

Abstract
Background & Aims
African Americans (AA) are an admixed population of West African (WA) and European ancestry (EA). Crohn's disease (CD) susceptibility genes have not been established. We therefore evaluated the contribution of European admixture and major established risk genes to AA CD.
Methods
Ninety-seven admixture informative markers were genotyped for ancestry estimates using STRUCTURE. 354 AA CD cases and 354 ethnicity-matched controls were genotyped for total 21 SNPs in ATG16L1, NOD2, IBD5, IL23R and IRGM by TaqMan or direct sequencing. Association was evaluated by logistic regression, adjusted for ancestry.
Results
Mean EA was similar among the CD cases and controls (20.9% and 20.4, respectively, p=0.58). No significant admixture differences were observed among cases (211 to 227) stratified by phenotypic sub-classifications including onset, surgery, site, and behavior. CD was associated with NOD2 carrier (6.93% CD, 2.15% Controls, p = 0.007), ATG16L1 Thr300Ala (36.1% CD, 29.3% Controls, p=0.003), SLC22A4 and SLC22A5 (IBD5 locus) functional SNPs (L503F [10.6% CD, 7.6% Controls, p=0.05] and g-207c [41.3% CD, 35.7% Controls, p=0.03], respectively) and IL23R rs2201841 (18.2% CD, 13.8% Controls, p=0.03), but not IRGM variants nor three African ancestral NOD2 nonsynonymous variants. IBD5 risk was recessive. An all-minor allele IBD5 haplotype from EA was associated (p=0.05), whereas a more common haplotype isolating g-207c was not.
Conclusions
Specific functional gene variations significantly contribute to AA CD risk. Established NOD2, SLC22A4-A5, and ATG16L1 variants show increased CD risk, with IBD5 in recessive. Although CD is more common in whites, European admixture is similar among AA cases and controls.
Keywords: genetics, epidemiology, Crohn's disease
Introduction
Historically, Crohn's disease (CD) incidence has been substantially lower in African Americans (AA) than white American populations. Some recent studies still show this, whereas two studies found hospitalization rates greatest among AA females.1, 2 AA CD prevalence in the population-based Kaiser Health Maintenance Organization study was two-thirds that of whites.2 Compared to most white populations, where ulcerative colitis (UC) occurs slightly more frequently than CD, in AA's CD incidence in children was observed 1.6-fold greater than UC suggesting potential etiological differences.3 CD phenotype features may also be different among races: we found AA's had significantly greater odds of colon-only and perianal disease, had lower odds of penetrating disease, and (for both CD and UC) greater odds of uveitis and sacroiliitis (2.1, 1.6, 0.52, 3.7 and 5.8-fold respectively).4 CD risk to AA siblings was much greater than population prevalence (2.5% vs. 45/100,000), suggesting that like white CD, AA CD likely has important genetic influences.4
The AA genome is an admixture of West African and European ancestry. When a phenotype is more common in one ancestral population compared to another, in an admixed population, the degree of genetic admixture in individuals with the phenotype often corresponds. For example, average BMI is greater in AA's than white Americans; correspondingly, BMI is inversely associated with degree of European ancestry in AA's.5 With CD more prevalent in whites than AA's, it is important to determine if overall CD risk arises from a relative increase in European admixture. It is also of interest to investigate whether phenotypic features more or less common in whites will correlate with the degree of European admixture.
Few studies have evaluated the influence of established CD molecular genetic risk factors in AA's. Nucleotide-binding oligomerization domain containing-2 (NOD2) mutations Arg702Trp, Gly908Arg and Leu1007fsinsC, the strongest, common CD risk factor in whites, was first evaluated in 58 AA children with CD and 124 healthy controls (HC's) from Wisconsin, but showed no association (carriers 3.8% CD vs. 4.3% HC's).6 We evaluated NOD2 in our initially recruited Mid-Atlantic (now Multicenter) African American IBD study (MAAIS – a sub-study of the NIDDK IBD Genetics Consortium [IBDGC]) of 183 CD and 143 HC's and found increased risk (8.2% vs. 2.1%, respectively, P=0.03).7 Given these discrepant findings, it is important that NOD2 AA risk be more substantiated. The IBD5 locus genes solute carrier family 22, member 4 and member 5 (SLC22A4 and SLC22A5) were the second genes having functional variations (Leu503Phe and g-207c, respectively) established as CD genetic risk factors. 8, 9 Whether both genes are required for CD risk and whether risk lies within variations that influence other proximal genes has not been discerned, primarily because of strong linkage disequilibrium (LD) within a single, common, minor allele haplotype found across a 600 kb region in whites.8 The first genome wide association study (GWAS) in whites established association for the interleukin (IL) 23 receptor gene, IL23R, the receptor product a key regulator of chronic inflammation in CD.10 Multiple IL23R single nucleotide polymorphisms (SNPs) have shown independent CD association with a poorly conserved albeit uncommon protective variant, Arg381Gln showing a greater than 3-fold protective effect.10 Autophagy 16 like-1 (ATG16L1) was the next CD gene established by GWAS; a Thr300Ala resulted in 1.5-fold risk.11, 12 300Ala reduces autophagasome elimination of bacteria13 further substantiating the concept, established with NOD2, that defective innate immunity results in CD risk. The Wellcome Trust GWAS identified variants in immunity-related GTPase family, M (IRGM ) that result in 1.4-fold CD risk. IRGM encodes a GTP-binding protein that induces autophagy likewise important to elimination of bacteria, including Mycobacterium tuberculosis.14, 15 IRGM association may be due to a 20 kb copy number variation (CNV) that influences gene expression16, 17 and/or a synonymous exon variant (rs10065172 encoding c.313C>T) that alters miRNA binding of miR-196 to IRGM resulting in loss of IRGM regulation in inflamed intestinal tissue.18 Of the established CD loci, NOD2, ATG16L1, SLC22A4-A5, IL23R and IRGM are among the highest risk CD genes (risk ≥ 1.4-fold) where functional variations have been identified and studied.
In view of the genetic heterogeneity of CD, the unique features of AA genetic ancestry and CD phenotype features, we performed a study to characterize the overall contributions of WA and European admixture to AA CD risk and to determine the genetic influences of the most well established, higher risk CD genes having characterized, functional polymorphisms.
Methods
Patient Populations (summarized in Supplementary Table 1)
Primary dataset
The study population included unrelated individuals, self-identified as non-Hispanic AA with a history of confirmed CD, recruited from 2003 to 2007 by the MAAIS. Similarly, unrelated, self-identified non-Hispanic AA's with no personal or family history of inflammatory bowel disease (IBD) and no history of chronic diarrhea, unexplained abdominal pain, rectal bleeding or irritable bowel syndrome were recruited as non-IBD, ethnically matched HC's. MAAIS recruitment was coordinated at the Johns Hopkins Genetics Research Center (GRC) of the IBDGC with satellite recruitment at Howard University, University of Florida, University of North Carolina, University of Pennsylvania, and the Washington Hospital Center. Twenty-six AA cases, all patients at Johns Hopkins Hospital, and 13 AA healthy spouse controls were also included.
IBD family history was defined as IBD in first- or second-degree relatives. Smoking was an average of at least 1 cigarette daily for at least 3 months prior to diagnosis. CD diagnosis was confirmed by medical records review per the IBDGC standards in an operations manual.19
Secondary dataset
This dataset was derived primarily from DNA samples of 66 adults recruited at the University of Chicago and 59 children recruited at the Medical College of Wisconsin with confirmed CD.6, 20 DNAs from 12 AA children with confirmed CD were also received from a Duke University IBD study.21 Lastly, 43 samples were obtained from non-Hispanic, AA (or African-Canadian) individuals recruited by IBDGC GRC's at University of Montreal, University of Pittsburgh, University of Toronto and Yale University. These DNA's were purified from individuals with confirmed diagnoses of CD (n=14), UC (n=19), indeterminate colitis (n=3) and from 7 controls.4 Additional HC's were from 152 AA's, without known asthma, common rheumatologic diseases or UC, recruited by the Baltimore Asthma Severity Study. 22
Phenotyping
Phenotype features for the primary dataset was determined using a previously validated protocol, largely in accordance with the Montreal classification.19, 23 Age at diagnosis was by chart review or if necessary, patient questionnaire. Disease location (maximal extent) was classified as L1 (ileum involvement only), L2 (colonic involvement only), or L3 (ileocolonic). L4 (esophageal, gastric, duodenal or jejunal) involvement was determined independently of L1-L3. Disease behavior (B1-B3) was per the Montreal classification.24 History of surgery was confirmed bowel resection or diversion for treatment of CD or complications. Extra-intestinal disease was determined as previously reported.4
Genotyping
Ancestry Estimation
Ninety-six well DNA plates containing case and control samples on each plate, unique positioned water controls, 13 blinded duplicates and 5 blinded white controls were genotyped using 97 ancestry informative markers (AIM's) by the Kittles laboratory as described.25, 26
Candidate SNPs
Samples from cases and HC's were plated together, and laboratory personnel were blinded to disease status. Seventeen SNPs were genotyped using Applied Biosystem's Taqman 5'-exonuclease assays, with alleles determined on an Applied Biosystems 7900HT Fast Real-Time PCR System analyzer. Three NOD2 SNPs (R702W, R708H, R790Q) were determined by direct sequencing of PCR products, and P268S was genotyped by PCR-RFLP.
Statistical analysis
Admixture determination
AIM genotypes from cases, HC's, duplicates and unknown samples were compared with the same AIM genotypes from DNA samples of WA's and Europeans. The data was used to determine fraction of European vs. WA ancestry using the program STRUCTURE 2.3 as described.26, 27 Each individual's estimated percentage of WA and European ancestry was coded as a continuous variable. Individuals with more than 85% European ancestry were excluded from analyses.5 Power to observe a significant difference of European ancestry between AA cases and matched controls was calculated by assuming European ancestry differences from 1% to 5%, and alpha level 0.05, using SAS 9.1 package program (SAS Institute, Cary, NC).
Phenotype and demographic comparisons with admixture
Within cases, associations between WA ancestry and CD disease behaviors, sites of involvement, extra-intestinal disease, IBD family history and age at diagnosis were assessed by T-test, ANOVA, and logistic regression.
Genotype association analyses
Analyses were performed by SAS 9.1/Genetics package program (SAS Institute, Cary, NC). Two sample t-tests were used for continuous variables. Nominal data were analyzed using the χ2 test. A two-tailed P <0.05 was considered significant. Logistic regression model was used to calculate genetic association after adjusting calculated WA ancestry. Population attributable risk (PAR) was calculated as reported previously.28 For haplotype analysis, pairwise LD was calculated by PROC ALLELE using SAS 9.1. Association between each haplotype and risk of CD was estimated by regression substitution, a function implemented in the R program HaploStat.29 We applied Benjamini and Hochberg false discovery rate (FDR) to correct for multiple testing.30
Results
West African/European admixture analysis among MAAIS participants
We genotyped 227 confirmed CD cases and 201 HC's from the primary dataset. Two AIMs were dropped because genotyping rates less than 20%. For the remaining 95 AIM's, genotyping call rate was between 96.3% and 100%. Among the 13 blinded duplicate control paired samples, one DNA failed to genotype completely. For the remaining 12 pairs of 24 samples, genotyping call rate was 97%, and 99.6% of those genotypes called in both pairs were identical. For the five blinded white control subjects WA ancestry ranged from 2.1% to 5.0%. Among all MAAIS study subjects, calculated WA ancestry ranged from 28.6% to 97.5%, excluding one self-identified AA HC with 2.2% WA ancestry (i.e. 97.8% European ancestry). We had 29%, 56%, and 80% power to identify a significant difference in admixture of 2%, 3% and 4% based on a sample of 200 pairs of case and control.
Characteristics of the 227 MAAIS CD and 200 matched control patients with satisfactory AIM's genotyping and WA ancestry greater than 15% are presented in Table 1. CD Study subjects showed a non-significant trend for younger age at study entry and female gender. There was no significant difference in tobacco use. Mean age at CD diagnosis was 26.8 (Standard Deviation [SD] 12.9) years and IBD family history 15.9%. Similar to our initial report, colon-only disease was relatively frequent (30.8%).4 Disease behavior was inflammatory (B1) in 40.5%, stricturing (B2) in 29.5%, and penetrating (B3) in 22.9%, with average length of observation between diagnosis and phenotype evaluation 10.8 years (SD 8.6 years).
Table 1. Baseline and clinical characteristics of cases (Crohn's disease) and controls (HC) in primary dataset (MAAIS).
| Characteristics | CD (n=227) | HC (n=200) | P value* | |
|---|---|---|---|---|
| Age at enrollment (years) | 37.4 ± 14.3 | 39.8 ± 12.7 | 0.07 | |
| Age at diagnosis | 26.7 ± 12.9 | -- | ||
| Gender (Male %) | 34.2% | 42.6% | 0.08 | |
| Family history of IBD (%)§ | 15.9% | 0.0% | <0.001 | |
| Cigarette smoking status: | Never (%) | 136 (65%) | 91 (51.7%) | 0.05 |
| Former (%) | 15 (7.1%) | 20 (11.3%) | ||
| Current (%) | 58 (27.9%) | 65 (37%) | ||
| CD disease behavior | B1:Inflammatory | 92 (40.5%) | - | |
| B2: Stricture | 67 (29.5%) | - | ||
| B3: Penetrating (fistulizing) | 52 (22.9%) | - | ||
| Disease location | L1: Ileal | 49 (21.6%) | - | |
| L2: Colonic | 70 (30.8 %) | - | ||
| L3: ileocolonic | 103 (45.3%) | - | ||
| L4: isolated upper disease | 2 (0.9%) | - | ||
| Any Perianal Disease§ | 70 (30.7%) | - | ||
| Surgical Resection for CD§ | 104 (46.0%) | - | ||
| West African ancestry estimates (%) | 0.796 +/- 0.116 | 0.798 +/- 0.119 | 0.86 |
Unknowns were 8 for family history, 13 for perianal, and 4 for surgery
Admixture and CD disease and phenotypes
There were no differences in overall mean estimated WA ancestry: 79.6% (SD 11.6%) in cases and 79.8% (SD11.9%) in HC's (p=0.86). WA ancestry was not different by disease site: L1 80% (SD 12.4%), L2 80% (SD 10.1%), and L3 79% (SD 12.4%), p=0.13, and there was no difference in WA ancestry in ileal involvement (L1 and L3) vs. colon-only disease (L2) (p=0.55). WA ancestry did not differ by disease behavior: B1 81.2% (SD11.1%), B2 77.7% (SD12.6%) and B3 78.8% (SD 11.8%), p=0.17. There was a trend towards greater WA ancestry among persons with inflammatory disease (B1) relative to complicated disease (B2 or B3), WA ancestry 78.1% (SD 12.2) p=0.07. However, after excluding B1 with short length of disease (< 6 years), association evidence weakened, but most directly because of smaller sample size as WA ancestry percentage remained the same: WA ancestry B1≥6 years of disease 81.1% (SD 11.4) vs. B2 or B3 78.1% (SD 12.2), p=0.11. Mean WA ancestry did not differ by perianal disease (present 79.9%, absent 79.7%, p=0.7), by extra-intestinal disease (present 80.8%, absent 79%; p=0.3), history of CD intestinal surgery (80% surgery, 79.3% no surgery; p=0.7), IBD family history (present 79%; absent 79.4%; p=0.9), or younger age at diagnosis (< age 40 at diagnosis 79.4%, ≥ age 40 at diagnosis 80.9%; p=0.5). Plots of cumulative proportion of WA ancestry for CD vs. HC's, B1 vs. B2 or B3 behaviors, and L1 or L3 vs. L2 sites are shown in Figure 1.
Figure 1.
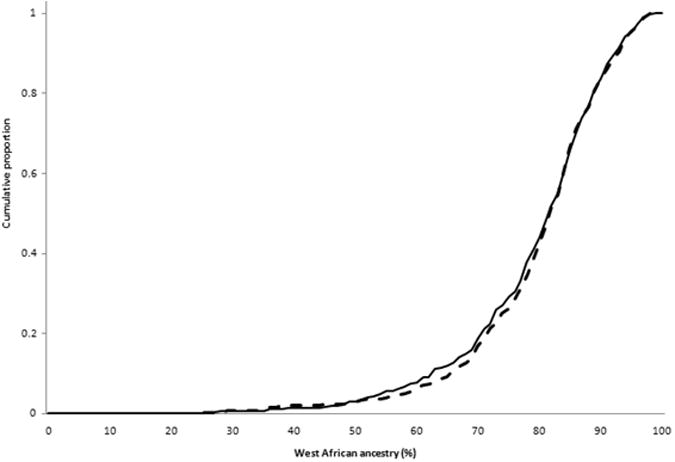
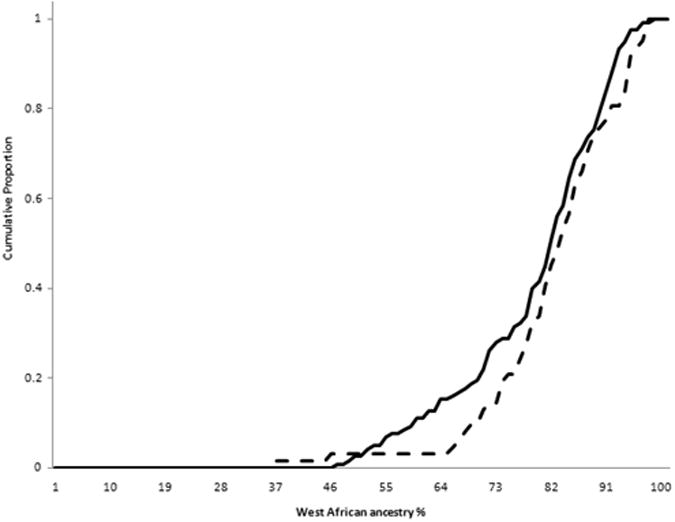
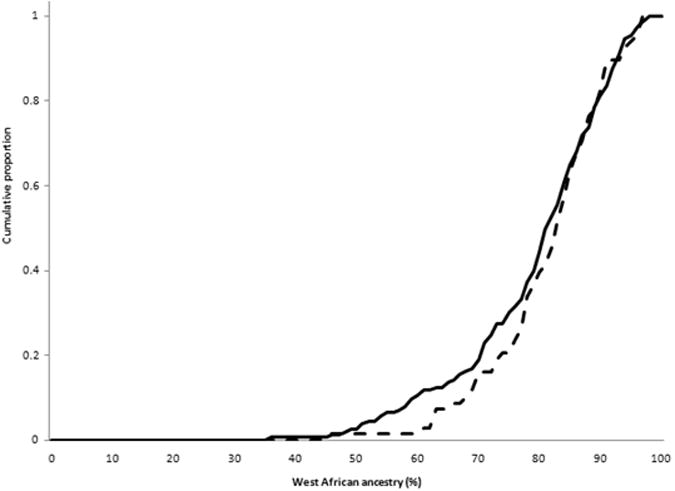
Cumulative proportion of persons with increment of West African ancestry for (a) CD (solid) vs. controls (dashed); (b)complicated (B2 or B3) (solid) vs. inflammatory behavior (dashed); and (c) ileal (L1 or L3) (solid) vs. colon-only (L2) disease (dashed) sites. Linear regression analyses treating WA as a continuous trait observed a borderline difference in complicated vs. inflammatory behavior (p=0.07), and no differences in CD vs. controls (p=0.57) or ileal vs. colon-only sites (p=0.55).
West African/European ancestry among non-MAAIS study subjects (secondary dataset)
Mean WA ancestry was 78 to 80% for each subset of study subjects with the exception of the 27 IBD (10 CD) African Canadian cases and controls (n=4) from the Toronto and Montreal GRCs (mean WA ancestry 90%, consistent with the major origin of African Canadians from the Caribbean), and therefore these subjects were excluded for further genotyping. One study subject from University of Chicago, Medical College of Wisconsin and BASS recruitments each had more than 85% European ancestry and thus excluded from further analysis. Of the remaining study subjects, mean WA ancestry was 77.9% for the cases and 79.2% for controls (p=0.41). SNP genotyping quality using DNA's from ten cases was poor and were excluded from further analysis. This left a total of 127 CD cases and 154 HC's in the secondary cohort for further analysis (Supplementary Table 1). For the combined primary and secondary datasets of 354 AA CD cases and 354 controls, mean EA remained highly similar (20.9% and 20.4%, p=0.58 respectively). We had 47%, 80% and 96% power to identify a significant difference in admixture of 2%, 3% and 4%, respectively, for this sample size.
Genotypic association studies
Genotype call rates were 97% or greater in both datasets, with the exception of the NOD2 SNPs genotyped by direct sequencing (95% call rate). Among HC's, Hardy-Weinberg Equilibrium (HWE) was ≥ 0.07 for all SNPs except NOD2 R790Q (HWE 0.04) (Supplementary Table 2). Since there was no significant heterogeneity for any genotyped SNPs between primary and secondary datasets (Supplementary Table 3), to maximize power we analyzed genetic association for both datasets combined. (However, because we recruited cases and controls uniformly in the primary data set, we made their genotyping results available [Supplementary Table 4]).
NOD2
In the combined dataset (Table 2), presence of any NOD2 mutation (all simple heterozygotes) was significantly associated with CD (p= 0.007; FDR adjusted p= 0.02). NOD2 carriage rate was 7.1% CD, 2.2% HC's with OR 3.28 (95%CI: 1.39 – 7.74). For the individual NOD2 risk mutations, only Leu1007fsinsC was significantly associated with CD (OR 8.08, 95%CI: 1.01-64.8, p=0.02; adjusted p= 0.04). Pro268Ser (“SNP5”), a common variant from European admixture, was significantly more frequent among cases (7.4% CD, 4.4% HCs, p=0.02). Using this marker to adjust for local (NOD2 region) admixture in AA's, we still observed significant risk for the three NOD2 mutations (NOD2 carrier OR after adjusting SNP5 3.15, 95%CI: 1.16 – 8.56, p=0.01). We also observed that the three NOD2 mutations were more common among individuals with overall lower WA ancestry (76% WA in NOD2 carriers vs. 79.9% WA in NOD2 non-carriers, p=0.07), although not meeting criteria of statistical significance.
Table 2. Association of SNPs for established CD genes among 354 CD cases and 354 controls (combined cohort dataset).
| Candidate Gene | SNP no. | 0 copy of variant allele | 1 copy of variant allele | 2 copies of variant allele | Carrier | MAF case/control | Minor Allelic test | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||
| Case/control OR (ref) | Case/control | OR (95%CI) p value | Case/control | OR (95%CI) P value | OR (95%CI) P value | OR (95%CI) P value | FDR adjusted p value | |||
| NOD2 C_insertion | rs2066847 L1007fs-insC (SNP13) | 343/350 (ref) | 8/1 | 8.16 (1.02– 65.6 ) 0.05 | 0/0 | -- | 8.16 (1.02 – 65.6 ) 0.05 | 1.14% / 0.14% | 8.08 (1.01-64.8) 0.02 | 0.04 |
|
| ||||||||||
| NOD2 (G->C, variant C) | rs2066845 G908R (SNP12) | 347/347 (ref) | 4/2 | 2.00 (0.36-10.9) 0.42 | 0/0 | -- | 2.00 (0.36-10.9) 0.42 | 0.57% / 0.29% | 1.99 (0.36-10.9) 0.42 | 0.62 |
|
| ||||||||||
| NOD2 (C->T, variant T) | rs2066844 R702W (SNP8) | 334/327 (ref) | 11/4 | 2.69 (0.85-8.54) 0.09 | 0/0 | -- | 2.69 (0.85-8.54) 0.09 | 1.59% / 0.60% | 2.66 (0.84-8.41) 0.08 | 0.15 |
|
| ||||||||||
| NOD2 carrier (any 1007fs, G908R, R702W) | 322/321 (ref) | 23/7 | 3.28 (1.39-7.74) 0.007 | 0/0 | -- | 3.28 (1.39-7.74) 0.007 | carrier rate 6.93% / 2.15% | 0.02 | ||
|
| ||||||||||
| NOD2(C->T, variant T) | rs2066842 P268S (SNP5) | 304/319 (ref) | 40/31 | 1.35 (0.83-2.22) 0.22 | 6/0 | 8.53 (1.22- ∼) 0.03 | 1.56 (0.94-2.61) 0.09 | 7.43% / 4.43% | 1.73 (1.10-2.74) 0.02 | 0.04 |
|
| ||||||||||
| NOD2 (G->A, variant A) | rs35285618 R708H | 333/322 (ref) | 12/9 | 1.29 (0.54-3.10) 0.57 | 0/0 | -- | 1.29 (0.54-3.10) 0.57 | 1.74% / 1.36% | 1.28 (0.54-3.07) 0.57 | 0.72 |
|
| ||||||||||
| NOD2(G->A, variant A) | rs5743279 R790Q | 318/306 (ref) | 26/23 | 1.09 (0.61-1.95) 0.78 | 1/2 | 0.48 (0.01-9.30) 0.97 | 1.04 (0.59-1.83) 0.89 | 4.06% / 4.08% | 0.99 (0.58-1.71) 0.98 | 0.98 |
|
| ||||||||||
| NOD2 (C->G, variant G) | rs5743278 A725G | 318/303 (ref) | 26/27 | 0.92 (0.50-1.68) 0.87 | 1/1 | 0.95 (0.01-75.0) 1.00 | 0.92 (0.53-1.60) 0.76 | 4.06% / 4.38% | 0.92 (0.54-1.57) 0.77 | 0.82 |
|
| ||||||||||
| ATG16L1 (A->G, variant G) | rs2241880 Thr300Ala | 141/179 (ref) | 164/140 | 1.49 (1.09-2.04) 0.01 | 44/33 | 1.86 (1.11-3.11) 0.02 | 1.55 (1.15-2.10) 0.004 | 36.1% / 29.3% | 1.41 (1.12-1.76) 0.003 | 0.02 |
|
| ||||||||||
| IBD5_OCTN1 SLC22A4 (C->T, variant T) | rs1050152 Leu503Phe | 285/297 (ref) | 58/51 | 1.19 (0.79-1.79) 0.42 | 8/1 | 8.34 (1.04-67.1) 0.04 | 1.32 (0.89-1.97) 0.17 | 10.5% / 7.59% | 1.43 (1.00-2.07) 0.05 | 0.09 |
|
| ||||||||||
| IBD5_OCTN2 SLC22A5 (G->C, variant C) | rs2631367 | 124/134 (ref) | 163/173 | 1.02 (0.74-1.41) 0.91 | 63/36 | 1.89 (1.17-3.05) 0.009 | 1.17 (0.86-1.59) 0.32 | 41.3% / 35.7% | 1.26 (1.02-1.57) 0.03 | 0.06 |
|
| ||||||||||
| IBD5_OCTN2 IGR2230 (G->A, variant A) | rs17622208 | 216/224 (ref) | 108/113 | 1.00 (0.71-1.39) 1.00 | 22/8 | 2.85 (1.19-7.56) 0.01 | 1.11 (0.81-1.54) 0.54 | 21.9% / 18.7% | 1.22 (0.94-1.59) 0.13 | 0.20 |
|
| ||||||||||
| IBD5_IGR2198 (C->G, variant G) | rs11739135 | 262/266 (ref) | 67/71 | 0.96 (0.65-1.42) 0.89 | 15/3 | 5.01 (1.41-27.6) 0.008 | 1.13 (0.77-1.64) 0.58 | 14.1% / 11.3% | 1.28 (0.93-1.77) 0.12 | 0.20 |
|
| ||||||||||
| IBD5_IGR2096 (G->T, variant T) | rs12521868 | 260/263 (ref) | 72/76 | 0.96 (0.65-1.40) 0.89 | 15/2 | 7.56 (1.73-68.8) 0.003 | 1.13 (0.78-1.63) 0.56 | 14.7% / 11.7% | 1.27 (0.95-1.77) 0.10 | 0.18 |
|
| ||||||||||
| IL23R (T->G, variant G) | Rs7517847 | 227/212 (ref) | 100/126 | 0.74 (0.54-1.02) 0.07 | 21/12 | 1.63 (0.79-3.40) 0.19 | 0.82 (0.60-1.11) 0.20 | 20.4% / 21.4% | 0.94 (0.73-1.22) 0.64 | 0.78 |
|
| ||||||||||
| IL23R (A->G, variant G) | rs2201841 | 231/263 (ref) | 101/81 | 1.42 (1.00-2.03) 0.05 | 12/8 | 1.71 (0.63-4.90) 0.35 | 1.45 (1.03-2.04) 0.03 | 18.2% / 13.8% | 1.39 (1.04-1.95) 0.03 | 0.06 |
|
| ||||||||||
| IL23R (G->A, variant A) | rs11209026 Arg381Gln | 333/333 (ref) | 14/18 | 0.78 (0.35-1.69) 0.61 | 0/0 | -- | 0.78 (0.35-1.69) 0.61 | 2.02% / 2.56% | 0.78 (0.39-1.59) 0.49 | 0.69 |
|
| ||||||||||
| IL23R (T->C, variant C) | rs1495965 | 98/103 (ref) | 175/183 | 1.01 (0.71-1.42) 0.98 | 72/65 | 1.16 (0.75-1.80) 0.49 | 1.05 (0.75-1.45) 0.78 | 46.2% / 44.6% | 1.07 (0.87-1.32) 0.54 | 0.78 |
|
| ||||||||||
| IL23R (C->A, variant A) | rs10889677 | 234/254 (ref) | 102/89 | 1.24 (0.88-1.77) 0.23 | 9/7 | 1.40 (0.45-4.48) 0.69 | 1.26 (0.91-1.74) 0.17 | 17.4% / 14.7% | 1.22 (0.92-1.63) 0.17 | 0.30 |
|
| ||||||||||
| IRGM (G->A, variant A) | Rs4958847 | 102/96 (ref) | 163/167 | 0.92 (0.65-1.31) 0.64 | 83/87 | 0.90 (0.60-1.35) 0.61 | 0.91 (0.66-1.27) 0.58 | 47.3% / 48.7% | 0.94 (0.76-1.16) 0.59 | 0.76 |
|
| ||||||||||
| IRGM (T->C, variant C) | rs13361189 | 112/110 (ref) | 153/165 | 0.97 (0.69-1.36) 0.86 | 73/75 | 0.96 (0.63-1.45) 0.83 | 0.97 (0.70-1.33) 0.83 | 44.2% / 45.0% | 0.98 (0.79-1.20) 0.82 | 0.86 |
|
| ||||||||||
| IRGM (C->T, variant T) | rs10065172 | 114/109 (ref) | 164/166 | 0.95 (0.67-1.33) 0.74 | 71/75 | 0.91 (0.60-1.37) 0.64 | 0.93 (0.68-1.28) 0.67 | 43.8% / 45.1% | 0.95 (0.77-1.17) 0.62 | 0.74 |
We sequenced the entire NOD2 coding exons and intron-exon boundaries in 31 AA CD cases and observed only three additional non-synonymous SNPs (in exon 4) that are uncommon or non-existent in Europeans (Supplementary Table 2): Arg708His, Ala725Gly and Arg790Gln. Since these 3 SNPs were proximal to Arg702Trp, and Arg702Trp genotyped poorly by TaqMan, we genotyped exon 4 SNPs by direct sequencing. However, we did not find association evidence for any of these three apparently African ancestral SNPs (Table 2).
ATG16L1
In AA's, unlike Europeans, the variant G allele that encodes the300Ala CD risk residue, is the less common allele. Its frequency was 36.1% in CD compared with 29.3% in controls (p=0.003, adjusted p= 0.02). Analysis of genotypic relative risks showed that CD risk was significantly increased in both Thr/Ala heterozygotes (OR 1.49, 95%CI: 1.09-2.04, p=0.01) as well as Ala/Ala homozygotes (OR 1.86, 95%CI: 1.11-3.11, p=0.02) (Table 2).
IBD5
We genotyped five SNPs, which included functional variants Leu503Phe in SLC22A4 and g-207c promoter variant in SLC22A5, and three marker SNPs more centromeric on the IBD5 risk haplotype.31 Allelic association was found for Leu503Phe 10.5% CD, 7.6% HC, p=0.05 and g-207c 41.3% CD, 35.7% HC, p=0.03, with only g-207c marginally significant after correction for multiple testing (adjusted p=0.06; Table 2). Interestingly, genotypic analysis showed significant CD risk for all IBD5 SNPs tested for homozygote but not heterozygote genotypes (Table 2). All five IBD5 SNPs remained significantly associated with CD for recessive mode of inheritance after FDR correction for multiple testing: SLC22A4 L503F (adjusted p=0.05), SLC22A5 g-207c (adjusted p = 0.02), IGR2230 (adjusted p=0.03), IGR2198 (adjusted p=0.02), IGR2096 (adjusted p=0.02).
IL23R
Analysis of five genotyped SNPs in IL23R suggested association only for variant alleles in intron7 SNP rs2201841 OR 1.39 (95%CI: 1.04-1.95, p=0.03, adjusted p=0.06). (Table 2). Only 4.0% of cases and 5.1% of HC's carried the 381Gln protective variant (p=0.61).
IRGM
Analysis of variant allele frequency and genotypic relative risks for three IRGM SNPs, that included rs13361189 (in tight LD with the 20kb CNV) and rs10065172 (miRNA binding variant), showed no associations with CD (Table 2).
Haplotype and Diplotype Analysis
Strong LD was observed between IBD5 variants but less between IL23R variants (Pairwise D′ values >0.65, r2 value > 0.1; D′ values > 0.02, r2 value > 0.004, respectively). When the most common IBD5 haplotype, consisting of the five major alleles CGGCG is considered as reference, no association was observed for the second most common haplotype, consisting of four major alleles and the SLC22A5 – g207c minor allele (CCGCG; 20.6% CD, 19.2% HC, p=0.24, Table 3). Similar to that observed in whites28, significant risk was observed for the all-minor-allele haplotype TCAGT relative to the reference haplotype (9.8% CD, 7.1% HC, p=0.05). Diplotype association analysis found the only significant association was for individuals carrying homozygote variants in all five SNPs (TCAGT/TCAGT, OR 8.14, P=0.03) relative to the wild type diplotype (CGGCG/CGGCG), further supporting the recessive risk of IBD5 (Supplementary Table 5). For 5-marker haplotype of IL23R, compared to the most common association haplotype TCGAT, there were two haplotypes suggesting association with CD (CAGGT P = 0.05, CCGAG P = 0.06, Table 3).
Table 3. Haplotype analysis of IBD5 and IL23R.
| Candidate gene | Haplotype | Cases | Controls | OR (95% CI) | P |
|---|---|---|---|---|---|
| IBD5 (rs1050152 C>T, rs2631367 G>C, rs17622208 G>A, rs11739135 C>G, rs12521868 G>T) | Global P=0.43 | ||||
| CGGCG | 55.9% | 61.1% | ref | ||
| -C--- | 20.6% | 19.2% | 1.18 (0.89-1.57) | 0.24 | |
| TCAGT | 9.8% | 7.1% | 1.47 (0.99-2.18) | 0.05 | |
| -CA-- | 8.0% | 7.1% | 1.26 (0.83-1.90) | 0.28 | |
| --AGT | 1.6% | 2.4% | 0.75 (0.33-1.71) | 0.49 | |
| -CAGT | 2.0% | 1.9% | 1.27 (0.53-3.04) | 0.60 | |
| IL23R (rs1495965 T>C, rs10889677 C>A, rs11209026 G>A, rs2201841 A>G, rs7517847 T>G) | Global p=0.03 | ||||
| TCGAT | 34.5% | 35.2% | ref | ||
| C---- | 27.8% | 31.3% | 0.88 (0.66-1.16) | 0.37 | |
| ----G | 13.9% | 15.6% | 0.85 (0.60-1.22) | 0.39 | |
| CA-G- | 14.8% | 9.8% | 1.45 (1.00-2.09) | 0.05 | |
| -A-GG | 1.2% | 2.1% | 0.67 (0.25-1.82) | 0.43 | |
| C---G | 3.3% | 1.6% | 2.59 (0.96-6.98) | 0.06 | |
| --A-G | 1.3% | 0.6% | 1.87 (0.44-8.02) | 0.40 |
Discussion
NOD2, SLC22A4, SLC22A5, IL23R, ATG16L1 and IRGM are among the most highly investigated CD risk genes in white populations. This study, the first to evaluate in AA's SLC22A4 and SLC22A5, IL23R, ATG16L1, and IRGM and, with more than double the study population and adjustment for ancestral admixture, to re-evaluate NOD2, found significant association evidence for established risk alleles for all loci but IRGM. All studies in white populations have found NOD2 mutations more frequent among CD cases than controls, with a meta-analysis of 29 studies finding overall NOD2 carrier risk of 3.2.32 However, NOD2 mutations are rare or absent among non-white populations, the three NOD2 mutations assumed to have arisen in European ancestral populations or by admixture.33, 34 Our expanded study showed robust association evidence for carriage of any NOD2 mutation (OR 3.28, p=0.007; FDR adjusted p=0.02), with risk similar to whites,32 and significant association remained even after adjustment for individual admixture. However, because of lower frequency of mutations in AA's, the estimated AA PAR was only 4.6 % vs. 22 % in whites.32 We also screened for and genotyped non-synonymous SNPs common in AA's, to determine if any African ancestral NOD2 mutations contribute to CD risk, but found no association. Our study showed that in the AA population as in whites, common NOD2 risk only arises from the 3 major European ancestral loss of function mutations and for disease prediction purposes only these need to be genotyped. In white populations, NOD2 mutation homozygotes result in approximately 20-fold CD risk and are especially predictive of ileal involvement and complicated disease course.35 Indeed, NOD2 is now widely commercially available for these uses. However, we did not observe any NOD2 homozygotes in our AA CD study population (as opposed to 12% in a study of white CD patients from Baltimore, Chicago and Pittsburgh IBD centers),35 and with only 0.01% of the AA population predicted to be NOD2 homozygotes (as opposed to 0.5% homozygote rate found among combined white controls),36 NOD2's clinical utility in AA's should be specifically assessed prior to its routine use in this population.
Prior to GWAS, the IBD5 risk haplotype was the only other established genetic risk factor, with functional variants in SLC22A4 and SLC22A5 as the leading candidate risk variants, with risk present for both heterozygote and homozygote carriers, although numerous studies (including a recent meta-analysis) observed a gene-dosage effect, as did the original IBD5 discovery.8, 37 We observed significant association only for homozygote carriers, maximal for Leu503Phe with point OR estimates of 8.4, far greater than meta-analysis point OR observed in whites. In contrast to studies in whites, frequency of heterozygotes among AA's for four IBD5 SNPs were actually lower among cases relative to controls, and for Leu503Phe heterozygotes had only a slight and non-significant increased presence relative to wild types (p=0.39).
The SLC22A5 –g207c functional variant, is frequently found in AA's on a haplotype isolated from the other IBD5 minor alleles variants including the SLC22A4 Leu507Phe functional variant, However, SLC22A5 showed no evidence that it alone could cause CD risk. Rather, CD risk was only present in haplotype and diplotype analyses when both SLC22A4 and SLC22A5 functional alleles were found together and with the other minor alleles in the extended, apparently European origin IBD5 haplotype compatible with the finding from Fisher SA et al.38 Similar to the three established NOD2 risk mutations, SLC22A4 Leu503Phe appears to originate completely from European admixture, not being found in the Yoruban genome. One unifying hypothesis to explain risk in AA's restricted to homozygotes vs. risk in whites only modestly increased in homozygotes relative to heterozygotes, is that within persons of EA, there might exist multiple albeit less frequent, additional SNPs besides Leu503Phe that similarly influence SLC22A4 epithelial transport activity, and thus some Leu/Leu wild types may be heterozygotes for less common SLC22A4 risk alleles, and some Leu/Phe heterozygotes may actually be compound homozygotes. A recent evolutionary analysis is consistent with this hypothesis.39 Thus, the true nature of IBD5 risk may be recessive for both white and AA populations. Interestingly, IBD5 risk for UC was found significant in meta-analysis only for recessive and not for dominant inheritance.37
IL23R was the first gene identified by European GWAS and proven as a significant risk for CD. We observed significant association in AA's with CD for intron7 IL23R SNP (rs2201841) (p=0.03), but no significant association was observed for other SNPs including the highly protective Arg381Gln variant – although power was limited given the 2.6% 381Gln allele frequency. In whites, rs2201841 has been shown as a risk factor independent of Arg381, and thus association evidence for rs2201841 may be important.28 Our haplotype analysis supported a role for IL23R in AA's showing nominal evidence for association for 2 of the 5 variant haplotypes with HC frequencies greater than 1% (Table 3). Given the number of SNPs and haplotypes evaluated, larger studies are required to substantiate IL23R as a risk for AA CD.
Although frequency of the ATG16L1 functional 300Ala variant is lower in AA's than observed in our population-based study in Canadian whites (29% AA HC vs. 52% white HC's), we observed similar and highly significant association (OR 1.41, p=0.003) with CD.28 Given that 300Ala is relatively frequent in Yorubans (27.5%), unlike NOD2 and SLC22A4 – in AA's the origin of the ATG16L1 functional variant risk is likely contributed by both ancestral populations.
IRGM, which encodes a GTP-binding protein that induces autophagy and plays an important role against intracellular pathogens, was recognized as CD risk gene by the Wellcome Trust GWAS.16 A 20 kb promoter deletion in whites has been associated with reduced IRGM expression in several cell types.16 We genotyped this 20 kb CNV in 96 AA's and observed that similar to whites, the rs13361189 “c” allele was in perfect LD with the 20kb deletion (data not shown), these minor alleles are nearly 20-fold more frequent in AA's relative to whites. There is some controversy with regard to the influence of the 20 kb CNV: in a well-powered Japanese study the CNV deletion polymorphism, although as in AA's also found at relatively high frequency, also observed nearly identical presence among CD cases and controls (37% and 38%, respectively).17 Hence, the background ancestry appears to have a controlling influence on the 20kb CNV risk. However, our power to exclude risk was more limited (51%-78% [Supplementary Table 6] vs. 90% in the Japanese study).17 Similarly, neither our study nor the Japanese study observed risk for rs10065172, reported to influence miRNA regulation of IRGM expression, and likewise found at high frequency in both Japanese and AA's. One possible explanation for this observed lack of IRGM association with CD in Japanese and AA is that, as proposed by Prescott et al., the true causal variant is neither the CNV nor rs10065172, but arose on a haplotype in Europeans after the split with African and Asian populations.17
AAs are a recently admixed population with an average of ∼80% of African ancestry but significant variation exists at individual level.40, 41 Studies of disease or phenotype patterns, including obesity, skin pigmentation, psychiatric disorders, and asthma in AAs have shown that African ancestry is different between cases and ethnicity matched controls from less than 1% to 4%.5, 42-44 This study is the first large population-based study to investigate the association between African ancestry and CD risk in AAs. Nonetheless, admixture was essentially identical. This was despite the fact that NOD2 and IBD5 loci– both with greater than 3-fold risks in our study - clearly arise only from European admixture. One explanation may be the relatively low protection from CD – perhaps counterbalancing the NOD2 and IBD5 risks – of IL23R 381Gln (present in only 5% of healthy AA's vs. 14% of healthy whites). Other considerations include the possibility that there may exist high risk, African ancestry variants yet to be discovered, and that our study had suboptimal power to identify modest admixture differences below 3%. Admixture analysis did not identify any significant associations within CD sub-phenotypes and demographic features. Power was even more limited for these analyses, however, and it will be of great interest to evaluate potential admixture-phenotype expression features in a larger study population, especially for features where a trend was observed, notably inflammatory vs. complicated disease behavior.
We previously provided the first evidence that family history of CD in AA's was significant,4 suggesting that as in whites, AA CD would likely have genetic origins. In the present study, we have provided strong evidence to support our previous modest evidence that established NOD2 mutations contribute to CD risk in AA's, and provide evidence that established functional polymorphisms in SLC22A4 and ATG16L1, and possibly an intronic polymorphism in IL23R also contribute to AA CD risk. Unexpected findings of our study include the nature of IBD5 risk as most likely recessive, the ability to isolate – likely from the greater diversity of the African genome – the –g207c SLC22A5 risk allele and by haplotype analysis provide evidence against it as a CD risk factor alone, and that the degree of European admixture in CD and HC's would be so similar. This study represents an initial characterization of CD genetic risk in AA's. This study shows that the established CD risk genes in whites, NOD2, IBD5, and ATG16L1, also contribute to the risk of CD in AAs without influence by their African ancestry. This finding could become increasingly more critical with commercial laboratories now incorporating CD gene testing to predict disease risk. Future studies evaluating additional established CD genes, with larger study populations and more dense genotyping, as well as AA GWAS's to identify unique African ancestral risk as well as protective variants are greatly anticipated to further elucidate the nature of CD genetics in this under-investigated population.
Supplementary Material
(Supplementary Table 1) Study Subjects
(Supplementary Table 2) Characteristics of 21 SNPs genotyped (combined dataset)
(Supplementary Table 3) Heterogeneity test between primary and secondary replicate dataset
(Supplementary Table 4) Association of SNPs among 227 CD and 200 controls in primary dataset
(Supplementary Table 5) Diplotype association test for IBD5
(Supplementary Table 6) Power calculation: Sample size 354 cases and 1:1 control; CD Population risk: 50/100,000; 2-sided p value 0.05;
Acknowledgments
Grant Support: This study is supported primarily by NIH grant DK062431 (SRB) with a supplement to assist with patient recruitment from the NCMHD. Additional funding was from the Harvey M. and Lynn P. Meyerhoff Inflammatory Bowel Disease Center, the Morton Hyatt Family, the Atran Foundation, and the Buford and Linda Lewis family. MHW also received support from St Agnes Hospital and Geoffrey Nguyen was supported by an AGA Research Scholar Award Additional IBDGC support was from DK062429 and DK062422 (JHC), DK062420 (RHD), DK062432 (JDR), DK062423 (MSS), and DK062413 (KT).
We thank the patients and the controls for participating in this study. We thank Patricia Ushry and Vivian Abadom, Johns Hopkins University; Dolly Walkup, RN and Mikki Sandridge, University of North Carolina; Susan Parrott, RN, University of Pennsylvania; Dr. Michelle Mendoza, Washington Hospital Center; Amy Gunnett, RN and Gretchen Simpkins, RN, University of Florida; and Melvin Hall, Howard University Hospital for coordinating recruitment at each site. We thank Dr. Michael S. Gold and Dr. Averell Sherker (now at NIDDK) for organizing recruitment at Washington Hospital Center, Washington, DC. We thank Theodore M. Bayless, MD, John Kwon, MD, Sharon Dudley-Brown, PhD, LNP, Mark Lazarev, MD, Florin Selaru, MD, and Tina Ha, MD from the Meyerhoff IBD Center for their patient referrals. We thank Dr. Gerald Canakis, Atlantic General Hospital, Berlin, MD; Dr. Ray Cross, University of Maryland, Baltimore, MD; and Dr. Carlton Greene and Dr. Edward Wolf, Baltimore, MD for patient referrals. We thank the Maryland, D.C. and Northern Virginia Chapter of the Crohn's Colitis Foundation of America for advertisement of our study. The database was developed with assistance by Phil Schumm (University of Chicago).
Abbreviations used in this paper
- AA
African American
- WA
West African
- EA
European ancestry
- CD
Crohn's disease
- UC
ulcerative colitis
- HC
healthy controls
- MAAIS
Multicenter African American IBD Study
- IBDGC
IBD Genetics Consortium
- LD
linkage disequilibrium
- NOD2
Nucleotide-binding oligomerization domain containing-2
- SLC22A4
Solute carrier family 22, member 4
- SLC22A5
Solute carrier family 22, member 5
- GWAS
genome wide association study
- IL23R
interleukin (IL) 23 receptor
- SNP
single nucleotide polymorphism
- ATG16L1
autophagy 16 like-1
- IRGM
immunity-related GTPase family, M
- CNV
copy number variation
- IBD
inflammatory bowel disease
- GRC
Genetics Research Center
- AIM
admixture informative marker
- PAR
population attributable risk
- HWE
Hardy-Weinberg Equilibrium
Footnotes
Disclosures for all authors: No relevant potential conflicts
References
- 1.Calkins BM, Lilienfeld AM, Garland CF, Mendeloff AI. Trends in incidence rates of ulcerative colitis and Crohn's disease. DigDisSci. 1984;29:913–920. doi: 10.1007/BF01312480. [DOI] [PubMed] [Google Scholar]
- 2.Kurata JH, Kantor-Fish S, Frankl H, Godby P, Vadheim CM. Crohn's disease among ethnic groups in a large health maintenance organization. Gastroenterology. 1992;102:1940–1948. doi: 10.1016/0016-5085(92)90317-r. [DOI] [PubMed] [Google Scholar]
- 3.Abramson O, Durant M, Mow W, Finley A, Kodali P, Wong A, Tavares V, McCroskey E, Liu L, Lewis JD, Allison JE, Flowers N, Hutfless S, Velayos FS, Perry GS, Cannon R, Herrinton LJ. Incidence, prevalence, and time trends of pediatric inflammatory bowel disease in Northern California, 1996 to 2006. J Pediatr. 2006;157:233–239. doi: 10.1016/j.jpeds.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 4.Nguyen GC, Torres EA, Regueiro M, Bromfield G, Bitton A, Stempak J, Dassopoulos T, Schumm P, Gregory FJ, Griffiths AM, Hanauer SB, Hanson J, Harris ML, Kane SV, Orkwis HK, Lahaie R, Oliva-Hemker M, Pare P, Wild GE, Rioux JD, Yang H, Duerr RH, Cho JH, Steinhart AH, Brant SR, Silverberg MS. Inflammatory bowel disease characteristics among African Americans, Hispanics, and non-Hispanic Whites: characterization of a large North American cohort. Am J Gastroenterol. 2006;101:1012–1023. doi: 10.1111/j.1572-0241.2006.00504.x. [DOI] [PubMed] [Google Scholar]
- 5.Cheng CY, Kao WH, Patterson N, Tandon A, Haiman CA, Harris TB, Xing C, John EM, Ambrosone CB, Brancati FL, Coresh J, Press MF, Parekh RS, Klag MJ, Meoni LA, Hsueh WC, Fejerman L, Pawlikowska L, Freedman ML, Jandorf LH, Bandera EV, Ciupak GL, Nalls MA, Akylbekova EL, Orwoll ES, Leak TS, Miljkovic I, Li R, Ursin G, Bernstein L, Ardlie K, Taylor HA, Boerwinckle E, Zmuda JM, Henderson BE, Wilson JG, Reich D. Admixture mapping of 15,280 African Americans identifies obesity susceptibility loci on chromosomes 5 and X. PLoS Genet. 2009;5:e1000490. doi: 10.1371/journal.pgen.1000490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kugathasan S, Loizides A, Babusukumar U, McGuire E, Wang T, Hooper P, Nebel J, Kofman G, Noel R, Broeckel U, Tolia V. Comparative phenotypic and CARD15 mutational analysis among African American, Hispanic, and White children with Crohn's disease. InflammBowelDis. 2005;11:631–638. doi: 10.1097/01.mib.0000171279.05471.21. [DOI] [PubMed] [Google Scholar]
- 7.Dassopoulos T, Nguyen GC, Talor MV, Datta LW, Isaacs KL, Lewis JD, Gold MS, Valentine JF, Smoot DT, Harris ML, Oliva-Hemker M, Bayless TM, Burek CL, Brant SR. NOD2 Mutations and Anti-Saccharomyces cerevisiae Antibodies Are Risk Factors for Crohn's Disease in African Americans. Am J Gastroenterol. 2009;105:378–386. doi: 10.1038/ajg.2009.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rioux JD, Daly MJ, Silverberg MS, Lindblad K, Steinhart H, Cohen Z, Delmonte T, Kocher K, Miller K, Guschwan S, Kulbokas EJ, O'Leary S, Winchester E, Dewar K, Green T, Stone V, Chow C, Cohen A, Langelier D, Lapointe G, Gaudet D, Faith J, Branco N, Bull SB, McLeod RS, Griffiths AM, Bitton A, Greenberg GR, Lander ES, Siminovitch KA, Hudson TJ. Genetic variation in the 5q31 cytokine gene cluster confers susceptibility to Crohn disease. Nat Genet. 2001;29:223–228. doi: 10.1038/ng1001-223. [DOI] [PubMed] [Google Scholar]
- 9.Peltekova VD, Wintle RF, Rubin LA, Amos CI, Huang Q, Gu X, Newman B, Van Oene M, Cescon D, Greenberg G, Griffiths AM, George-Hyslop PH, Siminovitch KA. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat Genet. 2004;36:471–475. doi: 10.1038/ng1339. [DOI] [PubMed] [Google Scholar]
- 10.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, Dassopoulos T, Bitton A, Yang H, Targan S, Datta LW, Kistner EO, Schumm LP, Lee AT, Gregersen PK, Barmada MM, Rotter JI, Nicolae DL, Cho JH. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, Gunther S, Prescott NJ, Onnie CM, Hasler R, Sipos B, Folsch UR, Lengauer T, Platzer M, Mathew CG, Krawczak M, Schreiber S. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 12.Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, Shugart YY, Griffiths AM, Targan SR, Ippoliti AF, Bernard EJ, Mei L, Nicolae DL, Regueiro M, Schumm LP, Steinhart AH, Rotter JI, Duerr RH, Cho JH, Daly MJ, Brant SR. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, Stone CD, Brunt EM, Xavier RJ, Sleckman BP, Li E, Mizushima N, Stappenbeck TS, Virgin HWt. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 16.McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P, Zody MC, Hall JL, Brant SR, Cho JH, Duerr RH, Silverberg MS, Taylor KD, Rioux JD, Altshuler D, Daly MJ, Xavier RJ. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nat Genet. 2008;40:1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prescott NJ, Dominy KM, Kubo M, Lewis CM, Fisher SA, Redon R, Huang N, Stranger BE, Blaszczyk K, Hudspith B, Parkes G, Hosono N, Yamazaki K, Onnie CM, Forbes A, Dermitzakis ET, Nakamura Y, Mansfield JC, Sanderson J, Hurles ME, Roberts RG, Mathew CG. Independent and population-specific association of risk variants at the IRGM locus with Crohn's disease. Hum Mol Genet. 2010;19:1828–1839. doi: 10.1093/hmg/ddq041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brest P, Lapaquette P, Souidi M, Lebrigand K, Cesaro A, Vouret-Craviari V, Mari B, Barbry P, Mosnier JF, Hebuterne X, Harel-Bellan A, Mograbi B, Darfeuille-Michaud A, Hofman P. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn's disease. Nat Genet. 43:242–245. doi: 10.1038/ng.762. [DOI] [PubMed] [Google Scholar]
- 19.Dassopoulos T, Nguyen GC, Bitton A, Bromfield GP, Schumm LP, Wu Y, Elkadri A, Regueiro M, Siemanowski B, Torres EA, Gregory FJ, Kane SV, Harrell LE, Franchimont D, Achkar JP, Griffiths A, Brant SR, Rioux JD, Taylor KD, Duerr RH, Silverberg MS, Cho JH, Steinhart AH. Assessment of reliability and validity of IBD phenotyping within the National Institutes of Diabetes and Digestive and Kidney Diseases (NIDDK) IBD Genetics Consortium (IBDGC) Inflammatory bowel diseases. 2007;13:975–983. doi: 10.1002/ibd.20144. [DOI] [PubMed] [Google Scholar]
- 20.Bonen DK, Ogura Y, Nicolae DL, Inohara N, Saab L, Tanabe T, Chen FF, Foster SJ, Duerr RH, Brant SR, Cho JH, Nunez G. Crohn's disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology. 2003;124:140–146. doi: 10.1053/gast.2003.50019. [DOI] [PubMed] [Google Scholar]
- 21.Kader HA, Berman WF, Al-Seraihy AS, Ware RE, Ulshen MH, Treem WR. Prevalence of factor V G1691A (Leiden), prothrombin G20210A, and methylene tetrahydrofolate reductase C677T thrombophilic mutations in children with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2002;35:629–635. doi: 10.1097/00005176-200211000-00008. [DOI] [PubMed] [Google Scholar]
- 22.Ford JG, Meyer IH, Sternfels P, Findley SE, McLean DE, Fagan JK, Richardson L. Patterns and predictors of asthma-related emergency department use in Harlem. Chest. 2001;120:1129–1135. doi: 10.1378/chest.120.4.1129. [DOI] [PubMed] [Google Scholar]
- 23.Satsangi J, Silverberg MS, Vermeire S, Colombel JF. The Montreal classification of inflammatory bowel disease: controversies, consensus, and implications. Gut. 2006;55:749–753. doi: 10.1136/gut.2005.082909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Silverberg M, Satsangi J, Ahmad T, Arnott ID, Bernstein CN, Brant SR, Caprilli R, Colombel JF, Gasche C, Geboes K, Jewell D, Karban A, Loftus EV, Jr, Pena S, Riddell RH, Sachar DB, Schreiber S, Steinhart AH, Targan SR, Vermeire S, Warren BF. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: Report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. CanJGastroenterol. 2005;19:5A–36A. doi: 10.1155/2005/269076. [DOI] [PubMed] [Google Scholar]
- 25.Tian C, Hinds DA, Shigeta R, Kittles R, Ballinger DG, Seldin MF. A genomewide single-nucleotide-polymorphism panel with high ancestry information for African American admixture mapping. Am J Hum Genet. 2006;79:640–649. doi: 10.1086/507954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giri VN, Egleston B, Ruth K, Uzzo RG, Chen DY, Buyyounouski M, Raysor S, Hooker S, Torres JB, Ramike T, Mastalski K, Kim TY, Kittles R. Race, genetic West African ancestry, and prostate cancer prediction by prostate-specific antigen in prospectively screened high-risk men. Cancer Prev Res (Phila) 2009;2:244–250. doi: 10.1158/1940-6207.CAPR-08-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okazaki T, Wang MH, Rawsthorne P, Sargent M, Datta LW, Shugart YY, Bernstein CN, Brant SR. Contributions of IBD5, IL23R, ATG16L1, and NOD2 to Crohn's disease risk in a population-based case-control study: Evidence of gene-gene interactions. Inflammatory bowel diseases. 2008;14:1528–1541. doi: 10.1002/ibd.20512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lake SL, Lyon H, Tantisira K, Silverman EK, Weiss ST, Laird NM, Schaid DJ. Estimation and tests of haplotype-environment interaction when linkage phase is ambiguous. Hum Hered. 2003;55:56–65. doi: 10.1159/000071811. [DOI] [PubMed] [Google Scholar]
- 30.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc. 1995;57:289–300. [Google Scholar]
- 31.Siminovitch KA. Advances in the molecular dissection of inflammatory bowel disease. Semin Immunol. 2006;18:244–253. doi: 10.1016/j.smim.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 32.Economou M, Trikalinos TA, Loizou KT, Tsianos EV, Ioannidis JP. Differential effects of NOD2 variants on Crohn's disease risk and phenotype in diverse populations: a metaanalysis. Am J Gastroenterol. 2004;99:2393–2404. doi: 10.1111/j.1572-0241.2004.40304.x. [DOI] [PubMed] [Google Scholar]
- 33.Croucher PJ, Mascheretti S, Hampe J, Huse K, Frenzel H, Stoll M, Lu T, Nikolaus S, Yang SK, Krawczak M, Kim WH, Schreiber S. Haplotype structure and association to Crohn's disease of CARD15 mutations in two ethnically divergent populations. Eur J Hum Genet. 2003;11:6–16. doi: 10.1038/sj.ejhg.5200897. [DOI] [PubMed] [Google Scholar]
- 34.Zaahl MG, Winter T, Warnich L, Kotze MJ. Analysis of the three common mutations in the CARD15 gene (R702W, G908R and 1007fs) in South African colored patients with inflammatory bowel disease. Mol Cell Probes. 2005;19:278–281. doi: 10.1016/j.mcp.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 35.Brant SR, Picco MF, Achkar JP, Bayless TM, Kane SV, Brzezinski A, Nouvet FJ, Bonen D, Karban A, Dassopoulos T, Karaliukas R, Beaty TH, Hanauer SB, Duerr RH, Cho JH. Defining complex contributions of NOD2/CARD15 gene mutations, age at onset, and tobacco use on Crohn's disease phenotypes. InflammBowel Dis. 2003;9:281–289. doi: 10.1097/00054725-200309000-00001. [DOI] [PubMed] [Google Scholar]
- 36.Hugot JP, Zaccaria I, Cavanaugh J, Yang H, Vermeire S, Lappalainen M, Schreiber S, Annese V, Jewell DP, Fowler EV, Brant SR, Silverberg MS, Cho J, Rioux JD, Satsangi J, Parkes M. Prevalence of CARD15/NOD2 mutations in Caucasian healthy people. Am J Gastroenterol. 2007;102:1259–1267. doi: 10.1111/j.1572-0241.2007.01149.x. [DOI] [PubMed] [Google Scholar]
- 37.Wang J, Wang X, Yang H, Wu D, Wang L, Qian J. Contribution of the IBD5 locus to inflammatory bowel disease: a meta-analysis. Human genetics. doi: 10.1007/s00439-011-0952-6. Epub. [DOI] [PubMed] [Google Scholar]
- 38.Fisher SA, Hampe J, Onnie CM, Daly MJ, Curley C, Purcell S, Sanderson J, Mansfield J, Annese V, Forbes A, Lewis CM, Schreiber S, Rioux JD, Mathew CG. Direct or indirect association in a complex disease: the role of SLC22A4 and SLC22A5 functional variants in Crohn disease. Human mutation. 2006;27:778–785. doi: 10.1002/humu.20358. [DOI] [PubMed] [Google Scholar]
- 39.Huff CD, Witherspoon D, Zhang C, et al. Crohn’s disease and genetic hitchhiking at IBD5. Mol Biol Evol. 2012;29:101–111. doi: 10.1093/molbev/msr151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tishkoff SA, Reed FA, Friedlaender FR, Ehret C, Ranciaro A, Froment A, Hirbo JB, Awomoyi AA, Bodo JM, Doumbo O, Ibrahim M, Juma AT, Kotze MJ, Lema G, Moore JH, Mortensen H, Nyambo TB, Omar SA, Powell K, Pretorius GS, Smith MW, Thera MA, Wambebe C, Weber JL, Williams SM. The genetic structure and history of Africans and African Americans. Science. 2009;324:1035–1044. doi: 10.1126/science.1172257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zakharia F, Basu A, Absher D, Assimes TL, Go AS, Hlatky MA, Iribarren C, Knowles JW, Li J, Narasimhan B, Sidney S, Southwick A, Myers RM, Quertermous T, Risch N, Tang H. Characterizing the admixed African ancestry of African Americans. Genome biology. 2009;10:R141. doi: 10.1186/gb-2009-10-12-r141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoggart CJ, Parra EJ, Shriver MD, Bonilla C, Kittles RA, Clayton DG, McKeigue PM. Control of confounding of genetic associations in stratified populations. Am J Hum Genet. 2003;72:1492–1504. doi: 10.1086/375613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zuo L, Luo X, Listman JB, Kranzler HR, Wang S, Anton RF, Blumberg HP, Stein MB, Pearlson GD, Covault J, Charney DS, van Kammen DP, Price LH, Lappalainen J, Cramer J, Krystal JH, Gelernter J. Population admixture modulates risk for alcohol dependence. Human genetics. 2009;125:605–613. doi: 10.1007/s00439-009-0647-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flores C, Ma SF, Pino-Yanes M, Wade MS, Perez-Mendez L, Kittles RA, Wang D, Papaiahgari S, Ford JG, Kumar R, Garcia JG. African ancestry is associated with asthma risk in african americans. PloS one. 7:e26807. doi: 10.1371/journal.pone.0026807. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(Supplementary Table 1) Study Subjects
(Supplementary Table 2) Characteristics of 21 SNPs genotyped (combined dataset)
(Supplementary Table 3) Heterogeneity test between primary and secondary replicate dataset
(Supplementary Table 4) Association of SNPs among 227 CD and 200 controls in primary dataset
(Supplementary Table 5) Diplotype association test for IBD5
(Supplementary Table 6) Power calculation: Sample size 354 cases and 1:1 control; CD Population risk: 50/100,000; 2-sided p value 0.05;