

Abstract
The objective of this study was to develop an in vitro pharmacodynamic (PD) system to test the impact of protein binding on antiretroviral (ARV) drug effect and intracellular ARV distribution. CD4+ T cells were isolated from peripheral blood mononuclear cells (PBMCs) and exposed to varying and physiologically relevant concentrations of human serum albumin (HSA) and the ARV drugs efavirenz (EFV), raltegravir (RAL), etravirine (ETR), and enfuvirtide (ENF). The effect of varying extracellular protein concentration on the intracellular distribution of EFV, RAL, and ETR was assessed using ultraperformance liquid chromatography tandem mass spectrometry. HIV infectivity was assessed using an HIV-1 reporter virus expressing an Env-green fluorescent protein (GFP) and quantified using flow cytometry. Increasing extracellular HSA concentration was associated with increased relative infectivity for all drugs tested as well as decreased intracellular concentrations for EFV, RAL, and ETR. Median-effect plots indicate linearity between log10 antiviral effect (fraction of virus affected divided by fraction unaffected) and log10 intracellular drug concentration. The median [interquartile range (IQR)] slope (m) of the median-effect plots was 2.97 (2.26–5.85) for EFV, 3.52 (3.11–3.74) for ETR, and 2.39 (2.15–3.74) for RAL. The intracellular ARV concentrations associated with half-maximal antiviral effect (IC50) of EFV, ETR, and RAL were 1.2 (0.51–5.39), 39.06 (30.10–51.76), and 4.67 (3.91–5.02) ng/ml, respectively. This study demonstrates a significant reduction in cell penetration and antiviral effect of highly bound ARVs due to increasing extracellular concentration of HSA. This study is therefore the first to demonstrate experimentally how protein binding impacts intracellular distribution and the efficacy of ARVs.
Introduction
Antiretroviral drugs (ARVs) are highly effective in treating HIV infection through suppression of viral replication.1 Recent studies indicate that ARVs, administered either to the infected or susceptible uninfected sexual partner, are also efficacious in preventing HIV transmission.2–5 For many ARVs, however, there is minimal understanding of drug distribution beyond the blood plasma into cells and other extravascular compartments. This limits our ability to understand drug efficacy or toxicity in cells and distant anatomic sites.
The free drug hypothesis states that (1) protein-free (unbound) drug concentration is in equilibrium on both sides of a biological membrane at steady state, and (2) free drug concentration is what exerts a pharmacological effect.6 Protein binding has been demonstrated to influence the distribution of many ARV drugs and may impact local efficacy7,8; however, the pharmacodynamic effect of protein binding has yet to be examined directly for ARVs. Protein binding's effect on drug clearance, volume of distribution, and elimination9,10 has been established. In contrast, the pharmacodynamic effects of protein binding are not as well demonstrated experimentally. The focus of our study is therefore to examine the effect of varying extracellular free drug concentration on the antiviral effect of ARVs, some with an intracellular, and one with a cell surface site of action.11
Concentration-response relationships in vitro are often determined using methods that do not directly assess extracellular protein-free drug concentrations or intracellular drug concentrations, both of which may be heavily influenced by culture conditions and are not necessarily reflected in extracellular total drug concentration. This is a potential source of imprecision in estimating effective drug concentrations and may be a source of discordance between effective in vitro and in vivo concentrations. Drugs most commonly bind to circulating proteins including albumin, glycoproteins, globulins, and lipoproteins. Because these proteins vary in concentration across diverse anatomic locations, this can also affect the local free drug concentrations and, theoretically, the pharmacological effect.
For ARVs, it is valuable to experimentally confirm this free drug hypothesis, as the site of action for nearly all ARVs is intracellular, which should be highly influenced by extracellular free drug concentrations. Furthermore, HIV replicates in diverse anatomic locations, each with a unique concentration of binding proteins that may also affect the local pharmacological effect. For ARV drugs, HIV-1 nonnucleoside reverse transcriptase inhibitors (nNRTIs) predominantly bind to human serum albumin (HSA) while protease inhibitors predominantly bind to α1-acid glycoprotein.12
HSA, the predominant plasma binding protein, exists at median [interquartile range (IQR)] concentrations of 58.1 mg/ml (52.6–64.1) in the blood plasma, 4.2 mg/ml (3.7–4.9) in seminal plasma,7 and 0.3 mg/ml (0.1–0.6) in cerebrospinal fluid.13 We have previously demonstrated the equilibrium of the protein-free drug concentration of the ARV efavirenz (EFV) in the blood plasma and seminal plasma despite a 20-fold total EFV blood plasma:seminal plasma gradient.7 Protein binding was the primary determinant of EFV distribution, and our findings support the first postulate of the free drug hypothesis. The second postulate of the free drug hypothesis—free drug exerts the pharmacological effect—has yet to be determined experimentally for ARVs. Drug transporters may also heavily influence intracellular drug concentration and may vary among extravascular compartments. However, the focus of this study is to examine the role that protein binding plays in the intracellular distribution and antiviral effect of ARVs.
One reason a standardized method has yet to be developed is because of the difficulty simulating in vivo conditions in vitro. Serum shift assays are commonly used in drug discovery and development to assess the impact of plasma protein binding on efficacy.6 The difficulty with this assay is that it does not take into account the many dynamic aspects of protein binding, such as intracellular (IC) distribution, and in vitro results do not always correlate well with in vivo efficacy.6 Two in vitro approaches are often used to simulate in vivo conditions for protein binding: adding serum to media or supplementing media with binding proteins.14 Addition of serum to media involves a dilution of binding proteins in serum, resulting in less than physiologically relevant concentrations of binding proteins. Supplementing media with binding proteins may alter the binding capacity of many proteins, resulting in different in vitro and in vivo results.
We sought to create in vitro conditions appropriate to the binding properties of ARVs and to simultaneously analyze the impact of protein binding on intracellular distribution and HIV infectivity. We developed this pharmacodynamic model system using EFV, a highly protein-bound ARV used commonly in the treatment of HIV, because we have previously shown how protein binding determines the distribution of EFV.7 We also examined other classes of ARVs to evaluate the generalizability of our findings.
Materials and Methods
Subjects and demographic characteristics
Research participants provided informed consent prior to screening or study participation and were recruited from the general population of Baltimore, Maryland. All were healthy adult men and women, between the ages of 18 and 65 years old, who were not taking any medications (prescription or over the counter) or herbal supplements. Study participation involved collection of up to 100 ml of whole blood for in vitro testing. Analysis of the intracellular distribution of EFV, etravirine (ETR), and raltegravir (RAL) with varying HSA concentration was performed with six individual subjects for each ARV. Simultaneous analysis of intracellular distribution and HIV infectivity was performed with six individual subjects for EFV and three individual subjects each for RAL, ETR, and enfuvirtide (ENF). The study was approved by the Johns Hopkins Medicine Institutional Review Board.
Experimental plan
Assessment of the effect of protein binding on intracellular drug distribution and HIV infectivity was made using combined analytical and biochemical techniques. EFV, ETR, RAL, and ENF were selected to provide a range of protein binding and mechanisms of action with both intracellular (IC) and extracellular (EC) sites of action. Protein binding in various anatomic compartments was simulated by in vitro variation of HSA concentration. To explore the relationship between these drugs, protein binding, and the impact on infectivity, we used a single-round infectivity assay using an HIV-green fluorescent protein (GFP) reporter virus to infect the cell of interest, CD4+ T cells. EFV is an nNRTI, used widely in antiretroviral therapy. It is highly bound to plasma proteins, at >99.75% bound, primarily to HSA.15 This principle was further tested with three distinctly different ARV drugs, all primary bound to HSA. ETR is also an nNRTI, and is 99.99% bound to plasma proteins.16 RAL is an integrase inhibitor, and is 83% bound to plasma proteins.17 ENF is an EC acting fusion inhibitor, and is 92% bound to plasma proteins.18 Because ENF is an EC acting drug, only infectivity with varying HSA concentrations, not IC concentrations, was assessed. Using these ARVs of varying protein binding and mechanism of action, we have efficiently demonstrated how protein binding affects IC ARV distribution and pharmacological effect.
Antiretroviral drugs
EFV, ETR, RAL, and ENF for in vitro testing were obtained through the NIH AIDS Research and Reference Reagent Program (Germantown, MD). EFV, ETR, and RAL were dissolved in DMSO. ENF was dissolved in molecular biology grade water. Stocks were aliquoted and stored at −20°C. Concentrations of solvent vehicles were less than 0.1% in infectivity assays.
Virus stocks
HIV-GFP, single round infection pseudovirus, was prepared as previously described.19 Briefly, HEK-293 T cells (ATCC; Manassas, VA) were propagated in DMEM medium supplemented with 10% fetal bovine serum (FBS), and transfected with an envelope defective HIV-1 vector (pNL43-ΔE-EGFP)20,21 containing GFP in the env ORF followed by a KDEL stop sequence retaining GFP in the endoplasmic reticulum.22,23 Cotransfection of cells with pNL43-ΔE-EGFP and a CXCR4 envelope expression vector was performed using Lipofectamine 2000, according to the manufacturer's instructions (Invitrogen, Grand Island, NY). Six hours after transfection, culture medium was replaced with RMPI1640 (Invitrogen, Grand Island, NY) supplemented with 10% FBS (Invitrogen, Grand Island, NY), and incubated at 37°C for 48 h for the production of the virus. Virus stocks were concentrated and standardized according to Jilek et al.19 Concentrated virus was reconstituted in RPMI+10% FBS, and stored in aliquots at −80°C until use. Each lot was titrated to determine the quantity of virus sufficient to infect approximately 20% of cells in an infectivity assay.
CD4+ T cell preparation
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood using Ficoll-paque PLUS (GE Healthcare Biosciences), according to the manufacturer's instructions. Isolated cells were resuspended at 1×106 cells per ml in a stimulation medium of RPMI 1640 supplemented with 10% FBS (Invitrogen, Grand Island, NY), 100 U/ml interleukin-2 (IL-2, Proleukin), 0.5 μg/ml phytohemagglutinin (PHA) (Fisher Scientific; Pittsburgh, PA), and penicillin/streptomycin. Cell suspensions were incubated for 3 days at 37°C and 5% CO2. CD4+ T cells were isolated using a magnetic activated cell sorting (MACS) CD4+ T cell isolation kit (Miltenyi Biotec; Cambridge, MA) according to the manufacturer's instructions.
Infectivity assay
Inhibition of HIV-1 infection was measured as adapted from previously established methods.19–21 Infections were performed in an assay medium containing RPMI 1640+5% FBS, 100 U/ml IL-2, and penicillin/streptomycin. Baseline assay medium prepared with 5% FBS contains 1 mg/ml of bovine serum albumin (BSA), the minimum amount of BSA necessary to prevent nonspecific binding of EFV7 to the test apparatus. Different lots of FBS were preselected for BSA concentration to be at 1 mg/ml in a 5% FBS baseline medium. Lyophilized HSA (Sigma Aldrich, St. Louis, MO) was dissolved into baseline assay medium to final concentrations of 80, 60, 40, 20, 10, 5, and 1 mg/ml HSA. These HSA concentrations span the physiologically relevant concentrations of HSA in various anatomic compartments, including blood plasma, seminal plasma, and cerebrospinal fluid. HSA was quantified using a colorimetric bromocresol green assay (BioAssay Systems, Hayward, CA) according to the manufacturer's instructions. CD4+ T cells from each individual donor were seeded at 1×105 cells per well for infectivity assays, and at 2×106 cells per well for experiments that simultaneously measured intracellular drug concentrations. Cells were incubated in HSA and ARV conditions for 3 h prior to infection. Infection was performed via spinoculation, incubated for 3 days, and analyzed for infection by flow cytometry of GFP expression. For each subject and experiment, viable cells were gated for expression of GFP. Analysis and gating for infectivity were determined using analysis software, FlowJo version 7.6.5 (Tree Star, Inc., Ashland, OR).
Infectivity for each individual sample (fu (SAMPLE)) is characterized as the fraction of virus infection events affected by the drug (fa) relative to the fraction of virus infection events unaffected by the drug (fu). Infectivity for each research participant is determined relative to the positive control (maximal infection–HIV-GFP virus and solvent vehicle only, no drug) and negative control (background—solvent vehicle only, no virus, no drug). All samples within each assay for a given subject were analyzed in triplicate to control for intraindividual variability in infectivity. The data from infectivity experiments are calculated as follows:
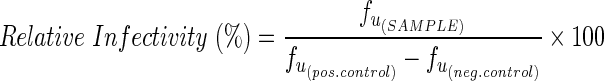 |
where fu=1 – fa.
Quantification of intracellular concentrations by UPLC-MS/MS
IC concentrations of EFV, ETR, and RAL, were determined using individual ultraperformance liquid chromatography tandem mass spectrometry (UPLC-MS/MS) assays performed using an AB Sciex QTRAP 5500 (AB Sciex, Foster City, CA) interfaced with an Acquity UPLC (Waters Inc., Milford, MA). Cells were isolated by centrifuging for 10 min at 300×g, 4°C, washed two times with ice cold phosphate-buffered saline (PBS) (no FBS), and resuspended in PBS containing 2% FBS and 0.5 mM EDTA for cell counting and UPLC-MS/MS analysis. Cell counts were performed using Guava ViaCount (Millipore, Billerica, MA) according to the manufacturer's instructions. IC concentrations were calculated using 0.28 pl as the average mean cell volume of a CD4+ PBMC.24
EFV samples were analyzed by modifications of a previous method.25 Briefly, samples were lysed and extracted using a liquid:liquid extraction method of 600 μl of 50 mM ammonium formate and 900 μl of hexanes:ethyl acetate (1:1). The organic layer was decanted and evaporated to dryness. Samples were reconstituted in methanol for analysis. EFV was resolved using a reverse-phase UPLC column (2.1×50 mm Acquity UPLC BEH C18) with a flow rate of 0.5 ml/min by gradient elution (mobile phase A of 0.1% formic acid in water; mobile phase B of 0.1% formic acid in acetonitrile). EFV was detected via negative-ion multiple reaction monitoring (MRM). The assay was linear from 0.125 to 500 ng/ml, employing a fluorinated analog of EFV (F-EFV) as the internal standard. EFV and F-EFV were detected via the MRM transitions m/z 314.0>244.1 and m/z 298.0>227.9, respectively.
ETR samples were lysed and extracted using a procedure identical to that used for EFV extraction. ETR was resolved using a reverse-phase UPLC column (2.1×50 mm, 2.5μm, Waters XTerra MS C18) with a flow rate of 0.5 ml/min by gradient elution (mobile phase A of 0.1% formic acid in water; mobile phase B of 0.1% formic acid in acetonitrile). ETR was detected via positive-ion MRM. The assay was linear from 0.0625 ng/ml to 500 ng/ml, employing 2H6-etravirine (2H6-ETR) as the internal standard. ETR and 2H6-ETR were detected via the MRM transitions m/z 435.1>304.0 and m/z 443.3>304.0, respectively.
For RAL analysis, samples were lysed and extracted using protein precipitation with 600 μl of methanol. The methanol layer was decanted and evaporated to dryness for analysis. RAL samples were reconstituted in water containing 0.1% formic acid for analysis. RAL was resolved using a reverse-phase HPLC column (3.0×50 mm, 3.5 μm, Waters XBridge MS C18) with a flow rate of 0.5 ml/min by isocratic elution (47.5% mobile phase A of 0.1% formic acid in water; 52.5% mobile phase B of 0.1% acetic acid in methanol). RAL was detected via positive-ion MRM. The assay was linear from 0.125 ng/ml to 500 ng/ml, employing 13C6-raltegravir (13C6-RAL) as the internal standard. RAL and 13C6-RAL were detected via the MRM transitions m/z 446.1>109.9 and m/z 451.1>114.9, respectively.
Pharmacodynamic relationships
The relationship between drug concentration and antiviral effect was determined using a median-effect estimation. The slope of the median-effect curve, m, was determined by least-squares regression for the line described in Equation 2.
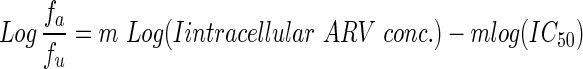 |
We calculated the IC50 to estimate drug concentration at half-maximal drug effect (fa=fu) where log(fa/fu) equals 0. Goodness of fit was evaluated with coefficient of determination (r2).
Results
Effect of extracellular HSA on intracellular drug concentrations
At a 5 and 10 ng/ml total concentration of EFV (Fig. 1A), increasing concentrations of EC HSA result in a decreasing IC penetration of EFV (Fig. 1A). Decreasing IC penetration with increasing EC HSA is also observed for RAL at 5 and 10 ng/ml total concentration (Fig. 1B), and for ETR at 2 and 10 ng/ml total concentration (Fig. 1C). The total concentrations were chosen to be able to simultaneously assay within this system changes in infectivity due to increased EC HSA.
FIG. 1.

Impact of extracellular human serum albumin (HSA) on intracellular antiretroviral (ARV) concentration. Effect of extracellular HSA on intracellular ARV penetration is measured by incubating CD4+ T cells with increasing total concentrations of HSA at 5 ng/ml (●) or 10 ng/ml (○) of (A) efavirenz (EFV) and (B) raltegravir (RAL), and 2 ng/ml (●) or 10 ng/ml (○) of (C) etravirine (ETR). Data are presented as the mean and standard deviation of six subjects.
Effect of protein binding on infectivity
Increasing the extracellular concentration of HSA decreases the inhibitory drug effect of EFV (Fig. 2A). This effect was seen over the total EFV concentration range of 0.5–10 ng/ml (Fig. 2A). Although the EC EFV-HSA relationship is observed at concentrations above and below this range, the effect of IC distribution was most prominent at the depicted concentration range for each ARV studied. This same relationship—increasing EC HSA concentration associated with decreasing drug effect—was also observed over a range of concentrations for RAL (total concentration range of 0.5–10 ng/ml, Fig. 2B), ETR (0.1–10 ng/ml, Fig. 2C), and ENF (100–500 ng/ml, Fig. 2D).
FIG. 2.

Increased extracellular HSA leads to increased relative infection. Relative infection is quantified following exposure of CD4+ T cells to varying concentrations of HSA, HIV-green fluorescent protein (GFP) single replication-competent virus, and ARV concentrations of (A) 0.5–10 ng/ml for EFV, (B) 0.5–10 ng/ml for RAL, (C) 0.1–10 ng/ml for ETR, and (D) 100–500 ng/ml for enfuvirtide (ENF). Data are presented as the mean and standard deviation of six subjects for EFV, and three subjects for RAL, ETR, and ENF.
Relationship between intracellular ARV concentration and infectivity
The relationship between log10 antiviral effect (fa/fu) and log10 intracellular drug EFV concentration was linear for each individual as seen in median-effect plots (Fig. 3; coefficient of determination, Table 1). This linear relationship was also seen for ETR and RAL (Supplementary Figs. S1 and S2; Supplementary Data are available online at www.liebertpub.com/aid). There are differences among the drugs for median slope (m) and IC50 as well as differences in interindividual variability for a given drug, especially EFV IC50 estimates, among these research participants (Table 1).
FIG. 3.

The impact of intracellular EFV on antiviral effect. The intracellular ARV concentrations and relative impact on antiviral effect were assessed using a median-effect model. (A–F) The intracellular dose-response relationship for six individual subjects CD4+ T cells following dosing with EFV. Response is measured as the log10 of the fraction of virus affected by drug divided by the fraction of virus unaffected by drug (fa/fu).
Table 1.
Dose-Response Relationship Parameter Estimates
| Efavirenz (EFV) | Etravirine (ETR) | Raltegravir (RAL) | |
|---|---|---|---|
| IC50 (ng/ml) | 1.2 (0.51–5.39) | 39.06 (30.10–51.76) | 4.67 (3.91–5.02) |
| Slope (m) | 2.97 (2.26–5.85) | 3.52 (3.11–3.74) | 2.39 (2.15–3.74) |
| R2 | 0.88 (0.85–0.89) | 0.84 (0.78–0.87) | 0.62 (0.52–0.78) |
The median (IQR) of parameter estimates was established using a median effect plot to determine the dose-response relationship for intracellular antiretroviral (ARV) concentrations and antiviral effect, reported as intracellular ARV concentrations associated with half-maximal antiviral effect (IC50), and slope of the dose-response curve (m), and r2 values for EFV, ETR, and RAL.
The curvilinear relationships between all three variables (HSA concentration, IC concentration, and infectivity) for EFV are combined in the three-dimensional Fig. 4, thus demonstrating the impact of increasing HSA concentration resulting in increasing relative infection and decreasing IC EFV concentration.
FIG. 4.

Summary of protein binding for EFV. Three-dimensional representation of the impact of increasing HSA concentration on increasing relative infection and decreasing intracellular EFV concentration.
Discussion
Because we have previously demonstrated how the distribution of EFV is determined by protein binding, we used EFV as an example of a highly protein-bound ARV to demonstrate that increased EC HSA concentration reduces both IC EFV concentration and antiviral effect. To generalize this concept, it was further demonstrated in three additional antiretroviral drugs, of varied protein binding and mechanisms of action. This concept is important to understanding both the pharmacokinetics and pharmacodynamics of ARV drugs. Since HIV is capable of permeating many extravascular physiological compartments, it is necessary that ARVs also penetrate into these compartments at suppressive antiviral concentrations to avoid generating pharmacological sanctuaries and permitting viral replication and/or generation of resistant variants. These findings are consistent with the free drug hypothesis, which has yet to be demonstrated experimentally for ARVs.
Highly protein-bound drugs can often fail in development, lacking in vivo efficacy despite promising in vitro results. For example, Fischel et al.14 described the development of ARV protease inhibitor SC-52151, which upon phase I/II development failed due to a lack of antiviral activity. This failure resulted from high protein binding and impaired intracellular uptake. Several reports have addressed the need to understand both the in vitro and in vivo pharmacodynamic impact of protein binding.6,14 Preliminary studies have begun to explain the impact of protein binding on antibacterial activity; however, there has not been any work to date describing the impact of protein binding on antiviral effect. Garrison et al.26 and Cha et al.27 showed the effects of vancomycin and daptomycin killing of Staphylococcus aureus in the presence and absence of protein. While these results showed protein binding affected the pharmacodynamic effect of the drugs, they simply tested the presence or absence of protein on antibacterial effect without any examination of clinically relevant ranges of protein concentration.
Zeitlinger et al.28 showed the effect of protein on fluoroquinolone killing of Staphylococcus aureus by varying the percentage of human serum or albumin added to the media to analyze the effect on bacterial killing. This methodology does not allow isolation of the variable of interest, protein binding, particularly if a drug is bound to additional proteins. In addition, this method also causes a dilution of binding proteins to concentrations less than those clinically relevant. Because drug binding proteins, including HSA, exist at vastly different concentrations in anatomic compartments (blood plasma, seminal plasma, cerebrospinal fluid), it is best to examine a range of concentrations relative to the distribution and disposition of the drug of study. Dissolution of lyophilized HSA does not always demonstrate the same degree of binding to drugs as HSA in serum, and therefore should be assessed for the drug of interest prior to using this method for protein binding analysis.14
HSA is particularly susceptible to conformational change resulting from an imbalance in fatty acid or electrolytes, or pH changes.29–31 We have previously demonstrated this was not an issue for EFV.7 Our method takes into account protein heterogeneity in a dynamic system to better understand the impact of protein binding on ARVs. While no in vitro assay perfectly replicates in vivo results, the development of this model system more closely approximates clinical conditions to better understand the impact of protein binding on ARVs among varied anatomic compartments.
Since most laboratories simply measure total drug concentration (bound and unbound) in making in vitro assessment of drug efficacy, these results can be highly misleading for the prediction of efficacy in different anatomic compartments with varying binding protein concentrations. It is believed that the variation in unbound drug concentration determines the effect in various anatomic compartments, but this has rarely been assessed given the difficulty in assessing the unbound drug fraction in extravascular compartments that are not readily accessible.32 The site of action for most ARVs is intracellular; however, IC pharmacokinetics and pharmacodynamics of ARVs are poorly understood. In addition, there is still large methodological uncertainty in measuring intracellular drug concentrations. For our extraction procedure, washing the cells with ice cold PBS (without serum) did not result in drug loss in the wash procedure. Addition of FBS or serum to a wash medium would, however, result in the loss of drug throughout a wash step, particularly for highly protein-bound drugs that have a high affinity for binding extracellular proteins. In regard to drug binding to intracellular proteins or lipids, there is both a lack of knowledge and a lack of reliable assays to accurately quantify the contribution of intracellular binding and the ability to differentiate between drug localized in cell membranes, within the cytoplasm, or bound to intracellular components.32 While we have not yet resolved protein binding within the cell, we have successfully demonstrated the effect of varying HSA outside the cell on IC distribution into the cell and the resulting antiviral effect.
Dose-response parameters were estimated in this study to describe the relationship between infectivity and intracellular drug concentration. These parameter estimates demonstrated modest interindividual variability for IC50 and m; however, the EFV IC50 varies more than 10-fold between upper and lower quartiles (largely due to having only a few data points below the IC50 estimate limited by UPLC-MS/MS sensitivity to detect these very low concentrations). This is the first study to examine an intracellular IC50 estimate as opposed to a whole system IC50 estimate. These data suggest differences unique to each drug with variation among individuals that affect both intracellular distribution and concentration-response for these ARVs that may impact a drug's overall antiviral efficacy.
A more traditional sigmoid Emax model also fits the data well and IC50 values were comparable between the two methods.33 We chose the median effect approach because it clearly demonstrated the variability observed in m, which has a potentially large impact on antiviral effect.20 Our m values, based on intracellular ARV concentrations, were roughly two times higher than previously published m values (EFV 1.69±0.08, ETR 1.81±0.23, and RAL 1.10±0.05) using the same methods.20 Our intracellular IC50 values were comparable to prior IC50 values for EFV (1.71±0.28 ng/ml) and RAL (6.62±0.8 ng/ml), but 20 times higher than prior values for ETR (1.87±0.3 ng/ml).20 In a recent analysis of the dose-response curve slope, it was demonstrated how heterogeneity largely affects slope.34 In particular, when heterogeneity is limited, the dose-response curves were steeper, which reflects a greater response of the drug to the target. In our system, because we are assessing only the intracellular concentration, it minimizes the heterogeneity as compared to a whole assay system that encompasses both intracellular and extracellular concentration in the dose-response analysis. This may be a possible explanation for our observed steep slopes. It may be of future value to analyze the relationship between intracellular antiviral effect and total system antiviral effect relative to the median effect slope (m). Understanding the relationship between HSA protein binding, intracellular distribution, and antiviral effect among diverse anatomic compartments could lead to a better understanding of individual ARV effects in these anatomic locations.
In summary, we have successfully developed a novel in vitro model system for evaluating the pharmacodynamic effect of HSA concentration on both intracellular ARV concentration and antiviral effect. We then applied this to quantitatively describe the impact of HSA on the concentration-response relationships of IC drug concentration and antiviral effect. These results provide experimental confirmation of the free-drug hypothesis for several ARV drugs over a range of clinically relevant protein concentrations, supporting the commonly held assumption that free drug concentration determines drug distribution and effect.
Supplementary Material
Acknowledgments
This work was supported, in part, by the National Institutes of Health (NIH) grant 5UM1AI068613. The contents of this article are solely the responsibility of the authors and do not necessarily represent the official view of the NIH. The authors are grateful to the study participants and to the staff of the Johns Hopkins Drug Development Unit.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Hammer SM. Eron JJ. Reiss P, et al. Antiretroviral treatment of adult HIV infection: 2008 recommendations of the international aids society–USA panel. JAMA. 2008;300(5):555–570. doi: 10.1001/jama.300.5.555. [DOI] [PubMed] [Google Scholar]
- 2.Grant RM. Lama JR. Anderson PL, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363(27):2587–2599. doi: 10.1056/NEJMoa1011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen MS. Chen YQ. McCauley M, et al. Prevention of HIV-1 infection with early antiretroviral therapy. N Engl J Med. 2011;365(6):493–505. doi: 10.1056/NEJMoa1105243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mujugira A. Baeten JM. Donnell D, et al. Characteristics of HIV-1 serodiscordant couples enrolled in a clinical trial of antiretroviral pre-exposure prophylaxis for HIV-1 prevention. PLoS ONE. 2011;6(10):e25828. doi: 10.1371/journal.pone.0025828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abdool Karim Q. Abdool Karim SS. Frohlich JA, et al. Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science. 2010;329(5996):1168–1174. doi: 10.1126/science.1193748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith DA. Di L. Kerns EH. The effect of plasma protein binding on in vivo efficacy: Misconceptions in drug discovery. Nat Rev. 2010;9(12):929–939. doi: 10.1038/nrd3287. [DOI] [PubMed] [Google Scholar]
- 7.Avery LB. Bakshi RP. Cao YJ. Hendrix CW. The male genital tract is not a pharmacological sanctuary from efavirenz. Clin Pharmacol Ther. 2011;90(1):151–156. doi: 10.1038/clpt.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischl MA. Richman DD. Flexner C, et al. Phase I/II study of the toxicity, pharmacokinetics, and activity of the HIV protease inhibitor SC-52151. JAIDS J Acquir Immune Defic Syndr. 1997;15(1):28–34. doi: 10.1097/00042560-199705010-00005. [DOI] [PubMed] [Google Scholar]
- 9.Benet LZ. Hoener BA. Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther. 2002;71(3):115–121. doi: 10.1067/mcp.2002.121829. [DOI] [PubMed] [Google Scholar]
- 10.Craig WA. Kunin CM. Significance of serum protein and tissue binding of antimicrobial agents. Annu Rev Med. 1976;27(1):287–300. doi: 10.1146/annurev.me.27.020176.001443. [DOI] [PubMed] [Google Scholar]
- 11.Avery LB. Sacktor N. McArthur JC. Hendrix CW. Protein-free efavirenz concentrations in cerebrospinal fluid and blood plasma are equivalent: Applying the law of mass action to predict protein-free drug concentration. Antimicrob Agents Chemother. 2013;57(3):1409–1414. doi: 10.1128/AAC.02329-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boffito M. Back DJ. Blaschke TF, et al. Protein binding in antiretroviral therapies. AIDS Res Hum Retroviruses. 2003;19(9):825–835. doi: 10.1089/088922203769232629. [DOI] [PubMed] [Google Scholar]
- 13.Ganrot K. Laurell C-B. Measurement of IgG and albumin content of cerebrospinal fluid, and its interpretation. Clin Chem. 1974;20(5):571–573. [PubMed] [Google Scholar]
- 14.Zeitlinger MA. Derendorf H. Mouton JW, et al. Protein binding: Do we ever learn? Antimicrob Agents Chemother. 2011;55(7):3067–3074. doi: 10.1128/AAC.01433-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Company. BMS. Sustiva Package Insert; Princeton, NJ: 2012. [Google Scholar]
- 16.Pharmaceuticals. T. Intelence Package Insert; Titusville, NJ: 2012. [Google Scholar]
- 17.Merck & Co. I. Isentress Package Insert; 2011. [Google Scholar]
- 18.Genentech I. 2012. Trimeris, Inc. Fuzeon Package Insert.
- 19.Jilek BL. Zarr M. Sampah ME, et al. A quantitative basis for antiretroviral therapy for HIV-1 infection. Nat Med. 2012;18(3):446–451. doi: 10.1038/nm.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen L. Peterson S. Sedaghat AR, et al. Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat Med. 2008;14(7):762–766. doi: 10.1038/nm1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sampah MES. Shen L. Jilek BL. Siliciano RF. Dose–response curve slope is a missing dimension in the analysis of HIV-1 drug resistance. Proc Natl Acad Sci USA. 2011;108(18):7613–7618. doi: 10.1073/pnas.1018360108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang H. Zhou Y. Alcock C, et al. Novel single-cell-level phenotypic assay for residual drug susceptibility and reduced replication capacity of drug-resistant human immunodeficiency virus type 1. J Virol. 2004;78(4):1718–1729. doi: 10.1128/JVI.78.4.1718-1729.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pierson TC. Zhou Y. Kieffer TL. Ruff CT. Buck C. Siliciano RF. Molecular characterization of preintegration latency in human immunodeficiency virus type 1 infection. J Virol. 2002;76(17):8518–8531. doi: 10.1128/JVI.76.17.8518-8531.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simiele M. D'Avolio A. Baietto L, et al. Evaluation of the mean corpuscular volume of peripheral blood mononuclear cells of HIV patients by a Coulter counter to determine intracellular drug concentrations. Antimicrob Agents Chemother. 2011;55(6):2976–2978. doi: 10.1128/AAC.01236-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Avery LB. Parsons TL. Meyers DJ. Hubbard WC. A highly sensitive ultra performance liquid chromatography–tandem mass spectrometric (UPLC–MS/MS) technique for quantitation of protein free and bound efavirenz (EFV) in human seminal and blood plasma. J Chromatogr B. 2010;878(31):3217–3224. doi: 10.1016/j.jchromb.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garrison M. Vance-Bryan K. Larson T. Toscano J. Rotschafer J. Assessment of effects of protein binding on daptomycin and vancomycin killing of Staphylococcus aureus by using an in vitro pharmacodynamic model. Antimicrob Agents Chemother. 1990;34(10):1925–1931. doi: 10.1128/aac.34.10.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cha R. Rybak MJ. Influence of protein binding under controlled conditions on the bactericidal activity of daptomycin in an in vitro pharmacodynamic model. J Antimicrob Chemother. 2004;54(1):259–262. doi: 10.1093/jac/dkh259. [DOI] [PubMed] [Google Scholar]
- 28.Zeitlinger M. Sauermann R. Fille M. Hausdorfer J. Leitner I. Müller M. Plasma protein binding of fluoroquinolones affects antimicrobial activity. J Antimicrob Chemother. 2008;61(3):561–567. doi: 10.1093/jac/dkm524. [DOI] [PubMed] [Google Scholar]
- 29.Bojko B. Sułkowska A. Maciążek-Jurczyk M. Równicka J. Pentak D. Sułkowski W. Alterations of furosemide binding to serum albumin induced by increased level of fatty acid. J Pharm Biomed Anal. 2010;51(1):273–277. doi: 10.1016/j.jpba.2009.07.025. [DOI] [PubMed] [Google Scholar]
- 30.Dockal M. Carter DC. Rüker F. Conformational transitions of the three recombinant domains of human serum albumin depending on pH. J Biol Chem. 2000;275(5):3042–3050. doi: 10.1074/jbc.275.5.3042. [DOI] [PubMed] [Google Scholar]
- 31.Matsuyama K. Sen AC. Perrin JH. The effects of pH, calcium and chloride ions on the binding of tolmetin to human serum albumin: Circular dichroic, dialysis and fluorometric measurements. J Pharm Pharmacol. 1987;39(3):190–195. doi: 10.1111/j.2042-7158.1987.tb06247.x. [DOI] [PubMed] [Google Scholar]
- 32.Bazzoli C. Jullien V. Le Tiec C. Rey E. Mentré F. Taburet AM. Intracellular pharmacokinetics of antiretroviral drugs in HIV-infected patients, and their correlation with drug action. Clin Pharmacokinet. 2010;49(1):17–45. doi: 10.2165/11318110-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 33.Goutelle S. Maurin M. Rougier F, et al. The Hill equation: A review of its capabilities in pharmacological modelling. Fund Clin Pharmacol. 2008;22(6):633–648. doi: 10.1111/j.1472-8206.2008.00633.x. [DOI] [PubMed] [Google Scholar]
- 34.Shen L. Rabi SA. Sedaghat AR, et al. A critical subset model provides a conceptual basis for the high antiviral activity of major HIV drugs. Sci Transl Med. 2011;3:91ra63. doi: 10.1126/scitranslmed.3002304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.