

Abstract
Defective control of the alternative pathway of complement is an important risk factor for several renal diseases, including atypical hemolytic uremic syndrome. Infections, drugs, pregnancy, and hemodynamic insults can trigger episodes of atypical hemolytic uremic syndrome in susceptible patients. Although the mechanisms linking these clinical events with disease flares are unknown, recent work has revealed that each of these clinical conditions causes cells to release microparticles. We hypothesized that microparticles released from injured endothelial cells promote intrarenal complement activation. Calcineurin inhibitors cause vascular and renal injury and can trigger hemolytic uremic syndrome. Here, we show that endothelial cells exposed to cyclosporine in vitro and in vivo release microparticles that activate the alternative pathway of complement. Cyclosporine-induced microparticles caused injury to bystander endothelial cells and are associated with complement-mediated injury of the kidneys and vasculature in cyclosporine-treated mice. Cyclosporine-induced microparticles did not bind factor H, an alternative pathway regulatory protein present in plasma, explaining their complement-activating phenotype. Finally, we found that in renal transplant patients, the number of endothelial microparticles in plasma increases 2 weeks after starting tacrolimus, and treatment with tacrolimus associated with increased C3 deposition on endothelial microparticles in the plasma of some patients. These results suggest that injury-associated release of endothelial microparticles is an important mechanism by which systemic insults trigger intravascular complement activation and complement-dependent renal diseases.
Hemolytic uremic syndrome (HUS) is a disease characterized by microangiopathic hemolytic anemia, thrombocytopenia, and renal failure.1 Recent work demonstrates that defects in regulation of the alternative pathway of complement are major risk factors for developing atypical HUS (aHUS), and mutations in numerous complement proteins have been identified in these patients.2–5 Even in patients with mutations in complement regulatory proteins, however, the disease is episodic and is frequently triggered by a clinical illness or stressor.4 Disease flares have been associated with the use of calcineurin inhibitors, infection, pregnancy, and malignant hypertension.4 It has been proposed that these insults trigger intrarenal complement activation, but the mechanisms by which these different conditions activate the alternative pathway within the kidney are not known.
Microparticles are submicrometer-sized membrane vesicles (0.05–1 μm) that are actively shed from cells in response to activation or injury.6,7 Complement activation on the cell membrane can induce cells to release microparticles.8,9 Although complement proteins can be detected on the surface of microparticles released from apoptotic and injured cells,10 less is known regarding whether microparticles from specific cell types themselves can cause complement activation. We hypothesized that injury of endothelial cells induces the release of complement-activating microparticles into the circulation. Because endothelial cells are in contact with complement proteins in the plasma, the release of complement-activating microparticles by endothelial cells could have a profound effect on intravascular complement activation.
Cyclosporine (CsA) is a calcineurin inhibitor used as an immunosuppressive agent for the prevention of organ allograft rejection and as a treatment for autoimmune diseases. However, the use of CsA is associated with the development of vascular injury, nephrotoxicity, and hypertension, and aHUS.11 Because of the association of CsA with renal toxicity, vascular injury, and aHUS, we examined whether exposure of endothelial cells to CsA could induce the release of microparticles, and we examined whether microparticles from CsA-exposed endothelial cells activate the complement system within the kidney.
Results
CsA Causes Complement-Mediated Renal and Vascular Injury
We treated wild-type and “factor B deficient mice (fB−/− mice)” mice with high-dose CsA for 2 weeks. At the end of that time we measured the serum urea nitrogen (SUN) level as a marker of renal function. SUN levels in wild-type mice treated with CsA were higher than those of vehicle-treated animals (Figure 1A). Treatment of fB−/− mice with CsA did not cause an increase in the SUN levels. We measured serum asymmetric dimethylarginine (ADMA) as a marker of systemic vascular injury and found that ADMA levels rose in mice treated with CsA (Figure 1B). Levels were not significantly higher in fB−/− mice treated with CsA than in fB−/− control mice, although there was a trend toward higher levels in the CsA-treated mice. The glomeruli in mice treated with cyclosporine demonstrated areas of mesangial proliferation and expansion (Figure 1C), and hyaline deposition was observed at the vascular pole of mice treated with cyclosporine (Figure 1D). We next examined the kidneys of these mice for evidence of complement activation (Figure 2). Increased C3 deposits were seen in the glomeruli of wild-type mice treated with CsA (Figure 2, B and E). Glomerular C3 did not change in fB−/− mice treated with CsA compared with vehicle controls (Figure 2, D and E), indicating that intraglomerular complement activation in CsA-treated mice proceeded via the alternative pathway.
Figure 1.

Cyclosporine causes alternative pathway-dependent injury of the kidneys and vasculature. Wild-type and fB−/− mice were injected daily with CsA for 2 weeks. (A) SUN was measured as a marker of renal dysfunction. The SUN levels in CsA-injected wild-type (WT) mice were significantly higher than those in vehicle (veh)-treated wild-type mice and in CsA-treated fB−/− mice. (B) ADMA was measured as a marker of vascular injury. The levels in CsA-treated wild-type mice were significantly higher than those in vehicle-treated controls, whereas levels in CsA-treated fB−/− mice did not significantly increase compared with those in vehicle-treated fB−/− mice. *P<0.05, **P<0.01. Light microscopy of periodic acid-Schiff–stained sections from CsA-treated mice demonstrated areas of (C) mesangial expansion and proliferation (arrow) and (D) hyaline deposition within the vascular pole (arrow). Original magnification ×400.
Figure 2.

Cyclosporine causes intraglomerular activation of the alternative pathway of complement. Wild-type (WT) and fB−/− mice were injected daily with CsA for 2 weeks, and intrarenal complement activation was assessed by immunofluorescence microscopy for C3. Glomeruli are indicated with arrows. (A) Wild-type mice treated with vehicle (veh) demonstrated a low level of glomerular C3. (B) Wild-type mice treated with CsA demonstrated C3 deposition throughout the glomeruli. (C) Low-level glomerular C3 was seen in vehicle-treated fB−/− mice, similar to that seen in wild-type mice. (D) Treatment of fB−/− mice with CsA did not cause a significant increase in glomerular C3. (E) Glomerular complement activation was determined by quantitative immunofluorescence of glomerular C3 in the different experimental groups. Original magnification ×200. ***P<0.001.
CsA Causes Endothelial Cells to Release Complement-Activating Microparticles In Vivo
Microparticles were purified from the plasma of CsA- and vehicle-treated mice (Figure 3A). We used flow cytometry to identify populations of microparticles from endothelial cells (CD31+) and platelets (CD41+). Treatment of wild-type mice with CsA caused an increase in the number of endothelial and platelet microparticles in plasma (Figure 3, B and C). Leukocyte microparticles (CD144+) did not increase (Figure 3D). C3 deposition on the surface of the endothelial microparticles was measured by flow cytometry, and we found that more C3 was present on the surface of microparticles isolated from CsA-treated wild-type mice than on those isolated from mice that received vehicle or on microparticles from CsA-treated fB−/− mice. (Figure 3, E and F). These results indicate that treatment of the mice with CsA increases the overall number of endothelial microparticles released and also increases the complement-activating activity of the endothelial microparticles. Furthermore, the increase in complement activation by CsA-induced microparticles requires an intact alternative complement pathway.
Figure 3.

Cyclosporine induces release of complement-activating endothelial microparticles in vivo. Wild-type (WT) and fB−/− mice were injected daily with CsA for 2 weeks. Microparticles were isolated from the plasma and evaluated by flow cytometry. (A) Beads were used to determine the size distribution of small particles in the samples, and the gate used for microparticle analysis is shown. Treatment with CsA increased the number of (B) endothelial (CD31+) and (C) platelet (CD41+) microparticles in the circulation. (D) The number of leukocyte microparticles (CD144+) did not increase in mice treated with CsA. (E and F) C3 deposition on the microparticles in the plasma of CsA treated mice was greater than that on microparticles from vehicle (veh)-treated mice. Dashed line, isotype control; black line, control-treated; shaded curve, CsA-treated. *P<0.05.
CsA Causes Endothelial Cells to Shed Microparticles In Vitro
We next tested whether CsA causes endothelial cells to increase their generation of microparticles in vitro. Because cyclosporine is poorly soluble in water, we used a liquid chromatography-mass spectrometry assay to measure intracellular concentrations of CsA in endothelial cells treated with various concentrations of CsA (Supplemental Figure 1). We found that exposure of endothelial cells to CsA, 250 μg/ml, caused a reproducible uptake of the drug by the cells, and this concentration caused lysis of >50% of the cells by 24 hours (Supplemental Figure 1). After 16 hours of exposure to this concentration of CsA (when apoptosis and necrosis are not yet evident by flow cytometry), the number of microparticles in the cell supernatant increased significantly (Figure 4A).
Figure 4.

Treatment of endothelial cells with cyclosporine in vitro causes generation of complement-activating microparticles (MPs). (A) Endothelial cells were treated with CsA, 50 or 250 μg/ml, for 16 hours, and the number of microparticles in the supernatant was measured by flow cytometry. (B) Microparticles from the supernatant of unmanipulated or CsA-treated cells were exposed to serum, and C3a was measured by ELISA. Microparticles from unmanipulated cells increased generation of C3a above that seen in serum alone, but microparticles from CsA-treated cells caused a significantly greater degree of C3a generation. (C) Microparticles from the supernatants of unmanipulated or CsA-treated cells were exposed to serum, and C3 deposition on the microparticles was assessed by flow cytometry. A representative graph from three independent experiments is shown. Dashed line, isotype control; black line, unmanipulated cells; shaded curve, CsA-treated cells. (D) Microparticles from the supernatant of unmanipulated or CsA-treated cells were exposed to serum, and C3 deposition on the microparticles was assessed by Western blot analysis using a polyclonal antibody to C3 and a monoclonal antibody to C3d. The α′2 band of C3 (representing the iC3b form) and C3d were readily detected on microparticles from CsA-treated cells that were exposed to serum (MP, microparticle; NMS, normal mouse serum; S, serum). (E) Unmanipulated endothelial cells were exposed to 10% mouse serum with the addition of variable quantities of microparticles from CsA-treated cells (CsA-MP + S) or an equal number of microparticles from unmanipulated cells (Control-MP + S). Addition of microparticles from the CsA-treated cells caused an increase in lysis of the endothelial cells compared with cells treated with microparticles from unmanipulated cells. (F) Unmanipulated endothelial cells were exposed to 10% mouse serum and microparticles from CsA-treated cells. Exposure of the cells to microparticles and serum caused lysis of the endothelial cells, but this effect was attenuated when serum from factor B–deficient mice was used or mAb 1379 (an inhibitor of alternative pathway activation) was added to the reaction. *P<0.05, **P<0.01, ***P<0.001.
CsA-Induced Endothelial Microparticles Activate Complement Proteins in Serum and Injure Endothelial Cells
Microparticles from unmanipulated endothelial cells or CsA-treated endothelial cells were purified and exposed to 10% serum. We measured C3a in the supernatants of microparticles exposed to serum (Figure 4B) and found that CsA-induced microparticles cause greater C3a generation than microparticles from untreated cells. Flow cytometry demonstrated more C3 deposited on the surface microparticles generated from CsA-treated cells than on the surface of control-treated cells (Figure 4C). Western blot analysis of microparticles for C3 demonstrated that much of the C3 on the CsA-induced microparticles was in the iC3b form, and C3dg and C3d were also detected (Figure 4D), demonstrating that the deposited C3 is processed by complement regulatory proteins. Exposure of CsA-treated endothelial cells to serum also caused C3 deposition on the cells, C3a generation, and complement-mediated cell lysis (Supplemental Figure 2).
Complement activation on the CsA-microparticles generates C3 activation fragments that can potentially react with nearby cells. To determine whether complement activation by microparticles within the plasma can cause bystander injury to nearby cells, we generated and purified microparticles from CsA-treated endothelial cells. We next incubated unmanipulated endothelial cells with 10% serum and added 12,000, 50,000, or 200,000 CsA-microparticles to each well. The addition of the microparticles to the reaction caused a small but measurable increase in lysis of the untreated endothelial cells (Figure 4E). Serum alone did not cause detectable lysis of the cells in this assay. For some of the samples, the percentage lysis calculation yielded negative numbers due to correction factors used in the calculation (see Concise Methods). We repeated this experiment and used serum from factor B–deficient mice (which lack alternative pathway activity) and serum treated with a monoclonal antibody (mAb) to mouse factor B (mAb 1379, an alternative pathway inhibitor12). Treatment of the serum with mAb 1379 reduced the degree of cell lysis (Figure 4F), demonstrating that injury of bystander endothelial cells by the microparticles involves activation of the alternative pathway of complement.
Injection of Mice with CsA-Induced Endothelial Microparticles Causes Mesangial Proliferation and Mesangial Complement Activation
To confirm that CsA-induced endothelial microparticles promote glomerular complement activation, we generated endothelial microparticles in vitro by treating confluent endothelial cells with CsA, 250 μg/ml, for 16 hours and then purified microparticles from the supernatant. Control microparticles were purified from the supernatant of untreated endothelial cells. Mice were injected via the tail vein with approximately 1.2×106 microparticles daily for 4 days, and on the fifth day the kidneys were harvested. Areas of mesangial proliferation and expansion were seen in mice injected with control and CsA-induced microparticles (Figure 5, A and B), but more glomeruli were affected in mice injected with CsA-induced microparticles (Figure 5C). ADMA levels in the plasma were measured as a marker of vascular injury (Figure 5D), and there was a trend toward higher levels in the mice injected with CsA-induced microparticles, although this did not achieve significance (P<0.1). Mesangial C3 deposition was also seen in mice injected with CsA-induced microparticles (Figure 5, D and E). Thus, passive transfer of CsA-induced microparticles into untreated mice is associated with markers of glomerular and vascular injury similar to those seen in mice treated with CsA.
Figure 5.

Cyclosporine-induced endothelial microparticles (MPs) cause mesangial expansion and complement activation. Wild-type mice were injected intravenously with microparticles isolated from untreated endothelial cells or endothelial cells treated with cyclosporine. (A) Mice injected with microparticles from untreated endothelial cells demonstrated areas of mesangial expansion (arrow). (B) Mice injected with microparticles from CsA-treated endothelial cells also showed areas of mesangial expansion (arrows), and (C) the percentage of glomeruli showing mesangial expansion was greater in the mice injected with microparticles from CsA-treated cells. *P<0.05. (D) ADMA levels were higher in mice injected with CsA-induced microparticles (P<0.1). Immunofluorescence microscopy was used to detect C3 in the kidneys. (E) Kidneys from mice injected with microparticles from untreated cells showed C3 deposition on Bowman capsule and along the tubular basement membranes. (F) Mice injected with microparticles from CsA-treated endothelial cells showed mesangial C3 deposition. Glomeruli are indicated with arrowheads. Original magnification ×400 for A, B, E, and F.
CsA-Induced Endothelial Microparticles Activate Complement Due to Decreased Binding of Factor H
We next measured expression of cell-surface complement regulatory proteins on the surface of the endothelial cell microparticles by flow cytometry. We found that endothelial cells express Crry and DAF, two membrane-bound complement regulatory proteins, and treatment of the cells with cyclosporine did not alter their overall expression of these proteins (Supplemental Figure 3). Microparticles purified from cells treated with CsA demonstrated a slight decrease in surface levels of the complement regulatory proteins Crry (Figure 6A) and DAF (Figure 6B) compared with microparticles purified from control-treated cells. Factor H is a circulating inhibitor of the alternative pathway. We found that factor H bound to the surface of microparticles released from untreated endothelial cells but did not bind to the surface of microparticles released from CsA-treated cells (Figure 6C). A recombinant form of the carboxy terminus of factor H (referred to as rH 19–20) competitively blocks complement regulation on the surface of cells by the full-sized factor H protein.13 Treatment of microparticles from unmanipulated cells with rH 19–20 increased complement activation on the microparticles (Figure 6D), confirming that factor H in the serum functionally limits complement activation on their surface. However, when rH 19–20 was added to microparticles purified from the supernatant of CsA-treated cells, it did not increase the degree of C3 deposition, further supporting the finding that factor H does not regulate complement activation on the surface of CsA-induced endothelial microparticles.
Figure 6.

Factor H does not control complement activation on the surface of cyclosporine-induced microparticles (MPs). Endothelial cells were treated with CsA, 250 μg/ml, for 16 hours. Microparticles were purified from cell supernatants, and the expression of complement regulatory proteins on the surface of the microparticles was examined by flow cytometry. (A) Expression of Crry was slightly decreased on microparticles from CsA-treated cells, and (B) DAF levels were similar to those on microparticles from unmanipulated cells. Dashed line, isotype control; black line, control-treated; shaded curve, CsA-treated. (C) Binding of factor H in serum to the microparticles was evaluated by flow cytometry. Factor H bound to microparticles from unmanipulated cells (left panel), but it did not bind to microparticles from CsA-treated cells (right panel). Black line, isotype control; shaded curve, CsA-treated. (D) Microparticles from unmanipulated cells from CsA-treated cells were treated with rH19–20 to block factor H function and were then exposed to serum. C3 deposition on the particles was assessed by flow cytometry. Treatment with rH19–20 increased C3 deposition on microparticles from unmanipulated cells (left panel) but did not increase deposition on microparticles from CsA-treated cells (right panel). Dashed line, isotype control; black line, serum only; shaded curve, rH19–20 and serum.
The Alternative Pathway of Complement Is Activated on Microparticles from CsA-Treated Cells
To investigate the pathway by which complement is activated on microparticles from CsA-treated endothelial cells, microparticles from unmanipulated and CsA-treated endothelial cells were exposed to serum. Slightly higher levels of IgG and IgM bound to the surface of microparticles from CsA-treated cells (Figure 7, A and B). Mannose-binding lectins (MBL) A and C also bound to the CsA microparticles (Figure 7, C and D). Levels of bound C1q (Figure 7E) and C4 (Figure 7F) did not increase, however.
Figure 7.

Immunoglobuline and MBLs bind to microparticles. Endothelial cells were treated with CsA, 250 μg/ml, for 16 hours. Microparticles were purified and exposed to 10% mouse serum, then immunoglobulin and complement proteins on the surface of the microparticles were assessed by flow cytometry. (A) Bound IgM was detected on microparticles, but levels did not increase on microparticles from CsA-treated cells. (B) IgG levels on microparticles from CsA-treated cells were greater than on microparticles from unmanipulated cells. (C) MBL-A and (D) MBL-C levels were greater on microparticles from CsA-treated cells than they were on microparticles from unmanipulated calls. (E) C4 and (F) C1q were detected on the microparticles, but levels did not increase on microparticles from CsA-treated cells. Dashed line, isotype control; black line, control-treated; shaded curve, CsA-treated.
To further explore the roles of the specific complement pathways, we used serum deficient in immunoglobulin, C1q, MBL A/C, and serum treated with mAb 1379. When the microparticles were incubated with serum from μMT mice (a strain of mice that does not generate IgG or IgM), no immunoglobulin bound to the microparticles, but C3 deposition was similar to that seen using wild-type serum (Figure 8A). Similarly, C1q deficiency did not decrease and C3 deposition on the microparticles was observed (Figure 8B). Thus, even though immunoglobulin and C1q bind to the microparticles, complement activation in the microparticle surface does not require the classic pathway. C3 deposition on the CsA microparticles was significantly reduced when alternative pathway activation was blocked with mAb 1379 (Figure 8C), indicating that the alternative pathway of complement is activated on the microparticle surface. Slightly less C3 was deposited on microparticles when they were incubated with serum deficient in MBL A/C compared with complement-sufficient serum (Figure 8D), suggesting that the MBL pathway contributes to complement activation on constitutively released and CsA-induced endothelial microparticles.
Figure 8.

Complement activation on cyclosporine-induced endothelial microparticles involves the alternative pathway. Endothelial cells were treated with CsA, 250 μg/ml, for 16 hours. Microparticles were purified and exposed to 10% mouse serum, then C3 on the surface of the microparticles was assessed by flow cytometry. (A) Microparticles from unmanipulated (left panel) and CsA-treated (right panel) endothelial cells were exposed to wild-type serum or immunoglobulin-deficient serum (from B cell–deficient μMT mice). No differences in the amount of C3 deposited on the cells were detected. (B) Microparticles were purified and exposed to wild-type serum or C1q-deficient serum. No differences in the amount of C3 deposited on the cells were detected. (C) Microparticles were purified and exposed to 10% normal serum or serum treated with an inhibitory antibody to factor B (mAb 1379). Treatment with mAb 1379 reduced C3 deposition on microparticles from unmanipulated and CsA-treated cells. (D) Microparticles were purified and exposed to wild-type serum or serum deficient in both MBL-A and MBL-C. No differences in the amount of C3 deposited on the cells were detected. For each graph: dashed line, isotype control; shaded curve, wild-type serum; black line, complement-deficient serum.
Complement-Activating Endothelial Microparticles Are Produced in Renal Transplant Patients Treated with Tacrolimus
To determine whether treatment of patients with calcineurin inhibitors is associated with the generation of endothelial microparticles, we first confirmed that treatment of human endothelial cells with tacrolimus in vitro induced release of microparticles (data not shown). We then obtained plasma from renal transplant patients before transplant and 2 weeks after starting treatment with tacrolimus. Microparticles were isolated from 250 μl of EDTA plasma (Figure 9A), and endothelial microparticles were identified by flow cytometry using antibodies to CD31 and CD41a. The number of endothelial microparticles was greater in all of the individual patients after starting tacrolimus (Figure 9B), and the mean for all patients was also significantly increased. C3 deposition on the particles increased in four of the five patients (Figure 8C).
Figure 9.

Treatment of patients with calcineurin inhibitors is associated with increased production of complement-activating endothelial microparticles. EDTA-plasma was obtained from five patients before renal transplantation and 14 days after initiation of treatment with tacrolimus. (A) Beads were used to determine the size distribution of small particles in the samples, and the gate used for microparticle analysis is shown. (B) Endothelial cell microparticles were identified by flow cytometry using antibodies for CD31 and CD41a. The number of endothelial microparticles isolated from 250 μl of plasma was greater in samples obtained after the initiation of tacrolimus (P<0.05 for pretacrolimus versus post-tacrolimus when the mean values from each time-point were compared). (C) C3 deposition on the surface of endothelial microparticles was examined by flow cytometry and is expressed in random fluorescence units. The abundance of C3 varied among the patients. The amount of C3 deposited on endothelial microparticles increased after initiation of tacrolimus in four of the patients.
Discussion
The results of these studies demonstrate that calcineurin inhibitors cause endothelial cells to increase the number of microparticles released into the circulation from the cell surface. Calcineurin inhibitors also alter the composition of the microparticles such that they become complement activating. We found that alternative pathway–deficient mice are protected from CsA-induced renal and vascular injury. We also found that endothelial microparticles cause injury of unmanipulated endothelial cells in vitro, and they cause mesangial proliferation and complement activation when passively transferred into wild-type mice in vivo. These results demonstrate that CsA-induced endothelial microparticles can cause bystander injury of endothelial cells, and they promote glomerular complement activation and mesangial expansion. Furthermore, preliminary data from human transplant patients indicate that treatment of these patients with tacrolimus is also associated with generation of endothelial microparticles.
The microparticles released from CsA-exposed endothelial cells express similar levels of membrane-bound complement regulatory proteins as do microparticles constitutively released from endothelial cells. However, factor H in serum does not bind as well to the surface of CsA-microparticles as it does to those from healthy cells, and complement regulation by factor H on the CsA-microparticles is reduced. Factor H is an alternative pathway regulator, and the decrease in the binding of factor H to the microparticle surface is consistent with the observation that complement activation by the CsA-microparticles involves the alternative pathway of complement.
Complement activation has previously been linked with the generation of microparticles,8,9 but the results of the current studies also demonstrate that microparticles themselves can directly activate the complement system. Although immunoglobulin and complement proteins have been detected on the surface of microparticles from apoptotic cells14 and microparticles isolated from patients with inflammatory diseases,15–17 these previous studies did not make clear whether the microparticles were the cause or the consequence of complement activation.
Our results also reveal a novel mechanism by which endothelial injury throughout the body can promote endovascular and intrarenal complement activation. This may not, in itself, be sufficient to cause renal injury. Mice injected with CsA-induced microparticles did not have impaired renal function (data not shown), even though there was a trend toward higher ADMA levels and mesangial expansion was seen. However, in susceptible hosts, these effects of calcineurin inhibitors may induce a flare of complement-mediated disease. It is also worth noting that this is a proinflammatory effect of a drug that is commonly used as an immunosuppressive agent for treatment of renal diseases and protection of renal allografts. These studies demonstrate that CsA-microparticles trigger alternative pathway activation within the glomerulus even in the presence of normal levels of circulating factor H, suggesting that these effects may be more severe in patients who already have compromised control of the alternative pathway of complement (e.g., patients mutations in the gene for factor H). Finally, the results presented above demonstrate that an intact alternative pathway contributes to the development of vascular and renal injury in mice treated with high-dose CsA. It has previously been reported that treatment of mice with CsA induces intrarenal complement activation, although a pathogenic role for the complement system was not confirmed in that study.18
Our findings are similar to previously published observations regarding platelet microparticles in Shiga toxin–induced HUS and aHUS.16,19 A study of the effects of Shiga toxin on platelets showed that Shiga toxin induces platelets to release microparticles in vitro, and patients with Shiga toxin–induced HUS have increased numbers of platelet microparticles during the acute phase of disease.16 Similar to the CsA-induced endothelial microparticles, the Shiga-induced platelet microparticles became complement activating even though the levels of membrane-bound complement regulatory proteins did not change. Another study by the same group demonstrated that incubation of platelets with serum from patients with aHUS caused the release of platelet microparticles, and that this effect was reversed by the addition of functional factor H.16 Our results and these previously published studies demonstrate that several of the factors associated with the development of HUS (Shiga toxin, mutations in factor H, and CsA) are associated with the generation of complement-activating microparticles. Furthermore, in all three cases complement activation on the microparticles involves activation of the alternative pathway of complement and is due to insufficient complement regulation on the microparticle surface by factor H.
Microparticles have several known physiologic effects. For example, microparticles released from several cell types are prothrombotic.20,21 Platelet microparticles were recently demonstrated to elicit the production of IL-1 by synovial fibroblasts.22 This innate immune effect was demonstrated to be an important cause of joint injury in inflammatory arthritis. Microparticles can also be immunogenic and modulate the adaptive immune response to antigens.23,24 It is possible that complement activation by endothelial microparticles and microparticles from other cell types contributes to these functions. If so, microparticles may represent an important mechanism by which cellular injury affects complement regulation and inflammation at distant sites, such as the kidney.
Other vascular insults may cause generation of endothelial microparticles with similar complement-activating activity. Renal disease,25–27 hypertension,28 thrombotic thrombocytopenic purpura,29 and pregnancy30 promote the release of endothelial microparticles. A wide range of diseases and insults are also associated with the generation of platelet and leukocyte microparticles.6,20 Injury-associated microparticles provide a surface that may enable initiation of complement activation, and the small size of microparticles may enable them to enter most body fluids.31,32 Microparticles are present in normal serum and circulate in continuous contact with complement proteins. Thus, the nature of the microparticles and their abundance in plasma may have a profound influence on complement homeostasis throughout the body.
We have used the toxic effects of high-dose CsA on endothelial cells as a means of studying the interaction of endothelial microparticles and the complement system, but these findings also advance our understanding of the toxic effects of CsA on the kidneys and vasculature. CsA is currently used to treat a wide range of diseases, yet the benefits of this drug are offset by its well known toxicity.11 In some diseases, the beneficial effects of the drug are mediated by its immunosuppressive effects on T cells. In other diseases, such as various forms of the nephrotic syndrome, CsA may directly exert beneficial effects on target cells outside of the immune system.33 Agents that could evoke the desired biologic response in a more targeted fashion, without causing off-target toxicity, would be of great benefit. In addition to possibly mediating CsA toxicity, microparticles may be a valuable biomarker of this toxicity.
Our results indicate that CsA-induced endothelial microparticles trigger injury of nearby endothelial cells. Although the dose used in our in vivo experiment is higher than the dose used in human patients, it is similar to doses used in other preclinical studies.18,33 Furthermore, we have found that treatment of transplant patients with tacrolimus is associated with an increase in the number of endothelial microparticles in plasma. In four of the patients, more C3 was deposited on the post-tacrolimus sample, but the abundance of C3 deposited on endothelial microparticles from different patients varied considerably. This may be due to differences in the patients’ underlying diseases or to variation in other factors that contribute to alternative pathway regulation in each patient. There was also variability in C3 deposition on endothelial microparticles from healthy, genetically identical mice, further suggesting that other factors influence the characteristics of endothelial microparticles.
In conclusion, we have found that exposure of endothelial cells to CsA causes the cells to release microparticles that activate the alternative pathway of complement. Renal and vascular injury in mice treated with high-dose CsA was greater in mice with an intact alternative pathway, demonstrating that therapeutic complement inhibitors may reduce the toxicity of CsA. More generally, injury-associated microparticles may represent an important mechanism by which vascular injury can initiate a systemic inflammatory response. Microparticles may be the trigger by which various insults lead to flares of complement-dependent diseases, such as aHUS. The detection of complement-activating microparticles may also provide an important biomarker of disease activity. Therapeutic complement inhibitors have now entered clinical use.34 Improved understanding of the mechanisms by which the complement system becomes activated in disease will improve our ability to treat and monitor patients with complement-dependent diseases.
Concise Methods
Materials
Antibodies used for flow cytometry include FITC-labeled goat antimouse C3 (MP Biomedicals, Santa Ana, CA; 12 μg/ml), FITC-labeled goat IgG used at the same concentration as an isotype control, biotinylated antimouse DAF antibody (Cedarlane, Burlington, NC; 0.6 μg/ml), biotinylated rat antimouse Crry antibody (BD Pharmingen, San Jose, CA; 1.5 μg/ml), antisera to human factor H (Quidel, San Diego, CA; 1:25 dilution), Cy5-labeled goat antimouse IgG, and FITC-labeled goat antimouse IgM (Jackson ImmunoResearch, Inc.; 4.5 μg/ml). We also used anti-C1q, anti–MBL-A, anti–MBL-C, and anti-C4 antibodies (Hycult Biotechnology, Plymouth Meeting, PA; 0.6 μg/ml). Other experiments used AlexaFluor 647 antimouse CD144 (BioLegend, San Diego, CA), AlexaFluor 647 rat IgG1 (BioLegend), R-phycoerythrin (PE)–labeled rat antimouse CD41 (BD Biosciences), PE-labeled rat IgG (BD Biosciences), FITC-labeled rat antimouse CD31 (BD Biosciences), or FITC-labeled rat IgG (Southern Biotech, Birmingham, AL), all at final concentration of 1.5 μg/ml. Streptavadin-PE (BD Pharmingen; 1.5 μg/ml) was used to detect the biotinylated labels. To detect endothelial microparticles in samples from human patients, antihuman CD31 and antihuman CD41a (both from BD Pharmingen) were used at a final concentration of 1 μg/ml. Normal goat serum (Jackson ImmunoResearch, Inc., West Grove, PA) and rat IgG (Jackson ImmunoResearch, Inc.) were used as isotype controls.
In Vivo Experiments
Eight- to 12-week-old male C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME) were injected subcutaneously with CsA (Bedford Labs, Bedford, OH; 30 mg/kg final dose) mixed in DPBS, or they were injected with an equal volume of DPBS as a vehicle control. To examine the role of the complement system in CsA-induced injury, mice with a targeted deletion of the gene for factor B (fB−/−) mice were also used.35 These mice have selective deficiency in the alternative pathway, but the classic and lectin complement pathways are intact. The mice received daily injections for 14 days. At the end of the study the mice were euthanized, and serum, plasma, and tissues were collected. To specifically examine the role of CsA-induced microparticles, C57BL/6 mice were injected via the tail vein on four successive days with approximately 1.2×106 microparticles generated in vitro by treating endothelial cells with CsA. After approval of the animal protocol by the University of Colorado Institutional Animal Care and Use Committee, mice were housed and maintained in the University of Colorado Center for Laboratory Animal Care in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals, and all animal procedures were performed in adherence to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. C3 deposition in the kidneys of these mice was examined by immunofluorescence microscopy (see Supplemental Material for detailed methods).
Human Samples
EDTA plasma was obtained from five renal transplant patients. Samples were obtained before transplantation and approximately 2 weeks after initiation of treatment with tacrolimus. The original cause of renal failure in these patients was hypertension, FSGS (two patients), IgA nephropathy, and polycystic kidney disease. An institutional review board approved the use of patient samples, and informed consent was obtained from patients in accordance with the Declaration of Helsinki.
Microparticle Isolation
For in vitro microparticle generation, early passage MS1 cells (a murine endothelial cell line) were obtained from ATCC (Manassas, VA). When cells were 90% confluent, the media were changed to fresh media with or without CsA at a concentration of 250 μg/ml. The CsA was left on the cells for 16 hours. To isolate the microparticles, supernatants were collected and centrifuged at 3000 g for 15 minutes at 4°C to remove any dead cells. Supernatants were then centrifuged at 20,000 g for 2.5 hours. The pellet was resuspended in MP buffer (Hank’s buffered saline solution containing 20 mM HEPES and 5 mM glucose). The sample was resuspended by vortexing for 1 minute, and 250 μl of buffer was added for every 10 ml raw supernatant initially collected. To isolate microparticles from plasma samples generated in the in vivo experiments, mice were bled by cardiac puncture into tubes containing EDTA) (10 mM final) and placed on ice until it was centrifuged at 3000 g for 15 minutes at 4°C to separate the plasma. Plasma was collected and stored at −80°C. On the day of analysis the samples were thawed in a 37°C water bath then centrifuged at 20,000 g for 2 hours. The pellets were resuspended at a volume of 40% of the initial plasma volume. See the Supplemental Material for detailed methods of Western blot analysis of microparticle proteins.
Complement Activation Experiments
To assess complement activation on the surface of isolated microparticles, they were incubated with 10% mouse serum for 30 minutes at 37°C. Serum from C57BL/6, μMT, C1q−/−, and MBL A/C−/− mice was used for these experiments.36 The microparticles were centrifuged at 20,000 g for 1 hour at 4°C and resuspended in 50 μl DPBS.
In some experiments, 2.88 μg of a peptide that blocks binding of factor H to cell membranes (rH19–20)13 was added to the reaction mix and incubated at room temperature for 15 minutes before the addition of serum. In other experiments, an inhibitory antibody to mouse factor B was used to block alternative pathway activation.12 Six μg of mAb 1379 or purified mouse IgG (as a control) was added to 5.5 μl undiluted C57BL/6 mouse serum on ice and incubated for 30 minutes. The 1379/serum was then added to 50 μl of microparticles and incubated at 37°C for 30 minutes. Samples were then placed on ice and processed for flow cytometry to assess deposition of C3 on the particle surface. In some reactions, C3a generation was measured by ELISA (see Supplemental Material for detailed methods).
Flow Cytometry
After microparticles were isolated and resuspended, they were stained using the antibodies described above. Antibody labels were detected using a FACSCalibur flow cytometer (BD Biosciences).
Microparticle-Mediated Lysis of Bystander Cells
MS1 cells were grown to approximately 90% confluence in 96 well plates. Microparticles were isolated from the supernatants of CsA-treated MS1 cells and were added to the 96 well plates of cells with and without the addition of 10% C57BL/6 mouse serum in microparticle buffer. Serum from fB−/− mice or wild-type serum to which mAb 1379 had been added were used to test the role of the alternative pathway in bystander cell lysis. The cells were incubated at 37°C for 15 minutes. The plates were allowed to cool to 23°C and total lysis buffer was added to some cells as a control. Equivalent volumes of Cyto-tox ONE buffer (Promega, Madison, WI) were added and incubated for 10 minutes. Fifty microliters of stop solution was added, and the reaction was evaluated using a fluorescent plate reader set to an excitation wavelength of 560 nm and an emission wavelength of 590 nm. Background LDH was measured in unmanipulated cells, the serum added to the reactions, the microparticles added to the reactions, and also in cells after full lysis with 9% Triton X-100. The percentage of lysis of cells in each sample was then calculated according to the following equation:
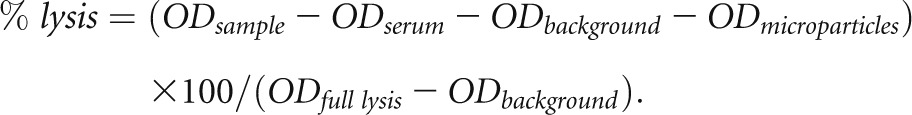 |
Renal Function
Renal function was assessed by measurement of the SUN using an Alfa Wasserman ACE Chemistry Analyzer (West Caldwell, NJ).
Measurement of ADMA and Intracellular Cyclosporine Levels
ADMA was quantified using a validated HPLC-tandem mass spectrometry assay. For CsA measurement, a validated online extraction HPLC-tandem mass spectrometry assay was used.37 See Supplemental Material for detailed methods.
Statistical Analyses
Multiple group comparisons were performed using ANOVA with post-test according to Tukey. Comparison between two groups was performed with a two-tailed t test. A P value <0.05 was considered to represent a statistically significant finding. Results are reported as mean ± SEM.
Disclosures
J.M.T. is a consultant for Alexion Pharmaceuticals, Inc.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grant DK076690 (J.M.T.) and grant HD070511 (J.M.T. and U.C.).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2012111064/-/DCSupplemental.
References
- 1.Noris M, Remuzzi G: Hemolytic uremic syndrome. J Am Soc Nephrol 16: 1035–1050, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Delvaeye M, Noris M, De Vriese A, Esmon CT, Esmon NL, Ferrell G, Del-Favero J, Plaisance S, Claes B, Lambrechts D, Zoja C, Remuzzi G, Conway EM: Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med 361: 345–357, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fang CJ, Fremeaux-Bacchi V, Liszewski MK, Pianetti G, Noris M, Goodship TH, Atkinson JP: Membrane cofactor protein mutations in atypical hemolytic uremic syndrome (aHUS), fatal Stx-HUS, C3 glomerulonephritis, and the HELLP syndrome. Blood 111: 624–632, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, Daina E, Fenili C, Castelletti F, Sorosina A, Piras R, Donadelli R, Maranta R, van der Meer I, Conway EM, Zipfel PF, Goodship TH, Remuzzi G: Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 5: 1844–1859, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noris M, Remuzzi G: Atypical hemolytic-uremic syndrome. N Engl J Med 361: 1676–1687, 2009 [DOI] [PubMed] [Google Scholar]
- 6.Anderson HC, Mulhall D, Garimella R: Role of extracellular membrane vesicles in the pathogenesis of various diseases, including cancer, renal diseases, atherosclerosis, and arthritis. Laboratory investigation; a journal of technical methods and pathology, 90: 1549-1557, 2010 [DOI] [PubMed] [Google Scholar]
- 7.Beyer C, Pisetsky DS: The role of microparticles in the pathogenesis of rheumatic diseases. Nat Rev Rheumatol 6: 21–29, 2010 [DOI] [PubMed] [Google Scholar]
- 8.Sims PJ, Faioni EM, Wiedmer T, Shattil SJ: Complement proteins C5b-9 cause release of membrane vesicles from the platelet surface that are enriched in the membrane receptor for coagulation factor Va and express prothrombinase activity. J Biol Chem 263: 18205–18212, 1988 [PubMed] [Google Scholar]
- 9.Moskovich O, Fishelson Z: Live cell imaging of outward and inward vesiculation induced by the complement c5b-9 complex. J Biol Chem 282: 29977–29986, 2007 [DOI] [PubMed] [Google Scholar]
- 10.Nauta AJ, Trouw LA, Daha MR, Tijsma O, Nieuwland R, Schwaeble WJ, Gingras AR, Mantovani A, Hack EC, Roos A: Direct binding of C1q to apoptotic cells and cell blebs induces complement activation. Eur J Immunol 32: 1726–1736, 2002 [DOI] [PubMed] [Google Scholar]
- 11.Naesens M, Kuypers DR, Sarwal M: Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol 4: 481–508, 2009 [DOI] [PubMed] [Google Scholar]
- 12.Thurman JM, Kraus DM, Girardi G, Hourcade D, Kang HJ, Royer PA, Mitchell LM, Giclas PC, Salmon J, Gilkeson G, Holers VM: A novel inhibitor of the alternative complement pathway prevents antiphospholipid antibody-induced pregnancy loss in mice. Mol Immunol 42: 87–97, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Renner B, Coleman K, Goldberg R, Amura C, Holland-Neidermyer A, Pierce K, Orth HN, Molina H, Ferreira VP, Cortes C, Pangburn MK, Holers VM, Thurman JM: The complement inhibitors Crry and factor H are critical for preventing autologous complement activation on renal tubular epithelial cells. J Immunol 185: 3086–3094, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nauta AJ, Castellano G, Xu W, Woltman AM, Borrias MC, Daha MR, van Kooten C, Roos A: Opsonization with C1q and mannose-binding lectin targets apoptotic cells to dendritic cells. J Immunol 173: 3044–3050, 2004 [DOI] [PubMed] [Google Scholar]
- 15.Biró E, Nieuwland R, Tak PP, Pronk LM, Schaap MC, Sturk A, Hack CE: Activated complement components and complement activator molecules on the surface of cell-derived microparticles in patients with rheumatoid arthritis and healthy individuals. Ann Rheum Dis 66: 1085–1092, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ståhl AL, Sartz L, Karpman D: Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood 117: 5503–5513, 2011 [DOI] [PubMed] [Google Scholar]
- 17.Nielsen CT, Østergaard O, Stener L, Iversen LV, Truedsson L, Gullstrand B, Jacobsen S, Heegaard NH: Increased IgG on cell-derived plasma microparticles in systemic lupus erythematosus is associated with autoantibodies and complement activation. Arthritis Rheum 64: 1227–1236, 2012 [DOI] [PubMed] [Google Scholar]
- 18.Kim YO, Lim SW, Li C, Kang HJ, Ahn KO, Yang HJ, Ghee JY, Kim SH, Kim JY, Choi BS, Kim J, Yang CW: Activation of intrarenal complement system in mouse model for chronic cyclosporine nephrotoxicity. Yonsei Med J 48: 517–525, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ståhl AL, Vaziri-Sani F, Heinen S, Kristoffersson AC, Gydell KH, Raafat R, Gutierrez A, Beringer O, Zipfel PF, Karpman D: Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood 111: 5307–5315, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Mackman N: On the trail of microparticles. Circ Res 104: 925–927, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brodsky SV, Malinowski K, Golightly M, Jesty J, Goligorsky MS: Plasminogen activator inhibitor-1 promotes formation of endothelial microparticles with procoagulant potential. Circulation 106: 2372–2378, 2002 [DOI] [PubMed] [Google Scholar]
- 22.Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, Massarotti EM, Remold-O’Donnell E, Farndale RW, Ware J, Lee DM: Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science 327: 580–583, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andre F, Schartz NE, Movassagh M, Flament C, Pautier P, Morice P, Pomel C, Lhomme C, Escudier B, Le Chevalier T, Tursz T, Amigorena S, Raposo G, Angevin E, Zitvogel L: Malignant effusions and immunogenic tumour-derived exosomes. Lancet 360: 295–305, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Bianco NR, Kim SH, Morelli AE, Robbins PD: Modulation of the immune response using dendritic cell-derived exosomes. Methods Mol Biol 380: 443–455, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Boulanger CM, Amabile N, Guérin AP, Pannier B, Leroyer AS, Mallat CN, Tedgui A, London GM: In vivo shear stress determines circulating levels of endothelial microparticles in end-stage renal disease. Hypertension 49: 902–908, 2007 [DOI] [PubMed] [Google Scholar]
- 26.Jourde-Chiche N, Dou L, Sabatier F, Calaf R, Cerini C, Robert S, Camoin-Jau L, Charpiot P, Argiles A, Dignat-George F, Brunet P: Levels of circulating endothelial progenitor cells are related to uremic toxins and vascular injury in hemodialysis patients. J Thromb Haemost 7: 1576–1584, 2009 [DOI] [PubMed] [Google Scholar]
- 27.Trappenburg MC, van Schilfgaarde M, Frerichs FC, Spronk HM, Ten Cate H, de Fijter CW, Terpstra WE, Leyte A: Chronic renal failure is accompanied by endothelial activation and a large increase in microparticle numbers with reduced procoagulant capacity. Nephrol Dial Transplant 27: 1446–1453, 2012 [DOI] [PubMed] [Google Scholar]
- 28.Huang PH, Huang SS, Chen YH, Lin CP, Chiang KH, Chen JS, Tsai HY, Lin FY, Chen JW, Lin SJ: Increased circulating CD31+/annexin V+ apoptotic microparticles and decreased circulating endothelial progenitor cell levels in hypertensive patients with microalbuminuria. J Hypertens 28: 1655–1665, 2010 [DOI] [PubMed] [Google Scholar]
- 29.Jimenez JJ, Jy W, Mauro LM, Horstman LL, Soderland C, Ahn YS: Endothelial microparticles released in thrombotic thrombocytopenic purpura express von Willebrand factor and markers of endothelial activation. Br J Haematol 123: 896–902, 2003 [DOI] [PubMed] [Google Scholar]
- 30.Bretelle F, Sabatier F, Desprez D, Camoin L, Grunebaum L, Combes V, D’Ercole C, Dignat-George F: Circulating microparticles: A marker of procoagulant state in normal pregnancy and pregnancy complicated by preeclampsia or intrauterine growth restriction. Thromb Haemost 89: 486–492, 2003 [PubMed] [Google Scholar]
- 31.Berckmans RJ, Nieuwland R, Tak PP, Böing AN, Romijn FP, Kraan MC, Breedveld FC, Hack CE, Sturk A: Cell-derived microparticles in synovial fluid from inflamed arthritic joints support coagulation exclusively via a factor VII-dependent mechanism. Arthritis Rheum 46: 2857–2866, 2002 [DOI] [PubMed] [Google Scholar]
- 32.Mrvar-Brecko A, Sustar V, Jansa V, Stukelj R, Jansa R, Mujagić E, Kruljc P, Iglic A, Hägerstrand H, Kralj-Iglic V: Isolated microvesicles from peripheral blood and body fluids as observed by scanning electron microscope. Blood Cells Mol Dis 44: 307–312, 2010 [DOI] [PubMed] [Google Scholar]
- 33.Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K, Reiser J, Mundel P: The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med 14: 931–938, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L: Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol 25: 1256–1264, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Matsumoto M, Fukuda W, Circolo A, Goellner J, Strauss-Schoenberger J, Wang X, Fujita S, Hidvegi T, Chaplin DD, Colten HR: Abrogation of the alternative complement pathway by targeted deletion of murine factor B. Proc Natl Acad Sci U S A 94: 8720–8725, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Renner B, Strassheim D, Amura CR, Kulik L, Ljubanovic D, Glogowska MJ, Takahashi K, Carroll MC, Holers VM, Thurman JM: B cell subsets contribute to renal injury and renal protection after ischemia/reperfusion. J Immunol 185: 4393–4400, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christians U, Jacobsen W, Serkova N, Benet LZ, Vidal C, Sewing KF, Manns MP, Kirchner GI: Automated, fast and sensitive quantification of drugs in blood by liquid chromatography-mass spectrometry with on-line extraction: immunosuppressants. J Chromatogr B Biomed Sci Appl 748: 41–53, 2000 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.