

Abstract
Scope
Rotenone is a toxicant believed to contribute to the development of Parkinson's disease.
Methods and results
Using human peripheral blood lymphocytes we demonstrated that exposure to rotenone resulted in disruption of electron transport accompanied by the production of reactive oxygen species, development of apoptosis and elevation of peroxidase activity of mitochondria. Employing LC/MS based lipidomics/oxidative lipidomics we characterized molecular species of cardiolipin (CL) and its oxidation/hydrolysis products formed early in apoptosis and associated with the rotenone-induced mitochondrial dysfunction.
Conclusions
The major oxidized CL species - tetra-linoleoyl-CL – underwent oxidation to yield epoxy-C18:2 and dihydroxy-C18:2 derivatives predominantly localized in sn-1 and sn-2 positions, respectively. In addition, accumulation of mono-lyso-CL species and oxygenated free C18:2 were detected in rotenone-treated lymphocytes. These oxidation/hydrolysis products may be useful for the development of new biomarkers of mitochondrial dysfunction.
Keywords: Cardiolipin peroxidation, lymphocytes, apoptosis, mitochondrial dysfunction, Parkinson's disease biomarkers
Introduction
Parkinson's disease (PD) is a neurodegenerative disorder in the elderly resulting in the damage and death of dopaminergic neurons in the brain [1]. Oxidative stress and mitochondrial dysfunction have been implicated as important contributors to neuronal death induced in substantia nigra of patients with PD [2–4]. In particular, increased lipid peroxidation products have been found in PD brains [5]. During the last three decades, epidemiological and toxicological studies provided data that pesticides are potential toxicants for dopamine-producing neurons and contribute to the development of PD [6]. Accordingly, exposure to pesticides has been associated with increased incidence of PD [7, 8]. Mitochondria are targets for a number of environmental compounds including pesticides [6, 7] such as rotenone [9, 10]. Rotenone is highly lipophillic, easily crosses the blood-brain barrier and accumulates in mitochondria where it binds to complex I, inhibits the flow of electrons and results in generation of superoxide [11]. However, the mechanistic links between the oxidative stress, lipid peroxidation, neuronal death, mitochondrial impairments induced by pesticides have not been identified.
Blood cells and circulating lymphocytes have been often used to study the pathogenic mechanisms of neurodegenerative diseases, including PD. Mitochondrial complex deficit [12, 13] and up-regulation of the α-synuclein gene [14] that render these cells susceptible to apoptosis were detected in lymphocytes from PD patients [15, 16]. Recently, we demonstrated that oxidation of a mitochondria-specific phospholipid, cardiolipin (CL), is essential for the execution of apoptosis (release of proapoptotic factors from mitochondria into the cytosol) in primary rat cortical neurons in vitro [17, 18] and in rat brain in vivo [19, 20]. Intermembrane space hemoprotein, cytochrome c (cyt c) has been identified as a catalyst of CL peroxidation [21]. CL is a negatively charged phospholipid with four fatty acid residues [21]. It is found exclusively in the inner-mitochondrial membrane where it accounts for 25% of all phospholipids [22] and plays a significant role in mitochondria bioenergetics [23–26]. Taking into account that lymphocytes express the catecholaminergic system and, similar to neuronal cells, molecular death machinery, including the release of mitochondrial pro-apoptotic factors into the cytosol, leading to the typical morphological and biochemical characteristics of apoptosis [27–29], we suggested that cyt c can be involved in the generation of specifically oxygenated molecular species of CL in circulating lymphocytes exposed to rotenone.
In this paper, by using lipidomics/oxidative lipidomics approaches we identified and characterized molecular species of CL in human peripheral blood lymphocytes, evaluated specific profiles of rotenone-induced CL oxidation products and established their association with the production of H2O2, impairments of mitochondrial dysfunction and apoptosis in human lymphocytes exposed to rotenone.
Material and Methods
Reagents
Tetra-linoleyl-cardiolipin (TLCL) and tetra-myristoyl-cardiolipin (TMCL) were purchased from Avanti Polar Lipids Inc (Alabaster, AL). Cytochrome c (cyt c), diethylenetriaminepentaacetic acid (DTPA), PLA1 from Thermomyces lanuginosus, PLA2 from porcine pancreas, H2O2 and all organic solvents (HPLC grade), Hystopaque 1077, PBS (Ca2+, Mg2+ free), rotenone, DMSO were purchased from Sigma-Aldrich (St. Louis, MO). HPTLC silica G plates were purchased from Whatman (Schleicher & Schuell, England). Fetal bovine serum, RPMI 1640 medium Penicillin/Streptomycin were from Life Technologies (Grand Island, NY). Heptadecanoic acid (C17:0) was obtained from Matreya LLC (Pleasant Gap, PA). 9S-hydroperoxy-10E,12Z-octadecadienoic acid, 9S-hydroxy-10E,12Z-octadecadienoic acid, 13-oxo-9Z,11E-octadecadienoic acid, 13S-hydroxy-9Z,11E-octadecadienoic acid, 13S-hydroperoxy-9Z,11E-octadecadienoic acid, 9S-hydroxy-10E,12Z-octadecadienoic-9,10,12,13-d4 acid, 9(10)epoxy-12Z-octadecenoic acid, 12(13)epoxy-9Z-octadecenoic acid were purchased from Cayman Chemical Co (Ann Arbor, Michigan, USA).
Isolation of human peripheral blood lymphocytes
Lymphocytes were isolated from buffy coat obtained from Central Blood Bank by differential centrifugation using Hystopaque 1077 as described [30].
Production of reactive oxygen species
Superoxide and hydrogen peroxide were detected by using dihydroethidium (DHE) and 2',7'-dichlorfluorescein-diacetate (DCFH-DA) assays, respectively, as previously described [31]. The data are presented as fold change of the mean intensity of either ethidium or DCF fluorescence compared with DHE or DCFH-DA loaded controls for superoxide and hydrogen peroxide, respectively.
Detection of apoptosis
Apoptosis was evaluated by phosphatidylserine (PS) externalization using Annexin V–FITC apoptosis detection kit (Biovision, Mountain View, CA) and caspase 3/7 with a luminescence Caspase–Glo™ 3/7 assay kit (Promega, Madison, WI).
Assessments of mitochondrial functional state
Mitochondrial membrane potential (MMP) was determined by JC-1 staining. Briefly, lymphocytes were stained with 10 μg/ml of JC-1 at 37 °C for 15 min and then washed twice with PBS. The samples were analyzed immediately by using RF-5301 PC spectrofluorometer (Shimadzu. Japan) (excitation 485 nm, slits 5 nm). The ratio of red (aggregates, 590 nm) and green (monomer, 529 nm) fluorescence was used as a relative measure of MMP. In addition the lymphocytes were examined under a Nikon ECLIPSE TE 200 fluorescence microscope (Tokyo, Japan) equipped with a digital Hamamatsu CCD camera (C4742-95-12NBR) and analyzed using the MetaImaging Series™ software version 4.6 (Universal Imaging Corp., Downingtown, PA). Cellular ATP content was measured by using an adenosine 5'-triphosphate bioluminescence somatic cell assay kit (Sigma) according to the manufacturer's instructions. Complex I activity was measured by consumption of NADH at 340nm as described [32].
Isolation of mitochondria and detection of peroxidase activity
Mitochondria were isolated from human peripheral blood lymphocytes as described [33]. Peroxidase activity was detected as previously described [34]. Briefly mitochondria (2 mg of protein/ml) were incubated with alamethicin (0.1 mg/ml) on ice for 15 min in 150 mM KCl, 0.5 mM EGTA, 25 mM KH2PO4 (pH 7.0). After that mitochondria were centrifuged (15,000g for 15 min) and re-suspended in 25 mM HEPES (pH 7.4) containing 100 μM DTPA, Amplex Red (50 μM) and tert-BOOH (2 mM). Fluorescence of resorufin, a product of Ample Red oxidation, was measured using Shimadzu RF5301–PC spectrofluorometer (λex and λem − 575 and 585 nm, respectively).
Analysis of CL and its oxygenated molecular species
Lipids were extracted using Folch procedure [35]. Lipid phosphorus was determined by a micro-method [36]. LC/MS was performed using a Dionex Ultimate™ 3000 HPLC system coupled on-line to a linear ion trap mass spectrometer (LXQ Thermo-Fisher) as described [37]. To fully characterize oxygenated free fatty acids and diversified CL oxidation products we chose to pre-separate them from other phospholipids by 2D-HPTLC as previously described [38]. Corresponding spots were scraped-off and lipids extracted [35]. Thus obtained CLs were treated either with phospholipase A1 (PLA1) from Thermomyces lanuginosus or PLA2 from porcine pancreatic phospholipase A2 (PLA2) to liberate fatty acids from sn-1 and sn-2 positions and analyzed by LC-MS. Briefly, CLs were treated with PLA1 (10 μl/μmol CL) or PLA2 (10U/μmol of CL) in 0.5 M borate buffer, pH 9.0 containing 20 mM cholic acid, 2 mM CaCl2 and 100 μM DTPA for 30 min. Under these conditions, almost 99% of CLs were hydrolyzed. At the end of incubation, lipids were extracted and fatty acids were analyzed by LC/MS using reverse phase C18 column. The differentiation between isobaric epoxy-C18:2 CL species from hydroxy-C18:2-containing species was achieved via i) treatment of CLs by exogenous PLA1/PLA2 resulting in the release of LA residues and ii) their subsequent separation and analysis by LC-MS using C18 column and two gradient solvent systems (system A: tetrahydrofuran/methanol/water/CH3COOH, 25:30:44.9:0.1 (v/v) and System B: methanol/water, 90:10 (v/v)) as previously described [39]. Under these conditions, the retention times for epoxy-C18:2 (m/z 295) and hydroxy-C18:2 (m/z 295) were 21.16 and 17.04 min, respectively. This was confirmed by comparison with the standards of epoxy-C18:2 (m/z 295) and hydroxy-C18:2 (m/z 295) available from Cayman Chemicals. Additionally, several major classes of phospholipids, including CLs, were separated and analyzed by LC-MS as described [37]. For quantitative assessments TMCL and oxygenated fatty acids were used as internal standards. Mono-lyso-CL was prepared from TMCL as described [40].
Statistics
The results are presented as mean ± S.D. values from at least three experiments, and statistical analyses were performed by either paired/unpaired Student's t-test or one-way ANOVA. The statistical significance of differences was set at p< 0.05.
Results
Rotenone induces mitochondria dysfunction in human lymphocytes
After exposure of human peripheral blood lymphocytes to rotenone (100 and 250 μM, 12 and 18 hrs at 37°C), the mitochondrial functions were assessed by measurements of MMP, determination of complex I activity and ATP levels. Rotenone induced significant inhibition of complex I activity in a dose- and time dependent manners (Fig 1A). Significant reductions of the MMP and ATP contents were detected in lymphocytes treated with rotenone (Fig.1B, C). Thus exposure of human lymphocytes to rotenone resulted in disruption of electron transport and mitochondrial dysfunction.
Figure 1. Effects of rotenone on mitochondrial functions in human peripheral blood lymphocytes.

Complex I activity (A), MMP (B) and content of ATP (C) in human lymphocytes exposed to rotenone (100, 250 μM, 12 and 18 hrs at 37°C). Typical fluorescent images of control and rotenone-treated lymphocytes (B,a) and assessment of MMP by using flow cytometry (B,b). Data are means ± S.E., n=6, *p<0.05 vs control.
Generation of reactive oxygen species in human lymphocytes exposed to rotenone
Interrupted mitochondrial electron transport, particularly at the level of complex I, is known to cause a massive production of superoxide (53). To determine whether superoxide, indeed, has been generated in rotenone-treated cells we used DHE assay. We found that rotenone caused concentration-dependent production of superoxide (Fig. 2A). Given that mitochondrial superoxide is rapidly converted to membrane permeable and relatively stable hydrogen peroxide we examined the intracellular concentration of hydrogen peroxide using DCFH-DA assay. A significant increase of DCF fluorescence was observed in human lymphocytes following rotenone exposure (Fig. 2B). Thus, rotenone-induced mitochondria dysfunction in lymphocytes was accompanied by the production of ROS.
Figure 2. Rotenone induced generation of reactive oxygen species generated in human peropheral blood lymphocytes.

Superoxide (A) and hydrogen peroxide (B) formation in lymphocytes exposed to rotenone (100 and 250 μM for 12 and 18 hrs at 37°C). Data are means ± S.E., n=6, *p<0.05 vs control.
Rotenone induces apoptosis in human lymphocytes
Assuming that generation of ROS is one of the pre-requisites for triggering apoptosis, we further determined whether rotenone-induced production of superoxide and H2O2 was accompanied by apoptosis. Indeed, we found that caspase 3/7 activity was significantly increased in lymphocytes exposed to rotenone as compared with control non-exposed lymphocytes (Fig. 3A). A 2.5 and 2.8-fold increase in caspase 3/7 activity was detected either for 12 or 18 hrs after the exposure to 100 and 250 μM of rotenone, respectively. In addition, using Annexin V binding assay we were able to detect a significant number of lymphocytes with externalized PS on the cell surface after their exposure to 100 and 250 μM of rotenone for 12 and 18 hrs (Fig. 3B). Thus, treatment of lymphocytes to rotenone resulted in development of apoptotic cell death pathway.
Figure 3. Apoptosis induced by rotenone in human peripheral blood lymphocytes.

Caspase 3/7 activation (A) and PS externalization (B) in lymphocytes exposed to rotenone (100 and 250 μM for 12 and 18 hrs, at 37°C). Data are means ± S.E., n=6, *p<0.05 vs control.
Rotenone stimulates peroxidase activity in mitochondria
We suggested that in lymphocytes, during rotenone-induced apoptosis, mitochondrial phospholipid CL interacts with cyt c to form a complex with peroxidase activity that consequently results in selective oxidation of CL polyunsaturated molecular species. To detect peroxidase activity of cyt c/CL complexes we isolated mitochondria from lymphocytes exposed to rotenone. To remove free cyt c we treated mitochondria with a channel-forming antibiotic, alamethicin (9). Notably, mitochondria isolated from lymphocytes exposed to rotenone at concentrations of 100 and 250 μM for 18 hrs exhibited significantly elevated levels of peroxidase activity as compared to mitochondria from control, non-treated lymphocytes (Fig. 4).
Figure 4. Peroxidase activity of mitochondria isolated from human peripheral blood lymphocytes.
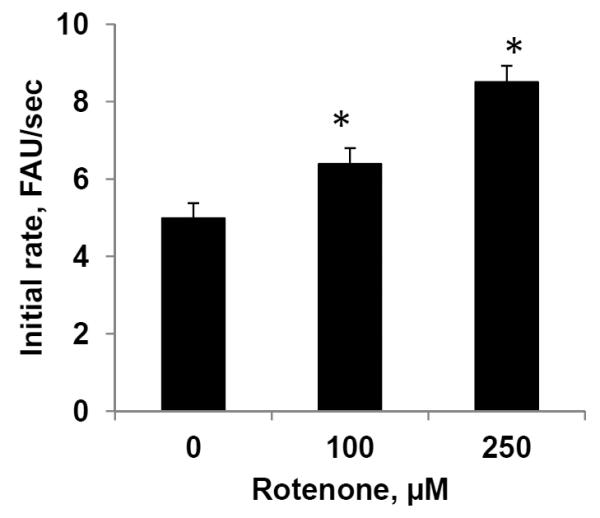
Mitochondria were isolated from lymphocytes exposed to rotenone (100 and 250 μM for 18 hrs, at 37°C) treated with alamethicin and peroxidase activity was detected. Data are means ± S.E., n=3, *p<0.05 vs control.
Identification of CL molecular species in human lymphocytes
As CL oxidation is required for the execution of apoptotic program, we further employed LC/MS to analyze CL molecular species and their oxidation products in lymphocytes. In a typical negative mode MS spectrum of CL, two major clusters were detected (Fig. 5A). MS/MS analysis was performed (Supporting Information Fig. S1) showed that CLs were represented by seven major molecular species predominantly containing readily peroxidizable linoleic acid residues (C18:2) (Table 1).
Figure 5. Rotenone induced oxidation of CL in human peripheral blood lymphocytes.

Typical negative mode ESI-MS spectrum of CL obtained from human lymphocytes (A), LC/MS quantitative assessment of TLCL (B) and oxidized (C) TLCL molecular species. Note: the decrease of TLCL and accumulation of its oxygenated species were dependent on rotenone concentration. Oxygenated molecular species of TLCL with m/z 1464 (plus 1 oxygen), 1480 (plus two oxygens) and 1496 (plus three oxygens) were detected in rotenone treated lymphocytes (18 hrs). Data are means ± S.E., n=5, *p<0.03 vs control.
Table 1.
Identification and quantitative assessment of major CL molecular species in human peripheral blood lymphocytes by LC/MS
| m/z | Molecular species | pmol/nmol CL |
|---|---|---|
| 1421.9 | C18:2/C18:2/C18:2/C16:1 | 30.5 ± 7.4 |
| 1447.9 | C18:2/C18:2/C18:2/C18:2 | 632.0 ± 24.0 |
| 1449.9 | C18:2/C18:2/C18:2/C18:1 | 147.1 ± 6.8 |
| 1469.9 | C18:2/C18:2/C18:2/C20:5 | 46.8 ± 3.9 |
| 1471.9 | C18:2/C18:2/C18:2/C20:4 | 49.0 ± 7.6 |
| 1473.9 | C18:2/C18:2/C18:2/C20:3 | 96.2 ± 11.0 |
| 1475.9 | C18:2/C18:2/C18:2/C20:2 | 54.3 ± 5.5 |
Data are means ± SD, n=5.
Identification of CL molecular species in human lymphocytes exposed to rotenone
Next, we performed detailed structural characterization of oxygenated CL molecular species in human peripheral blood lymphocytes. Quantitative assessments of rotenone-induced changes in CL revealed a significant reduction of highly unsaturated species of CL, particularly TLCL (Fig. 5B). The loss of “oxidizable” TLCL was dependent on concentration of rotenone and accompanied by the appearance of its oxygenated species with one, two and three oxygen functionalities whereby mono-oxygenated derivatives were predominant (Fig. 5C). The structure of oxygenated CLs was confirmed by MS/MS analysis as exemplified by a typical fragmentation pattern of mono-oxygenated TLCL molecular (Supporting Information Fig. S2).
Stereo-specificity of TLCL oxygenation in sn-1 and sn-2 positions was examined by LC/ESI-MS using hydrolysis of CL with PLA1 and/or PLA2, respectively. Quantitative MS analysis of fatty acids liberated from sn-1 position revealed a significantly decreased C18:2 content (Fig. 6Aa) and elevated levels of its oxygenated product with 1–3 oxygens (Fig. 6Ab) that markedly exceeded oxidative loss of C18:2 from the sn-2 position (Fig. 6Ba). Notably, the major oxygenated differences between oxygenated products generated in sn-1 and sn-2 positions were not only quantitative but also qualitative with epoxy-C18:2 derivatives (Fig. 6Ab and Supporting Information Fig.S3) and dihydroxy-C18:2 derivatives (Fig. 6Bb and Supporting Information Fig. S4) accumulating in sn-1 and sn-2 positions, respectively.
Figure 6. Identification of esterified oxygenated fatty acids in human peripheral blood lymphocytes esposed to rotenone.

Quantitative LC/MS assessment of oxygenated C18:2 localized and sn-1 (A) and sn-2 (B) positions of CL from rotenone-exposed lymphocytes (100 and 250 μM, for 18 hrs at 37°C. (a) Decrease of oxidizable C18:2 and (b) formation of oxygenated C18:2 hydrolyzed from sn-1 and sn-2 positions of CL, respectively. Data are means ± S.E., n=5, *p<0.03 vs control.
To test whether cyt c could be a candidate catalyst involved in rotenone-induced generation of oxygenated molecular species of CL, we performed a model oxidation experiment using TLCL, the major molecular species of CL present in human peripheral blood lymphocytes (Fig. 4A, Table 1). When TLCL was incubated in the presence of cyt c and hydrogen peroxide for 30 min at 37°C we found that a decreased content of TLCL was accompanied by the accumulation of its oxygenated products with 1–3 oxygens with similar composition and stereo-specificity as those detected in rotenone-exposed lymphocytes (Fig. 7A, B). These results are compatible with involvement of cyt c in catalysis of TLCL oxidation in lymphocytes exposed to rotenone.
Figure 7. Cyt c induced oxidation of TLCL in the presence of H2O2.

Quantitative LC/MS assessment of TLCL and its oxygenated species (A) formed in cyt c driven reaction. Oxygenated products of C18:2 (B) formed in sn-1 (a) and sn-2 (b) positions of CL upon treatment of TLCL with cyt c/H2O2. After incubation with cyt c/H2O2, TLCL was treated either with PLA1 or PLA2. Liberated oxygenated and non-oxygenated C18:2 were analyzed by reverse phase LC/MS. Data are means ± S.E., n=5, *p<0.05 vs control.
Rotenone induced accumulation of mono-lyso-CL in human lymphocytes
It is possible that the loss of CL in rotenone-challenged lymphocytes may be associated, at least in part, with activation of endogenous PLA2. The presence of Ca2+-independent iPLA2 in capable of utilizing (oxidatively modified) CL species in mitochondria has been reported [41–43]. Indeed, treatment of lymphocytes with rotenone resulted in increased content of mono-lyso-CL species (Fig. 8A). In line with this accumulation of oxygenated C18:2 containing from one to three oxygens in the fraction of free fatty acids was detected (Fig. 8B). After rotenone exposure, the endogenous contents of mono-lyso-CL and free fatty acids – likely released from phospholipids by endogenous PLA2 - are shown in Figs. 8Ab and 8Bb. We found that the content of free C18:2 with two oxygens (m/z 311) was higher than that of mono-oxygenated C18:2. This suggests that endogenous PLA2 – likely mitochondrial iPLA2 – hydrolyzed peroxidized TLCLs with two oxygens (m/z 311) more effectively than CLs with mono-oxygenated C18:2 (m/z 295). No significant accumulation of other lyso-phospholipids in lymphocytes in response to rotenone was detected (data not shown). This suggests that accumulated mono-lyso-CL molecular species originated from oxygenated TLCL formed in lymphocytes upon rotenone exposure.
Figure 8. Rotenone induced accumulation of mono-lyso-CL and oxygenated free fatty acids in human peripheral blood lymphocytes.

(A) LC/MS base profile (a) and quantitative assessment (b) of mono-lyso-CL. (B) Content of free C18:2 containing one (a) two (b) and three (c) oxygens in lymphocytes treated with rotenone (100 and 250 μM for 18 hrs at 37°C. Note: the increase of mono-lyso-CL was accompanied by accumulation of oxygenated free C18:2. Data are means ± S.E., n=5, *p<0.05 vs control.
Discussion
Although oxygenated fatty acids and phospholipids are critical signaling molecules (and/or biomarkers) in several neurological disorders [44], essential information on molecular targets, particularly specific polyunsaturated molecular species of phospholipids undergoing oxidation and leading to mitochondrial dysfunction and their association with neurodegenerative disease such as PD, is lacking. While the general association of oxidative stress with PD has been emphasized in numerous studies (reviewed in [45–48], attempts to link the products of oxidative modification of different biomolecules to specific pathogenetic pathways of PD were not victorious [5, 49]. This may be due, at least in part, to insufficient information on the diversity and structure of oxidized biomolecules generated in mitochondria – the major metabolic candidate as a source of impaired and oxidatively modified PD-associated molecular species. Among those, CLs – unique and functionally essential phospholipids of mitochondria – may be of particular importance [50–52]. In this paper by using LC/MS-based oxidative lipidomics, we characterized all major molecular species of CL and its oxidized molecular species formed in rotenone-associated dysfunctional mitochondria in human peripheral blood lymphocytes.
Changes in the CL content as well as its composition have been shown to be responsible for mitochondrial dysfunction associated with several pathological conditions [53–56]. CL profile in mitochondria can be changed due to: i) loss of CL as a result of alteration in CL synthase activity [2, 31, 57], ii) altered fatty acid composition of CL as a result of disruption of CL remodeling process [58–60] and, iii) CL oxidation due to ROS generation [55]. Generation of ROS [8, 61] and activation of the intrinsic apoptotic cell death pathway [62] have been associated with rotenone-induced neuron degeneration in vitro and in vivo [53–55], dissipation of MMP, release cyt c from mitochondria into the cytosol and apoptosis [63]. Chronic and systemic inhibition of complex I leads to selective degeneration of dopaminergic neurons and produces neuro-pathological features of PD [4, 64].
Having in mind potential detection of mitochondrial phospholipid biomarkers of PD, we examined human peripheral blood lymphocytes that are often used to identify the mechanism leading to development of neurodegenerative diseases such as PD and Alzheimer disease [8, 65, 66]. In fact, increased apoptosis of lymphocytes in patients with PD has been documented [15, 16]. Both extrinsic and intrinsic apoptotic pathways were recognized in lymphocytes of PD patients [16, 67, 68]. Notably, rotenone induced apoptosis in lymphocytes has been linked to its ability to generate ROS () leading to mitochondrial damage [10]. Further rotenone-induced inhibition of complex I activity and impairment of electron transport leading to massive production of ROS [69], and possibly protein and lipid peroxidation have been documented in dopaminergic cell line using BODYPI oxidation assays [70]. However, specific features of CL peroxidation and identification of CL oxidation products as essential factors in mitochondrial stages of lymphocyte apoptosis, have not been studied so far.
Here, we demonstrated that exposure of human lymphocytes to rotenone is associated with mitochondria dysfunction, ROS production, development of apoptotic cell death pathway and accumulation of oxygenated species of highly unsaturated CL containing four C18:2 residues as well as its metabolite mono-lyso-CL. Detailed structural analysis of CL oxidation products revealed unusual features of rotenone-triggered peroxidation: i) predominant peroxidation of TLCL, CL containing four C18:2 residues ii) preferential accumulation of oxygenated C18:2 in sn-1 rather than sn-2 position; iii) quantitative abundance of mono-oxygenated species vs species with two and three oxygen functionalities. Further studies will determine whether these specific features are uniquely associated with the rotenone-driven inhibition of respiratory complex I or may be common to other oxidative routes leading to the execution of apoptotic program in lymphocytes.
Our previous work has identified cyt c, an intermembrane space electron carrier, as a catalyst of the reaction during which it binds CL to yield a complex with CL-specific peroxidase activity [34]. The complex generates oxygenated CL species at the early stage of apoptosis in vitro and in vivo [17, 18, 20, 37, 71, 72]. Moreover, cyt c/CL complexes can interact with α-synuclein to form oligomers with high peroxidase activity [52] thus contributing to the formation of Lewis bodies – a morphological hallmark of PD. These results provided direct evidence for previously suggested conversion of cyt c into peroxidase and its possible role in neurodegenerative process [73], including pathogenesis of PD [74, 75]. Notably, while peroxidase activity cyclooxygenase-2 and peroxidation of non-esterified fatty acids have been linked to the pathogenesis of PD [74, 75] the oxidation of esterified lipids has not been yet investigated. Our data indicate that in rotenone treated lymphocytes, cyt c utilizes ROS, particularly hydrogen peroxide, formed during apoptotic cell death, to cause peroxidation of C18:2-containing CL species in mitochondria (Fig. 9). In addition, rotenone-induced oxidative stress and ROS production can cause activation of Ca2+-independent iPLA2 [76] resulting in the accumulation of CL hydrolysis products such as mono-lyso-CL and oxygenated fatty acids. iPLA2 has been identified as the major endogenous type of PLA2 capable of hydrolyzing peroxidized phospholipids in mitochondria [41, 77].
Figure 9. Proposed mechanism of rotenone-induced CL oxidation and its hydrolysis in human lymphocytes mitochondria.

IMM-inner mitochondrial membrane; cyt c, cytochrome c; iPLA2- Ca2+-independent phospholipase A2; CL, cardiolipin; CLox, oxygenated cardiolipin; mCL, monolyso-cardiolipin; FAox, oxygenated C18:2.
We found that the most predominant molecular species of CL that underwent oxidative modification after exposure of human lymphocytes to rotenone was tetralinoleoyl-CL with four symmetric C18:2-residues in both sn-1 and sn-2 positions. In addition we were able to detect endogenously formed TLCLox, non-ox-mono-lyso-CL and C18:2-ox. To determine which of C18:2 residues was a preferred substrate of rotenone-induced peroxidation, we treated isolated CL fraction with either PLA1 or PLA2 – to produce lyso-CLs and release C18:2 from the respective sn-1 or sn-2 positions. This resulted in liberation of oxygenated C18:2 species and di-lyso-CLs whereby higher contents of oxygenated C18:2 was produced by PLA1 (as compared to PLA2). Because 99% of total CLs were hydrolyzed and converted into di-lyso-CLs and FAs, we further analyzed oxygenated species of C18:2. We found that only one out of four C18:2 in TLCL molecule (in either sn-1 or sn-2 positions) underwent oxidative modification upon rotenone exposure. Assuming that endogenous iPLA2 cleaves predominantly C18:2-ox in sn-2 but not non-ox-C18:2, C18:2-ox plus non-ox-mono-lyso-CL should be expected hydrolysis products – in line with our observations. Thus not only CL peroxidation products but also mono-lyso-CL in lymphocytes may be used as biomarkers of PD-associated metabolic disturbances – similar to recent finding in patients with Barth syndrome, a disease associated with mitochondrial dysfunction [78].
While this study has been focused on CLs as well as their oxidized and hydrolyzed metabolites, we have also analyzed several other major classes of phospholipids, such as phosphatidylcholine, phosphatidylethanolamine, phopshatidylinositol, phosphatidylserine and sphingomyelin. We found that exposure of lymphocytes to rotenone resulted in significant accumulation of several oxygenated molecular species of TLCL as well as its hydrolysis products – mono-lyso-CL and oxygenated free C18:2. These rotenone-induced changes were CL-specific: neither oxygenated products nor hydrolysis products were detected in other examined classes of phospholipids. Thus no rotenone-induced remodeling of other phospholipids took place under experimental conditions used.
Apoptotic cell death pathway is activated in lymphocytes of PD patients [15, 16]. Therefore, peripheral blood lymphocytes are considered as potential candidate-biomarkers of mitochondrial dysfunction in PD. Given a recently established role of selective peroxidation of a mitochondria-specific phospholipid, CL, in execution of mitochondrial stages of apoptosis, one can assume that detailed analysis of CL peroxidation products may lead to the development of useful biomarkers. It has been reported that micromolar concentrations of rotenone (10–250 μM) induce apoptosis in isolated human lymphocytes [10]. However, specific role of CL peroxidation as an essential factor in mitochondrial stages of lymphocyte apoptosis, has not been yet studied. In the current work, we found that rotenone (at concentrations of 100 and 250 μM) was effective in stimulating selective accumulation of CL oxidation products and induction of apoptosis in isolated human lymphocytes. While the concentrations of rotenone used may seem to be relatively high, one should consider them in the context of its known toxicity and exposure doses. The estimates of rotenone toxicity for humans are commonly based on animal studies. In rat rotenone PD model with administration of pesticide through I.V. route (3–18 mg/kg/day) [79, 80], its concentration in circulation is within micromolar range. Of note, rotenone is usually sold as 1 – 5% formulation that corresponds to approximately its 30–150 mM solution. Thus agricultural workers can be occupationally exposed to relatively high doses of rotenone.
In conclusion, we demonstrated that exposure of human peripheral blood lymphocytes to a pesticide, rotenone, causes time- and dose-dependent selective oxidation of TLCL, accumulation of its hydrolysis products - mono-lyso-CLs - as well as generation of TLCL oxygenated molecular species associated with mitochondrial dysfunction likely through enzymatic cyt c catalyzed reactions triggered early in apoptosis. We believe that characterization of oxidatively modified CL molecular species as well as identification its hydrolysis products are important for better understanding of PD pathogenesis and may lead to the development of new biomarkers of mitochondrial dysfunction associated with PD.
Supplementary Material
Acknowledgments
Supported by NIH: ES020693, HL70755, U19AIO68021; by NIOSH OH008282.
Abbreviations
- CL
cardiolipin
- cyt c
cytochrome c
- DCFH-DA
2',7'-dichlorfluorescein-diacetate
- DHE
dihydroethidium
- DTPA
diethylenetriaminepentaacetic acid
- MMP
mitochondrial membrane potential
- PD
Parkinson's disease
- PS
phosphatidylserine
- PLA2
phospholipase A2
- PLA1
phospholipase A1
- ROS
reactive oxygen species
- TLCL
tetra-linoleyl-cardiolipin
- TMCL
tetra-myristoyl-cardiolipin
Footnotes
The authors have declared no conflict of interest.
References
- [1].Thomas B, Beal MF. Parkinson's disease. Human molecular genetics. 2007;16(Spec No. 2):R183–194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- [2].Schapira AH. Evidence for mitochondrial dysfunction in Parkinson's disease--a critical appraisal. Mov Disord. 1994;9:125–138. doi: 10.1002/mds.870090202. [DOI] [PubMed] [Google Scholar]
- [3].Fukae J, Mizuno Y, Hattori N. Mitochondrial dysfunction in Parkinson's disease. Mitochondrion. 2007;7:58–62. doi: 10.1016/j.mito.2006.12.002. [DOI] [PubMed] [Google Scholar]
- [4].Winklhofer KF, Haass C. Mitochondrial dysfunction in Parkinson's disease. Biochim Biophys Acta. 2010;1802:29–44. doi: 10.1016/j.bbadis.2009.08.013. [DOI] [PubMed] [Google Scholar]
- [5].Mariani E, Polidori MC, Cherubini A, Mecocci P. Oxidative stress in brain aging, neurodegenerative and vascular diseases: an overview. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;827:65–75. doi: 10.1016/j.jchromb.2005.04.023. [DOI] [PubMed] [Google Scholar]
- [6].Brown RC, Lockwood AH, Sonawane BR. Neurodegenerative diseases: an overview of environmental risk factors. Environ Health Perspect. 2005;113:1250–1256. doi: 10.1289/ehp.7567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Berry C, La Vecchia C, Nicotera P. Paraquat and Parkinson's disease. Cell Death Differ. 2010;17:1115–1125. doi: 10.1038/cdd.2009.217. [DOI] [PubMed] [Google Scholar]
- [8].Migliore L, Petrozzi L, Lucetti C, Gambaccini G, et al. Oxidative damage and cytogenetic analysis in leukocytes of Parkinson's disease patients. Neurology. 2002;58:1809–1815. doi: 10.1212/wnl.58.12.1809. [DOI] [PubMed] [Google Scholar]
- [9].Gomez C, Bandez MJ, Navarro A. Pesticides and impairment of mitochondrial function in relation with the parkinsonian syndrome. Front Biosci. 2007;12:1079–1093. doi: 10.2741/2128. [DOI] [PubMed] [Google Scholar]
- [10].Avila-Gomez IC, Velez-Pardo C, Jimenez-Del-Rio M. Effects of insulin-like growth factor-1 on rotenone-induced apoptosis in human lymphocyte cells. Basic Clin Pharmacol Toxicol. 2010;106:53–61. doi: 10.1111/j.1742-7843.2009.00472.x. [DOI] [PubMed] [Google Scholar]
- [11].Drechsel DA, Patel M. Role of reactive oxygen species in the neurotoxicity of environmental agents implicated in Parkinson's disease. Free Radic Biol Med. 2008;44:1873–1886. doi: 10.1016/j.freeradbiomed.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Schulz JB, Beal MF. Mitochondrial dysfunction in movement disorders. Curr Opin Neurol. 1994;7:333–339. doi: 10.1097/00019052-199408000-00010. [DOI] [PubMed] [Google Scholar]
- [13].Shinde S, Pasupathy K. Respiratory-chain enzyme activities in isolated mitochondria of lymphocytes from patients with Parkinson's disease: preliminary study. Neurol India. 2006;54:390–393. doi: 10.4103/0028-3886.28112. [DOI] [PubMed] [Google Scholar]
- [14].Kim S, Jeon BS, Heo C, Im PS, et al. Alpha-synuclein induces apoptosis by altered expression in human peripheral lymphocyte in Parkinson's disease. FASEB J. 2004;18:1615–1617. doi: 10.1096/fj.04-1917fje. [DOI] [PubMed] [Google Scholar]
- [15].Blandini F, Sinforiani E, Pacchetti C, Samuele A, et al. Peripheral proteasome and caspase activity in Parkinson disease and Alzheimer disease. Neurology. 2006;66:529–534. doi: 10.1212/01.wnl.0000198511.09968.b3. [DOI] [PubMed] [Google Scholar]
- [16].Calopa M, Bas J, Callen A, Mestre M. Apoptosis of peripheral blood lymphocytes in Parkinson patients. Neurobiol Dis. 2010;38:1–7. doi: 10.1016/j.nbd.2009.12.017. [DOI] [PubMed] [Google Scholar]
- [17].Tyurin VA, Tyurina YY, Feng W, Mnuskin A, et al. Mass-spectrometric characterization of phospholipids and their primary peroxidation products in rat cortical neurons during staurosporine-induced apoptosis. Journal of neurochemistry. 2008;107:1614–1633. doi: 10.1111/j.1471-4159.2008.05728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ji J, Tyurina YY, Tang M, Feng W, et al. Mitochondrial injury after mechanical stretch of cortical neurons in vitro: biomarkers of apoptosis and selective peroxidation of anionic phospholipids. Journal of neurotrauma. 2012;29:776–788. doi: 10.1089/neu.2010.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bayir H, Tyurin VA, Tyurina YY, Viner R, et al. Selective early cardiolipin peroxidation after traumatic brain injury: an oxidative lipidomics analysis. Annals of neurology. 2007;62:154–169. doi: 10.1002/ana.21168. [DOI] [PubMed] [Google Scholar]
- [20].Ji J, Kline AE, Amoscato A, Samhan-Arias AK, et al. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nature neuroscience. 2012;15:1407–1413. doi: 10.1038/nn.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Schlame M, Ren M. The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim Biophys Acta. 2009;1788:2080–2083. doi: 10.1016/j.bbamem.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Daum G, Lees ND, Bard M, Dickson R. Biochemistry, cell biology and molecular biology of lipids of Saccharomyces cerevisiae. Yeast. 1998;14:1471–1510. doi: 10.1002/(SICI)1097-0061(199812)14:16<1471::AID-YEA353>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- [23].Fry M, Green DE. Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. J Biol Chem. 1981;256:1874–1880. [PubMed] [Google Scholar]
- [24].Sharpley MS, Shannon RJ, Draghi F, Hirst J. Interactions between phospholipids and NADH:ubiquinone oxidoreductase (complex I) from bovine mitochondria. Biochemistry. 2006;45:241–248. doi: 10.1021/bi051809x. [DOI] [PubMed] [Google Scholar]
- [25].Robinson NC. Functional binding of cardiolipin to cytochrome c oxidase. J Bioenerg Biomembr. 1993;25:153–163. doi: 10.1007/BF00762857. [DOI] [PubMed] [Google Scholar]
- [26].Eble KS, Coleman WB, Hantgan RR, Cunningham CC. Tightly associated cardiolipin in the bovine heart mitochondrial ATP synthase as analyzed by 31P nuclear magnetic resonance spectroscopy. J Biol Chem. 1990;265:19434–19440. [PubMed] [Google Scholar]
- [27].Checkoway H, Costa LG, Woods JS, Castoldi AF, et al. Peripheral blood cell activities of monoamine oxidase B and superoxide dismutase in Parkinson's disease. J Neural Transm Park Dis Dement Sect. 1992;4:283–290. doi: 10.1007/BF02260077. [DOI] [PubMed] [Google Scholar]
- [28].Colombo C, Cosentino M, Marino F, Rasini E, et al. Dopaminergic modulation of apoptosis in human peripheral blood mononuclear cells: possible relevance for Parkinson's disease. Ann N Y Acad Sci. 2003;1010:679–682. doi: 10.1196/annals.1299.124. [DOI] [PubMed] [Google Scholar]
- [29].Amenta F, Bronzetti E, Cantalamessa F, El-Assouad D, et al. Identification of dopamine plasma membrane and vesicular transporters in human peripheral blood lymphocytes. J Neuroimmunol. 2001;117:133–142. doi: 10.1016/s0165-5728(01)00317-4. [DOI] [PubMed] [Google Scholar]
- [30].Ridings J, Weedon H, Ioannou C, Flego L, et al. Purification of cord blood lymphocytes. Journal of immunological methods. 1996;195:43–48. doi: 10.1016/0022-1759(96)00089-0. [DOI] [PubMed] [Google Scholar]
- [31].Huang Z, Jiang J, Tyurin VA, Zhao Q, et al. Cardiolipin deficiency leads to decreased cardiolipin peroxidation and increased resistance of cells to apoptosis. Free Radic Biol Med. 2008;44:1935–1944. doi: 10.1016/j.freeradbiomed.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].de Wit LE, Spruijt L, Schoonderwoerd GC, de Coo IF, et al. A simplified and reliable assay for complex I in human blood lymphocytes. Journal of immunological methods. 2007;326:76–82. doi: 10.1016/j.jim.2007.07.009. [DOI] [PubMed] [Google Scholar]
- [33].Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nature protocols. 2009;4:1582–1590. doi: 10.1038/nprot.2009.151. [DOI] [PubMed] [Google Scholar]
- [34].Kagan VE, Tyurin VA, Jiang J, Tyurina YY, et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- [35].Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- [36].Boettcher C, Pries C, Vangent CM. A Rapid and Sensitive Sub-Micro Phosphorus Determination. Anal Chim Acta. 1961;24:203. [Google Scholar]
- [37].Tyurin VA, Tyurina YY, Ritov VB, Lysytsya A, et al. Oxidative lipidomics of apoptosis: quantitative assessment of phospholipid hydroperoxides in cells and tissues. Methods Mol Biol. 2010;610:353–374. doi: 10.1007/978-1-60327-029-8_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rouser G, Fkeischer S, Yamamoto A. Two dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids. 1970;5:494–496. doi: 10.1007/BF02531316. [DOI] [PubMed] [Google Scholar]
- [39].Tyurina YY, Kisin ER, Murray A, Tyurin VA, et al. Global phospholipidomics analysis reveals selective pulmonary peroxidation profiles upon inhalation of single-walled carbon nanotubes. ACS nano. 2011;5:7342–7353. doi: 10.1021/nn202201j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kim J, Hoppel CL. Monolysocardiolipin: improved preparation with high yield. J Lipid Res. 2011;52:389–392. doi: 10.1194/jlr.D010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Moon SH, Jenkins CM, Liu X, Guan S, et al. Activation of mitochondrial calcium-independent phospholipase A2gamma (iPLA2gamma) by divalent cations mediating arachidonate release and production of downstream eicosanoids. J Biol Chem. 2012;287:14880–14895. doi: 10.1074/jbc.M111.336776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zachman DK, Chicco AJ, McCune SA, Murphy RC, et al. The role of calcium-independent phospholipase A2 in cardiolipin remodeling in the spontaneously hypertensive heart failure rat heart. J Lipid Res. 2010;51:525–534. doi: 10.1194/jlr.M000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Seleznev K, Zhao C, Zhang XH, Song K, Ma ZA. Calcium-independent phospholipase A2 localizes in and protects mitochondria during apoptotic induction by staurosporine. J Biol Chem. 2006;281:22275–22288. doi: 10.1074/jbc.M604330200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hu C, van der Heijden R, Wang M, van der Greef J, et al. Analytical strategies in lipidomics and applications in disease biomarker discovery. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:2836–2846. doi: 10.1016/j.jchromb.2009.01.038. [DOI] [PubMed] [Google Scholar]
- [45].Tsang AH, Chung KK. Oxidative and nitrosative stress in Parkinson's disease. Biochim Biophys Acta. 2009;1792:643–650. doi: 10.1016/j.bbadis.2008.12.006. [DOI] [PubMed] [Google Scholar]
- [46].Navarro A, Boveris A. Brain mitochondrial dysfunction and oxidative damage in Parkinson's disease. J Bioenerg Biomembr. 2009;41:517–521. doi: 10.1007/s10863-009-9250-6. [DOI] [PubMed] [Google Scholar]
- [47].Surendran S, Rajasankar S. Parkinson's disease: oxidative stress and therapeutic approaches. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 2010;31:531–540. doi: 10.1007/s10072-010-0245-1. [DOI] [PubMed] [Google Scholar]
- [48].Ruiperez V, Darios F, Davletov B. Alpha-synuclein, lipids and Parkinson's disease. Progress in lipid research. 2010;49:420–428. doi: 10.1016/j.plipres.2010.05.004. [DOI] [PubMed] [Google Scholar]
- [49].Breusing N, Grune T. Biomarkers of protein oxidation from a chemical, biological and medical point of view. Exp Gerontol. 2010;45:733–737. doi: 10.1016/j.exger.2010.04.004. [DOI] [PubMed] [Google Scholar]
- [50].Nakamura K, Nemani VM, Azarbal F, Skibinski G, et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J Biol Chem. 2011;286:20710–20726. doi: 10.1074/jbc.M110.213538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Benkler M, Agmon-Levin N, Hassin-Baer S, Cohen OS, et al. Immunology, autoimmunity, and autoantibodies in Parkinson's disease. Clinical reviews in allergy & immunology. 2012;42:164–171. doi: 10.1007/s12016-010-8242-y. [DOI] [PubMed] [Google Scholar]
- [52].Bayir H, Kapralov AA, Jiang J, Huang Z, et al. Peroxidase mechanism of lipid-dependent cross-linking of synuclein with cytochrome C: protection against apoptosis versus delayed oxidative stress in Parkinson disease. J Biol Chem. 2009;284:15951–15969. doi: 10.1074/jbc.M900418200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Paradies G, Ruggiero FM, Dinoi P, Petrosillo G, Quagliariello E. Decreased cytochrome oxidase activity and changes in phospholipids in heart mitochondria from hypothyroid rats. Arch Biochem Biophys. 1993;307:91–95. doi: 10.1006/abbi.1993.1565. [DOI] [PubMed] [Google Scholar]
- [54].Chicco AJ, Sparagna GC. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am J Physiol Cell Physiol. 2007;292:C33–44. doi: 10.1152/ajpcell.00243.2006. [DOI] [PubMed] [Google Scholar]
- [55].Lesnefsky EJ, Hoppel CL. Cardiolipin as an oxidative target in cardiac mitochondria in the aged rat. Biochim Biophys Acta. 2008;1777:1020–1027. doi: 10.1016/j.bbabio.2008.05.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Paradies G, Petrosillo G, Paradies V, Ruggiero FM. Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium. 2009;45:643–650. doi: 10.1016/j.ceca.2009.03.012. [DOI] [PubMed] [Google Scholar]
- [57].Xu FY, Taylor WA, Hurd JA, Hatch GM. Etomoxir mediates differential metabolic channeling of fatty acid and glycerol precursors into cardiolipin in H9c2 cells. J Lipid Res. 2003;44:415–423. doi: 10.1194/jlr.M200335-JLR200. [DOI] [PubMed] [Google Scholar]
- [58].Xu Y, Malhotra A, Ren M, Schlame M. The enzymatic function of tafazzin. J Biol Chem. 2006;281:39217–39224. doi: 10.1074/jbc.M606100200. [DOI] [PubMed] [Google Scholar]
- [59].Li J, Romestaing C, Han X, Li Y, et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 2010;12:154–165. doi: 10.1016/j.cmet.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Taylor WA, Xu FY, Ma BJ, Mutter TC, et al. Expression of monolysocardiolipin acyltransferase activity is regulated in concert with the level of cardiolipin and cardiolipin biosynthesis in the mammalian heart. BMC Biochem. 2002;3:9. doi: 10.1186/1471-2091-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Testa CM, Sherer TB, Greenamyre JT. Rotenone induces oxidative stress and dopaminergic neuron damage in organotypic substantia nigra cultures. Brain Res Mol Brain Res. 2005;134:109–118. doi: 10.1016/j.molbrainres.2004.11.007. [DOI] [PubMed] [Google Scholar]
- [62].Yao Z, Wood NW. Cell death pathways in Parkinson's disease: role of mitochondria. Antioxid Redox Signal. 2009;11:2135–2149. doi: 10.1089/ars.2009.2624. [DOI] [PubMed] [Google Scholar]
- [63].Fiskum G, Starkov A, Polster BM, Chinopoulos C. Mitochondrial mechanisms of neural cell death and neuroprotective interventions in Parkinson's disease. Ann N Y Acad Sci. 2003;991:111–119. doi: 10.1111/j.1749-6632.2003.tb07469.x. [DOI] [PubMed] [Google Scholar]
- [64].Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol. 2008;4:600–609. doi: 10.1038/ncpneuro0924. [DOI] [PubMed] [Google Scholar]
- [65].Chen CM, Liu JL, Wu YR, Chen YC, et al. Increased oxidative damage in peripheral blood correlates with severity of Parkinson's disease. Neurobiol Dis. 2009;33:429–435. doi: 10.1016/j.nbd.2008.11.011. [DOI] [PubMed] [Google Scholar]
- [66].Leuner K, Schulz K, Schutt T, Pantel J, et al. Peripheral mitochondrial dysfunction in Alzheimer's disease: focus on lymphocytes. Molecular neurobiology. 2012;46:194–204. doi: 10.1007/s12035-012-8300-y. [DOI] [PubMed] [Google Scholar]
- [67].Gatta L, Cardinale A, Wannenes F, Consoli C, et al. Peripheral blood mononuclear cells from mild cognitive impairment patients show deregulation of Bax and Sod1 mRNAs. Neurosci Lett. 2009;453:36–40. doi: 10.1016/j.neulet.2009.02.003. [DOI] [PubMed] [Google Scholar]
- [68].Xu G, Shi Y. Apoptosis signaling pathways and lymphocyte homeostasis. Cell Res. 2007;17:759–771. doi: 10.1038/cr.2007.52. [DOI] [PubMed] [Google Scholar]
- [69].Genova ML, Pich MM, Bernacchia A, Bianchi C, et al. The mitochondrial production of reactive oxygen species in relation to aging and pathology. Ann N Y Acad Sci. 2004;1011:86–100. doi: 10.1007/978-3-662-41088-2_10. [DOI] [PubMed] [Google Scholar]
- [70].Seo BB, Marella M, Yagi T, Matsuno-Yagi A. The single subunit NADH dehydrogenase reduces generation of reactive oxygen species from complex I. FEBS letters. 2006;580:6105–6108. doi: 10.1016/j.febslet.2006.10.008. [DOI] [PubMed] [Google Scholar]
- [71].Tyurina YY, Tyurin VA, Epperly MW, Greenberger JS, Kagan VE. Oxidative lipidomics of gamma-irradiation-induced intestinal injury. Free Radic Biol Med. 2008;44:299–314. doi: 10.1016/j.freeradbiomed.2007.08.021. [DOI] [PubMed] [Google Scholar]
- [72].Tyurina YY, Tyurin VA, Kaynar AM, Kapralova VI, et al. Oxidative lipidomics of hyperoxic acute lung injury: mass spectrometric characterization of cardiolipin and phosphatidylserine peroxidation. Am J Physiol Lung Cell Mol Physiol. 2010;299:L73–85. doi: 10.1152/ajplung.00035.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Patriarca A, Polticelli F, Piro MC, Sinibaldi F, et al. Conversion of cytochrome c into a peroxidase: inhibitory mechanisms and implication for neurodegenerative diseases. Arch Biochem Biophys. 2012;522:62–69. doi: 10.1016/j.abb.2012.03.028. [DOI] [PubMed] [Google Scholar]
- [74].Everse J, Coates PW. Neurodegeneration and peroxidases. Neurobiology of aging. 2009;30:1011–1025. doi: 10.1016/j.neurobiolaging.2007.10.007. [DOI] [PubMed] [Google Scholar]
- [75].Teismann P. COX-2 in the neurodegenerative process of Parkinson's disease. BioFactors. 2012 doi: 10.1002/biof.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Jezek J, Jaburek M, Zelenka J, Jezek P. Mitochondrial phospholipase A2 activated by reactive oxygen species in heart mitochondria induces mild uncoupling. Physiological research / Academia Scientiarum Bohemoslovaca. 2010;59:737–747. doi: 10.33549/physiolres.931905. [DOI] [PubMed] [Google Scholar]
- [77].Brustovetsky T, Antonsson B, Jemmerson R, Dubinsky JM, Brustovetsky N. Activation of calcium-independent phospholipase A (iPLA) in brain mitochondria and release of apoptogenic factors by BAX and truncated BID. Journal of neurochemistry. 2005;94:980–994. doi: 10.1111/j.1471-4159.2005.03248.x. [DOI] [PubMed] [Google Scholar]
- [78].Bowron A, Frost R, Powers VE, Thomas PH, et al. Diagnosis of Barth syndrome using a novel LC-MS/MS method for leukocyte cardiolipin analysis. Journal of inherited metabolic disease. 2012 doi: 10.1007/s10545-012-9552-4. [DOI] [PubMed] [Google Scholar]
- [79].Ferrante RJ, Schulz JB, Kowall NW, Beal MF. Systemic administration of rotenone produces selective damage in the striatum and globus pallidus, but not in the substantia nigra. Brain research. 1997;753:157–162. doi: 10.1016/s0006-8993(97)00008-5. [DOI] [PubMed] [Google Scholar]
- [80].Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, et al. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nature neuroscience. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.