

Summary
Interleukin-33 (IL-33) and its receptor ST2 are over-expressed in clinical colitis tissue. However, the significance of these observations is at present unknown. Significantly, we demonstrate here that IL33 and ST2 are the primary early genes induced in the inflamed colon of BALB/c mice following dextran sulphate sodium (DSS)-induced experimental ulcerative colitis. Accordingly diarrhoea and DSS-induced colon inflammation were impaired in ST2−/− BALB/c mice and exacerbated in wild-type mice by treatment with exogenous recombinant IL-33, associated respectively with reduced and enhanced expression of chemokines (CXCL9 and CXCL10), and inflammatory (IL-4, IL-13, IL-1, IL-6, IL-17) and angiogenic (vascular endothelial growth factor) cytokines in vivo. The exacerbation effect of treatment with recombinant IL-33 on DSS-induced acute colitis was abolished in IL-4−/− BALB/c mice. Hence, IL-33 signalling via ST2, by inducing an IL-4-dependent immune response, may be a major pathogenic factor in the exacerbation of ulcerative colitis.
Keywords: colitis, early interleukin-33 expression, interleukin-4 deficiency, ST2 deficiency
Introduction
Ulcerative colitis (UC) is an inflammatory disease of the colon associated with recurring inflammation and the formation of ulcers.1 This leads to clinical symptoms and signs including diarrhoea and serious complications, such as peritonitis and increased risk of colorectal cancer.1 The aetiology of UC is largely unknown, which is the main reason why current therapeutic options are limited. Environmental and infectious disease factor-mediated barrier dysfunction and abnormal angiogenesis in gut epithelium are thought to play a critical role in the initiation and perpetuation of the disease.1–2
Dextran sulphate sodium (DSS) -induced colitis in mice is a well-established model for human UC.3 Mice fed with DSS polymers develop disease similar to human UC, characterized by diarrhoea, colonic inflammation and ulceration. This is a result of direct toxic effects of DSS on the gut epithelial cells of the basal crypts.3–4 The induction of acute DSS-induced colitis does not depend on lymphocytes;4 therefore it is a particularly useful model to study innate immune mechanisms of the intestinal epithelium in the pathogenesis of colitis.
The pathogenesis of ulcerative colitis in humans and animal models is primarily associated with dysregulation of type II cytokines [interleukin-4 (IL-4), IL-5 and IL-13],2–7 whereas type I [interferon-γ (IFN-γ)], and pro-inflammatory [IL-1, IL-6, IL-17 and tumour necrosis factor-α (TNF-α)] cytokines may also contribute to the pathogenesis, probably in the chronic phase of UC.2–10 The early innate inflammatory signal(s) that coordinate the engagement of these cytokines are unresolved although IL-33, a new member of the IL-1 family, is a potential candidate.11
Interleukin-33 is a pleiotropic cytokine that signals via its receptor ST2 and can elicit different immune responses depending on context.11–12 It is expressed primarily in the epithelium and endothelium and can be released when cells sense inflammatory signals or undergo necrosis.11–12 The IL-33 receptor, ST2, is expressed by almost all innate cells but only by selected adaptive immune cells.11–17 Interleukin-33 signalling via ST2 can induce both antigen-dependent and antigen-independent type II immune responses by directly activating a wide-range of innate immune cells including eosinophils, macrophages, nuocytes, mast cells or T helper type 2 (Th2) and IL-5+ Th cells in vitro and in vivo.11–17 In addition, IL-33 can also promote Th1 and/or Th17 type responses in pro-inflammatory disorders in mice, by as yet undefined mechanisms.18–19 Increasing evidence suggests that IL-33 and ST2 play a pathogenic role in inflammatory bowel disease.20–23 Interleukin-33 and ST2 expression is increased in inflamed colonic mucosa and in the serum of patients with inflammatory bowel disease.20–23 Experimental IL-33 gene-deletion impairs pathogenesis of colitis,24 although the mechanisms by which the IL-33/ST2 system exacerbates colitis are unresolved.
The aims of this study were to elucidate the mechanisms by which IL-33 exacerbates experimental colitis in mice. Our study demonstrated that IL-33 and ST2 are the genes early induced in the colonic tissue during DSS-induced colitis. Furthermore, IL-33 exacerbates acute colitis in association with the induction of pro-inflammatory and angiogenic cytokines as well as chemokine production in an ST2-dependent and IL-4-dependent manner.
Materials and methods
Mice
BALB/c mice were purchased from Harlan Olac (Bicester, UK), and ST2−/−, IL-4−/− and IL-4R−/− mice on a BALB/c background were generated as described previously.13–17 Mice were housed in specific pathogen-free conditions at the University of Glasgow in accordance with the UK Home Office animal welfare guidelines.
The induction of DSS colitis
For the induction of acute colitis, female mice were given 3·5% (weight/volume) DSS (ICN Biomedicals, Aurora, OH) in their drinking water from day 0 for 12 consecutive days. Some mice received recombinant IL-33 (1 μg/mouse/day) or PBS intraperitoneally daily from day 0 for 19 days. The IL-33 was produced and purified as previously described.13 The body weight and stool consistency were monitored daily. Diarrhoea was scored as follows: 0 (normal); 2 (loose stools); 4 (watery diarrhoea).25 Body weight loss was calculated as the difference between the baseline weight on day 0 and the body weight on a particular day.
Cytokine/chemokine measurements
Colons were opened longitudinally and washed in sterile PBS supplemented with 1% penicillin/streptomycin (Life Technologies, Carlsbad, CA). Three segments from the distal colon of 1 cm in length were placed in 24 flat-bottom well culture plates (Costar, Cambridge, MA) containing fresh RPMI-1640 (Life Technologies) supplemented with 1% penicillin/streptomycin and incubated at 37° for 24 hr. Culture supernatants were then harvested, centrifuged at 13 000 g, and stored at − 20°. Cytokine/chemokine concentrations were detected by a multi-cytokine/chemokine (20-plex) bead fluorescence assay (Invitrogen, Paisley, UK) according to the manufacturer's instructions, using a Luminex platform.
Histological analysis
Colon specimens were fixed in 10% neutral formalin, embedded in paraffin and stained with haematoxylin & eosin. Histological examination was performed on three serial sections at six different sites of the colon and was scored blind using a standard histological scoring system.25
Meta-analysis of high-throughput transcriptomics data
Raw RNA microarray (Affymetrix CEL) files in the public domain derived from mouse colon tissue response to DSS induction at days 0, 2, 4 and 6 were downloaded from the Gene Expression Omnibus (GEO, GSE22307 and ref 26) and analysed as previously described.27 Briefly, the analysis of the differential gene expression patterns used Affymetrix Gene Chip Mouse Genome 430 2.0 Array.26 The CEL files were normalized with the RMA algorithm and subjected to a highly stringent statistical analysis using one-way analysis of variance, followed by the Tukey honestly significant difference post-hoc test and multiple testing correction by applying the Benjamini–Hochberg false discovery rate (P < 0·05) and using genespring gx11 software (Agilent, Santa Clara, CA). The significantly expressed genes were selected by a standard cut-off at twofold increased expression compared with the values on day 0. These differentially expressed genes were then classified based on Gene Ontology (GO) software specifically for genes implicated in the ‘regulation of inflammatory response’ as well as the ‘cytokines and chemokines’ in the colonic epithelium of DSS-induced colitis in mice.
Statistical analysis
Analysis using Student's t-test was applied to in vitro studies. Analysis between individuals in groups in vivo was by analysis of variance followed by Student's t-test. Results are expressed as mean ± SEM, and are representative of at least two individual experiments. P < 0·05, was considered significant.
Results
IL-33 and ST2 are the major genes early induced in the colonic tissue in DSS colitis
While it has been suggested that IL33 and ST2 are expressed in colonic tissue and in epithelial cells in clinical colitis,20–23 the kinetics of their expression and relative expression compared with other DSS-induced genes in inflamed colonic tissue is unknown. To understand the inflammatory process associated with the initiation of colitis, we systematically studied the early colon gene expression profile of DSS-induced colitis by analysing the publicly available microarray datasets deposited in the GEO using a meta-analysis approach.26–27
We specifically focused on the expression of cytokines and chemokines, and genes implicated in the regulation of inflammation using the Gene Ontology Analysis module in genespring gx11. Hierarchical clustering analysis showed that IL33 was the strongest of the 40 differentially expressed cytokine and chemokine genes expressed early in the colonic tissue (see Supplementary material, Fig. S1A). Furthermore, IL33 and its receptor; the ST2 gene (IL1RL1) were the most highly induced genes, among the 28 genes, involved in the regulation of the inflammatory response (Fig. S1B). The induced IL33 message in colonic tissue was detectable from day 4, and ST2 from day 6 after DSS administration (Fig. 1a and Fig. S1A,B). The expression levels of several other key inflammatory cytokine and chemokines, including IL-1β, IL-6, CXCL9 and CXCL10 were also significantly up-regulated (> 2-log fold) by DSS in the acute inflamed colonic tissue (Fig. 1a). However, Th2 (IL-4 and IL-5), Th1 (IFN-γ), IL-17 and the ‘alarmin’ (IL-1β and HMGB1) cytokine genes were not significantly induced (Fig. S1A,B, and data not shown).
Figure 1.
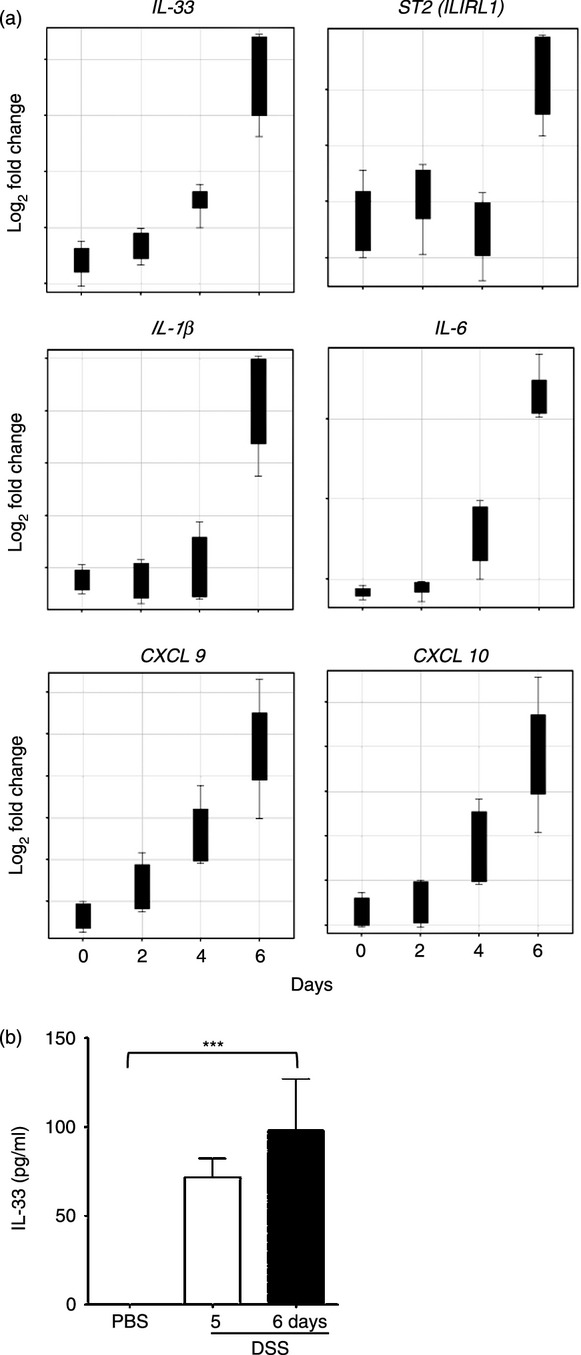
Interleukin-33 (IL-33) and ST2 are the early induced genes in colonic tissue of colitic mice. (a) Genespring GX11 analysis of Affymetrix Gene-Chip Expression Data (GSE22307) from the colonic tissue of dextran sulphate sodium (DSS) -induced colitis on Days 0, 2, 4 and 6, respectively, in mice. The differentially expressed cytokines and chemokines and genes implicated in the regulation of inflammation has been obtained from the hierarchical clusters and displayed as Box Plots (Fig. S1). (b) In vitro IL-33 protein levels in cultured colonic tissues from mice five or 6 days after DSS or PBS administration, respectively.
We further determined IL-33 protein levels in vitro in the cultured colonic tissue from mice that had received DSS or PBS as control as described in the Materials and methods. Consistent with the induction of IL33 message (Fig. 1a), IL-33 secretion in cultured colonic tissue from mice 5 days after DSS administration was also significantly enhanced compared with that from PBS-administered control mice (Fig. 1b).
These results therefore demonstrated that IL-33 and ST2 are key genes induced early in the inflamed colon of DSS-treated mice, suggesting that this cytokine/receptor system may be associated with the development of acute colitis.
ST2 deficiency impairs, and exogenous IL-33 exacerbates DSS-induced colitis
We next defined the importance of IL-33 and ST2 in the pathogenesis of colitis in wild-type (WT) and ST2−/− mice in vivo. Groups of WT and ST2−/− BALB/c mice were given either PBS, DSS, IL-33 alone or DSS plus IL-33 and the development of clinical signs of colitis was monitored up to day 20. As shown in Fig. 2(a), WT mice that received DSS but not PBS or IL-33 alone developed diarrhoea from day 10, which was markedly delayed by 10 days in ST2−/− mice. In addition, exogenous IL-33 significantly exacerbated diarrhoea particularly on day 20 in the WT but not ST2−/− DSS colitis mice (Fig. 2a). However, as reported,24 the injection of IL-33 or ST2 deficiency had no significant effect on body weight changes in the acute stage of colitis in mice (see Supplementary material, Fig. S2A,B).
Figure 2.
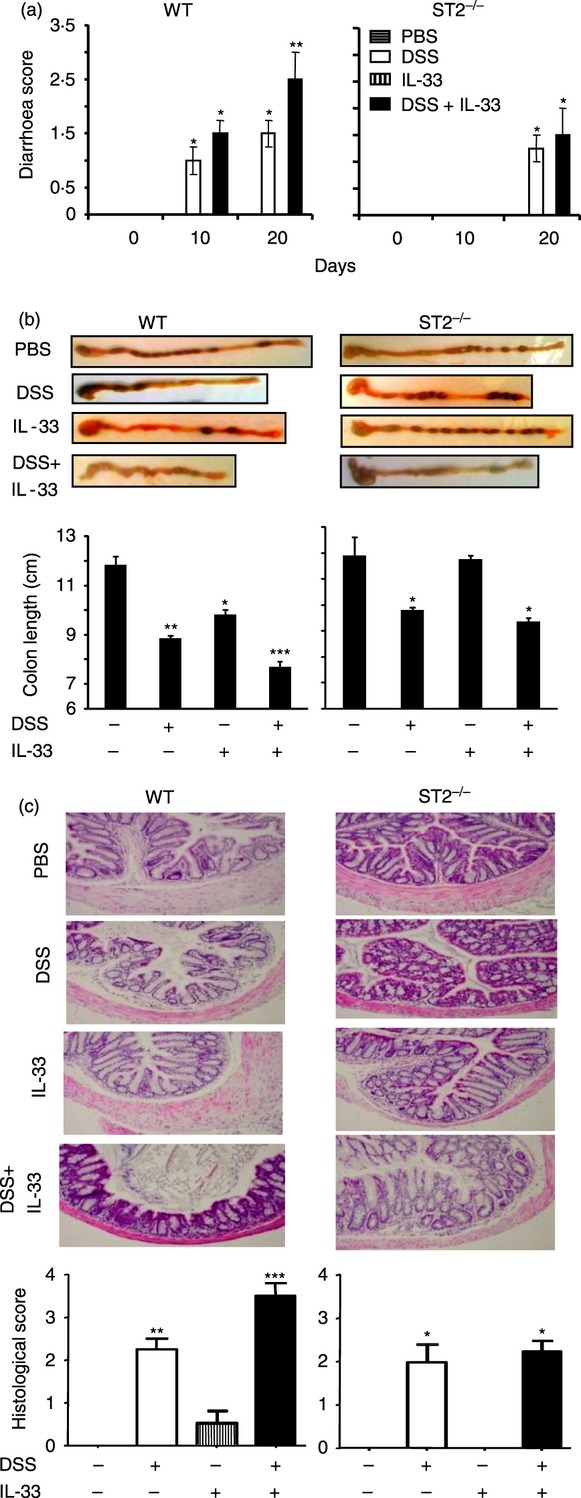
ST2 deficiency impairs and interleukin-33 (IL-33) injection exacerbates dextran sulphate sodium (DSS) colitis. Groups of wild-type (WT) and ST2−/− mice were either fed or not fed with DSS and injected with IL-33 (1 μg/mouse/day) or PBS. (a) Diarrhoea score and (b) colon length (on day 20) in the mice were determined as described in the Materials and methods. (c) The colon sections were stained with haematoxylin & eosin and scored. Data are representative of two experiments, n = 5 mice per group, *P < 0·05, **P < 0·01 compared with PBS controls.
Consistent with these clinical parameters, compared with PBS control, the IL-33 alone group had slightly shortened, and the DSS, but in particular the DSS plus IL-33-treated group had markedly shortened, colon lengths (Fig. 2b) and colon inflammation (Fig. 2c) that persisted for at least 8 days after DSS was withdrawn. These pathogenic changes examined in groups of similarly treated ST2−/− mice were significantly reduced (Fig. 2b,c).
These results demonstrated that IL-33/ST2 signals have a pathogenic role in the early development and exacerbation of acute colitis.
IL-33 enhances inflammatory cytokine and chemokine productions in colitis
Pro-inflammatory and angiogenic cytokines and inflammatory chemokines are closely associated with the pathogenesis of colitis.2–30 We further assessed the serum cytokine/chemokine profile in colitis mice by 20-plex Luminex (see Materials and methods). Experimental colitis was induced in naive WT and ST2−/− mice, which were then treated with or without IL-33 or PBS as described above. The experiment was terminated on day 20 and serum samples were collected for multi-cytokine/chemokine analysis. Interleukin-33 given alone significantly enhanced IL-13 and CXCL9 but reduced IFN-γ and IL-10 production in WT mice but not ST2−/− mice, compared with PBS control serum (Fig. 3). The group treated with DSS alone had no significant effect on serum cytokine concentration, except for increased IL-12 expression in WT and ST2−/− mice at this time-point. However, treatment with DSS plus IL-33 markedly enhanced most of the key pro-inflammatory cytokines and chemokines, including IL-4, IL-13, IL-6, IL-17, vascular endothelial growth factor (VEGF), CXCL9 and CXCL10 but reduced IL-10 and IFN-γ production in WT mice but not ST2−/− mice compared with control mice treated with PBS, DSS or IL-33 alone.
Figure 3.
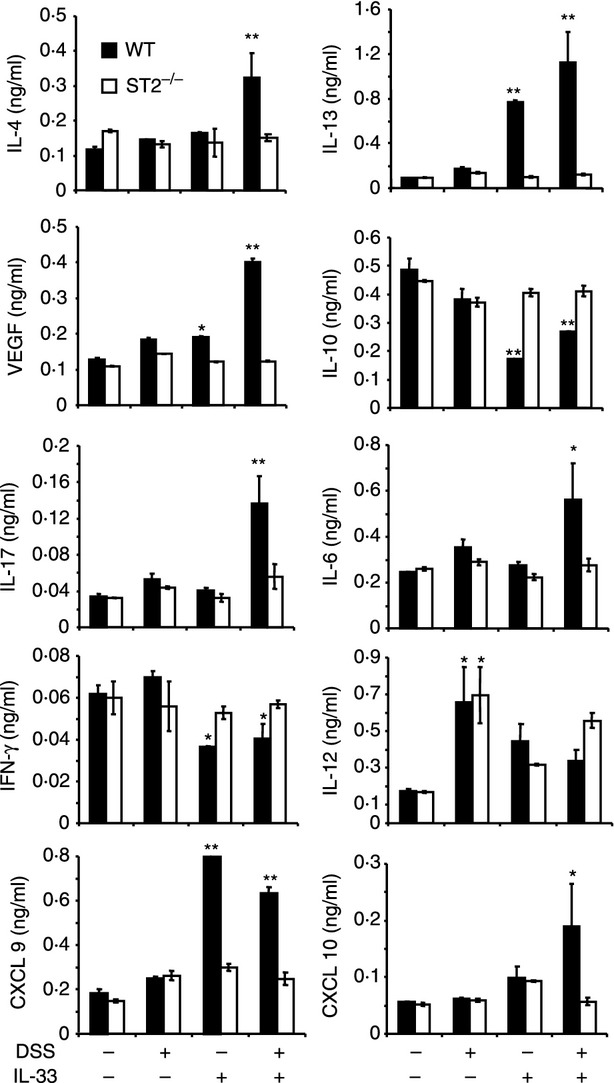
Interleukin-33 (IL-33) enhances key inflammatory cytokines and chemokines in colitis. Serum samples were collected on day 20 from the groups of wild-type (WT) and ST2−/− mice in Fig. 2 Total serum cytokine and chemokine concentrations were measured by luminex. Data are representative of two experiments, n = 5 mice per group, *P < 0·05, **P < 0·01 compared with the PBS control.
Together these results suggest that IL-33 may promote colitis by inducing an ST2-dependent production of inflammatory type II (IL-4, IL-13), type 17 (IL-6, IL-17) and angiogenic (VEGF) cytokines and chemokines (CXCL9 and CXCL10) as well as by reducing type I (IFN-γ) and immuno-suppressive (IL-10) cytokine production in mice with DSS-induced colitis.
IL-33 exacerbates colitis via IL-4
Type II cytokines (IL-4 and IL-13), in particular IL-4, have been reported to have a critical role in the initiation of DSS-induced colitis5,7 and we found, above, that IL-33 can induce serum type II cytokines in mice with colitis (Fig. 3). To define the requirement of IL-4 in colitis exacerbation and type II cytokine induction by IL-33, IL-4−/− mice were given the same treatments of PBS, IL-33, DSS or DSS plus IL-33 as described in Fig. 2. As reported,27 IL-4−/− mice that received DSS to induce colitis showed a delayed appearance of diarrhoea on day 10 and had attenuated pathogenic changes in the colon compared with WT mice (Fig. 4a,b). More importantly, similar to ST2−/− mice, IL-33 failed to exacerbate these clinical and pathological parameters of colitis in the IL-4−/− mice. Compared with WT controls, changes in colon length and histological score associated with administration of IL-33 were also not apparent in IL-4−/− mice (Fig. 4b).
Figure 4.
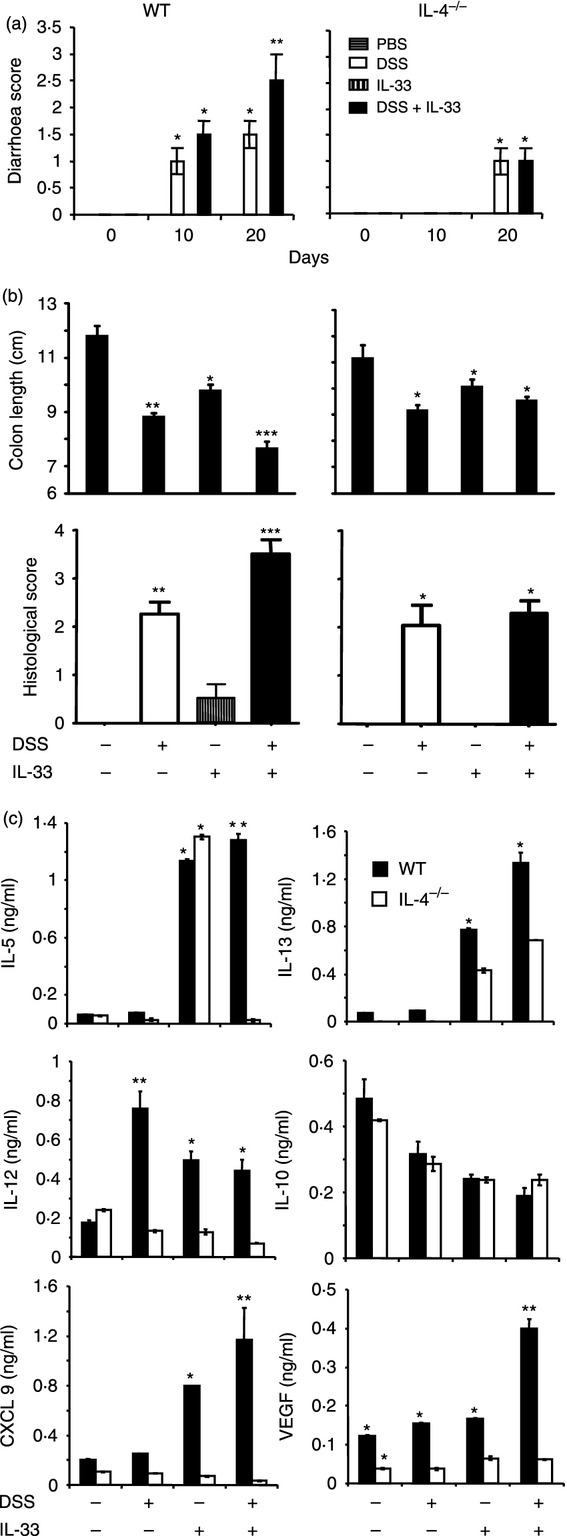
Interleukin-33 (IL-33) exacerbates colitis via IL-4. Groups of wild-type (WT) and IL-4−/− mice were injected with IL-33 as above. (a) Diarrhoea score, (b) colon length and histological score in the mice were determined as described in the Materials and methods. (c) The levels of serum cytokine were measured by luminex. Data are representative of two experiments, n = 5 mice per group, *P < 0·05, **P < 0·01 compared with the PBS group.
In addition, IL-4 deficiency abolished the production of IL-13, IL-12, CXCL9 and VEGF in the IL-33-treated group, IL-12 and VEGF in the DSS-treated group and IL-5, IL-13, IL-12, CXCL9 and VEGF in the DSS plus IL-33-treated group compared with cytokine and chemokine induction in similarly treated WT mice on day 20 (Fig. 4c). However, the serum concentrations of IL-10 were not affected by IL-4 deficiency.
We further investigated the importance of IL-4 receptor (IL-4R) in the context, which is required for both IL-4 and IL-13 signalling. We found that similar to ST2−/− and IL-4−/− mice, the shortened colon lengths in DSS or DSS plus IL-33 treated WT mice were also prevented in the groups of similarly treated IL-4R−/− mice (see Supplementary material, Fig. S3A). The reduced colon pathogenic change was accompanied by reduced IFN-γ and TNF-α, but enhanced IL-4 and IL-13 production in colon cultures in IL-4R−/− mice groups compared with the groups of similarly treated WT mice (Fig. S3B). The enhanced IL-4 and IL-13 may be a result of the loss of consumption of these cytokines in the IL-4R−/− mice tissues.
Therefore, these results suggest that IL-33 exacerbates colitis primarily via IL-4.
Discussion
Data reported in this comprehensive study reveal a hitherto unrecognized effect and mechanism by which the IL-33/ST2 axis exacerbates DSS-induced colitis. Increasing evidence suggests that the development of UC may be attributed to intestinal epithelial barrier dysfunction and abnormal angiogenesis.1–4 Our results contribute to this evidence and suggest that colon-derived IL-33 may be an additional key pathogenic factor that links epithelial damage and the initiation of colitis for several reasons:
(i) Interleukin-33 may function as a novel epithelial ‘alarmin’, similar to high-mobility group box 1 (HMGB1) and IL-1β, that initiate early inflammatory immune responses.31 Interestingly, we found that IL-33, but not IL-1β and HMGB1, is the earliest inflammatory cytokine induced in inflamed colonic epithelium in colitis (Fig. 1 and data not shown). Hence, colon-derived IL-33 may be a critical initiator of pathogenesis of DSS colitis. (ii) ST2−/− mice have impaired colitis (Fig. 2). (iii) IL-33 is capable of specifically inducing the key pathogenic cytokines (IL-4, IL-5, IL-13, IL-6, IL-17, IFN-γ, TNF-α and VEGF) and chemokines but reducing immunosuppressive (IL-10) cytokines in DSS-induced colitis via ST2 (Fig. 3).
Although it is recognized that type II cytokines, IL-4, IL-5 and IL-13 play a pathogenic role in the development of UC,5,7 until now, it was unknown how these typical Th2 cytokines were induced in the innate context of colitis and whether these cytokines contributed to the IL-33-mediated effect. Our mechanistic studies suggest that IL-33 can induce these type II cytokines and directly via IL-4 and IL-4R in colitis. It is well documented that IL-33 can induce all these type II cytokines by an array of innate cells, including eosinophils, basophils, mast cells, but not nuocytes which only produce IL-5 and IL-13, not IL-412–17 and data not shown). In contrast, T cells, which are the key cells expressing type II cytokines in allergy and asthma, are not the main IL-4 producers in this innate immune UC model, because naive T cells do not express ST2 in the absence of T-cell receptor activation and are thus unresponsive to IL-33.14–15
Our results also show for the first time that IL-4 is required for IL-33-mediated exacerbation of colitis, and for subsequent VEGF and CXCL9 production (Figs. 3 and 4). VEGF is a major pro-angiogenic cytokine and plays an important role in the pathogenesis of colitis by enhancing colonic permeability and facilitating migration of inflammatory cells.29 CXCL9 and CXCL10 are the key chemokines for the recruitment of monocytes and macrophages, and these are intimately associated with the pathogenesis of colitis.30–32 Together, these results provide a possible mechanism underlying the IL-33 / IL-4 pathogenic pathway in colitis.
Interleukin-12 and IL-17 are the key cytokines for type I and 17 responses and are also thought to play pathogenic roles in UC, Crohn's disease and the chronic stage of DSS-induced colitis.2,8 We noted in this study that IL-33 can also induce serum IL-12 and IL-17, at the later stages of the disease, 20 days after DSS administration (Fig. 3). This suggests that in addition to its role in the early stages of disease, IL-33 may also contribute to the switching of the early type II to late type I and IL-17 responses in the chronic stages of UC and Crohn's disease. Whereas it is still unclear how IL-33 induces IL-12 and IL-17 in colitis, as Th1 and Th17 cells do not express ST2, it is likely that IL-33 may promote these responses via innate cells.18–33
It is noteworthy that changes in the severity of colitis caused by IL-33 injection or ST2 deficiency were not significantly associated with a change in body weight in the mice (Fig. S2A,B). This is consistent with a previous study showing identical body weight loss in WT C57BL/6 and IL-33−/− mice when fed with DSS.24 Intriguingly, compared with WT mice, the IL-33−/− mice had a delayed recovery in body weight after withdrawal of DSS.24 However, this was not the case in ST2−/− mice in the present study and the reason is currently unclear. It may be because of the differences in genetic background of the mice and experimental conditions or the ST2-independent bioactivity of full-length IL-33 as previously suggested.34
Furthermore, recent evidence suggests that injection of IL-33 may have a beneficial effect on chronic DSS-induced colitis or trinitrobenzene sulphonic acid-induced colitis, a model of Crohn's disease in mice,35–36 suggesting that IL-33 may play a complex role in different types and throughout the duration of colitis. More studies are needed to clarify this issue.
Interleukin-33 is clearly expressed in the inflamed mucosa of patients with inflammatory bowel disease, particularly in UC, and is reduced after anti-TNF-α therapy.20–23 In these cases mucosal expression of IL-33 is also mostly localized to intestinal epithelial cells20,21 and in activated sub-epithelial myofibroblasts.22 However, the clinical relevance of the IL-33/ST2 system in inflammatory bowel disease is unknown. Our results have extended these clinical findings with a putative mechanism and suggest that colon-derived IL-33 may represent an important factor for the development and exacerbation of UC.
Acknowledgments
This study received financial support from the Arthritis Research UK, Medical Research Council UK and the Wellcome Trust, UK.
Disclosure
The authors have no financial conflicts of interest.
Supporting Information
Figure S1. The early gene profile in colonic epithelia in dextran sulphate sodium (DSS) colitis mice. Genespring GX11 analysis of Affymetrix Gene-Chip Expression Data (GSE22307) from the colonic epithelium of DSS-induced mouse colitis on Days 0, 2, 4 and 6, respectively. The differentially expressed genes were then classified and clustered based on Gene Ontology (GO) Analysis to decode the differentially expressed genes in (A) Cytokines and Chemokines and (B) Regulation of Inflammatory Response.
Figure S2. Interleukin-33 (IL-33) injection and ST2 deficiency do not significantly affect body weight in dextran sulphate sodium (DSS) colitis mice. Groups of wild-type (WT) and ST2−/− mice were fed with or without 3·5% DSS and/or injected with or without IL-33. (A) Body weight in WT colitis mice with or without IL-33 (B) Body weight in WT and ST2−/− colitic mice. Data are representative of two experiments, n = 8 mice per group.
Figure S3. Role of interleukin-4 receptor (IL-4R) in IL-33 exacerbates colitis. Groups of wild-type (WT) and IL-4R−/− mice were either fed or not fed with dextran sulphate sodium (DSS) and injected with IL-33 as above. (A) Colon length score in the mice was determined. (B) The cytokine levels in cultured colonic tissues from PBS, DSS or DSS plus IL-33-treated mice were measured by luminex as described in Materials and methods. n = 5 mice per group, *P < 0·05 compared with PBS group.
References
- Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–29. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. Novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- Dieleman LA, Ridwan BU, Tennyson GS, Beagley KW, Bucy RP, Elson CO. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology. 1994;107:1643–52. doi: 10.1016/0016-5085(94)90803-6. [DOI] [PubMed] [Google Scholar]
- Mizoguchi A, Mizoguchi E, Bhan AK. The critical role of interleukin 4 but not interferon γ in the pathogenesis of colitis in T-cell receptor α mutant mice. Gastroenterology. 1999;116:320–6. doi: 10.1016/s0016-5085(99)70128-9. [DOI] [PubMed] [Google Scholar]
- Heller F, Florian P, Bojarski C, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–64. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, Strong SA, Fiocchi C, Strober W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN-γ, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157:1261–70. [PubMed] [Google Scholar]
- Obermeier F, Kojouharoff G, Hans W, Scholmerich J, Gross V, Falk W. Interferon-γ (IFN-γ)- and tumour necrosis factor (TNF)-induced nitric oxide as toxic effector molecule in chronic dextran sulphate sodium (DSS)-induced colitis in mice. Clin Exp Immunol. 1999;116:238–45. doi: 10.1046/j.1365-2249.1999.00878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer C, Duewell P, Mayer C, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut. 2010;59:1192–9. doi: 10.1136/gut.2009.197822. [DOI] [PubMed] [Google Scholar]
- Sanchez-Munoz F, Dominguez-Lopez A, Yamamoto-Furusho JK. Role of cytokines in inflammatory bowel disease. World J Gastroenterol. 2008;14:4280–8. doi: 10.3748/wjg.14.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz J, Owyang A, Oldham E, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–90. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- Liew FY, Pitman NI, McInnes IB. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat Rev Immunol. 2010;10:103–10. doi: 10.1038/nri2692. [DOI] [PubMed] [Google Scholar]
- Kurowska-Stolarska M, Kewin P, Murphy G, et al. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol. 2008;181:4780–90. doi: 10.4049/jimmunol.181.7.4780. [DOI] [PubMed] [Google Scholar]
- Xu D, Chan WL, Leung BP, Huang F, Wheeler R, Piedrafita D, Robinson JH, Liew FY. Selective expression of a stable cell surface molecule on type 2 but not type 1 helper T cells. J Exp Med. 1998;187:787–94. doi: 10.1084/jem.187.5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolarski B, Kurowska-Stolarska M, Kewin P, Xu D, Liew FY. IL-33 exacerbates eosinophil-mediated airway inflammation. J Immunol. 2010;185:3472–80. doi: 10.4049/jimmunol.1000730. [DOI] [PubMed] [Google Scholar]
- Neill DR, Wong SH, Bellosi A, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464:1367–70. doi: 10.1038/nature08900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komai-Koma M, Brombacher F, Pushparaj PN, et al. Interleukin-33 amplifies IgE synthesis and triggers mast cell degranulation via interleukin-4 in naive mice. Allergy. 2012;67:1118–26. doi: 10.1111/j.1398-9995.2012.02859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Jiang HR, Kewin P, et al. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc Natl Acad Sci U S A. 2008;105:10913–8. doi: 10.1073/pnas.0801898105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verri WA, Jr, Guerrero AT, Fukada SY, et al. IL-33 mediates antigen-induced cutaneous and articular hypernociception in mice. Proc Natl Acad Sci U S A. 2008;105:2723–8. doi: 10.1073/pnas.0712116105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorelli L, Garg RR, Hoang SB, et al. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc Natl Acad Sci U S A. 2010;107:8017–22. doi: 10.1073/pnas.0912678107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran CJ, Nunez LE, Diaz-Jimenez D, et al. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:1097–107. doi: 10.1002/ibd.21175. [DOI] [PubMed] [Google Scholar]
- Kobori A, Yagi Y, Imaeda H, et al. Interleukin-33 expression is specifically enhanced in inflamed mucosa of ulcerative colitis. J Gastroenterol. 2010;45:999–1007. doi: 10.1007/s00535-010-0245-1. [DOI] [PubMed] [Google Scholar]
- Seidelin JB, Rogler G, Nielsen OH. A role for interleukin-33 in TH2-polarized intestinal inflammation? Mucosal Immunol. 2011;4:496–502. doi: 10.1038/mi.2011.22. [DOI] [PubMed] [Google Scholar]
- Oboki K, Ohno T, Kajiwara N, et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc Natl Acad Sci U S A. 2010;107:18581–6. doi: 10.1073/pnas.1003059107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara K, Yajima T, Kubo C, Yoshikai Y. Role of interleukin 15 in colitis induced by dextran sulphate sodium in mice. Gut. 2006;55:334–41. doi: 10.1136/gut.2005.076000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang K, Bruce M, Pattillo CB, Zhang S, Stone R, 2nd, Clifford J, Kevil CG. Temporal genomewide expression profiling of DSS colitis reveals novel inflammatory and angiogenesis genes similar to ulcerative colitis. Physiol Genomics. 2011;43:43–56. doi: 10.1152/physiolgenomics.00138.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar SD, Krishnamurthy K, Manikandan J, Pakeerappa PN, Pushparaj PN. Deciphering the key molecular and cellular events in neutrophil transmigration during acute inflammation. Bioinformation. 2011;6:111–4. doi: 10.6026/97320630006111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevceva L, Pavli P, Husband A, Ramsay A, Doe WF. Dextran sulphate sodium-induced colitis is ameliorated in interleukin 4 deficient mice. Genes Immun. 2001;2:309–16. doi: 10.1038/sj.gene.6363782. [DOI] [PubMed] [Google Scholar]
- Scaldaferri F, Vetrano S, Sans M, et al. VEGF-A links angiogenesis and inflammation in inflammatory bowel disease pathogenesis. Gastroenterology. 2009;136:585–95. doi: 10.1053/j.gastro.2008.09.064. [DOI] [PubMed] [Google Scholar]
- Tokuyama H, Ueha S, Kurachi M, Matsushima K, Moriyasu F, Blumberg RS, Kakimi K. The simultaneous blockade of chemokine receptors CCR2, CCR5 and CXCR3 by a non-peptide chemokine receptor antagonist protects mice from dextran sodium sulfate-mediated colitis. Int Immunol. 2005;17:1023–34. doi: 10.1093/intimm/dxh284. [DOI] [PubMed] [Google Scholar]
- Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS ONE. 2008;3:e3331. doi: 10.1371/journal.pone.0003331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S, Baumgart DC. Targeting leukocyte migration and adhesion in Crohn's disease and ulcerative colitis. Inflammopharmacology. 2012;20:1–18. doi: 10.1007/s10787-011-0104-6. [DOI] [PubMed] [Google Scholar]
- Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, Powrie F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010;464:1371–5. doi: 10.1038/nature08949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzina IG, Pickering EM, Kopach P, et al. Full-length IL-33 promotes inflammation but not Th2 response in vivo in an ST2-independent fashion. J Immunol. 2012;189:403–10. doi: 10.4049/jimmunol.1200259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobeta P, Doser K, Falk W, Obermeier F, Hofmann C. IL-33 attenuates development and perpetuation of chronic intestinal inflammation. Inflamm Bowel Dis. 2012;18:1900–9. doi: 10.1002/ibd.22900. [DOI] [PubMed] [Google Scholar]
- Duan L, Chen J, Zhang H, et al. Interleukin-33 ameliorates experimental colitis through promoting Th2/Foxp3+ regulatory T-cell responses in mice. Mol Med. 2012;18:753–61. doi: 10.2119/molmed.2011.00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The early gene profile in colonic epithelia in dextran sulphate sodium (DSS) colitis mice. Genespring GX11 analysis of Affymetrix Gene-Chip Expression Data (GSE22307) from the colonic epithelium of DSS-induced mouse colitis on Days 0, 2, 4 and 6, respectively. The differentially expressed genes were then classified and clustered based on Gene Ontology (GO) Analysis to decode the differentially expressed genes in (A) Cytokines and Chemokines and (B) Regulation of Inflammatory Response.
Figure S2. Interleukin-33 (IL-33) injection and ST2 deficiency do not significantly affect body weight in dextran sulphate sodium (DSS) colitis mice. Groups of wild-type (WT) and ST2−/− mice were fed with or without 3·5% DSS and/or injected with or without IL-33. (A) Body weight in WT colitis mice with or without IL-33 (B) Body weight in WT and ST2−/− colitic mice. Data are representative of two experiments, n = 8 mice per group.
Figure S3. Role of interleukin-4 receptor (IL-4R) in IL-33 exacerbates colitis. Groups of wild-type (WT) and IL-4R−/− mice were either fed or not fed with dextran sulphate sodium (DSS) and injected with IL-33 as above. (A) Colon length score in the mice was determined. (B) The cytokine levels in cultured colonic tissues from PBS, DSS or DSS plus IL-33-treated mice were measured by luminex as described in Materials and methods. n = 5 mice per group, *P < 0·05 compared with PBS group.