

Summary
The current study sought to delineate the gene expression profile of the host response in the caecum and colon during acute infection with Clostridium difficile in a mouse model of infection, and to investigate the nature of the unfolded protein response in this process. The infected mice displayed a significant up-regulation in the expression of chemokines (Cxcl1, Cxcl2 and Ccl2), numerous pro-inflammatory cytokines (Ifng, Il1b, Il6, and Il17f), as well as Il22 and a number of anti-microbial peptides (Defa1, Defa28, Defb1, Slpi and Reg3g) at the site(s) of infection. This was accompanied by a significant influx of neutrophils, dendritic cells, cells of the monocyte/macrophage lineage and all major subsets of lymphocytes to these site(s). However, CD4 T cells of the untreated and C. difficile-infected mice expressed similar levels of CD69 and CD25. Neither tissue had up-regulated levels of Tbx21, Gata3 or Rorc. The caeca and colons of the infected mice showed a significant increase in eukaryotic initiation factor 2α (eIF2α) phosphorylation, but neither the splicing of Xbp1 nor the up-regulation of endoplasmic reticulum chaperones, casting doubt on the full-fledged induction of the unfolded protein response by C. difficile. They also displayed significantly higher phosphorylation of AKT and signal transducer and activator of transcription 3 (STAT3), an indication of pro-survival signalling. These data underscore the local, innate, pro-inflammatory nature of the response to C. difficile and highlight eIF2α phosphorylation and the interleukin-22–pSTAT3–RegIIIγ axis as two of the pathways that could be used to contain and counteract the damage inflicted on the intestinal epithelium.
Keywords: Clostridium difficile, eIF2α, interleukin-22, pSTAT3, RegIIIγ
Introduction
Clostridium difficile is a Gram-positive, spore-forming, anaerobic bacterium.1 It is the most prevalent cause of infectious diarrhoea in antibiotic-treated patients in hospitals.2–3 Infection with C. difficile can lead to a broad range of clinical outcomes, including asymptomatic colonization, mild diarrhoea and severe pseudomembranous colitis. Clostridium difficile encodes a number of toxins. Of these, two exotoxins, TcdA and TcdB, are the bacterium's main virulence factors. Both toxins are glucosyltransferases that irreversibly inactivate small GTPases of the Rho family.4–5 This in turn leads to the depolymerization of the epithelial actin cytoskeleton, impaired function of tight junctions and severe epithelial cell damage.6,7 The use of ileal loop models has provided useful insights into the function of these toxins.9 Studies using mouse models of C. difficile infection have proven the higher susceptibility of MyD88−/−10 and Toll-like receptor 4−/−11 mice and the protective effect of Toll-like receptor 5 stimulation against acute C. difficile colitis.12 The higher susceptibility of MyD88−/− mice is at least in part due to impaired CXCL1 expression and the consequent reduction in neutrophil influx to the site of infection.13 Interestingly, NOD1−/− mice also have reduced neutrophil recruitment to the site of infection, but show similar levels of epithelial damage as wild-type mice.14 However, much remains to be determined about the host inflammatory and mucosal response to C. difficile infection, which serves as our rationale for analysing the expression of over 90 of the genes involved in mucosal biology (Table 1) at the peak of acute inflammation in this infection.
List of evaluated genes - The groupings of the genes are based on either the structural relationship of the particular set of genes, and/or on their established (or purported) functions
| Chemokines: Ccl2, Ccl3, Ccl4, Ccl5, Ccl7, Ccl11, Cxcl1, Cxcl2, Cxcl9, Cxcl10 |
| Cytokines and related molecules: April, Baff, Csf2, Csf3, Fgf7, Il1b, Il2, Il3, Il4, Il5, Il6, Il9, Il10, Il12a, Il13, Il15, Il17a, Il17f, Il18, Il21, Il22, Il23a, Il25, Il27, Il33, Ifng, Mif, Tgfb1, Tnfa, Tslp |
| Anti-microbial peptides and mucins: Camp, Defa1, Defa28, Defb1, Defb3, Lyz1, Muc2, Muc3, Reg3 g, Slpi |
| Nod- and toll-like receptors: Nod1, Nod2, Tlr2, Tlr4, Tlr5, Tlr9 |
| Short chain fatty acid receptors (SCFAs): Ffar2, Ffar3, Gpr35 |
| Tight junction and adhesion proteins: Cdh1, Cgn, Cldn1, Cldn2, Epcam, Tff2, Tjp1, Tjp2 |
| Transcription factors: Gata3, Rorc, Shh, Tbx21, Tcf4, Xbp1, Zfp148 |
| Prostaglandin and/or leukotriene pathway enzymes: Alox5, Pla2g2a, Ptgs1, Ptgs2 |
| Miscellaneous: Actb, Aldh1a1, Aldh1a2, Ang4, Ctfr, Gapdh, Gdc, H2-Aa, H2-Ea, Hspa1b, Hspb1, Hrh4, Pigr, Pparg, Retnla, Rtc |
The induction of the unfolded protein response (UPR) in C. difficile infection has not been investigated; nor has pro-survival signalling been a major focus of studies on this infection. A number of reports have implicated the UPR in pro-inflammatory responses in general,15–16 and in intestinal inflammation in particular.17,18 More specifically, X-box-binding protein 1 (XBP1),17 activating transcription factor 6 (ATF6)18 and eukaryotic initiation factor 2α (eIF2α) phosphorylation19 each play a protective role against dextran sodium sulphate-induced colitis. The UPR is a concerted adaptive programme that counters endoplasmic reticulum (ER) stress by down-regulating the synthesis of secreted proteins, up-regulating ER chaperone and foldase levels, and activating ER-associated degradation, hence easing the burden on the stressed ER by decreasing its protein load, increasing its folding capacity and eliminating irreparably misfolded proteins.20–21 In higher eukaryotes, PRKR-Like Endoplasmic Reticulum Kinase (PERK), Inositol-Requiring Enzyme 1 (IRE1) and ATF6 act as the proximal transducers of ER stress. Each of these serves a distinct role in the UPR. The most rapid outcome is translational attenuation. It is mediated by activated PERK through the phosphorylation of eIF2α and takes effect as early as 30 min after exposure to ER stress.22–23 The GADD34/PP1 complex provides feedback inhibition of this process by specifically promoting eIF2α dephosphorylation.24–25 IRE1 exerts its cytoprotective effect mainly by removing a 26-base intron from the mRNA encoding XBP1.26–27 The spliced Xbp1 encodes a potent transcription factor whose targets encode several proteins involved in ER protein folding and the degradation of misfolded ER proteins.28–29In response to ER stress, the transmembrane portion of ATF6 is cleaved by S1P and S2P proteases that reside in the Golgi apparatus.30 The cleaved fragment moves to the nucleus and, mainly in parallel with XBP1, up-regulates genes that increase ER chaperone activity and the degradation of misfolded proteins.31–32 The protective roles of eIF2α phosphorylation, XBP1 and ATF-6 in mouse models of chemically induced colitis,17,18 serve as our rationale for investigating the potential effect of C. difficile infection on different elements of the UPR.
Here we have used the mouse model of C. difficile infection originally reported by Chen et al.,33 and previously studied in our group,34,35 to address the following unanswered questions. First, how does the host expression of chemokines, cytokines, anti-microbial peptides and other epithelial-associated genes change during acute C. difficile infection? Second, does the induction of the UPR accompany the inflammatory response in the colon and caecum during acute C. difficile infection?
Materials and methods
Ethics statement
All animal experiments were conducted with the approval of the University Committee on Use and Care of Animals (UCUCA) at the University of Michigan (Protocol Number: 10212). The University's animal-care policies follow the Public Health Service policy on Humane Care and Use of Laboratory Animals. The mice were housed in an AAALAC-accredited facility. None of the conducted experiments involved the deliberate induction of discomfort or injury. The physical condition and behaviour of the mice were assessed on a daily basis. The mice were killed by CO2 asphyxiation in compliance with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association.
Animals
C57BL/6 mice obtained from Jackson Laboratories (Bar Harbor, ME) were used to establish a breeding colony at the University of Michigan Medical School. They were housed under specific pathogen-free conditions and consumed clean food and water ad libitum. Male mice at 5–8 weeks of age were used for the current set of experiments.
Administration of antibiotics and infecting mice with Clostridium difficile
The mouse model of C. difficile infection described by Chen et al.33 was used for this study. Male mice, 5–8 weeks old, were either left untreated or received an antibiotic mixture of colistin (850 U/ml), gentamicin (0.035 mg/ml), kanamycin (0.4 mg/ml), metronidazole (0.215 mg/ml) and vancomycin (0.045 mg/ml) in sterile drinking water for 3 days. The mice receiving the antibiotic cocktail were then switched to regular drinking water for 2 days. Afterwards, each of the treated mice was given a single intraperitoneal dose of clindamycin (10 mg/kg) a day before infection with C. difficile. The C. difficile strain used in this study was the reference strain VPI 10463 (ATCC 43255), which was grown and prepared for inoculation as previously described.35 Each mouse received 105 colony-forming units (CFU) of the bacterium in its vegetative state by oral gavage. All the animals were monitored for signs of disease including diarrhoea, hunched posture and weight loss. All untreated and C. difficile-infected mice were killed 42 h after the infection (Fig. 1).
Figure 1.
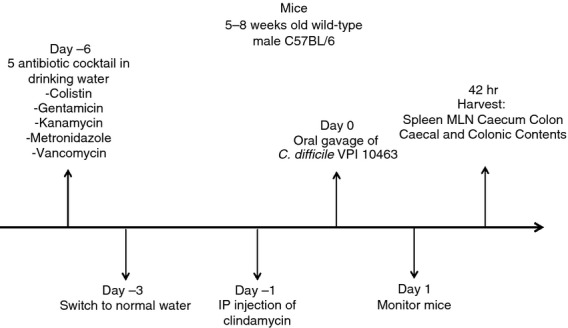
Experimental timeline of the Clostridium difficile infection.
Enrichment of intestinal leucocytes
Intestinal leucocyte enrichment was performed as previously described,14–37 with certain modifications. The caecum and colon of each mouse were excised, opened longitudinally and washed in PBS to remove the faecal content. Afterwards, each caecum or colon was incubated in calcium- and magnesium-free HBSS containing 2.5% fetal bovine serum and 1 mm DTT for 20 min at 37° to remove the mucus, washed three times and then incubated twice in calcium- and magnesium-free HBSS containing 2.5% fetal bovine serum and 1 mm EDTA for 20 min at 37° with one wash between the two incubations. Following the second incubation, the samples were washed three times. The tissues were then incubated in calcium- and magnesium-free HBSS containing 2.5% fetal bovine serum, 400 U/ml collagenase type 3 (Worthington Biochemical, Freehold, NJ) and 0.5 mg/ml DNase I (Roche, Indianapolis, IN) for 90 min at 37°. The digested samples were resuspended in a 20% Percoll solution (Sigma-Aldrich, Milwaukee, WI) and centrifuged at 900 g for 30 min at room temperature. The total number of cells obtained from each digest was counted in the presence of trypan blue using a haemocytometer.
Antibodies
The conjugated antibodies used for flow cytometry including those against B220 (clone RA3-6B2), CD4 (clone GK1.5), CD8 (clone 53-6.7), CD11b (clone M1/70), CD11c (clone HL3), CD19 (clone 1D3), CD25 (clone PC61), CD45 (clone 30-F11), CD69 (H1.2F3), FoxP3 (clone FJK-16s), Gr-1 (clone RB6-8C5) and MHC II (clone M5/114.15.2), as well as an unconjugated antibody against Fc RIII/II (clone 2.4G2) were purchased from BD Biosciences (San Diego, CA), eBioScience (San Diego, CA) and BioLegend (San Diego, CA). Immunoblotting antibodies against β-actin (clone 13E5), calreticulin, phospho-eIF2α (clone 119A11), eIF2α (clone L57A5), GAPDH (clone 14C10), P58IPK (clone C56E7), phospho-AKT (clone D9E), AKT (clone C67E7), phospho-STAT3 (clone D3A7) and STAT3 (clone 79D7) were obtained from Cell Signaling Technology (Danvers, MA). Anti-BiP (clone 40) was from BD Biosciences. Alkaline phosphatase-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Flow cytometry
Cell suspensions prepared from spleens and mesenteric lymph nodes,38 as well as caecal and colonic digests were washed in staining buffer [Hanks' balanced salt solution (HBSS) containing 0.5% BSA and 0.1% sodium azide), and pre-blocked with unlabelled anti-FcRIII/II antibody. Afterwards, the cells were stained in a final volume of 100 μl in 96-well round-bottom plates for 30 min. The cells were then washed (twice) in the staining buffer and resuspended in BD Biosciences' stabilizing fixative. Data on the samples were acquired on a three-laser Canto II flow cytometer using FACSDiva software (BD Biosciences). The acquired data were analysed with the FlowJo software (TreeStar, Ashland, OR). First, leucocytes were defined as cells with the surface expression of CD45. The following leucocyte subsets were then identified within this gate. Neutrophils were defined as Gr-1+ CD11c− MHC II− cells; CD11c+ MHC II+ cells were classified as dendritic cells; CD11b+ Gr-1− CD11c− cells were defined as members of the monocyte/macrophage lineage, with those expressing MHC II considered to be mature and/or activated; lymphocytes were subdivided by the surface expression of CD4, CD8 or B220 and CD19. CD4 T cells co-expressing FoxP3 and CD25 were defined as regulatory T cells.
Western blot analysis
Caecum and colon snips obtained from untreated and C. difficile-infected mice were homogenized on ice with a rotor/stator-type homogenizer (Biospec Products, Bartlesville, OK) while immersed in ice-cold modified RIPA buffer (50 mm Tris–HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS) supplemented with HALT protease and phosphatase inhibitor cocktail (Thermo Fisher, Rockford, IL). All tissue lysates were subjected to two rounds of centrifugation at 10 000 g for 10 min. The BCA protein assay (Thermo Fisher) was used to determine the protein concentration of each of the cleared lysates. A 30 μg sample of each caecum or colon lysate protein was boiled for 5 min in reducing sample buffer containing DTT and resolved by SDS–PAGE, transferred to PVDF membranes and probed with the indicated antibodies. The membranes were exposed to enhanced chemifluorescence substrate (GE Healthcare, Piscataway, NJ), followed by scanning on a Typhoon Trio+ imaging system (GE Healthcare) to obtain a digital image of the probed protein. The bands were then quantified with ImageQuant software (GE Healthcare).
Quantification of mRNA levels using custom-made quantitative RT-PCR cards
Caecum and colon snips obtained from untreated and C. difficile-infected mice were homogenized with a rotor/stator-type homogenizer while immersed in TRIzol RNA reagent (Life Technologies, Grand Island, NY). The TRIzol RNA reagent and the RNeasy Mini kit (Qiagen, Valencia, CA) were used in successive steps to isolate RNA from the caecum and colon samples, each according to its manufacturer's instructions. An Agilent Bioanalyser (Agilent Technologies, Palo Alto, CA) and a Nanodrop instrument (Thermo Fisher) were used to determine the quality and concentration of each RNA isolate, respectively. Complementary DNA (cDNA) was generated from each RNA sample using the RT2 First Strand kit (Qiagen). Expression levels of the genes under study were determined by using two different sets of mouse RT2 Profiler PCR cards (Qiagen), each custom-made to contain eight replicate sets of 48 primer pairs (Table 1). Each well of the replicate sets was loaded with 5 ng of cDNA reaction product. Each card was run on a LightCycler 480 real-time PCR system (Roche). The relative RNA expression levels were inferred from the Ct values.
Detection of Xbp1 mRNA splicing
Xbp1 splicing was assessed as previously described.39 Briefly, the Superscript III RT-PCR kit (Life Technologies) was used to amplify both unspliced and spliced Xbp1 in RNA samples obtained at the end of the experimental period. The primers used in the assay flanked the Xbp1 intron and had the following sequences: upstream: ttgtggttgagaaccagg; downstream: tccatgggaagatgttctgg.
Quantification of Gadd34 and Wars mRNA levels
Quantitative RT-PCR, including methods for verifying primer efficiency and specificity, were performed as previously described.40 The Ct value for each gene of each sample was normalized against the geometric mean of the Gapdh and Hprt for that sample.41
Statistical analyses
For the following assays, differences between untreated and C. difficile-infected mice were evaluated for significance by using paired t-tests at P ≤ 0.05: diversity of the bacterial community examined by pyrosequencing; cell numbers obtained by analysing the flow cytometric data; mRNA expression for the UPR genes Gadd34 and Wars obtained by single gene quantitative RT-PCR; and protein expression or phosphorylation assessed by immunoblotting. The quantitative RT-PCR data acquired with custom-made cards were normalized as previously described,39 with certain modifications. Briefly, the variation among the cards was adjusted by defining a normalization constant for each card based upon the mean Ct value of the 16 mRNAs that had the highest mRNA abundance (lowest Ct values) in each type of untreated tissue across the entire series of each custom-made set of RT2 Profiler PCR cards. Each individual Ct value was then adjusted by adding in this card-specific normalization factor, so that each card had the same average estimate of mRNA for the 16 most abundant mRNAs. The normalized numbers were used to calculate ΔCt values for each gene by deducting the geometric mean of the Actb and Gapdh Ct values of each sample from the Ct value of each gene in that sample.41 The SAM (Statistical Analysis for Microarray) software developed by Tusher and colleagues42 was then used to compare the expression levels of each gene between the caeca or colons of untreated and C. difficile-infected mice. In each case, genes for which false discovery rates ≤ 0.05 were considered significant. All the significant genes with at least a twofold increase in expression were defined as up-regulated.
Results
The timeline for the infection, as described in the Materials and methods section, is depicted in Fig. 1. Following pre-treatment with antibiotics, the mice received an oral gavage of 105 CFU of C. difficile strain VPI 10463 on day 0. At day 2, there was significantly lower bacterial species diversity in the caecum and colon (see Supplementary material, Figure S1 and Table S1), C. difficile infection was established, and detectable levels of toxin were present in the faeces (data not shown). At this time-point, the infected mice had lost weight, and their caeca and colons showed clear histopathological changes, which included neutrophilic inflammation in the mucosa and submucosa, varying degrees of submucosal oedema, epithelial hypertrophy and luminal exudates (see Supplementary material, Figure S2).
To study the mucosal host response to C. difficile infection, we used a quantitative RT-PCR approach to examine the expression patterns of > 90 genes in the caeca and colons of the infected mice, a scale of analysis not previously reported for this infection model. This was complemented with flow cytometric analysis to determine the type and number of different leucocyte subsets recruited to the sites of infection. The list of selected genes included chemokines, cytokines and related molecules, transcription factors, Nod- and Toll-like receptors, anti-microbial peptides, short-chain fatty acid receptors, tight junction and adhesion proteins, as well as others (see the full list in Table 1).
There was a significant up-regulation of the chemokines Ccl2, Ccl4, Cxcl1, Cxcl2, Cxcl9 and Cxcl10 in the caeca and colons in the aftermath of infection (Fig. 2a). There was also a significant up-regulation of Ccl3 in the colon. Both the caeca and colons of the infected mice displayed a significant increase in the expression levels of the pro-inflammatory cytokines Ifng, Il1b, Il6 and Il17f (Fig. 2b). The colons, in addition, had significantly higher levels of the cytokines Csf2, Csf3, Il9 and Tnfa.
Figure 2.
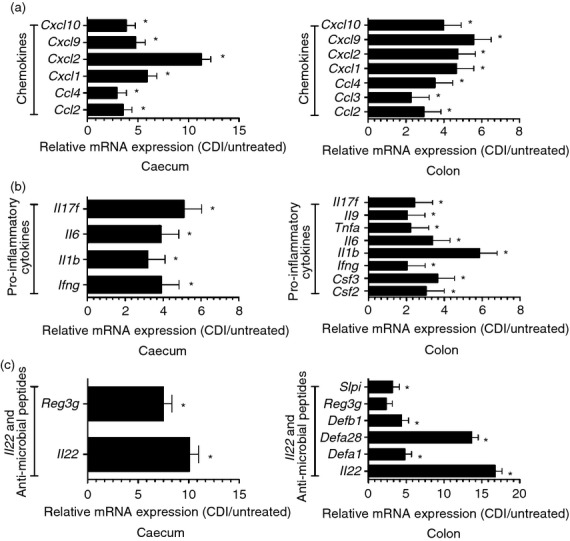
Significantly up-regulated genes in the caeca and colons of Clostridium difficile-infected (CDI) mice. Custom-made RT-PCR cards were used to evaluate gene expression levels in the caeca (n = 12 pairs) and colons (n = 12 pairs) of untreated and CDI mice from three sets of experiments. (a) significantly up-regulated chemokines in the caeca and colons of CDI mice; (b) pro-inflammatory cytokines that had significantly higher expression levels in these tissues; (c) significant up-regulation of Il22 and anti-microbial peptide mRNAs. Each bar represents the mean ± SEM of the relative expression of the depicted gene. ⋆ denotes a significant difference in expression levels between untreated and CDI mice, i.e. false discovery rate ≤ 0.05. The up-regulation of Reg3g in the colon did not reach statistical significance.
The observed chemokine and inflammatory gene expression pattern was clearly reflected in the composition of the inflammatory infiltrates in the caeca and colons of the C. difficile-infected mice. Both organs contained significantly higher numbers of neutrophils after the infection (Fig. 3a), a finding consistent with the significant up-regulation of Cxcl1, Cxcl2 and Il17f. In addition, there was a substantial increase in CD11b expression on the recruited neutrophils (Fig. 3b). Flow cytometric analysis showed a significant increase in the number of dendritic cells and cells of the monocyte/macrophage lineage in the caeca of the C. difficile-infected mice (Fig. 4a,b; compare with Supplementary material, Figure S3A and B); which was consistent with the increased expression levels of Ccl2. The infected colons showed a similar trend. A substantial fraction of the monocyte/macrophage lineage cells in the caeca and colons of the infected mice expressed low levels of MHC II (Fig. 4c), which was consistent with their recent recruitment. The significant increase in the number of lymphocytes (B cells, CD4 T cells and CD8 T cells) in the caeca and colons of the C. difficile-infected mice (Fig. 5a; compare with Supplementary material, Figure S4A) also correlated with the heightened expression of chemokines and pro-inflammatory genes. Nonetheless, the recruited CD4 T cells expressed levels of CD69 that were comparable with that found in their untreated counterparts (Fig. 5b; compare with Figure S4B) and had low levels of CD25 expression (assessed on CD4 T cells with gating to exclude the FoxP3+ subset) (Fig. 5c; compare with Figure S4C). These observations were in full biological concordance with the lack of any significant change in Tbx21, Gata3 or Rorc expression levels or in that of cytokines secreted by polarized T cells (data not shown).
Figure 3.
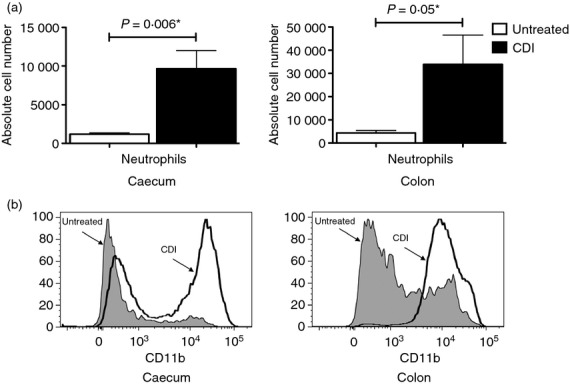
Recruitment of neutrophils to the caeca and colons of Clostridium difficile-infected (CDI) mice. (a) Bar graphs showing the increased number of neutrophils in the caeca and colons of CDI mice in comparison to untreated mice. The bars represent the mean ± SEM of nine pairs of caeca and colons. A P-value ≤ 0.05 indicates a significant difference between the untreated and CDI samples and is marked with a ⋆. (b) Histograms showing increased expression levels of CD11b on neutrophils in the caeca and colons of CDI mice.
Figure 4.
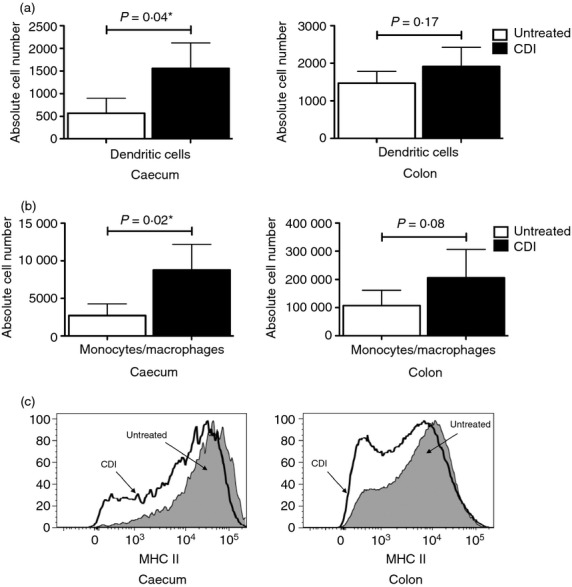
Increased number of dendritic cells and cells of the monocyte/macrophage lineage in the caeca of Clostridium difficile-infected (CDI) mice. Bar graphs showing the number of dendritic cells (a), and cells of the monocyte/macrophage lineage (b), in the caeca and colons of CDI mice in comparison to untreated mice. The bars represent the mean ± SEM of nine pairs of caeca and colons. A P-value ≤ 0.05 indicates a significant difference between the untreated and CDI samples and is marked with a ⋆. (c) Histograms showing the higher fraction of monocyte/macrophage lineage cells expressing low levels of MHC II in the caeca and colons of CDI mice.
Figure 5.
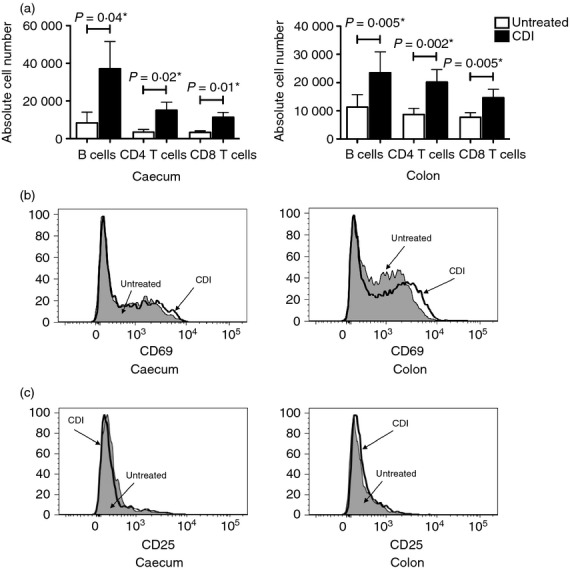
Recruitment of various lymphocyte subsets to the caeca and colons of Clostridium difficile-infected (CDI) mice. Bar graphs showing the number of B cells, CD4 T cells and CD8 T cells in the caeca and colons of CDI mice in comparison to untreated mice (a). The bars represent the mean ± SEM of nine pairs of caeca and colons. A P-value ≤ 0.05 indicates a significant difference between the untreated and CDI samples and is marked with a ⋆. (b) and (c) respectively show histograms of CD69 and CD25 expression levels in the CD4 T cells of caeca and colons of CDI mice in comparison to untreated mice.
There was a significant up-regulation of Il22 expression and a number of anti-microbial peptides in the caeca and colons of the infected mice. These included Defa1, Defa28, Defb1 and Slpi in the colon and Reg3g in the caecum (Fig. 2c). There was also an increase in Reg3g levels in the colons of infected mice; however, in these experiments, the increase did not reach statistical significance.
To assess the activation of the UPR in C. difficile infection in mice, caecal and colonic samples from untreated and C. difficile-infected mice were examined for their expression of numerous UPR markers. Immunoblotting showed a significant increase in the level of eIF2α phosphorylation, the most rapid aspect of the UPR, in the caeca and colons of the infected mice (Fig. 6a). This coincided with the significant up-regulation of Gadd34 and Wars mRNA expression levels, both downstream of eIF2α phosphorylation, in the infected samples (Fig. 6b). By contrast, the caecal and colonic tissues from the infected mice did not undergo Xbp1 splicing (Fig. 6c), thereby ruling out the activation of the IRE1 pathway. Up-regulation of ER chaperones is the hallmark of UPR activation. When assessed by immunoblotting, the caecal and colonic protein samples from the infected mice did not show the induction of BiP, P58IPK or calreticulin as a result of infection (Fig. 6d–f). There was no indication of ER chaperone up-regulation at the mRNA level either (data not shown).
Figure 6.
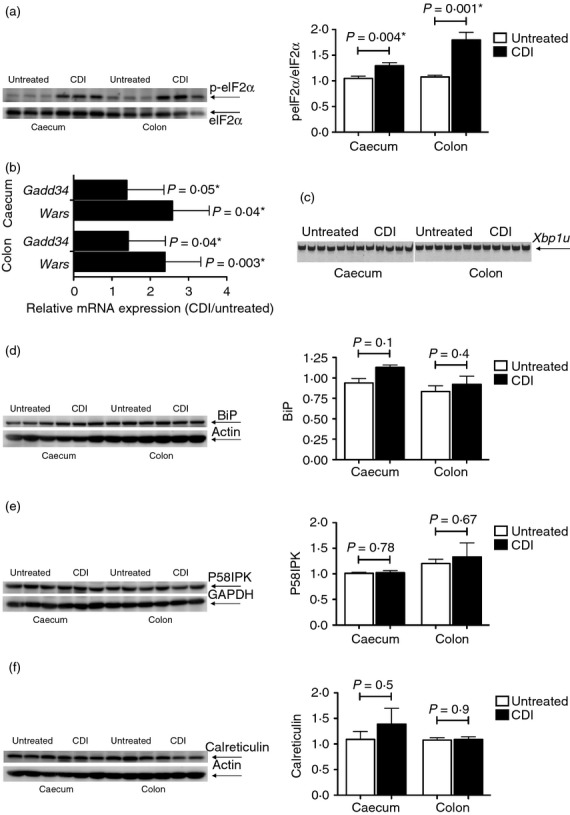
Assessment of the unfolded protein response in Clostridium difficile-infected (CDI) mice. Protein lysates from the caeca and colons of untreated and C. difficile-infected mice were used to evaluate the phosphorylation of eIF2α (n = 6 pairs) (a), and the expression levels of BiP (n = 6 pairs) (d), P58IPK (n = 6 pairs) (e) and calreticulin (n = 6 pairs) (f). In each case, the panel on the left shows the image of the immunoblot for the evaluated molecule for three pairs of caeca and colons and the bar graph on the right depicts the mean ± SEM of the response for all the six evaluated pairs. The expression levels of Gadd34 and Wars were determined by quantitative RT-PCR (n = 12 pairs). Each bar represents the mean ± SEM of the relative expression of the depicted gene (b). Conventional RT-PCR was used to determine the lack of Xbp1 splicing (n = 12 pairs, of which 6 are shown) (c). A P-value ≤ 0.05 indicates a significant difference between the untreated and CDI samples and is marked with a ⋆.
The phosphorylation of eIF2α and the up-regulation of Il22 in the caeca and colons of C. difficile-infected mice, as well as the up-regulation of Reg3g in their caeca, raises the prospect of pro-survival signalling in these tissues in response to infection. To investigate this possibility, caecal and colonic protein lysates from untreated and C. difficile-infected mice were probed for the phosphorylation levels of AKT and STAT3. Both the caeca and colons of the infected mice showed a significant increase in AKT (Fig. 7a) and STAT3 (Fig. 7b) phosphorylation levels in comparison to their untreated counterparts. These data support the induction of pro-survival signals in C. difficile-infected mice.
Figure 7.
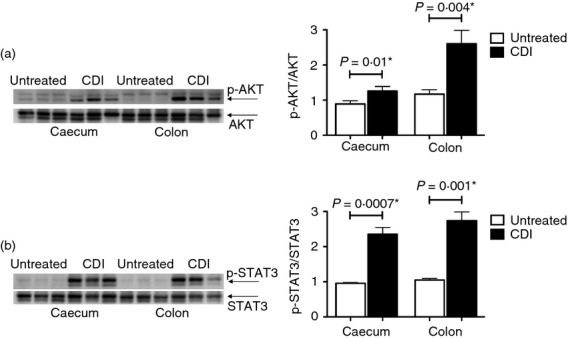
Phosphorylation of AKT and signal transducer and activator of transcription 3 (STAT3) in the caeca and colons of Clostridium difficile-infected (CDI) mice. Protein lysates from the caeca and colons of untreated and CDI mice were used to evaluate the phosphorylation of AKT (n = 6 pairs) (a), and STAT3 (n = 6 pairs) (b). In each case, the panel on the left shows the image of the immunoblot for the evaluated molecule for three pairs of caeca and colons and the bar graph on the right depicts the mean ± SEM of the response for all the six evaluated pairs. A P-value ≤ 0.05 indicates a significant difference between the untreated and CDI samples and is marked with a ⋆.
Discussion
This study contains two major novel elements. (i) It analyses the host response in the caeca and colons of C. difficile-infected mice with a panel of > 90 of the genes involved in mucosal biology, and correlates these changes with the cellular response at these sites of infection, as determined by flow cytometry. (ii) It examines the induction of the UPR and pro-survival signals at these sites in the aftermath of C. difficile infection.
Collectively, the gene expression and flow cytometric results point to four main trends in the local response to C. difficile infection.
First, they show an up-regulation of chemokine genes involved in recruiting effector cells of the innate immune response to the sites of infection. CXCL1 and CXCL2 are potent neutrophil chemoattractants and activators, and induce neutrophil mobilization from the bone marrow.43–44 CCL2 is in turn a chemoattractant for monocytes. Most nucleated cells express CCL2 in response to pro-inflammatory cytokines such as interleukin-1β (IL-1β)45 or upon engagement of innate immune receptors by a number of microbial products. Flow cytometric analysis had shown a substantial increase in the number of neutrophils in the caeca and colons of the infected mice and up-regulated levels of CD11b on the recruited neutrophils, an indication of their potential activation.46 It also documented that a higher fraction of cells of the monocyte/macrophage lineage express low levels of MHC II in the caeca and colons of the infected mice, further confirming monocyte recruitment to the site of infection and raising the prospect of their differentiation after exposure to cytokines and/or microbial products.47 The up-regulation of Cxcl1, Cxcl2 and Ccl2 in the caeca and colons of C. difficile-infected mice is consistent with the flow cytometric evidence of neutrophil and monocyte recruitment to the site(s) of infection, and is in agreement with previously published findings.13–50
Second, the quantitative PCR data document the induction of pro-inflammatory cytokine genes. Interleukin-1β, IL-6, tumour necrosis factor-α (TNF-α), Colony Stimulating Factor 2 (CSF2), Colony Stimulating Factor 3 (CSF3) and interferon-γ (IFN-γ) are all potent pro-inflammatory cytokines. Moreover, IFN-γ can induce both CXCL9 and CXCL10 expression, which explains the significant up-regulation of Cxcl9 and Cxcl10 in our quantitative PCR analysis. In synergy with IL-1β and TNF-α, IL-17F induces CCL2 and CXCL1 production in vitro51 and recruits neutrophils to the site of infection in vivo.52 The up-regulation of genes for this group of cytokines at the site(s) of C. difficile infection further underscores the innate nature of the response in this model.
Third, the quantitative PCR data do not show an increase in Tbx21, Gata3 or Rorc expression levels or the cytokines secreted by polarized T cells. CD69 and CD25 expression levels are used to assess early T-cell activation.53,54 Although flow cytometry confirmed the recruitment of lymphocytes to the sites of infection, CD4 T cells of the untreated and C. difficile-infected mice expressed comparable levels of CD69, and had low levels of CD25 expression on their surface. Our inference from the flow cytometric data is that the CD4 T cells recruited to the sites of infection are at best at the very early stages of activation and therefore unlikely to exert a polarized T cell's effector function(s). The absence of a significant increase in Tbx21, Gata3 or Rorc expression levels or that of cytokines secreted by polarized T cells gives further credence to this notion. It also indicates that any study of the adaptive immune response and potential polarization of the T-cell response should be undertaken in a protracted, chronic model of C. difficile infection.
Lastly, the quantitative PCR data demonstrate the higher expression of genes involved in containing the inflammation and restoring mucosal homeostasis and integrity. Interleukin-22 serves a crucial role in maintaining the barrier function of mucosal surfaces by promoting anti-microbial immunity and tissue repair.56–57 It plays a part in the expression of defensins in keratinocytes.58–59 More importantly, IL-22 has a direct role in the induction of RegIIIγ in the gut.60 RegIIIγ in turn, promotes a spatial separation between intestinal microbiota and the host, thereby minimizing the chance of harmful immune responses.61 The up-regulation of Il22 in the caeca and colons of the infected mice, as well as the significant increase in expression of anti-microbial peptides, particularly Reg3g, all point to the host's efforts to contain the inflicted damage and to restore epithelial homeostasis at the infected sites.
The previous use of C. difficile toxins alone in experimental ileal loop models has documented a small fraction of our findings in the current study. These include the ability of TcdA to induce the release of the pro-inflammatory mediators IL-1β,62 TNF-α,63 IFN-γ,64 CXCL1,48 CXCL249 and CCL3,65 as well as the fact that both IFN-γ−/−64 and CCR1−/−65 mice have a milder form of enteritis in response to TcdA injection. Despite the useful insights provided by the ileal loop model into the actions of C. difficile toxins, it should be noted that the model has some important shortcomings. First, it is a surgery-based model, which entails the injection of C. difficile toxin preparations into the animal and not infection with the bacterium itself; second, it targets the wrong organ for disease, i.e. ileum instead of the colon; and third, it does not reflect any interaction of C. difficile with the host's microbiota.
The current work is the first to assess the induction of the UPR during acute C. difficile infection. A number of recent studies have implicated the UPR in the response to different forms of intestinal inflammation. These include the protective role(s) of XBP1,17 ATF618 and eIF2α phosphorylation19 against dextran sodium sulphate-induced colitis. Despite the phosphorylation of eIF2α and the slight up-regulation of the phospho-eIF2α targets Wars and Gadd34 in the caeca and colons of C. difficile-infected mice (which serve as an early indication of phospho-eIF2α exerting its downstream effect), the lack of Xbp1 splicing and the absence of ER chaperone up-regulation in these tissues cast serious doubt on the activation of the UPR in this model of infection. Although numerous laboratories have shown that the UPR output can be modulated in a context-specific manner,66–67 a more likely explanation for the current set of findings is the phosphorylation of eIF2α by a kinase other than PERK. Of the four kinases that can phosphorylate eIF2α, Protein Kinase RNA-activated (PKR) is the most plausible candidate. The phosphorylation of AKT and STAT3, as well as eIF2α, in the C. difficile-infected mice gives further credence to this hypothesis because, in addition to phosphorylating eIF2α, PKR is an upstream inducer of both AKT and STAT3 phosphorylation.68
AKT plays an important role in promoting intestinal epithelial homeostasis and wound repair during intestinal inflammation.69 Furthermore, the protective effect of lysophosphatidic acid against C. difficile toxin-induced cell death in vitro is in part due to its induction of AKT phosphorylation.70 Therefore, the phosphorylation of AKT in the C. difficile-infected mice may be a pro-survival signal that aims to counteract and contain the inflicted epithelial damage.
The phosphorylation of STAT3 in the C. difficile-infected mice should be viewed from a broader perspective. First, the use of STAT3IEC-KO mice has shown that activation of intestinal epithelial STAT3 regulates immune homeostasis in the gut by promoting IL-22-dependent mucosal wound healing.71 The up-regulation of Il22 expression in the caeca and colons of C. difficile-infected mice, and the significantly higher expression of Reg3g, suggests a scenario where the recruitment of STAT3 to the IL-22 receptor72–73 and its consequent phosphorylation would initiate signalling pathways involved in epithelial repair and wound healing. Second, given the concurrent phosphorylation of eIF2α, AKT and STAT3 in the caeca and colons of the infected mice, STAT3 phosphorylation may be in part mediated by PKR. The phosphorylated STAT3 generated in this manner can then contribute to epithelial homeostasis and wound repair.19 Third, one can raise the possibility of STAT3 recruitment to, and its phosphorylation on, the IL-10 receptor. Interleukin-10 can inhibit the production of a distinct, yet diverse, set of inflammatory mediators. This is achieved by selectively inhibiting transcription and requires STAT3 activation on the IL-10 receptor.74 The pro-inflammatory genes Ccl2, Ccl3, Csf2, Cxcl1, Il1b, Il6 and Tnfa, that are up-regulated in the caeca and/or colons of the C. difficile-infected mice, belong to the subset of genes whose transcription is controlled in this manner. However, the fact that C. difficile-infected mice do not display an increase in Il10 expression as a result of the infection, makes this an unlikely scenario.
We contend that the concomitant induction of a local pro-inflammatory response, and the production of IL-22 and RegIIIγ, constitute the host's standard way of containing and counteracting an acute infection in the gut. Our study shows the phosphorylation of eIF2α in the infected mice, but not the full-fledged induction of the UPR. On the weight of evidence, it is plausible that PKR, and not PERK, is responsible for the phosphorylation of eIF2α. This prediction can be put to the test by using intestinal epithelial cell-specific PERK and PKR knockout mice. Our study also provides evidence for the induction of pro-survival signalling, which may contribute to the host's return to epithelial homeostasis. The phosphorylation of eIF2α as a result of infection raises the prospect that phosphorylated eIF2α confers the same protective effect in acute C. difficile infection as the one it confers against chemically induced colitis.19 This, in conjunction with the induction of pro-survival signals, can be used to argue that manipulation of common biochemical pathways such as those related to translational control and pro-survival signalling, rather than disease-specific and pathogen-specific approaches, could potentially be of therapeutic benefit across a spectrum of conditions with analogous and/or shared pathophysiologies.75
Acknowledgments
We thank David Adams and the staff of the University of Michigan Flow Cytometry Core for their invaluable help in performing the flow cytometry experiments; Sue Foltin for her help with the pyrosequencing experiments and Ryan Muraglia for his help in their analysis; and Dr Roderick McDonald for his help with the experiments and his advice on the manuscript. This work was in part supported by National Institutes of Health grants T32 HL007749 (CMT), U19 AI090871 (GBH and VBY), P30 DK034933 (GBH and VBY) and RO1 DK084058 (DTR).
Author contributions
AASA and GBH conceived, designed and interpreted the experiments; CMT, JRED, DTR and VBY contributed to the design and interpretation. AASA, CMT, AJM, NRF and HMT performed the experiments. AASA, JRED, DTR and GBH analysed the data. AASA and GBH wrote the manuscript and all the other authors provided comment and advice on the manuscript.
Disclosures
Vincent B. Young is on the advisory board of ViroPharma in relation to developing non-toxigenic C. difficile for the management of C. difficile infection. The other authors declare no conflict of interest.
Supporting Information
Table S1. The list of operational taxonomic units (OTUs) and their taxonomic identification.
References
- Hall I, O'Toole E. Intestinal flora in newborn infants with description of a new pathogenic anaerobe. Am J Dis Child. 1935;49:390–402. [Google Scholar]
- Kelly CP, LaMont JT. Clostridium difficile infection. Annu Rev Med. 1998;49:375–90. doi: 10.1146/annurev.med.49.1.375. [DOI] [PubMed] [Google Scholar]
- Rupnik M, Wilcox MH, Gerding DN. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol. 2009;7:526–36. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- Just I, Selzer J, von Eichel-Streiber C, Aktories K. The low molecular mass GTP-binding protein Rho is affected by toxin A from Clostridium difficile. J Clin Invest. 1995;95:1026–31. doi: 10.1172/JCI117747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just I, Selzer J, Wilm M, von Eichel-Streiber C, Mann M, Aktories K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature. 1995;375:500–3. doi: 10.1038/375500a0. [DOI] [PubMed] [Google Scholar]
- Hecht G, Pothoulakis C, LaMont JT, Madara JL. Clostridium difficile toxin A perturbs cytoskeletal structure and tight junction permeability of cultured human intestinal epithelial monolayers. J Clin Invest. 1988;82:1516–24. doi: 10.1172/JCI113760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht G, Koutsouris A, Pothoulakis C, LaMont JT, Madara JL. Clostridium difficile toxin B disrupts the barrier function of T84 monolayers. Gastroenterology. 1992;102:416–23. doi: 10.1016/0016-5085(92)90085-d. [DOI] [PubMed] [Google Scholar]
- Pothoulakis C, Lamont JT. Microbes and microbial toxins: paradigms for microbial–mucosal interactions II. The integrated response of the intestine to Clostridium difficile toxins. Am J Physiol Gastrointest Liver Physiol. 2001;280:G178–83. doi: 10.1152/ajpgi.2001.280.2.G178. [DOI] [PubMed] [Google Scholar]
- Best EL, Freeman J, Wilcox MH. Models for the study of Clostridium difficile infection. Gut Microbes. 2012;3:145–67. doi: 10.4161/gmic.19526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawley TD, Clare S, Walker AW, et al. Antibiotic treatment of Clostridium difficile carrier mice triggers a supershedder state, spore-mediated transmission, and severe disease in immunocompromised hosts. Infect Immun. 2009;77:3661–9. doi: 10.1128/IAI.00558-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan A, Lynch M, Smith SM, et al. A role for TLR4 in Clostridium difficile infection and the recognition of surface layer proteins. PLoS Pathog. 2011;7:e1002076. doi: 10.1371/journal.ppat.1002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarchum I, Liu M, Lipuma L, Pamer EG. Toll-like receptor 5 stimulation protects mice from acute Clostridium difficile colitis. Infect Immun. 2011;79:1498–503. doi: 10.1128/IAI.01196-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarchum I, Liu M, Shi C, Equinda M, Pamer EG. Critical role for MyD88-mediated neutrophil recruitment during C. difficile colitis. Infect Immun. 2012;80:2989–96. doi: 10.1128/IAI.00448-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Yamazaki T, Kamada N, Tawaratsumida K, Kim YG, Nunez G, Inohara N. Nucleotide-binding oligomerization domain 1 mediates recognition of Clostridium difficile and induces neutrophil recruitment and protection against the pathogen. J Immunol. 2011;186:4872–80. doi: 10.4049/jimmunol.1003761. [DOI] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–62. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Glimcher LH. Regulation of innate immunity by signaling pathways emerging from the endoplasmic reticulum. Curr Opin Immunol. 2011;23:35–40. doi: 10.1016/j.coi.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaser A, Lee AH, Franke A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–56. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandl K, Rutschmann S, Li X, Du X, Xiao N, Schnabl B, Brenner DA, Beutler B. Enhanced sensitivity to DSS colitis caused by a hypomorphic Mbtps1 mutation disrupting the ATF6-driven unfolded protein response. Proc Natl Acad Sci USA. 2009;106:3300–5. doi: 10.1073/pnas.0813036106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao SS, Song B, Kaufman RJ. PKR protects colonic epithelium against colitis through the unfolded protein response and prosurvival signaling. Inflamm Bowel Dis. 2012;18:1735–42. doi: 10.1002/ibd.22878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–89. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Lin JH, Walter P, Yen TS. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- Novoa I, Zhang Y, Zeng H, Jungreis R, Harding HP, Ron D. Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 2003;22:1180–7. doi: 10.1093/emboj/cdg112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J Cell Biol. 2001;153:1011–22. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce M, Bryant KF, Jousse C, et al. A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science. 2005;307:935–9. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- Sidrauski C, Walter P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell. 1997;90:1031–9. doi: 10.1016/s0092-8674(00)80369-4. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–91. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–59. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda Y, Okada T, Yoshida H, Kaufman RJ, Nagata K, Mori K. Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. J Cell Biol. 2006;172:383–93. doi: 10.1083/jcb.200507057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355–64. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- Wu J, Rutkowski DT, Dubois M, et al. ATF6α optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell. 2007;13:351–64. doi: 10.1016/j.devcel.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev Cell. 2007;13:365–76. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Chen X, Katchar K, Goldsmith JD, Nanthakumar N, Cheknis A, Gerding DN, Kelly CP. A mouse model of Clostridium difficile-associated disease. Gastroenterology. 2008;135:1984–92. doi: 10.1053/j.gastro.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Reeves AE, Theriot CM, Bergin IL, Huffnagle GB, Schloss PD, Young VB. The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile infection. Gut Microbes. 2011;2:145–58. doi: 10.4161/gmic.2.3.16333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot CM, Koumpouras CC, Carlson PE, Bergin II, Aronoff DM, Young VB. Cefoperazone-treated mice as an experimental platform to assess differential virulence of Clostridium difficile strains. Gut Microbes. 2011;2:326–34. doi: 10.4161/gmic.19142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves AE, Koenigsknecht MJ, Bergin IL, Young VB. Suppression of Clostridium difficile in the gastrointestinal tracts of germfree mice inoculated with a murine isolate from the family Lachnospiraceae. Infect Immun. 2012;80:3786–94. doi: 10.1128/IAI.00647-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigmann B, Tubbe I, Seidel D, Nicolaev A, Becker C, Neurath MF. Isolation and subsequent analysis of murine lamina propria mononuclear cells from colonic tissue. Nat Protoc. 2007;2:2307–11. doi: 10.1038/nprot.2007.315. [DOI] [PubMed] [Google Scholar]
- Hunt SV. Preparation of lymphocytes and accessory cells. In: Klaus GGB, editor. Lymphocytes: A Practical Approach. Oxford: IRL Press; 1987. pp. 1–34. [Google Scholar]
- Sadighi Akha AA, Harper JM, Salmon AB, Schroeder BA, Tyra HM, Rutkowski DT, Miller RA. Heightened induction of proapoptotic signals in response to endoplasmic reticulum stress in primary fibroblasts from a mouse model of longevity. J Biol Chem. 2011;286:30344–30351. doi: 10.1074/jbc.M111.220541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski DT, Arnold SM, Miller CN, et al. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. 2010;120:2423–31. doi: 10.1172/JCI41649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdon PC, Martin C, Rankin SM. The CXC chemokine MIP-2 stimulates neutrophil mobilization from the rat bone marrow in a CD49d-dependent manner. Blood. 2005;105:2543–8. doi: 10.1182/blood-2004-08-3193. [DOI] [PubMed] [Google Scholar]
- Struyf S, van Collie E, Paemen L, Put W, Lenaerts JP, Proost P, Opdenakker G, van Damme J. Synergistic induction of MCP-1 and -2 by IL-1β and interferons in fibroblasts and epithelial cells. J Leukoc Biol. 1998;63:364–72. doi: 10.1002/jlb.63.3.364. [DOI] [PubMed] [Google Scholar]
- Lundahl J, Jacobson SH, Paulsson JM. IL-8 from local subcutaneous wounds regulates CD11b activation. Scand J Immunol. 2012;75:419–25. doi: 10.1111/j.1365-3083.2012.02679.x. [DOI] [PubMed] [Google Scholar]
- Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol. 2009;27:669–92. doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- Hirota SA, Fines K, Ng J, et al. Hypoxia-inducible factor signaling provides protection in Clostridium difficile-induced intestinal injury. Gastroenterology. 2010;139(259–269):e253. doi: 10.1053/j.gastro.2010.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagliuolo I, Keates AC, Wang CC, et al. Clostridium difficile toxin A stimulates macrophage-inflammatory protein-2 production in rat intestinal epithelial cells. J Immunol. 1998;160:6039–45. [PubMed] [Google Scholar]
- Kim JM, Kim JS, Jun HC, Oh YK, Song IS, Kim CY. Differential expression and polarized secretion of CXC and CC chemokines by human intestinal epithelial cancer cell lines in response to Clostridium difficile toxin A. Microbiol Immunol. 2002;46:333–42. doi: 10.1111/j.1348-0421.2002.tb02704.x. [DOI] [PubMed] [Google Scholar]
- Iyoda M, Shibata T, Kawaguchi M, Hizawa N, Yamaoka T, Kokubu F, Akizawa T. IL-17A and IL-17F stimulate chemokines via MAPK pathways (ERK1/2 and p38 but not JNK) in mouse cultured mesangial cells: synergy with TNF-α and IL-1β. Am J Physiol Renal Physiol. 2010;298:F779–87. doi: 10.1152/ajprenal.00198.2009. [DOI] [PubMed] [Google Scholar]
- Hurst SD, Muchamuel T, Gorman DM, et al. New IL-17 family members promote Th1 or Th2 responses in the lung: in vivo function of the novel cytokine IL-25. J Immunol. 2002;169:443–53. doi: 10.4049/jimmunol.169.1.443. [DOI] [PubMed] [Google Scholar]
- Castellanos MC, Lopez-Giral S, Lopez-Cabrera M, de Landazuri MO. Multiple cis-acting elements regulate the expression of the early T cell activation antigen CD69. Eur J Immunol. 2002;32:3108–17. doi: 10.1002/1521-4141(200211)32:11<3108::AID-IMMU3108>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- John S, Reeves RB, Lin JX, Child R, Leiden JM, Thompson CB, Leonard WJ. Regulation of cell-type-specific interleukin-2 receptor α-chain gene expression: potential role of physical interactions between Elf-1, HMG-I(Y), and NF-κB family proteins. Mol Cell Biol. 1995;15:1786–96. doi: 10.1128/mcb.15.3.1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadighi Akha AA, Berger SB, Miller RA. Enhancement of CD8 T-cell function through modifying surface glycoproteins in young and old mice. Immunology. 2006;119:187–94. doi: 10.1111/j.1365-2567.2006.02420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol. 2011;29:71–109. doi: 10.1146/annurev-immunol-031210-101312. [DOI] [PubMed] [Google Scholar]
- Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol. 2011;12:383–90. doi: 10.1038/ni.2025. [DOI] [PubMed] [Google Scholar]
- Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. 2004;21:241–54. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–9. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–9. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- Vaishnava S, Yamamoto M, Severson KM, et al. The antibacterial lectin RegIIIγ promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334:255–8. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng J, Hirota SA, Gross O, et al. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology. 2010;139:542–52. doi: 10.1053/j.gastro.2010.04.005. . 552 e541-543. [DOI] [PubMed] [Google Scholar]
- Castagliuolo I, Kelly CP, Qiu BS, Nikulasson ST, LaMont JT, Pothoulakis C. IL-11 inhibits Clostridium difficile toxin A enterotoxicity in rat ileum. Am J Physiol. 1997;273:G333–41. doi: 10.1152/ajpgi.1997.273.2.G333. [DOI] [PubMed] [Google Scholar]
- Ishida Y, Maegawa T, Kondo T, Kimura A, Iwakura Y, Nakamura S, Mukaida N. Essential involvement of IFN-γ in Clostridium difficile toxin A-induced enteritis. J Immunol. 2004;172:3018–25. doi: 10.4049/jimmunol.172.5.3018. [DOI] [PubMed] [Google Scholar]
- Morteau O, Castagliuolo I, Mykoniatis A, et al. Genetic deficiency in the chemokine receptor CCR1 protects against acute Clostridium difficile toxin A enteritis in mice. Gastroenterology. 2002;122:725–33. doi: 10.1053/gast.2002.31873. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, Hegde RS. Regulation of basal cellular physiology by the homeostatic unfolded protein response. J Cell Biol. 2010;189:783–94. doi: 10.1083/jcb.201003138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- Garcia MA, Meurs EF, Esteban M. The dsRNA protein kinase PKR: virus and cell control. Biochimie. 2007;89:799–811. doi: 10.1016/j.biochi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Koch S, Nava P, Addis C, Kim W, Denning TL, Li L, Parkos CA, Nusrat A. The Wnt antagonist Dkk1 regulates intestinal epithelial homeostasis and wound repair. Gastroenterology. 2011;141:259–68. doi: 10.1053/j.gastro.2011.03.043. , 268 e251-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sautin YY, Crawford JM, Svetlov SI. Enhancement of survival by LPA via Erk1/Erk2 and PI 3-kinase/Akt pathways in a murine hepatocyte cell line. Am J Physiol Cell Physiol. 2001;281:C2010–9. doi: 10.1152/ajpcell.00077.2001. [DOI] [PubMed] [Google Scholar]
- Pickert G, Neufert C, Leppkes M, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465–72. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejeune D, Dumoutier L, Constantinescu S, Kruijer W, Schuringa JJ, Renauld JC. Interleukin-22 (IL-22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line. Pathways that are shared with and distinct from IL-10. J Biol Chem. 2002;277:33676–82. doi: 10.1074/jbc.M204204200. [DOI] [PubMed] [Google Scholar]
- Dumoutier L, de Meester C, Tavernier J, Renauld JC. New activation modus of STAT3: a tyrosine-less region of the interleukin-22 receptor recruits STAT3 by interacting with its coiled-coil domain. J Biol Chem. 2009;284:26377–84. doi: 10.1074/jbc.M109.007955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc Natl Acad Sci USA. 2005;102:8686–91. doi: 10.1073/pnas.0500419102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Radford H, Peretti D, et al. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature. 2012;485:507–11. doi: 10.1038/nature11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The list of operational taxonomic units (OTUs) and their taxonomic identification.