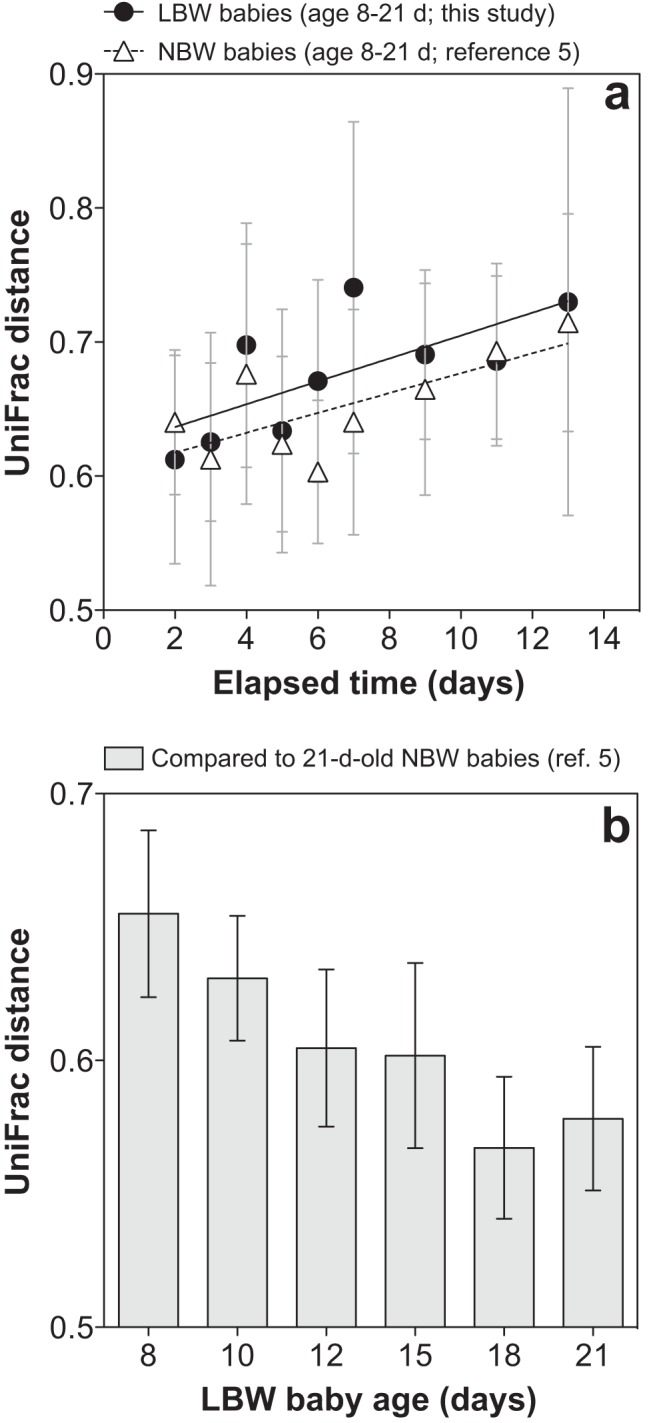
Relationship between neonatal stool microbiota composition and time. (a) Average (95% CI) within-subject, unweighted UniFrac distance plotted against the age difference (lag) in days for LBW (R2 = 0.5) and NBW (R2 = 0.5) infants. Lines indicate best fit linear regressions. The NBW infants were not sampled on day 18; thus, for this analysis, the corresponding age was excluded from the LBW data set. (b) For LBW infants at various ages, average (95% CI) unweighted UniFrac distance compared to a healthy reference cohort (the 21-day-old NBW infants).
Dynamics of particular taxa in LBW infants, including an uncultivated Mycoplasma.
Several noteworthy taxa were briefly abundant in LBW infant stool samples (Fig. 1c). On day 18, Clostridium perfringens represented ~40% of sequences from baby 2, but it was below the detection level on all other days. On day 15, Dysgonomonas capnocytophagoides comprised ~8% of sequences from baby 3; this fastidious organism (and opportunistic pathogen) has not, to our knowledge, been reported in pediatric clinical samples. Finally, on day 15, a Peptoniphilus sp. represented ~7% of sequences from baby 4, having been detected previously in his day 12 skin swab (1%; Fig. 1b)—a possible bellwether for the taxon’s appearance in the distal gut.
However, the most striking example emerged from the oral data set and involved taxa from baby 3’s saliva samples: specifically, the genera Mycoplasma (several species) and Pseudomonas (P. aeruginosa), which became dominant on days 15, 18, and 21 (Fig. 1a). Indeed, the sequences comprising one, highly abundant Mycoplasma-related OTU appeared to be phylogenetically novel. This finding prompted an in-depth analysis of these and related sequences belonging to the phylum Tenericutes.
Among the OTUs detected in the LBW infants, three were assigned to the phylum Tenericutes; together, they contained 788 sequences. Representatives of the first and second OTUs were >99% identical to Mycoplasma hominis and Ureaplasma parvum, respectively. However, the representative of the third OTU, which contained 771 sequences, was only 88% identical to the closest named species in GenBank (e.g., Mycoplasma iowae, Mycoplasma microti, and Mycoplasma muris). This novel OTU was virtually exclusive to baby 3, the only extremely LBW (ELBW) infant in the study (ELBW is defined as <1.0 kg). Its expansion in baby 3’s oral cavity, which peaked on DOL 18 at 47.2% of sequences, coincided with antibiotic treatment for suspected (but ultimately unconfirmed) sepsis (Fig. 5 and Table 2).
FIG 5 .
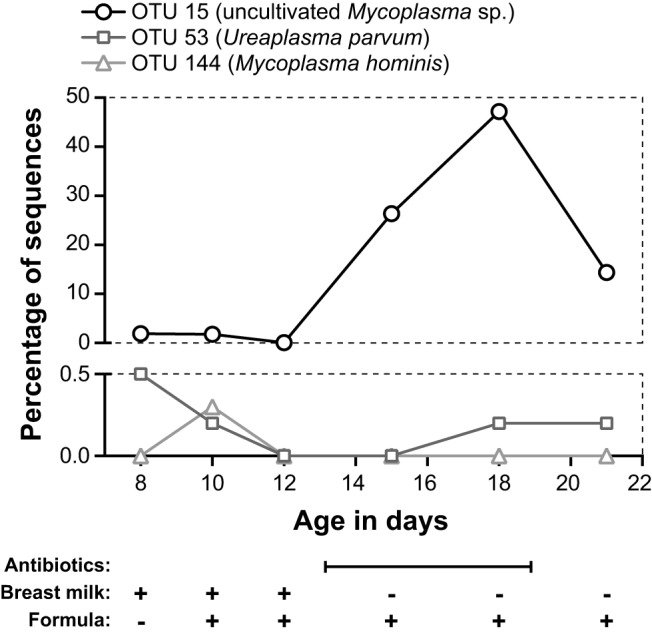
Relative abundances of three OTUs belonging to the Mycoplasmataceae from oral samples from extremely LBW baby 3. OTU 15, a novel, uncultivated Mycoplasma sp., is plotted against the upper y axis. OTUs 53 and 144, which are closely related to OTUs from cultivated strains, are plotted against the lower y axis. Expansion of OTU 15 coincided with antibiotic treatment from DOL 13 to 19 (see Table 2 for details). Feedings were delivered via naso- or orogastric tube. For antibiotics, the date range is indicated. The baby’s diet (breast milk or formula) is given for each sample date.
Phylogenetic analysis suggests that the novel OTU belongs to a single, well-supported clade comprising uncultivated lineages from cow rumen, which are among its closest relatives at 94.3 to 94.8% sequence identity, and termite gut (see Fig. S4 in the supplemental material). Interestingly, a recently deposited GenBank sequence (uncultured Mycoplasma sp. clone Mnola; accession no. JX508800) is 99% identical to our infant-derived OTU (Fig. S4); this clone was isolated from a vaginal swab from a Trichomonas vaginalis-infected patient (37). Finally, we amplified and cloned near-full-length 16S rRNA gene sequences from baby 3’s DOL 18 saliva (see Materials and Methods). This yielded sequences belonging to the novel OTU that confirmed the phylogenetic placement of the shorter pyrosequences (Fig. S4). To our knowledge, this is the first report of infant-derived (and second report of human-derived) sequences from this as-yet-uncultivated Mycoplasma-related clade.
DISCUSSION
In a small cohort of 8- to 21-day-old LBW infants, we found that microbiota composition was shaped primarily by body site and host individual; this is consistent with patterns observed in healthy adults (1–3). Minutes after delivery, the composition of the newborn microbiota is undifferentiated across body sites (4). Our results suggest that site-specific bacterial communities emerge relatively early—indeed, within the neonatal period—despite an overall dearth of microbes characteristic of healthy adults (see Fig. S1 in the supplemental material). To our knowledge, this is the first study to assess microbiota differentiation across multiple body sites in neonates; at the present time, there are no other data available from multiple body sites in the same baby, so we cannot directly evaluate whether similar patterns occur in, for example, NBW infants.
Among the three sites examined, LBW infant skin was the most adult-like in terms of microbiota composition (Fig. 3); this may result from infant skin being more selective for, and/or more heavily exposed to, the skin microbiota of adult caretakers in the NICU compared to other body sites (33), although we did not quantify the amount of time each infant spent in direct contact with mothers or other caregivers. (In the mouth and gut, the main difference between neonates and adults seems to be a relative lack of strict anaerobes [38].) While developmental changes over the first year of life have been reported for the infant skin microbiome (8), they were not apparent within the relatively short, neonatal time frame of the current study (Table 4).
Finally, delivery mode has been noted to exert a strong influence on the composition of the newborn microbiota (4); while this effect was conceivably manifest in our study (e.g., Ureaplasma in baby 3; Bacteroides in baby 6 [Fig. 1] [36, 39]), its pervasiveness and persistence will require examination in larger cohorts of high-risk infants.
We found that microbiota composition was relatively stable over time within LBW neonates. This small effect size for time, compared to those for body site and host individual, is also consistent with patterns observed in healthy adults (1–3, 40). Nonetheless, our comparative approach uncovered subtle yet important temporal changes that occurred over the 8- to 21-day age range: in particular, a gradual (i.e., delayed) compositional divergence of the oral and fecal microbiota (Table 3), largely driven by progressive temporal turnover in the distal gut (Table 4), the latter of which proceeded at a rate indistinguishable from that of age-matched NBW infants (Fig. 4a). Long recognized as a key process taking place in early infancy (38, 41–43), our study draws into focus the initiation phase of gut microbiome development, capturing, possibly, the time span over which the site begins to receive and select for gut-specific microbes, which may then grow to outnumber or outcompete transient or generalist immigrants from the oral cavity (or other sources shared by the two sites). However, given our small cohort of six infants for which there were a number of uncontrolled variables (e.g., gestational age at delivery, multiple gestation, medical treatment, delivery mode), we caution that our data are likely limited in terms of their generalizability and capacity to detect subtle effects. The biogeographic patterns we report warrant follow-up in larger, well-controlled, prospective cohort studies.
We also detected a novel, uncultivated lineage of Mycoplasma at high abundance in the oral cavity of ELBW baby 3. Mycoplasma and Ureaplasma spp. colonize the human respiratory and urogenital tracts, and some play roles as perinatal pathogens (39). M. hominis and Ureaplasma spp. can cause chorioamnionitis (a risk factor for preterm premature rupture of membranes [PPROM]) and pass from mother to newborn, and the latter organisms have been associated with preterm labor and low birthweight (39, 44). In neonates, they cause respiratory, blood, and central nervous system (CNS) infections (39). Lacking cell walls, these organisms are innately resistant to beta-lactam (e.g., ampicillin, cefotaxime) and glycopeptide (e.g., vancomycin) antibiotics (45). Although not innately resistant, their susceptibility to aminoglycosides (e.g., gentamicin) is variable (46).
Baby 3 was delivered vaginally after PPROM at ~24.5 weeks of completed gestation and was treated intravenously with ampicillin and gentamicin for the first 7 days of life. Thus, carriage of Mycoplasma- and Ureaplasma-related OTUs at low abundance at the start of the study, on DOL 8, may have been due to vertical transmission at delivery, followed by resistance to the initial course of antibiotics, although alternative scenarios are possible (e.g., later exposure in the NICU). Baby 3 was again treated with antibiotics (vancomycin, gentamicin, cefotaxime) from DOL 13 to 19 (Table 2), and this coincided with a marked increase in the proportional abundance of Pseudomonas aeruginosa (Fig. 1a) and OTU 15, a member of a novel, uncultivated clade belonging to the Mycoplasmataceae, in baby 3’s oral samples (Fig. 5; see Fig. S4 in the supplemental material). Intriguingly, a recent study found high abundances of this uncultivated Mycoplasma in the vaginal microbiota of Trichomonas vaginalis-infected women (detected in 19/30 T. vaginalis-infected and 1/29 uninfected individuals) (37), again raising the possibility that this organism too was transferred from mother to infant at delivery. Further investigation into the diversity, distribution, and clinical significance of this novel, uncultivated Mycoplasma in human hosts is warranted, particularly in pregnant women and premature infants.
Although the LBW infants in this study were relatively free of major medical problems, we found that their microbiomes were dominated at times by bacterial taxa that have been associated with neonatal infections and NEC, e.g., Staphylococcus, C. perfringens, P. aeruginosa, and others (28, 32, 47, 48). Yet, despite the abundance of taxa with pathogenic potential, it appears that certain normal processes were under way, including the development of body site-specific bacterial communities and progressive compositional turnover in the distal gut, as observed in healthy hosts (2, 38). Our analysis was cohort based; however, it might be useful to know whether individual infants vary in the precise timing of body site-associated compositional differentiation, and if so, whether such variation depends on gestational age at delivery or particular NICU management protocols. Unfortunately, our cohort was not well suited to this analysis because of its small size, but also because gestational age at delivery was confounded with delivery location and the amount of time spent in the NICU (Tables 1 and 2). This underscores a need for larger and distinct cohorts but also highlights a challenge: the smallest, most premature infants will almost always require the most intensive medical support, thus entangling factors such as gut and immune immaturity with, for example, the number of invasive procedures or days on antibiotics. Nevertheless, monitoring of oral and other potential source communities in the NICU might be particularly warranted during the time the gut microbiome remains “undifferentiated” and, possibly, more open to invasion.
MATERIALS AND METHODS
Patients and sample collection.
Six low-birthweight (LBW) infants were recruited from a level III NICU at the University of Chicago Comer Children’s Hospital. The infants were born within 1 week of each other in the summer of 2010. The cause of the low birthweight was preterm delivery in five of the infants (a singleton and two pairs of twins) and fetal growth restriction in the sixth. Birth weights ranged from 0.75 to 1.82 kg (see Table 1 for clinical details; <2.5 kg is considered low birthweight). Stool and saliva samples and skin swabs were obtained from each infant on postnatal days 8, 10, 12, 15, 18, and 21. The age range of 8 to 21 days was selected because it may represent a critical window for the colonization of the infant, and although it did not occur in the present cohort, for the onset of NEC. Stool sampling involved manual perineal stimulation with a lubricated cotton swab, which induced prompt defecation. Oral and skin samples were collected by gently swabbing the dorsum of the tongue and the anterior upper chest wall, respectively. For the oral samples, we simply call the collected materials “saliva,” because it is likely that multiple sites were contacted during the gentle swabbing. Samples were collected using sterile nylon or cotton swabs, placed in 3 ml of universal transport medium (UTM; EMD Millipore, Billerica, MA), and promptly frozen at −80°C. A total of 108 samples were collected for the study. Data pertaining to the care and location of the infants during the sampling period are presented in Table 2. All infants remained hospitalized throughout the study. The Institutional Review Board of the University of Chicago approved the study protocol, and the infants’ parents provided written informed consent.
DNA extraction, PCR amplification, and pyrosequencing.
Genomic DNA was isolated from each sample (1.5 ml UTM) using a QIAamp DNA stool minikit (Qiagen, Valencia, CA) with modifications, including bead beating (49). A fragment of the 16S rRNA gene spanning the V3-V5 hypervariable regions was amplified. The forward primer (5′ CGT ATC GCC TCC CTC GCG CCA TCA GNN NNN NNN NNN NGC ACT CCT ACG GGA GGC AGC A 3′) contained the 454 Life Sciences primer A sequence, a unique 12-nucleotide (nt) error-correcting Golay barcode used to label each amplicon (designated by the N’s) (50), the broad-range bacterial primer 338F (F stands for forward), and a two-base linker located between the bar code and the rRNA primer (GC). The reverse primer (5′ CTA TGC GCC TTG CCA GCC CGC TCA GAA CCG TCA ATT CCT TTG AGT TT 3′) contained the 454 Life Sciences primer B sequence, a two-base linker (AA), and the broad-range bacterial primer 906R (R stands for reverse). Amplifications were carried out in triplicate 25-µl reactions using 0.4 µM forward and reverse primers, 3-µl template DNA, and 1× HotMasterMix (5 PRIME, Gaithersburg, MD). Bovine serum albumin (BSA) was added at a final concentration of 0.1 µg/µl to reaction mixtures containing fecal DNA. Thermal cycling was carried out at 94°C for 2 min, followed by 35 cycles, with 1 cycle consisting of 94°C for 45 s, 50°C for 30 s, and 72°C for 90 s, with a final extension step of 10 min at 72°C. Replicate reactions were pooled and then purified using an Ultra-Clean-htp 96-well PCR clean-up kit according to the manufacturer’s instructions (MO BIO, Carlsbad, CA).
DNA concentrations were determined using a high-sensitivity Quant-iT double-stranded DNA (dsDNA) kit according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). Purified amplicons were combined in equimolar ratios into a single tube, ethanol precipitated, and resuspended in 100 µl of nuclease-free water. The pooled DNA was gel purified and recovered using a QIAquick gel extraction kit (Qiagen). Unidirectional amplicon sequencing was performed by the W. M. Keck Center for Comparative and Functional Genomics at the University of Illinois, Urbana-Champaign using a 454 Life Sciences genome sequencer FLX instrument, titanium (Ti) series reagents, primer A, and 6 regions of a 16-region gasket (Roche, Branford, CT). Sequencing generated 186,428 raw reads.
Sequence analysis.
Raw reads were filtered using the QIIME software package (51). Reads were removed from the analysis if they were <200 or >600 nt in length, contained an ambiguous base, had a mean quality score of <25 across the entire read, contained a homopolymer run >6 nt in length, did not contain the forward primer sequence, or contained an uncorrectable barcode. Remaining reads were truncated at the first base of the first 50-nt sliding window with a mean quality score of <25 (if found), and retained unless <200 nt in length after truncation. Filtered reads were assigned to samples by examining the 12-nt barcode. A total of 119,191 filtered reads were associated with samples at this step (mean read length, 535 nt).
Error correction, chimera detection (using UCHIME), and clustering of filtered reads into de novo operational taxonomic units (OTUs) at 97% sequence identity were performed in USEARCH using otupipe-like scripts enabled in QIIME (http://www.drive5.com/usearch/manual/otu_clustering.html) (52, 53). A representative sequence was chosen from each OTU by selecting the “first” sequence (i.e., the UCLUST cluster seed). Representative sequences were aligned against the Greengenes core set (54) using PyNAST (55) with a minimum alignment length of 150 nt and a minimum identity of 80%. Fifteen OTU representative sequences failed to align; BLASTn searches against GenBank’s nr/nt database revealed 13 human OTUs, 1 Candida albicans OTU (representing 276 reads from baby 6, day 8 stool), and 1 poor-quality OTU, all of which were excluded from further analysis. Taxonomic assignments were made using the Ribosomal Database Project (RDP) classifier version 2.2 with a minimum support threshold of 80% and the RDP taxonomic nomenclature (56). For the most abundant OTUs study-wide (here, those with >0.05% average abundance across all samples), RDP assignments were manually confirmed and, when possible, annotated with species-level information using BLASTn searches against the nr/nt database. A table of sequence counts per classified OTU × sample was generated in which the criteria for an OTU’s inclusion were that it contained at least 2 sequences and was assigned at least to the genus level. The final OTU table consisted of 321 OTUs containing a total of 105,462 sequences.
Sequences representing OTUs that did not make it into the final table were removed from the alignment. Hypervariable (i.e., uninformative) positions were then excluded using the PH Lane mask (57). A phylogeny was inferred using FastTree version 2.1.3 (58) with the Jukes-Cantor plus CAT model. The final OTU table and phylogeny served as inputs to subsequent analyses, including rarefaction, α and β diversity calculations, unweighted UniFrac-based principal coordinate analysis (PCoA), and phylum- and genus-level taxonomic summaries implemented in QIIME. Unweighted UniFrac-based permutational multivariate analysis of variance (PERMANOVA) was performed in PRIMER-E version 6 (59). Other statistical tests were performed in QIIME or Prism (GraphPad Software, Inc.).
Sequence analysis focused on a novel, uncultivated oral Mycoplasma.
Phylogenetic relationships among sequences belonging to OTUs assigned to the phylum Tenericutes (3 OTUs) were investigated in detail. This analysis was prompted by the identification of an OTU assigned to the genus Mycoplasma containing 771 reads (99% of which were from baby 3’s saliva) and exhibiting low sequence identity (~88%) to the most closely related cultivated strains represented in GenBank (http://www.ncbi.nlm.nih.gov/genbank/). Sequences were aligned against the Greengenes core set using the NAST algorithm (60) (http://greengenes.lbl.gov) and imported into ARB (version 08.08.27) (61). In ARB, the alignment was manually improved using secondary structure information and alignment to nearest neighbors in the context of an expanded, in-house database founded upon the Greengenes alignment. Phylogenetic relationships among the 3 OTUs found in the present study, their closest relatives (uncultivated mycoplasmas), and selected representatives of cultivated Tenericutes were inferred using bootstrapped maximum likelihood inference methods in RAxML (version 7.2.8) (62). In order to confirm and further explore the phylogenetic placement of the novel Mycoplasma-related OTU, a small number of near-full-length 16S rRNA gene sequences were recovered from baby 3’s day 18 saliva sample via amplification (with primers 8F/1391R), cloning, and Sanger sequencing using methods described elsewhere (63). Fifteen high-quality sequences were assembled (4 uncultivated Mycoplasma sequences and 11 Pseudomonas aeruginosa sequences). The near-full-length Mycoplasma sequences were analyzed using NAST, ARB, and RAxML as described above.
Comparison to microbiota of NBW infants via pyrosequencing of archived stool DNA.
Archived stool DNA samples from healthy, age-matched (i.e., time series spanning 8 to 21 days in age), normal birthweight (NBW) (>2.5 kg) infants enrolled in a prior study (5) were amplified, sequenced, and analyzed using the pyrosequencing and bioinformatics approaches described herein. The archived DNA had been isolated using the QIAamp stool DNA minikit (Qiagen) and stored at −80°C. The Stanford University Administrative Panel on Human Subjects in Medical Research approved this work, and the infants’ parents provided written informed consent.
Comparison to microbiota of healthy adults using publically available sequence data.
Sequence data from the LBW and NBW infants were compared to publically available sequence data from the corresponding body sites of healthy adults. Adult data were selected from two published studies that used pyrosequencing approaches similar to those used here (1, 2). From the first study (1), we selected samples from 7 adults (3 female), “days 1 and 2” (of 4 sampling dates), including dorsal tongue swabs, skin swabs (forehead and right forearm), and stool samples (56 samples in total). These 16S rRNA gene sequences were V2 region FLX reads originating from the distal primer (338R). From the second study (2), we selected samples from 6 adults (a subset chosen at random but matched for gender to the LBW infants), “visit 2” (of 2 sampling visits), including saliva samples, skin swabs (right retroauricular crease), and stool samples (18 samples in total). These were V3-V5 region Ti reads originating from the distal primer (926R). By comparison, the infant-derived sequences generated for the present study were V3-V5 region Ti reads originating from the proximal primer (338F). Thus, given the differences in sequence length and sequenced region among the data sets, the pooled sequences were trimmed to a length of not more than 300 nt and OTUs were picked against a set of reference sequences. This was accomplished in QIIME using uclust_ref-based OTU picking against the Greengenes gg_97_otus_4feb2011.fasta reference set at an identity threshold of 95% (relaxed from 97% to allow for greater recruitment), with reverse strand matching enabled and no new clusters allowed. A total of 3,158 reference OTUs were detected; these encompassed 96% of the 475,080 total sequences. Rarefied and unrarefied OTU tables, along with a reference tree (gg_97_otus_4feb2011.tre), were used to calculate unweighted UniFrac distance matrices, which served as inputs for PCoA in QIIME.
The DNA extraction method varied among the studies compared: studies of adults used a MO BIO kit, while studies of infants used a Qiagen kit. To investigate potential kit-associated bias, we pyrosequenced 16S rRNA genes amplified from archived adult stool DNA that had been isolated using a Qiagen kit (from the NBW infants’ fathers [5]; mothers were excluded due to possible pregnancy-associated shifts in microbiota composition [64]). These new adult sequences were filtered as described herein and trimmed to a length of not more than 300 nt, pooled with the other sequences, and analyzed as described in the preceding paragraph. Because the Qiagen-extracted adult stool samples (5) clustered with the MO BIO-extracted ones (1, 2) (see Fig. S1 in the supplemental material), we concluded that DNA extraction kit did not grossly bias the results of the unweighted UniFrac-based PCoA.
Nucleotide sequence accession numbers.
The sequence data generated for this study were deposited in the QIIME database (study identification numbers 2042 and 2046).
SUPPLEMENTAL MATERIAL
Unweighted UniFrac-based principal coordinate analysis (PCoA) of infant- and adult-associated bacterial communities profiled using 16S rRNA gene sequence surveys. Each point corresponds to a sample colored according to cohort and body site. Sites include the oral cavity (a), skin surface (b), and distal gut (stool samples) (c). Panels a to d represent the same analysis; panels a, b and c display each site individually and are overlaid in panel d. The percentage of the total variation explained by the plotted principal coordinate (PCo) is indicated on the axis. The OTU table was rarefied to 250 sequences per sample prior to analysis. See Materials and Methods for details on the selection of samples from published studies. In panel c, the 6 LBW infant samples clustering nearest to the adult samples are from Bacteroides-dominated baby 6. Download
Bacterial OTU richness at a depth of 200 sequences/sample. OTUs were defined at 97% sequence identity. (a) Observed OTU richness for body sites. The values for 50 rarefaction replicates are shown. The error bars represent 95% CI. Values that are significantly different (P < 0.001) by Tukey’s posthoc tests are indicated by bars and three asterisks. Values that are not significantly different (ns) are indicated. (b to d) Each symbol represents the value for an individual baby. The horizontal bars represent the means for the 6 LBW infants, respectively. Of 108 samples, 106 yielded at least 200 sequences and were included in the analysis. On average, communities on skin were richer in bacterial OTUs than those in saliva or stool. Download
Intersubject compositional variation plotted against infant age for each body site. Each point corresponds to a between-subject UniFrac distance. Horizontal bars represent means. Skin microbiota composition is least personalized on day 8; stool microbiota composition becomes slightly less personalized over time. Download
Phylogenetic relationships among 3 Tenericutes OTUs (gray boxes) and close relatives, inferred using a maximum likelihood approach. Node support was assessed using 100 bootstrap replicates; values of >80% are shown. The phylogeny was inferred using RAxML version 7.2.8 and the GTRCAT model. The near-full-length alignment contained 620 distinct alignment positions (columns). The alignment also included nine shorter pyrosequencing reads. Bar, 0.1 substitution per site. (Top left) OTU proportional abundances in baby 3’s saliva; ND, not detected. Download
Results of main and pairwise a posteriori tests of the effect of age (within body sites) on the compositional structure of bacterial communities associated with the LBW infants in this study using unweighted UniFrac-based permutational multivariate ANOVA and the t statistic, showing a posteriori tests for stool samples only.
Results of main and pairwise a posteriori tests of the effect of age (within body sites) on the compositional structure of bacterial communities associated with the LBW infants in this study using unweighted UniFrac-based permutational multivariate ANOVA and the t statistic, showing a posteriori tests for saliva and skin samples.
ACKNOWLEDGMENTS
We thank the subjects and their families for their participation and the NICU staff for their support. We also thank members of the Morowitz and Relman laboratories, and in particular Valeriy Poroyko (University of Chicago) for technical assistance and Diana Proctor (Stanford University) for critical review of the manuscript.
This work was supported in part by NIH grant 1R01AI092531-01 (M.J.M. and D.A.R.; Jill Banfield, principal investigator [PI]), a Walter V. and Idun Berry postdoctoral fellowship (E.K.C.), NIH Pioneer award DP1OD000964 (D.A.R.), March of Dimes Foundation research grant 5-FY10-103 (M.J.M.), the March of Dimes Prematurity Research Center at Stanford University School of Medicine (D.A.R.), and by the Thomas C. and Joan M. Merigan Endowment at Stanford University (D.A.R.).
Footnotes
Citation Costello EK, Carlisle EM, Bik EM, Morowitz MJ, Relman DA. 2013. Microbiome assembly across multiple body sites in low-birthweight infants. mBio 4(6):e00782-13. doi:10.1128/mBio.00782-13.
REFERENCES
- 1. Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. 2009. Bacterial community variation in human body habitats across space and time. Science 326:1694–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Microbiome Human, Project Consortium 2012. Structure, function and diversity of the healthy human microbiome. Nature 486:207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Caporaso JG, Lauber CL, Costello EK, Berg-Lyons D, Gonzalez A, Stombaugh J, Knights D, Gajer P, Ravel J, Fierer N, Gordon JI, Knight R. 2011. Moving pictures of the human microbiome. Genome Biol. 12:R50. 10.1186/gb-2011-12-5-r50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, Knight R. 2010. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. U. S. A. 107:11971–11975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. 2007. Development of the human infant intestinal microbiota. PLoS Biol. 5:e177. 10.1371/journal.pbio.0050177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, Angenent LT, Ley RE. 2011. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. U. S. A. 108(Suppl 1):4578–4585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, Heath AC, Warner B, Reeder J, Kuczynski J, Caporaso JG, Lozupone CA, Lauber C, Clemente JC, Knights D, Knight R, Gordon JI. 2012. Human gut microbiome viewed across age and geography. Nature 486:222–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Capone KA, Dowd SE, Stamatas GN, Nikolovski J. 2011. Diversity of the human skin microbiome early in life. J. Invest. Dermatol. 131:2026–2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crielaard W, Zaura E, Schuller AA, Huse SM, Montijn RC, Keijser BJ. 2011. Exploring the oral microbiota of children at various developmental stages of their dentition in the relation to their oral health. BMC Med. Genomics 4:22. 10.1186/1755-8794-4-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morowitz MJ, Poroyko V, Caplan M, Alverdy J, Liu DC. 2010. Redefining the role of intestinal microbes in the pathogenesis of necrotizing enterocolitis. Pediatrics 125:777–785 [DOI] [PubMed] [Google Scholar]
- 11. Neu J, Walker WA. 2011. Necrotizing enterocolitis. N. Engl. J. Med. 364:255–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stoll BJ, Hansen N, Fanaroff AA, Wright LL, Carlo WA, Ehrenkranz RA, Lemons JA, Donovan EF, Stark AR, Tyson JE, Oh W, Bauer CR, Korones SB, Shankaran S, Laptook AR, Stevenson DK, Papile LA, Poole WK. 2002. Late-onset sepsis in very low birth weight neonates: the experience of the NICHD Neonatal Research Network. Pediatrics 110:285–291 [DOI] [PubMed] [Google Scholar]
- 13. Yee WH, Soraisham AS, Shah VS, Aziz K, Yoon W, Lee SK, Canadian Neonatal Network 2012. Incidence and timing of presentation of necrotizing enterocolitis in preterm infants. Pediatrics 129:e298–e304 [DOI] [PubMed] [Google Scholar]
- 14. González-Rivera R, Culverhouse RC, Hamvas A, Tarr PI, Warner BB. 2011. The age of necrotizing enterocolitis onset: an application of Sartwell’s incubation period model. J. Perinatol. 31:519–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Levy O. 2007. Innate immunity of the newborn: basic mechanisms and clinical correlates. Nat. Rev. Immunol. 7:379–390 [DOI] [PubMed] [Google Scholar]
- 16. Hooper LV, Gordon JI. 2001. Commensal host-bacterial relationships in the gut. Science 292:1115–1118 [DOI] [PubMed] [Google Scholar]
- 17. Sekirov I, Finlay BB. 2009. The role of the intestinal microbiota in enteric infection. J. Physiol. (Lond.) 587:4159–4167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cho I, Yamanishi S, Cox L, Methé BA, Zavadil J, Li K, Gao Z, Mahana D, Raju K, Teitler I, Li H, Alekseyenko AV, Blaser MJ. 2012. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 488:621–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, Glickman JN, Siebert R, Baron RM, Kasper DL, Blumberg RS. 2012. Microbial exposure during early life has persistent effects on natural killer T cell function. Science 336:489–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carlisle EM, Morowitz MJ. 2011. Pediatric surgery and the human microbiome. J. Pediatr. Surg. 46:577–584 [DOI] [PubMed] [Google Scholar]
- 21. Manzoni P, De Luca D, Stronati M, Jacqz-Aigrain E, Ruffinazzi G, Luparia M, Tavella E, Boano E, Castagnola E, Mostert M, Farina D. 2013. Prevention of nosocomial infections in neonatal intensive care units. Am. J. Perinatol. 30:81–88 [DOI] [PubMed] [Google Scholar]
- 22. Millar MR, Linton CJ, Cade A, Glancy D, Hall M, Jalal H. 1996. Application of 16S rRNA gene PCR to study bowel flora of preterm infants with and without necrotizing enterocolitis. J. Clin. Microbiol. 34:2506–2510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang Y, Hoenig JD, Malin KJ, Qamar S, Petrof EO, Sun J, Antonopoulos DA, Chang EB, Claud EC. 2009. 16S rRNA gene-based analysis of fecal microbiota from preterm infants with and without necrotizing enterocolitis. ISME J. 3:944–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mshvildadze M, Neu J, Shuster J, Theriaque D, Li N, Mai V. 2010. Intestinal microbial ecology in premature infants assessed with non-culture-based techniques. J. Pediatr. 156:20–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang JY, Shin SM, Chun J, Lee JH, Seo JK. 2011. Pyrosequencing-based molecular monitoring of the intestinal bacterial colonization in preterm infants. J. Pediatr. Gastroenterol. Nutr. 53:512–519 [DOI] [PubMed] [Google Scholar]
- 26. Mai V, Young CM, Ukhanova M, Wang X, Sun Y, Casella G, Theriaque D, Li N, Sharma R, Hudak M, Neu J. 2011. Fecal microbiota in premature infants prior to necrotizing enterocolitis. PLoS One 6:e20647. 10.1371/journal.pone.0020647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morowitz MJ, Denef VJ, Costello EK, Thomas BC, Poroyko V, Relman DA, Banfield JF. 2011. Strain-resolved community genomic analysis of gut microbial colonization in a premature infant. Proc. Natl. Acad. Sci. U. S. A. 108:1128–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Madan JC, Salari RC, Saxena D, Davidson L, O’Toole GA, Moore JH, Sogin ML, Foster JA, Edwards WH, Palumbo P, Hibberd PL. 2012. Gut microbial colonisation in premature neonates predicts neonatal sepsis. Arch. Dis. Child. (Fetal Neonatal Ed.) 97:F456–F462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sharon I, Morowitz MJ, Thomas BC, Costello EK, Relman DA, Banfield JF. 2013. Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Res. 23:111–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Normann E, Fahlén A, Engstrand L, Lilja HE. 2013. Intestinal microbial profiles in extremely preterm infants with and without necrotizing enterocolitis. Acta Paediatr. 102:129–136 [DOI] [PubMed] [Google Scholar]
- 31. Mai V, Torrazza RM, Ukhanova M, Wang X, Sun Y, Li N, Shuster J, Sharma R, Hudak ML, Neu J. 2013. Distortions in development of intestinal microbiota associated with late onset sepsis in preterm infants. PLoS One 8:e52876. 10.1371/journal.pone.0052876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morrow AL, Lagomarcino AJ, Schibler KR, Taft DH, Yu Z, Wang B, Altaye M, Wagner M, Gevers D, Ward DV, Kennedy MA, Huttenhower C, Newburg DS. 2013. Early microbial and metabolomic signatures predict later onset of necrotizing enterocolitis in preterm infants. Microbiome 1:13. 10.1186/2049-2618-1-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Keyworth N, Millar MR, Holland KT. 1992. Development of cutaneous microflora in premature neonates. Arch. Dis. Child. 67:797–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71:8228–8235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bennet R, Nord CE. 1987. Development of the faecal anaerobic microflora after caesarean section and treatment with antibiotics in newborn infants. Infection 15:332–336 [DOI] [PubMed] [Google Scholar]
- 36. Penders J, Thijs C, Vink C, Stelma FF, Snijders B, Kummeling I, van den Brandt PA, Stobberingh EE. 2006. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 118:511–521 [DOI] [PubMed] [Google Scholar]
- 37. Martin DH, Zozaya M, Lillis RA, Myers L, Nsuami MJ, Ferris MJ. 2013. Unique vaginal microbiota which include an unknown Mycoplasma-like organism are associated with Trichomonas vaginalis infection. J. Infect. Dis. 207:1922–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mackie RI, Sghir A, Gaskins HR. 1999. Developmental microbial ecology of the neonatal gastrointestinal tract. Am. J. Clin. Nutr. 69:1035S–1045S [DOI] [PubMed] [Google Scholar]
- 39. Waites KB, Katz B, Schelonka RL. 2005. Mycoplasmas and ureaplasmas as neonatal pathogens. Clin. Microbiol. Rev. 18:757–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuczynski J, Costello EK, Nemergut DR, Zaneveld J, Lauber CL, Knights D, Koren O, Fierer N, Kelley ST, Ley RE, Gordon JI, Knight R. 2010. Direct sequencing of the human microbiome readily reveals community differences. Genome Biol. 11:210. 10.1186/gb-2010-11-5-210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schaedler RW, Dubos R, Costello R. 1965. The development of the bacterial flora in the gastrointestinal tract of mice. J. Exp. Med. 122:59–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Savage DC. 1977. Microbial ecology of the gastrointestinal tract. Annu. Rev. Microbiol. 31:107–133 [DOI] [PubMed] [Google Scholar]
- 43. Murgas Torrazza R, Neu J. 2011. The developing intestinal microbiome and its relationship to health and disease in the neonate. J. Perinatol. 31(Suppl 1):S29–S34 [DOI] [PubMed] [Google Scholar]
- 44. DiGiulio DB, Romero R, Amogan HP, Kusanovic JP, Bik EM, Gotsch F, Kim CJ, Erez O, Edwin S, Relman DA. 2008. Microbial prevalence, diversity and abundance in amniotic fluid during preterm labor: a molecular and culture-based investigation. PLoS One 3:e3056. 10.1371/journal.pone.0003056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Taylor-Robinson D, Bébéar C. 1997. Antibiotic susceptibilities of mycoplasmas and treatment of mycoplasmal infections. J. Antimicrob. Chemother. 40:622–630 [DOI] [PubMed] [Google Scholar]
- 46. Waites KB, Crouse DT, Cassell GH. 1992. Antibiotic susceptibilities and therapeutic options for Ureaplasma urealyticum infections in neonates. Pediatr. Infect. Dis. J. 11:23–29 [DOI] [PubMed] [Google Scholar]
- 47. de la Cochetiere MF, Piloquet H, des Robert C, Darmaun D, Galmiche JP, Rozé JC. 2004. Early intestinal bacterial colonization and necrotizing enterocolitis in premature infants: the putative role of Clostridium. Pediatr. Res. 56:366–370 [DOI] [PubMed] [Google Scholar]
- 48. Stewart CJ, Marrs EC, Magorrian S, Nelson A, Lanyon C, Perry JD, Embleton ND, Cummings SP, Berrington JE. 2012. The preterm gut microbiota: changes associated with necrotizing enterocolitis and infection. Acta Paediatr. 101:1121–1127 [DOI] [PubMed] [Google Scholar]
- 49. Zoetendal EG, Heilig HG, Klaassens ES, Booijink CC, Kleerebezem M, Smidt H, De Vos WM. 2006. Isolation of DNA from bacterial samples of the human gastrointestinal tract. Nat. Protoc. 1:870–873 [DOI] [PubMed] [Google Scholar]
- 50. Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. 2008. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat. Methods 5:235–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461 [DOI] [PubMed] [Google Scholar]
- 53. Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72:5069–5072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. 2010. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73:5261–5267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lane DJ. 1991. 16S/23S rRNA sequencing, p 115–148 In Stackebrandt E, Goodfellow M. (ed), Nucleic acid techniques in bacterial systematics. John Wiley & Sons, Chichester, United Kingdom [Google Scholar]
- 58. Price MN, Dehal PS, Arkin AP. 2010. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Clarke KR, Gorley RN. 2006. PRIMER v6: user manual/tutorial. PRIMER-E, Plymouth, United Kingdom [Google Scholar]
- 60. DeSantis TZ, Hugenholtz P, Keller K, Brodie EL, Larsen N, Piceno YM, Phan R, Andersen GL. 2006. NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res. 34:W394–W399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ludwig W, Strunk O, Westram R, Richter L, Meier H, Kumar Y, Buchner A, Lai T, Steppi S, Jobb G, Förster W, Brettske I, Gerber S, Ginhart AW, Gross O, Grumann S, Hermann S, Jost R, König A, Liss T, Lüssmann R, May M, Nonhoff B, Reichel B, Strehlow R, Stamatakis A, Stuckmann N, Vilbig A, Lenke M, Ludwig T, Bode A, Schleifer KH. 2004. ARB: a software environment for sequence data. Nucleic Acids Res. 32:1363–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stamatakis A, Hoover P, Rougemont J. 2008. A rapid bootstrap algorithm for the RAxML Web servers. Syst. Biol. 57:758–771 [DOI] [PubMed] [Google Scholar]
- 63. Bik EM, Long CD, Armitage GC, Loomer P, Emerson J, Mongodin EF, Nelson KE, Gill SR, Fraser-Liggett CM, Relman DA. 2010. Bacterial diversity in the oral cavity of 10 healthy individuals. ISME J. 4:962–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Koren O, Goodrich JK, Cullender TC, Spor A, Laitinen K, Bäckhed HK, Gonzalez A, Werner JJ, Angenent LT, Knight R, Bäckhed F, Isolauri E, Salminen S, Ley RE. 2012. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 150:470–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Unweighted UniFrac-based principal coordinate analysis (PCoA) of infant- and adult-associated bacterial communities profiled using 16S rRNA gene sequence surveys. Each point corresponds to a sample colored according to cohort and body site. Sites include the oral cavity (a), skin surface (b), and distal gut (stool samples) (c). Panels a to d represent the same analysis; panels a, b and c display each site individually and are overlaid in panel d. The percentage of the total variation explained by the plotted principal coordinate (PCo) is indicated on the axis. The OTU table was rarefied to 250 sequences per sample prior to analysis. See Materials and Methods for details on the selection of samples from published studies. In panel c, the 6 LBW infant samples clustering nearest to the adult samples are from Bacteroides-dominated baby 6. Download
Bacterial OTU richness at a depth of 200 sequences/sample. OTUs were defined at 97% sequence identity. (a) Observed OTU richness for body sites. The values for 50 rarefaction replicates are shown. The error bars represent 95% CI. Values that are significantly different (P < 0.001) by Tukey’s posthoc tests are indicated by bars and three asterisks. Values that are not significantly different (ns) are indicated. (b to d) Each symbol represents the value for an individual baby. The horizontal bars represent the means for the 6 LBW infants, respectively. Of 108 samples, 106 yielded at least 200 sequences and were included in the analysis. On average, communities on skin were richer in bacterial OTUs than those in saliva or stool. Download
Intersubject compositional variation plotted against infant age for each body site. Each point corresponds to a between-subject UniFrac distance. Horizontal bars represent means. Skin microbiota composition is least personalized on day 8; stool microbiota composition becomes slightly less personalized over time. Download
Phylogenetic relationships among 3 Tenericutes OTUs (gray boxes) and close relatives, inferred using a maximum likelihood approach. Node support was assessed using 100 bootstrap replicates; values of >80% are shown. The phylogeny was inferred using RAxML version 7.2.8 and the GTRCAT model. The near-full-length alignment contained 620 distinct alignment positions (columns). The alignment also included nine shorter pyrosequencing reads. Bar, 0.1 substitution per site. (Top left) OTU proportional abundances in baby 3’s saliva; ND, not detected. Download
Results of main and pairwise a posteriori tests of the effect of age (within body sites) on the compositional structure of bacterial communities associated with the LBW infants in this study using unweighted UniFrac-based permutational multivariate ANOVA and the t statistic, showing a posteriori tests for stool samples only.
Results of main and pairwise a posteriori tests of the effect of age (within body sites) on the compositional structure of bacterial communities associated with the LBW infants in this study using unweighted UniFrac-based permutational multivariate ANOVA and the t statistic, showing a posteriori tests for saliva and skin samples.