

Abstract
We investigated the derivation of non-natural peptide triazole dual receptor site antagonists of HIV-1 Env gp120 in order to establish a path for developing peptidomimetic antiviral agents. Previously, we found that the peptide triazole HNG-156 (R-I-N-N-I-X-W-S-E-A-M-M-CONH2, where X is ferrocenyltriazole-Pro (FtP)) had nanomolar binding affinity to gp120, inhibited gp120 binding to CD4 and the co-receptor surrogate mAb 17b and had potent antiviral activity in cell infection assays. Further, truncated variants of HNG-156, typified by UM-24 (Cit-N-N-I-X-W-S-CONH2) and containing the critical central stereospecific LX-LW cluster, retained the functional characteristics of the parent peptide triazole. In the current work, we examined the possibility to replace natural with unnatural residue components in UM-24 to the greatest extent possible. The analogue with the critical “hot spot” residue Trp 6 replaced with L-3-Benzothienylalanine (Bta) (KR-41), as well as a completely non-natural analogue containing D-amino acid substitutions outside the central cluster (KR-42, DCit-DN-DN-DI-X-Bta-DS-CONH2), retained the dual receptor site antagonism / antiviral activity signature. The results define differential functional roles of subdomains within the peptide triazole and provide a structural basis for designing metabolically stable peptidomimetic inhibitors of HIV-1 Env gp120.
Keywords: Acquired Immune Deficiency Virus, HIV entry Inhibitors, Synthetic non-natural peptide triazoles, Surface Plasmon Resonance, Isothermal Titration Calorimetry
Introduction
The number of people dying from AIDS-related illnesses worldwide has steadily decreased due to greater access to antiretroviral therapies.[1] Highly active anti-retroviral therapy (HAART) has allowed HIV-1-infected individuals to live longer by inhibiting various stages of the viral life cycle.[2] However, the active components of HAART therapy can cause harsh side effects and over time encounter increasing drug resistance.[3] Therefore the discovery of new inhibitors remains an urgent global need.[4] One target for next generation inhibitors is the virus cell entry step. Peptide fusion inhibitors such as Enfuvirtide (T20, Fuzeon™)[5] and small-molecule co-receptor inhibitors such as Maraviroc (Selzentry™)[6] have become clinically available but suffer from resistance and injection site effects for the former, limitations to CCR5 cell infection for the latter. The need for improved and effective entry inhibitors remains a goal of current investigation.
HIV-1 cell entry leading to AIDS pathogenesis is mediated by a single type of protein cluster on the virus surface denoted envelope spike, or Env.[7] Each virus Env spike consists of a trimer of two non-covalently associated glycoproteins, an inner gp41 transmembrane protein and a gp120 exterior protein.[8] In the host cell entry process, these two proteins participate in a sequence of cell receptor interactions initiated by binding of gp120 to cell surface CD4.[9] This binding event causes conformational rearrangements within gp120 that expose and stabilize a co-receptor binding site.[10] CD4 and consequent co-receptor binding result in further conformational changes in the spike assembly that expose fusogenic as well as initially cryptic helical domains of gp41.[11] The gp41 domains participate in conformational rearrangement and cell attachment processes leading to virus-cell membrane fusion and introduction of a viral genome into the cell.[12] The various steps in viral entry are relevant targets for anti-HIV intervention.
Previously, we reported a family of peptide triazoles which have nanomolar affinity for HIV-1 envelope protein gp120, and inhibit the interactions of gp120 at both host cell receptor and co-receptor binding sites.[13] These peptides were generated by modification of a central proline residue on a 12-mer peptide, denoted 12p1, which was originally identified as a micro-molar affinity gp120 binder from phage display library screening.[14] We found that replacement of the central Pro residue by azido-Pro and it’s [3+2] click conjugation with alkynes led to variants with several orders of magnitude greater affinity. The resulting peptide triazoles bind to gp120 with 1:1 stoichiometry, inhibit interactions at both the CD4 and co-receptor binding sites and are broadly active in neutralizing cell infection by virus subtypes from all major clades.[15] Some variants of the peptide triazoles can cause cell-independent inactivation and virolysis.[16] Investigation with the highly potent ferrocenyl-triazole containing HNG-156 (R-I-N-N-I-X-W-S-E-A-M-M-CONH2) showed a tolerance for truncation.[17] The shorter 7-mer UM-24 (Cit-N-N-I-X-W-S-CONH2) retained substantial binding and antiviral activity to gp120.
The finding of functional truncates and identification of a localized functional epitope on gp120[18] argues for the potential to identify size-minimized peptidomimetic compounds that retain the dual antagonist and antiviral signature of the peptide triazoles. Hence, in this work, we investigated the extent to which the natural residues of truncate-sized molecules could be replaced by non-natural variants. We indeed found that a fully non-natural variant, peptide KR-42, retained function. The results provide a basis for the design of Env gp120 inhibitors that will retain the important Env antagonist potential with improved metabolic stability.
Results
Design of non- natural peptides
We replaced Trp in the starting sequence UM-24 with the non-natural analog L-Bta (L-3-Benzothienylalanine) and L-amino acid residues at both N-terminal sequence Cit-Asn-Asn and C-terminal Ser, while retaining the triazolePro component. The resulting peptides KR-41 and KR-42 (Figure 1) were prepared by solid phase peptide synthesis (SPPS), purified to a level of 95–98% by RP-HPLC and validated by mass spectrometry (see Supporting Information Figures S1, S2 and S3). Peptide triazoles based on the UM-24 starting point were selected because the control peptide has comparable affinity to its 12-mer peptide HNG-156, and its reduced size vs HNG-156 makes synthesis more streamlined and interpretation of functional outcomes more straightforward.
Figure 1.
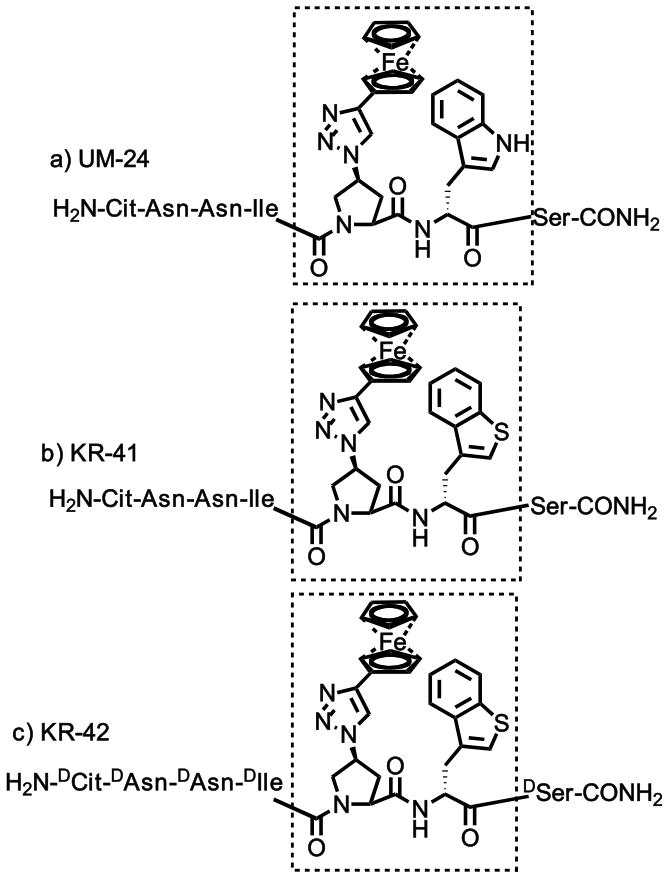
List of peptides designed and synthesized a) UM-24, b) KR-41 and c) KR-42. Residues are noted using 3 letter amino acid code, and the core groups and specific configuration are highlighted with Chemdraw.
Replacement of Trp 6 with non-natural analog
We produced KR-41 to contain the tryptophan analog L-Bt. To measure the inhibition of CD4 and 17b binding to gp120 by KR-41, we immobilized sCD4 (1200 RUs) and mAb 17b (1300 RUs) on separate sensor chips and used 2B6R (anti-interleukin 5 mAb[19] as a control surface. A fixed concentration of YU2-gp120 (100 nM) pre-incubated with serially diluted peptide was passed over the surfaces with a flow of 100 μL/min and about 150 and 300 sec association and dissociation time periods, respectively (Figure 2). Responses after 10 sec dissociation were taken as amount bound, and dose response of these values was evaluated by steady state analysis (Figure 3). Data analysis using Origin7 yielded IC50 values given in Table 1. We examined the antiviral activity of KR-41 using a single round infection assay by HIV-1BaL, where KR-41 displayed an EC50 of 14 μM in comparison to 6.2 μM for the UM-24 control peptide. HIV-1 envelope-pseudotyped, luciferase-reporter viruses were produced, and single round luciferase activity was detected in the presence of inhibitors using a luciferase assay system. The antiviral data were promising, in that this peptide showed similar potency towards HIV-1BaL as UM-24 (Figure 4, Table 1). Hence, one can conclude that Trp6 replacement is feasible with a non-natural Trp analog Bta without loss of potency.
Figure 2.

SPR-sensograms of the competition inhibition of UM-24 (1a, 2a), KR-41 (1b, 2b) and KR-42 (1c, 2c) immobilized 1) sCD4 and 2) mAb 17b surface, immobilized sCD4 (1200 RU) and mAb 17b (1300 RUs), reference cell Immobilized with a 2B6R (antibody to human IL-5) respectively. The sensorgram was collected by passing over serial diluted peptide with a constant 100 nM concentration of gp120 at a flow rate of 100 μL/min. The top, brightest black sensorgram line in all sensograms represents standard 100 nM gp120, without peptide; the top gray to bottom black lines represent peptide concentrations 3.4 nM to 5 μM for UM-24 and KR-41, and 12.2 nM to 25 μM for KR-42. The data point values at 160 sec were used to get an IC50 in Origin7 software after normalization.
Figure 3.
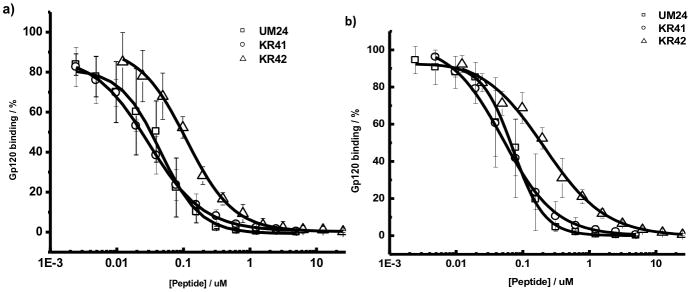
Surface Plasmon Resonance analyzed effects of peptides UM-24, KR-41 and KR-42 on a) sCD4 and b) mAb 17b interactions with HIV-1YU-2 gp120. Results are given as the percentage of gp120 bound relative to gp120 bound in the absence of peptide: 100 × [(RU−RUgp120)/RUgp120]. The normalized values were plotted in Origin7 to get IC50 values. The IC50 values were 45.0 nM, 30 nM and 118.77 nM for UM-24, KR-41 and KR-42 respectively for sCD4 inhibition. The IC50 values were 71.5 nM, 50.8 nM and 207.8 nM for UM-24, KR-41 and KR-42 respectively for mAb 17b inhibition.
Table 1.
Competition SPR and antiviral inhibition efficacies of UM-24, KR-41 and KR-42 peptides.
| Peptide | SPR[a] | EC50 (μM)[a] | |
|---|---|---|---|
| sCD4 | m17b | ||
| IC50 (nM) | |||
| UM-24 | 45.0 ± 0.4 | 71.5 ± 0.2 | 6.7 ± 1 |
| KR-41 | 30.0 ± 0.4 | 50.8 ± 0.1 | 14.0 ± 2 |
| KR-42 | 118.7 ± 0.6 | 207.8 ± 0.5 | 29.0 ± 4 |
Each data point shown is the average of triplicate experiments, and the data are reported as the mean with standard deviation. SPR IC50 values were determined using competition SPR. Antiviral IC50 values were obtained through a single-round infection assay and expressed as relative infection versus untreated (100%).
Figure 4.
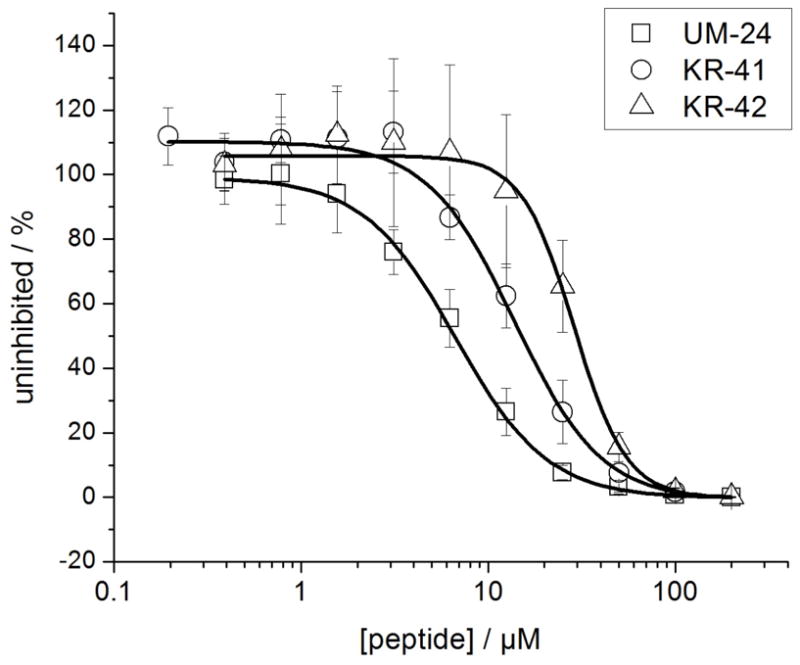
Analysis of antiviral potencies of UM-24, KR-41 and KR-42 using single-round cell infection assays. Recombinant HIV-1BaL was pre-incubated with serial dilution of peptides for 30 min at 37°C. The virus-inhibitor mixture was then added to HOS.CD4.CCR5 for 48h. Infection was determined based on luciferase activity. Data points were fit to a simple sigmoidal inhibition model using the Origin software package to derive the best-fit lines. The EC50 values were 6.7 ± 1 μM (UM-24), 14 ± 2 μM (KR-41) and 29 ± 4 μM (KR-42). Data represent a minimum of three repeats.
D-amino acid substitutions outside the central LFtP-LBta core
In order to achieve a more completely non-natural molecule, we replaced residues surrounding the “hot spot” LFtP-LBta grouping with D-amino acids. D-amino acids have previously been used to replace natural amino acids in HIV-1 peptide entry inhibitors by employing structure-guided mirror image phage display.[20] D-amino acid replacement allowed evaluation of the functional dependence on the stereochemical environment in residues around the central hydrophobic core, in particular the N-terminal domain. We investigated the UM-24 variant, denoted KR-42, which has the sequence DCit-DAsn-DAsn-DIle-LFtP-LBta-DSer-NH2. Intriguingly, replacement of core-surrounding residues with D-amino acids was tolerated to a significant extent as judged by both molecular and cell assays. Using a competitive surface plasmon resonance assay, we evaluated the ability of KR-42 to inhibit the binding of YU2 gp120 to CD4 and 17b (Figures 2 and 3). These analyses were carried out using addition of a constant concentration of gp120 with serially diluted KR-42, followed by flowing the mixtures over sensor chips containing immobilized CD4 or 17b. To assess the activity of KR-42 to inhibit cell infection, antiviral assays were performed using single round infection of target cells by HIV-1BaL. The EC50 data were compared with UM-24 and showed that KR-42 is five-fold less active (Figure 4, Table 1).
Direct binding analyses of non natural peptide triazoles using isothermal titration calorimetry
We measured the direct binding characteristics of non-natural peptide triazoles to gp120 using isothermal titration calorimetry; the results are shown in Figure 5 and Table 2. Titrations with KR-41, KR-42 and UM-24 control (Figure 5) all showed 1:1 binding stoichiometry (Table2). The KD values for the non-natural peptides, obtained obtained by ITC, showed a decline of affinity vs UM-24. Despite this decline, KR-42 retained a substantial affinity, consistent with the competition results presented above. Nonetheless, all of the peptides had similar thermodynamic signatures, namely, the pattern of a large negative ΔH and negative ΔS. This argues that the non-natural peptide triazoles retain the same fundamental mechanistic features as the natural UM-24. The large favorable change in enthalpy that is opposed by a large unfavorable entropy change suggests that the three peptide triazoles induce some degree of conformational structuring of gp120. Comparison of the binding energetics shows that the three peptide triazoles bind with similar enthalpy changes whereas the entropy contributions to Gibbs energy are more unfavorable for the non-natural peptide triazoles, which consequently bind with lower affinity to gp120.
Figure 5.
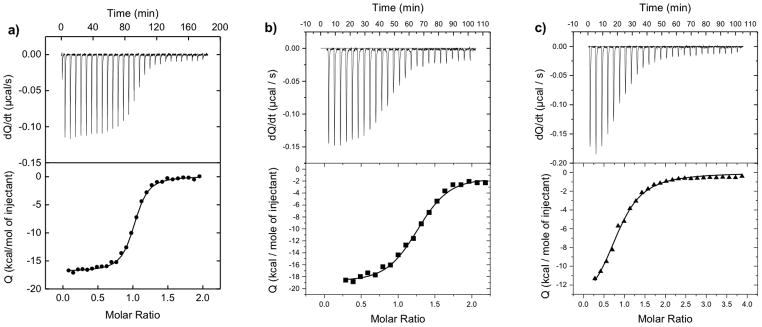
Calorimetric titrations of gp120 with a) UM-24, b) KR-41 and c) KR-42 at 25°C in PBS at pH 7.4. The concentration of gp120 was 2 μM; the UM-24 and KR-41 concentrations were 25 μM, and the KR-42 concentration was 50 μM. VP-ITC titration calorimetric system used from MicroCal/GE Healthcare (Northampton, MA, USA). The calorimetric cell (~1.4mL), containing gp120 from the YU2 strain dissolved in PBS (Roche Diagnostics GmbH, Mannheim, Germany), pH 7.4, was titrated with different peptides dissolved in the same buffer.
Table 2.
Thermodynamic values for the binding of UM-24, KR-41 and KR-42 to gp120.
| Peptide | ITC[b] (KD, nM) | ΔG (kcalmol−1) | ΔH (kcalmol−1) | −TΔS (kcalmol−1) |
|---|---|---|---|---|
| UM-24 | 21.1 ±1.4 | −10.5±0.8 | −14.1±0.6 | +3.6±1.6 |
| KR-41 | 70.0±1.6 | −9.8 ±1.7 | −14.9±7.5 | +5.1±2.6 |
| KR-42 | 282.1±2.2 | −8.9 ±2.9 | −14.6±1.7 | +5.7±2.2 |
Data collected in duplicate. ITC experiments were performed at 25°C in 1xPBS (pH 7.4). The dissociation constant (Kd) and the change in enthalpy (ΔH) were determined at 25°C by isothermal titration calorimetry using a VP-ITC titration calorimetric system from MicoCal/GE Healthcare (Northampton, MA, USA). ΔG and −TΔS were calculated using the equations: ΔG = −RTln(1/Kd), ΔG = ΔH − TΔS. The data are reported as the mean with standard deviation.
Discussion
We sought in the current work to establish the potential to form peptidomimetic variants of peptide triazoles. Previous studies have found that the class of broadly active peptide triazole inhibitors can bind specifically and with nanomolar affinity to HIV-1 gp120, dual antagonize the binding sites of Env for both host cell receptors CD4 and CCR5/CXCR4 co-receptor and inhibit cell infection by both X4 and R5 viruses.[21] All of the gp120 binding inhibition and antiviral activities of the peptide triazoles [13, 15–18] depend on specific binding to a highly conserved peptide triazole functional epitope in gp120.[18] Here we investigated the functions of increasingly non-natural peptide triazoles. We based the investigation of localized sub-domains in the sequence-minimized UM-24 peptide triazole as depicted in Figure 6. Here, the LFtP-LW sequence with a function-requiring specific stereochemistry is depicted as the central domain. In this depiction the W-S sequence at the C-terminus and the Cit-N-N-I sequence at the N-terminus are natural L-amino acids. We examined the outcome of conversion of these C- and N-terminal sequences into non-natural variants. The results of this work showed that such conversions were tolerated with significant retention of function.
Figure 6.
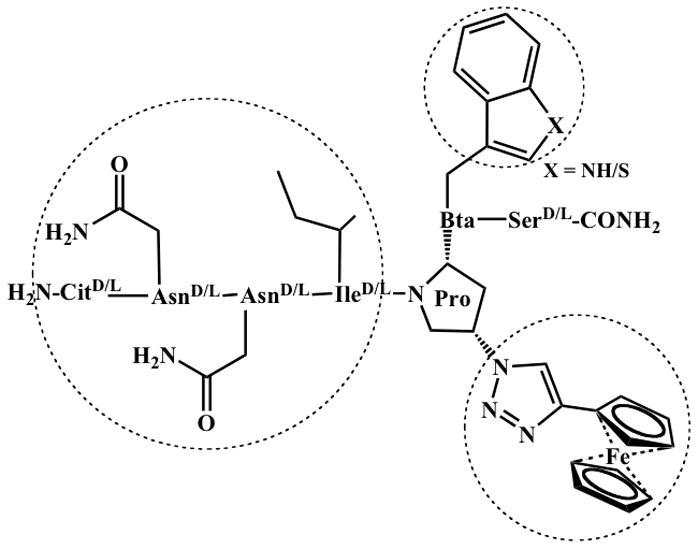
Tripartite model of ferrocenyl peptide triazoles emphasizing the functionally important structural domains around the central proline of triazole, hydrophobic residue, C-terminal to Pro, and N-terminal extension from Pro. The sequence variations embodied in conversion of UM-24 to KR-42 are indicated.
We found that the important core residue Trp of the truncated peptide triazole UM-24 could be replaced by Bta, benzothienyl-Ala. The resulting peptide, KR-41 as shown in Figure 1, varies from UM-24 by containing a sulfur atom of Bta in place of the NH of Trp. KR-41 is closely similar to UM-24 in competition of CD4 and 17b binding to gp120 (Figures 2 and 3, Table 1), direct binding affinity to gp120 (ITC, Figure 5, Table 2) and inhibition of virus infection (Figure 4, Table 1). That the replacement of Trp 6 with Bta was substantially tolerated functionally argues that the hydrogen bonding pattern of the indole NH can be varied at least to some extent without loss of productive interaction with gp120. On the other hand, hydrophobic space filling of the cavity where the side chain of residue 7 resides is likely to be important. This hypothesis is argued by the prior finding that replacement of Trp with Ala leads to loss of function.[14] This in turn opens up the possibility to replace the Trp indole of the original peptide triazole with other large hydrophobic side chains as along as they fit with steric constraints of gp120 binding surface (see below).
The most sweeping non-natural conversion of peptide triazoles in the current work was the replacement of all residues outside the core LFtP-LW with D-amino acids. Initially, this conversion was tested in a preliminary experiment with UM-24. Retention of function in this experiment (data not shown) led to the study of similar D-residue conversion with the KR-41 background, leading to KR-42. This peptide contains no natural amino acids, being comprised of D-residues at positions 1–4 and 7, FtP at position 5 and Bta at position 6. Pleasingly, KR-42 retains the hallmark of direct binding to gp120, dual receptor site antagonism and inhibition of virus cell infection. Furthermore, the ITC analysis argues that the conformational changes induced by the natural peptide and reflected in particular by the substantial −ΔH (Table 2) are triggered by KR-42. While the potency of KR-42 does suffer by comparison to KR-41, the results argue that the fundamental binding and functional signature of peptide triazoles is retained in KR-42.
The retention of significant function in KR-42 leads to the question of what role the L-residues have in peptide triazoles that can be mimicked by D-residues. Prior evaluation of peptide variations in the N-term residues 1–4 and C-term residue 7 suggest that these help stabilize binding to gp120 (Umashankara, et al unpublished data). For example, removal of these residues in more severe truncations greatly suppresses activity. The tolerance of D-residues suggests that residues outside of the hydrophobic FtP-W (or FtP-Bta) cluster may contribute important contacts with gp120 but that these likely are generic, such as H-bonding. The results of this work, together with those from prior SAR investigations,[13b, 17] are consistent with the notion that the tripartite model of Figure 6 can be used as a guide for developing peptidomimetic inhibitors with peptide triazole antiviral activities. Within the central Ile-AzP-Trp, synthetic variations can be used to refine the roles of hydrogen bonding and spatial complementarity. The successful replacement of Trp by Bta suggests that hydrogen bonding of indole NH is not critical. On the other hand, hydrophobic space filling of the cavity in which the side chain resides could be important, and alternative non-natural substitutions can be employed to improve efficacy and non-natural character. Synthetic non-natural variations of the isoleucine side chain in the central core may also improve steric fit onto the gp120 surface and hence improve efficacy. In addition, since the C- and N- termini around the central core can be modified completely with D-amino-acids with retention of function, these residues, especially those that can provide hydrogen bonds, may be replaceable by fully organic scaffolds with similar H-bonding potential.
Experimental Section
Reagents
All Fmoc-protected α-amino acids, O-Benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate (HBTU), 1-Hydroxybenzotriazole (HOBt), Rink amide resin {(4-(2′,4′-Dimethoxyphenyl-Fmoc-aminomethyl)phenoxy resin) with a 0.55 mmol/g substitution}, N,N-dimethylformamide (DMF), Pyridine and N,N-Diisopropylethylamine (DIPEA) were purchased from Chem-Impex International Inc, Ethynylferrocene and Cu(1)I was purchased from Sigma-Aldrich and used without further purification. Fmoc-cis-4-azidoproline was synthesized starting with commercially available trans-Hyp-OH. The monoclonal antibody, 17b IgG, was purchased from Strategic Biosolutions, USA. CHO-ST4.2 cells were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: CHO ST4.2 from Dr. Dan Littman. The expression plasmid HIV-1YU-2 gp120 was a kind gift from Dr. Joseph Sodroski.
Synthesis and purification of peptides
Peptides UM-24, KR-41, and KR-42 were synthesized using manual Fmoc chemistry-based SPPS from Rink Amide resin at 0.25 mmol scale. The residues were assembled on resin and a [3+2] cycloaddition reaction was completed using ethynylferrocene. The click reaction was stirred at room temperature overnight. Subsequently, the resin was filtered and taken in to cleavage cocktail of 95:2:2:1 trifluoroacetic acid (TFA)/1,2-ethanedithiol/water/thioanisole for 3 h. Separated resin from aliquot solution and precipitated in cold diethyl ether. The crude peptide was dissolved in 50% ACN/H2O and purified by the reversed-phase HPLC (Beckmann Coulter) on a preparative C18 column (100 Å, 2.14 × 25 cm) with a linear gradient of 5–95 % of ACN/water in 0.1% TFA. The final purified peptides were lyophilized and validated by MALDI-TOF-MS, m/z KR-41: MObs = 1153.47 Da (M calculated = 1152.6Da); KR-42: MObs = 1153.34 Da (M calculated =1152.6). The validation HPLC and MALDI-MS profiles for these peptides are given in the supporting information Figures S1, S2 and S3.
Recombinant Protein Production
HIV-1YU-2gp120 with a C-terminal hexa-histidine tag, were produced using the Invitrogen protocol in human embryonic kidney cells (HEK 293F). Briefly, cells were grown to 1×106 cells/ml and transfected with the pcDNA3.1+ expression plasmid containing full length YU-2 gp120. Cells were grown at 7.5% CO2 for 5 days and then harvested. Supernatant was spun down and filtered using 0.22 μM filters. The supernatant was then purified over a Nickel-NTA column (Amershem). The column was washed with buffer A (20 mM imidazole, 150 mM NaCl, 20 mM Tris-HCl, pH 7.4) and protein was eluted with buffer B (200 mM imidazole, 150 mM NaCl, 20 mM Tris-HCl, pH 7.4). The protein was concentrated with a Centriprep-30 (Amicon) and immediately fractionated on a Hi-load 26/60 Superdex 200 column in PBS buffer. Peak fractions were collected and analyzed by SDS-PAGE. Single band fractions corresponding to the correct protein size were collected and concentrated, and frozen at −80°C. Soluble four domain CD4 was expressed in CHO-ST4.s cells in a hollow fiber bioreactor. Supernatant was purified using a sulfopropyl-substituted ion exchange column (Amershem Biosciences, Piscataway, NJ). Bound fractions were fractionated on a quaternary ammonium substituted ion exchange column (Amerchem Biosciences, Piscataway, NJ). Unbound material was concentrated and analyzed by SDS-PAGE and ELISA.
Optical Biosensor Binding Assays
Surface plasmon resonance (SPR) assays similar to those reported previously were performed on a BIAcore 3000 optical biosensor instrument (GE Healthcare, USA). All experiments were done at 25°C using standard Phosphate Buffer Saline (PBS, pH 7.4, with 0.005% Surfactant Tween). A CM5 sensor chip (GE Healthcare) was used in all cases and was derivatized by using N-ethyl-N-(3-dimethylaminopropyl) carbodiimide/N-hydroxysuccinimide[22] with HIV-1YU-2gp120 (~3000 RUs) or, as a control surface, 2B6R (monoclonal antibody to human interleukin-5). For direct binding experiments, peptides UM-24, KR-41, and KR-42 peptide in PBS buffer (concentration range of UM-24 and KR-41, 5μM-3.4nM and KR-42, 25μM to 12.2 nM) were injected over the sensor surface at a flow rate of 100 μL/min with a 150 sec association phase and a 300 sec dissociation phase. Analysis of peptide-mediated sCD4 and mAb 17b inhibition was achieved by injecting a fixed concentration of HIV-1YU2gp120 (100nM), with increasing peptide concentration (range of UM-24 and KR-41, 5 μM-3.4 nM and KR-42, 25 μM to 12.2 nM), over sCD4 (~1200RU) and mAb 17b (~1300RU) surfaces for 150 sec association and 300 sec dissociation at a flow rate of 100 μL/min in PBS. Regeneration of the surface was achieved with a single 3 second pulse (5μl) of 10 mM glycine, pH 1.5 for all the experiments. All analyses were performed in triplicate. SPR data analyses were performed using BIAEvaluation 4.0 software (GE Healthcare). A double reference subtraction was performed for each data set to account for non-specific binding. Direct binding data were fitted to a simple 1:1 binding model with a parameter included for mass transport. The average kinetic parameters (association {ka} and dissociation {kd} rates) obtained from a minimum of 3 data sets were used to define equilibrium association (KA) and dissociation (KD) constants. The evaluation method for SPR inhibition data included a calculation of the inhibitor concentration at 50% of the maximal response (IC50). The inhibition curve was converted into a calibration curve by the use of a fitting function. The amount of gp120 bound (expressed as surface plasmon resonance units [RU]) was measured 10 sec after the end of the injection and by subtracting RUs bound for the same solution on a blank mock-coupled surface. Results are given as the percentage of gp120 bound relative to gp120 bound in the absence of peptide: 100 × [(RU−RUgp120)/RUgp120]. The normalized values were plotted in Origin7 to get IC50 values, shown in Figure 3.
Isothermal titration calorimetry (ITC)
Isothermal titration calorimetric experiments were performed at 25°C using a VP-ITC titration calorimetric system from MicroCal LLC/GE Healthcare (Northampton, MA). The calorimetric cell (~1.4 mL), containing YU2-gp120 at about 2 μM dissolved in PBS, pH 7.4 (Roche Diagnostics GmbH), was titrated with 25 μM of UM-24 and KR-41 and 50 μM of KR-42. The peptide solution was added in aliquots of 10 μL volume per injection. All experimental buffers were degassed to avoid any formation of air bubbles in the calorimeter during stirring. The heat evaluated upon each injection of the peptide solution was obtained from the integral of the calorimetric signal. The heat associated with a binding of ligand to the protein in the cell was obtained by subtracting the heat of dilution from the heat of reaction. Heat of dilution due to mismatch between the syringe and cell solution were negligible in all experiments. The individual binding heats were plotted against the molar ratio, and the values for the enthalpy change (ΔH) and association constant, Ka (KD = 1/Ka), were obtained by nonlinear regression of the data.
Preparation of Recombinant Luciferase Expressing Virus and Cell Infection Assays
Single round recombinant, luciferase-reporter viruses were produced in HEK293T cells using Fugene transfection reagent (Qiagen) according to the manufacturer’s protocol. Cells were seeded in T75 flasks (approximately 3 × 106 cells per flask) and transfected the following day with 4 μg of plasmid encoding the envelope (HIV-1BaL or VSV-G) together with 8 μg of the envelope-deficient pNL4-3-Fluc+env− provirus developed by N. Landau.[23] Culture supernatants containing viral particles were collected 48–72 hours after transfection, clarified by centrifugation, filtered, aliquoted and stored at −80°C until use. For inhibition experiments, the viral stocks were first incubated with serial dilutions of the inhibitor at 37 °C for 30 minutes. The mixture was added to human osteosarcoma cells that stably express CD4 and CCR5 (HOS.CD4.CCR5) for 48 hours. The cells were then lysed with passive lysis buffer (Promega) followed by freeze-thaw cycles. Luciferase assays were performed using 1 mM D-luciferin salt (Anaspec) as substrate and detected on a 1450 Microbeta Liquid Scintillation and Luminescence Counter (Wallac and Jet). IC50 values were estimated using non-linear regression analysis with Origin V.8.1 (Origin Lab). All experiments were performed at least in triplicate and results were expressed as relative infection with respect to cell infected with virus in the absence of inhibitor (100% infected).
Supplementary Material
Acknowledgments
This study was supported by National Institute of General Medical Sciences (NIGMS) with grant # 5P01GM056550.
References
- 1.a) A new report by the Joint United Nations Programme on HIV/AIDS (UNAIDS) released on 21 November, 2011 http://www.unaids.org/en/resources/publications/2011.; b) Engelman A, Cherepanov P. Nat Rev Microbiol. 2012;10:279–90. doi: 10.1038/nrmicro2747. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Teixeira C, Gomes JRB, Gomes P, Maurel F. Eur J Med Chem. 2011;46:979–92. doi: 10.1016/j.ejmech.2011.01.046. [DOI] [PubMed] [Google Scholar]
- 2.Montaner JS, Lima VD, Barrios R, Yip B, Wood E, Kerr T, Shannon S, Harrigan PR, Hogg RS, Daly P, Kendall P. Lancet. 2010;376:532–9. doi: 10.1016/S0140-6736(10)60936-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Lucas GM, Chaisson RE, Moore RD. Ann Int Med. 1999;131:81–87. doi: 10.7326/0003-4819-131-2-199907200-00002. [DOI] [PubMed] [Google Scholar]; b) Yerly S, Kaiser L, Race E, Bru JP, Clavel F, Perrin L. Lancet. 1999;354:729–733. doi: 10.1016/S0140-6736(98)12262-6. [DOI] [PubMed] [Google Scholar]; c) Greenberg ML, Cammack N. J Antimicrob Chemother. 2004;54:333–340. doi: 10.1093/jac/dkh330. [DOI] [PubMed] [Google Scholar]; d) Ray N, Harrison JE, Blackburn LA, Martin JN, Deeks SG, Doms RW, Virol J. 2007;81:3240–3250. doi: 10.1128/JVI.02413-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Shafer RW, Schapiro JM. AIDS Rev. 2008;10:67–84. [PMC free article] [PubMed] [Google Scholar]
- 4.a) Flexner C. Nat Rev Drug Discover. 2007;6:959–966. doi: 10.1038/nrd2336. [DOI] [PubMed] [Google Scholar]; b) Hertje M, Zhou M, Dietrich U. ChemMedChem. 2009;5:1825–1835. doi: 10.1002/cmdc.201000292. [DOI] [PubMed] [Google Scholar]
- 5.a) Dwyer JJ, Wilson KL, Davison DK, Freel SA, Seedorff JE, Wring SA, Tvermoes NA, Matthews TJ, Greenberg ML, Delmedico MK. Proc Natl Acad Sci USA. 2007;104:12772–12777. doi: 10.1073/pnas.0701478104. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Eggink D, Berkhout B, Sanders RW. Curr Pharm Des. 2010;16:3716–3728. doi: 10.2174/138161210794079218. [DOI] [PubMed] [Google Scholar]
- 6.a) Este JA, Telenti A. Lancet. 2007;370:81–88. doi: 10.1016/S0140-6736(07)61052-6. [DOI] [PubMed] [Google Scholar]; b) Perry CM. Drugs. 2010;70:1189–1213. doi: 10.2165/11203940-000000000-00000. [DOI] [PubMed] [Google Scholar]; c) Yost R, Pasquale TR, Sahloff EG. Am J Health Syst Pharm. 2009;66:715–26. doi: 10.2146/ajhp080206. [DOI] [PubMed] [Google Scholar]
- 7.a) Chan DC, Kim PS. Cell. 1998;93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]; b) Cai L, Jiang S. ChemMedChem. 2009;5:1813–1824. doi: 10.1002/cmdc.201000289. [DOI] [PubMed] [Google Scholar]; c) D’Souza MP, Cairns JS, Plaeger SF. JAMA. 2000;284:215–222. doi: 10.1001/jama.284.2.215. [DOI] [PubMed] [Google Scholar]; d) Pancera M, Lebowitz J, Schön A, Zhu P, Freire E, Kwong PD, Roux KH, Sodroski J, Wyatt R. J Virol. 2005;79:9954–9969. doi: 10.1128/JVI.79.15.9954-9969.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. Nature. 2008;455:109–113. doi: 10.1038/nature07159. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Borgnia HMJ, Shi D, Bartesaghi A, He H, Pejchal R, Kang YA, Depetris R, Marozsan AJ, Sanders RW, Klasse PJ, Milne JLS, Wilson IA, Olson WC, Moore JP, Subramaniam S. Proc Natl Acad Sci USA. 2011;108:11440–11445. doi: 10.1073/pnas.1101414108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klatzmann D, Champagne E, Chamaret S, Gruest J, Guetard D, Hercend T, Gluckman JC, Montagnier L. Nature. 1984;312:767–768. doi: 10.1038/312767a0. [DOI] [PubMed] [Google Scholar]
- 10.a) Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. Science. 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]; b) Feng Y, Broder CC, Kennedy PE, Berger EA. Science. 1996;272:872–877. [Google Scholar]
- 11.Wyatt R, Sodroski J. Science. 1998;280:1884–1888. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- 12.a) Doms RW, Moore JP. J Cell Biol. 2000;151:F9–14. doi: 10.1083/jcb.151.2.f9. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Eckert DM, Kim PS. Annual Rev Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 13.a) Gopi HN, Tirupula KC, Baxter S, Ajith S, Chaiken IM. ChemMedChem. 2006;1:54–57. doi: 10.1002/cmdc.200500037. [DOI] [PubMed] [Google Scholar]; b) Gopi HN, Umashankara M, Pirrone V, LaLonde J, Madani N, Tuzer F, Baxter S, Zentner I, Cocklin S, Jawanda N, Miller SR, Schon A, Klein JC, Freire E, Krebs FC, Smith AB, Sodroski J, Chaiken IM. J Med Chem. 2008;51:2638–2647. doi: 10.1021/jm070814r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gopi H, Cocklin S, Pirrone V, McFadden K, Tuzer F, Zentner I, Ajith S, Baxter S, Jawanda N, Krebs FC, Chaiken IM. J Mol Recognit. 2009;22:169–174. doi: 10.1002/jmr.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrer M, Harrison SC. J Virol. 1999;73:5795–5802. doi: 10.1128/jvi.73.7.5795-5802.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Cocklin S, Gopi H, Querido B, Nimmagadda M, Kuriakose S, Cicala C, Ajith S, Baxter S, Arthos J, Martin-Garcia J, Chaiken IM. J Virol. 2007;81:3645–3648. doi: 10.1128/JVI.01778-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) McFadden K, Fletcher P, Rossi F, Kantharaju, Umashankara M, Pirrone V, Rajagopal S, Gopi H, Krebs FC, Martin-Garcia J, Shattock RJ, Chaiken I. Antimicrob Agents Chemother. 2012;56:1073–1080. doi: 10.1128/AAC.05555-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bastian AR, Kantharaju, McFadden K, Duffy C, Rajagopal S, Contarino MR, Papazoglou E, Chaiken I. ChemMedChem. 2011;6:1335–1339. doi: 10.1002/cmdc.201100177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Umashankara M, McFadden K, Zentner I, Schon A, Rajagopal S, Tuzer F, Kuriakose SA, Contarino M, Lalonde J, Freire E, Chaiken I. ChemMedChem. 2010;5:1871–1879. doi: 10.1002/cmdc.201000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tuzer F, Madani N, Kantharaju K, Zentner I, Holmes A, Lalonde J, Sodroski J, Chaiken I. PROTEINS: Structure, Function, and Bioinformatics. Oct 26, 2012. [DOI] [Google Scholar]
- 19.Scibek JJ, Evergren E, Zahn S, Canziani GA, Van Ryk D, Chaiken IM. Anal Biochem. 2002;307:258–265. doi: 10.1016/s0003-2697(02)00043-x. [DOI] [PubMed] [Google Scholar]
- 20.a) Schumacher TN, Mayr LM, Minor DL, Jr, Milhollen MA, Burgess MW, Kim PS. Science. 1996;271:1854–1857. doi: 10.1126/science.271.5257.1854. [DOI] [PubMed] [Google Scholar]; b) Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. Cell. 1999;99:103–115. doi: 10.1016/s0092-8674(00)80066-5. [DOI] [PubMed] [Google Scholar]; c) Liu D, Madani N, Li Y, Cao R, Choi WT, Kawatkar SP, Lim MY, Kumar S, Dong CZ, Wang J, Russell JD, Lefebure CR, An J, Wilson S, Gao YG, Pallansch LA, Sodroski JG, Huang Z. J Virol. 2007;81:11489–11498. doi: 10.1128/JVI.02845-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. Proc Natl Acad Sci USA. 2007;104:16828–16833. doi: 10.1073/pnas.0708109104. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Choi WT, Kumar KS, Madani N, Han X, Tian S, Dong CZ, Liu D, Duggineni S, Yuan J, Sodroski JG, Huang Z, An J. Biochemistry. 2012 doi: 10.1021/bi2016712. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Francis JN, Redman JS, Eckert DM, Kay MS. Biocon Chem. 2012;23:1252–1258. doi: 10.1021/bc300076f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.a) Biorn AC, Cocklin S, Madani N, Si Z, Ivanovic T, Samanen J, Van Ryk DI, Pantophlet R, Burton DR, Freire E, Sodroski J, Chaiken IM. Biochemistry. 2004;43:1928–1938. doi: 10.1021/bi035088i. [DOI] [PubMed] [Google Scholar]; b) Cocklin S, Gopi HN, Querido B, Nimmagadda M, Kuriakose S, Cicala C, Ajith S, Baxter S, Arthos J, Martín-García J, Chaiken IM. J Virol. 2007;81:3645–8. doi: 10.1128/JVI.01778-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishino T, Pillalamarri U, Panarello D, Bhattacharya M, Urbina C, Horvat S, Sarkhel S, Jameson B, Chaiken I. Biochemistry. 2006;45:1106–1115. doi: 10.1021/bi0518038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Connor RI, Chen BK, Choe S, Landau NR. Virology. 1995;206:935. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.