

Abstract
The central nervous system (CNS) is a remarkably complex organ system, requiring an equally complex network of molecular pathways controlling the multitude of diverse, cellular activities. Gene expression is a critical node at which regulatory control of molecular networks is implemented. As such, elucidating the various mechanisms employed in the physiological regulation of gene expression in the CNS is important both for establishing a reference for comparison to the diseased state and for expanding the set of validated drug targets available for disease intervention. MicroRNAs (miRNAs) are an abundant class of small RNA that mediates potent inhibitory effects on global gene expression. Recent advances have been made in methods employed to study the contribution of these miRNAs to gene expression. Here we review these latest advances and present a methodological workflow from the perspective of an investigator studying the physiological regulation of a gene of interest. We discuss methods for identifying putative miRNA target sites in a transcript of interest, strategies for validating predicted target sites, assays for detecting miRNA expression, and approaches for disrupting endogenous miRNA function. We consider both advantages and limitations, highlighting certain caveats that inform the suitability of a given method for a specific application. Through careful implementation of the appropriate methodologies discussed herein, we are optimistic that important discoveries related to miRNA participation in CNS physiology and dysfunction are on the horizon.
Keywords: Alzheimer, Predictors, RIP-Chip, HITS-CLIP, PAR-CLIP, Non-coding RNA, Method, Post-transcriptional
Introduction
The anatomical and cytoarchitectural complexity of the central nervous system (CNS) is a function of an equally sophisticated molecular framework. Derangements in tightly regulated molecular pathways underlying this complexity are not surprisingly linked to many CNS disorders and are currently a focus of intense investigation. As an example, there is growing evidence that genetic and biochemical abnormalities may be shared among some neurodevelopmental, neurodegenerative and neuropsychiatric disorders across the lifespan (Ray et al., 2011b; Sokol et al., 2011).
Gene expression is a critical node at which regulatory influences on molecular pathways are exerted. As such, fleshing out the full complement of regulatory schemes responsible for control of CNS gene expression should be a priority in the search for novel points of intervention against CNS disorders. Elucidating these regulatory networks not only provides a reference for exploring underlying etiologies but also expands the number of functionally validated drug targets, the paucity of which is a current impediment for CNS drug discovery (Krause and Chenard, 2008).
Insufficiency of current drug targets for CNS disorders: Alzheimer disease (AD) as an example
AD is illustrative of the need for an expanded set of functionally validated drug targets in CNS drug discovery. The disease itself is characterized by extracellular amyloid plaques consisting primarily of fibrillar amyloid-β (Aβ) peptide and intraneuronal neurofibrillary tangles composed of paired helical filaments of hyperphosphorylated tau (Nelson et al., 2009). Synaptic loss occurs early in disease and appears to correlate with cognitive decline better than other pathological findings (Terry et al., 1991). There are also functional alterations in memory networks in the early phase of disease (Sperling et al., 2010) and significant influences from multiple factors, including genetics and environment (Bertram et al., 2010; Lahiri et al., 2009). Despite the focus on disease-modifying therapies, there have been several recent clinical trial setbacks for phase three drugs directed against well known AD targets (Becker and Greig, 2010; Sambamurti et al., 2011; Schneider and Lahiri, 2009; The Lancet, 2010). Currently approved medications are not that effective and only modify symptoms; there is still no approved medication that alters the progression of the disease (Lahiri et al., 2003; Mangialasche et al., 2010; Raina et al., 2008; Schneider et al., 2011). New putative biomarkers, therapeutic strategies and targets are being tested (Alley et al., 2010; Bailey et al., 2011; Cogswell et al., 2008; Ray et al., 2011a) and are needed for successful intervention (Lahiri, 2011).
Regulatory networks controlling expression of gene products implicated in AD
Our aim has been to clarify the regulatory networks that control expression of gene products implicated in AD. The goal is to fully characterize the normal, physiological pathways with the expectation they might serve as novel therapeutic targets for modulating disease progression. Given the hypothesized centrality of the amyloid-β (Aβ) peptide to AD etiology (Hardy and Selkoe, 2002), we have focused our efforts on those gene products involved in Aβ production, especially the Aβ precursor protein (APP). The regulation of APP messenger RNA (mRNA) expression has been extensively investigated. The promoter structure is complex and consists of proximal and distal elements (Lahiri and Robakis, 1991; Pollwein et al., 1992; Quitschke and Goldgaber, 1992; Song and Lahiri, 1998) that mediate both constitutive and dynamic transcriptional expression (Ge et al., 2004; Lahiri and Nall, 1995). Regulatory elements in the genomic 5′-UTR can drive APP promoter activity (Maloney et al., 2004; Vostrov et al., 2010). Elements in the APP mRNA 5′-UTR also mediate post-transcriptional regulation (Lahiri et al., 2005; Rogers et al., 1999, 2002). The APP 3′-UTR contains several stability control elements that regulate APP mRNA stability (Zaidi and Malter, 1994; Zaidi et al., 1994). Alternative polyadenylation of the APP 3′-UTR also regulates transcript stability (Mbella et al., 2000). Therefore, there is a complex regulatory network of trans-acting factors that act on a dense landscape of cis-elements located across the APP genomic and transcript sequence to regulate its expression. However, this landscape is still incomplete. We and others have begun to elucidate the contribution of microRNA (miRNA) to this physiological regulatory network (Fan et al., 2010; Hébert et al., 2009; Liu et al., 2010; Long and Lahiri, 2011a, b, c; Vilardo et al., 2010). As with so many other gene products, we are now realizing that miRNAs contribute integrally to the regulation of APP expression and that there is much regarding miRNA control of APP to discover. Further, studies have also demonstrated that miRNAs are dysregulated in neurons of the AD brain and in the periphery and may contribute to disease etiology (Cogswell et al., 2008; Hébert et al., 2008; Lukiw, 2007; Nelson and Wang, 2010; Nunez-Iglesias et al., 2010; Schipper et al., 2007; Wang et al., 2008a)
MiRNAs are an abundant class of small, non-coding RNA (18–25 nucleotides (nt)) that serve as potent mediators of post-transcriptional gene expression (Bartel, 2004). The current estimated number of human miRNAs now exceeds 1400 (miRBase release 17) with greater than 60% of all human mRNA predicted to contain conserved miRNA targets (Friedman et al., 2009). However, in relation to this number of probable miRNA-mRNA regulatory interactions, the number of known, validated targets is vanishingly small, along with the depth of our understanding regarding the nature of miRNA gene expression regulation in the CNS.
Scope of the review
Exploring the complement of miRNA that physiologically regulate AD- and other CNS-relevant gene products is likely to reveal novel targets for manipulating the pathological state. We will briefly review here a number of techniques that might be employed to study miRNA regulation, discussing their advantages and drawbacks. This review will address various experimental approaches from the perspective of an investigator studying the physiological regulation of a gene of interest (GOI). This approach complements the perspective of other miRNA studies attempting to identify targets and functional effects of a miRNA of interest [for a review with this focus see (van Rooij, 2011)].
Biology of miRNA
Biogenesis, processing and function
The founding miRNA members, lin-4 and let-7, were originally identified in Caenorrhabditis elegans (C. elegans) as regulators of developmental timing (Lee et al., 1993; Reinhart et al., 2000; Wightman et al., 1993). Large scale cloning studies later identified a plethora of miRNAs that are expressed across many species, including humans (Lagos-Quintana et al., 2001; Lau et al., 2001; Lee and Ambros, 2001). Most miRNAs are transcribed by RNA polymerase II as long primary transcripts (pri-miRNA) that form local stem-loop structures harboring the mature miRNA sequence (Fig. 1) (Lee et al., 2004). Following primiRNA transcription, the nuclear microprocessor complex, consisting of Drosha and DGCR8 in human cells, cleaves the pri-miRNA, releasing the stem-loop structure (pre-miRNA) from flanking RNA and leaving a 3′ 2-nt overhang (Han et al., 2004; Lee et al., 2003). This newly formed pre-miRNA is exported from the nucleus by exportin-5 (Lund et al., 2004). In the cytosol, the complex of Dicer and TRBP in human cells binds the pre-miRNA at the 2-nt overhang and cleaves the loop approximately 22-nt downstream releasing the double stranded miRNA (Chendrimada et al., 2005; Hutvágner et al., 2001). The miRNA is then incorporated into a microribonucleoprotein (miRNP) complex (Mourelatos et al., 2002), also termed the RNA induced silencing complex (RISC) (Pratt and MacRae, 2009) [summarized in Fig. 1].
Fig. 1.

miRNA biogenesis and RISC assembly in human cells. miRNA are initially transcribed as pri-miRNA, processed by the complex of Drosha and DGCR8 to pre-miRNA, exported to the cytosol by exportin-5, and cleaved to a mature miRNA guide strand and passenger strand (miRNA*) by the complex of Dicer and TRBP. For RISC assembly, the mature guide strand (red strand in illustration) is loaded onto an AGO protein along with other assembly proteins not illustrated. The passenger strand is degraded while the guide strand interacts with specific target sites in the 3′-UTR of transcripts. Near perfect complementarity between the target transcript and the seed sequence of the miRNA is required to effectuate an inhibitory response.
The central effector protein of RISC is a member of the AGO subfamily of Argonaute proteins (Peters and Meister, 2007). The guide strand of the double stranded miRNA is loaded onto AGO while the passenger strand is generally degraded. The guide strand guides the RISC to specific mRNA targets in animals by hybridizing with imperfect complementarity to target sites generally located in the 3′-UTR. However, target sites require a near perfect match between the 5′ end of the miRNA (seed sequence) and target transcript (Lewis et al., 2003). Less stringency is required at the 3′ end of the miRNA. AGO then mediates gene repression by either destabilizing the mRNA transcript via deadenylation or by repressing translation through numerous possible mechanisms (Filipowicz et al., 2008). Recent studies suggest that mRNA destabilization is the primary mechanism by which miRNA regulatory interactions reduce protein output (Guo et al., 2010; Hendrickson et al., 2009). The mechanism of miRNA-mediated gene repression contrasts to that of widely-used synthetic short interfering RNAs (siRNAs), which hybridize with perfect complementarity to mRNA targets and induce endonucleolytic cleavage and degradation of the transcript.
MicroRNA oxidation, turnover and stability
Control of miRNA steady-state levels is a function of pri-miRNA transcription, pri- and pre-miRNA processing by the processing complexes, and regulated turnover of mature miRNA. Unlike miRNAs, mRNAs are stabilized by the presence of a 5′ 7-methylguanylate cap and 3′ poly(A) tail that protect against exonuclease activity. Controls on mRNA stability are tightly regulated and mRNA turnover modulates miRNA efficacy. A shorter mRNA half-life generally correlates with reduced miRNA efficacy (Larsson et al., 2010). Conversely, miRNA termini are exposed and appear to rely on interactions with AGO proteins (Wang et al., 2008b) to stabilize against exonucleolytic degradation (Winter and Diederichs, 2011).
Recent studies have explored the half-life of miRNAs in various cell types and identified great variability. In one study, miRNA half-life was measured in mouse embryonic fibroblasts and bone marrow-derived macrophages following conditional Dicer ablation (Gantier et al., 2011). The predicted half-life after controlling for dilutional effects of cell proliferation was much longer than that of mRNAs and averaged around 119 h. Conversely, two recent studies have identified much shorter miRNA half-lives in neuronal cell types following inhibition of RNA polymerase II-based transcription. Analysis in primary neural cell (HN) cultures and frozen human brain specimens revealed miRNA half-lives in the range of 1–4 h (Sethi and Lukiw, 2009). Similarly short miRNA half-lives were observed in retinal photoreceptors and cultured hippocampal and cortical neurons (Krol et al., 2010). Together, these studies suggest that miRNAs are generally very stable in vivo but appear to be under regulatory control in neurons that promotes rapid turnover.
Specific mechanisms by which metazoan cells control miRNA degradation and turnover have been described but are just beginning to be explored (Kai and Pasquinelli, 2010; Siomi and Siomi, 2010). In C. elegans, mature miRNA degradation was recently shown to be mediated by the 5′-3′ exoribonuclease XRN-2 following dissociation of miRNA from RISC (Chatterjee and Grosshans, 2009). Interestingly, increased levels of target mRNA protected against XRN-2 mediated degradation suggesting that duplex formation in RISC may stabilize miRNA. An additional control mechanism that appears to influence miRNA stability is the addition of non-templated terminal nucleotides. The poly(A) polymerase (PAP) GLD-2 monoadenylates miR-122 at the 3′ terminus resulting in increased miR-122 stability (Katoh et al., 2009). Conversely, the non-canonical PAP Tut4 polyuridylates pre-let-7 at the 3′ terminus resulting in reduced pre-let-7 stability and reduced mature let-7 levels (Heo et al., 2008, 2009). This interaction requires Lin28 proteins that act as processivity factors for Tut4 (Yeom et al., 2011). Two recent studies have examined pre-miRNA modifications at the transcriptome level and identified extensive mono and polynucleotide additions and novel cleavage patterns indicating post-transcriptional nuclease modifications (Burroughs et al., 2011; Newman et al., 2011). These studies demonstrate that post-transcriptional modifications of pre-miRNA are widespread and suggest they may represent a widely employed mechanism for miRNA stability control.
RNAs are generally highly susceptible to oxidation, especially within highly oxidizing environments as occurs in neurodegenerative disorders such as AD (Nunomura et al., 2009). miRNA oxidation is an understudied phenomenon. As such, little is known as to whether miRNA oxidation might modulate miRNA function and/or stability and is a topic in need of additional study.
Experimental implications and quality control analysis
The rapid turnover of miRNAs in neuronal cell types has important implications for many of the miRNA analyses to be described here as utilized for CNS samples. This finding suggests a potent RNase pool active against miRNAs exists in neurons. In an intact cell, subcellular sequestration and post-translational modifications of RNase proteins likely prevent unwanted interactions with miRNAs. However, in most experimental miRNA analyses, the cellular structure is purposefully disrupted in order to extract RNA molecules. Therefore, cellular RNases are released from normal regulatory control of the cellular environment and are free to interact in solution with substrate cellular RNAs. Given the robust nature of RNase activity, most RNA extraction procedures employ chaotropic salts (i.e. guanidinium thiocyanate) and strongly reducing conditions to denature proteins and eliminate RNase activity following cellular disruption. However, if RNase activity is not completely eliminated, RNA degradation will proceed and total RNA integrity (including miRNA) will be low. Degradation of the miRNA pool may not occur in an unbiased manner and may therefore lead to results that do not reflect the initial biological state.
As a result, one of the most critical steps in any miRNA workflow (such as the one described below) is to ensure high quality of the RNA extract prior to performing any analyses that measure or otherwise require intact miRNA. Generally, the same quality control measures used for standard RNA analyses are applicable to extracts containing miRNAs. Quality control measures that must be assessed prior to any miRNA measurement include spectrophotometric analysis of RNA extract concentration and purity and electrophoretic analysis of RNA integrity. Ultraviolet absorbance spectra provide information on purity of extraction procedures with regard to contaminants such as protein, chaotropic salts and phenol. Agarose electrophoresis has been historically used to assess for RNA degradation by analyzing the ratio of 28S:18S ribosomal RNA levels. A ratio of approximately two is generally an indication of high RNA integrity. A more state-of-the-art approach utilizes capillary electrophoresis with a lab-on-chip design (e.g. Agilent Bioanalyzer 2100) to generate electropherograms and virtual gels that enable the assessment of 28S:18S ratios. A novel scoring algorithm, the RNA integrity number (RIN), based on multiple features of the electropherogram has been developed to provide a more nuanced assessment of RNA integrity and is widely used (Schroeder et al., 2006). One study has found that samples with RIN values less than five provide unreliable quantitative PCR results and should be discarded (Becker et al., 2010). However, the exact RIN threshold between high and low quality must be determined for a given application. Finally, for specific assessment of the small RNA component within an RNA extract, a small RNA chip is available for use in the Bioanalyzer that specifically resolves the small RNA fraction. However, a recent study has found that RNA degradation from large RNA species can significantly contaminate the analysis if the total RNA integrity is not very high (Becker et al., 2010).
An important point for CNS miRNA analyses of post-mortem human specimens is that post-mortem interval (PMI) is a crucial variable that must be controlled between groups. A longer PMI in an experimental group is likely to lead to enhanced RNA degradation in those samples. This may generate falsely low measured miRNA values relative to groups with shorter PMI. Therefore, short PMI is ideal and, at minimum, PMI should be controlled between experimental groups.
General approach for studying miRNA regulation of GOI
To elucidate the physiological regulation of a GOI by miRNA, we present the following workflow: i) identification of putative miRNA target sites within the transcript, ii) functional validation of these putative regulatory interactions, iii) confirmation of miRNA expression in the model of choice, iv) assessing the physiological relevance of these regulatory interactions by blocking the interaction between miRNA and target site, and v) testing the effect on a relevant downstream pathway of interest. A more unbiased workflow would make no assumptions about putative miRNA target sites and simply screen a complete library of all known miRNAs for regulatory effects on GOI using an optimized high throughput assay. These types of library screens are often employed to detect effects on a specific phenotype or pathway of interest (Lam et al., 2010) but could be modified with the proper adaptations to detect a specific GOI. However, the costs, logistics, and time required for assay optimization are extensive. In most cases, the standard workflow described below, with its biases and caveats in mind, is sufficient to identify novel regulatory controls on the GOI.
Identifying putative miRNA target sites
The first step in the workflow for elucidating miRNA networks that physiologically regulate gene products implicated in CNS disorders is to identify putative miRNA target sites in the transcript of interest. In lieu of performing unbiased miRNA screens as described above, this step is necessary to filter the list of miRNA that may contribute to the physiological regulation of the GOI to those with plausible recognition elements located within the transcript. There are two general approaches to identifying these putative target sites: i) computational predictions, and ii.) experimental methods for detecting physical interactions between miRNA and mRNA in a regulatory context. Computational predictions utilize algorithms to scan transcript sequences (generally limited to the 3′-UTR) for putative sites of interaction with miRNA, whereas the experimental methods utilize pull-down assays to detect physical interactions between miRNA, AGO proteins and specific transcript sites. Since neither of these approaches assesses the functionality of miRNA interactions with putative target sites, validation experiments must be pursued to confirm regulatory effects mediated by miRNA and the specificity of the putative target sites.
Bioinformatic predictions of miRNA target sites
Many bioinformatic predictor algorithms have been developed for identifying putative miRNA target sites located within mRNA transcripts and are implemented within web servers utilizing easy-to-use graphical interfaces for quick compilation of putative target sites. Most interfaces offer the flexibility of at least two different search formats: 1) predicted global mRNA targets for a miRNA of interest, or 2) predicted miRNA target sites located within a transcript of interest. For studies focused on CNS-relevant GOI, the latter format would be the search method of choice. Computational prediction of miRNA target sites in animals is not a trivial pursuit. The tolerance for imperfect complementarity between miRNAs and mRNA target sites means that simple complementarity scanning approaches are inadequate. In short, each algorithm is based upon unique principles or assumptions making direct comparisons between prediction sets sometimes difficult. There are some common parameters that a majority of the algorithms assess to varying degrees when predicting putative target sites. These include seed sequence complementarity, the free energy of duplex formation, cross-species conservation at the target site and local sequence context surrounding the target sites. There are too many algorithms than can possibly be reviewed here; we will focus on those predictors commonly used in the field and those demonstrated to produce the most accurate predictions (Baek et al., 2008; Selbach et al., 2008).
TargetScan algorithm
The era of computational miRNA target site prediction began in 2003 with the release of the TargetScan algorithm (Lewis et al., 2003). This algorithm was based upon empirical studies of early C. elegans miRNA-target site interactions and defined a 7-nt seed sequence at the 5′ end of the miRNA. Strict complementarity between target transcript and miRNA at this seed sequence site is the initial basis for making predictions in the algorithm. TargetScan then assesses for mRNA-miRNA duplex formation outside of the seed sequence using thermodynamic free energy considerations. The algorithm requires cross-species conservation at predicted target sites based on the assumption that physiologically relevant interactions would be under strict evolutionary pressures to maintain complementarity. Further refinements to the algorithm, based on favored sequence contexts (Grimson et al., 2007; Lewis et al., 2005), and to the quantitative methods for assessing cross-species conservation (Friedman et al., 2009), have improved algorithm reliability with each release.
PicTar algorithm
PicTar utilizes a similar seed sequence strategy to identify target sites but allows for small imperfections in the complementarity of the seed sequence (Krek et al., 2005). The algorithm then filters for duplex thermodynamic stability and cross-species conservation. Following filtering, the algorithm computes probabilities that a given site binds a miRNA and uses a maximum likelihood model to compute the likelihood that a given UTR is targeted by combinatorial interactions of miRNAs.
DIANA-microT algorithm
DIANA-microT 3.0 is the latest iteration of a predictor algorithm that, like TargetScan and PicTar, is based on seed sequence complementarity (Kiriakidou et al., 2004; Maragkakis et al., 2009a, b). The algorithm searches for perfect complementarity beginning at position 1 or 2 of the 5′ end of the miRNA and extending 7–9 nucleotides (7mer, 8mer, 9mer). A single G:U wobble pair and 6mer seed matches are permitted if the free energy of duplex formation is sufficiently compensated by sequence complementarity outside of the seed sequence. Cross-species conservation is assessed at the predicted target site but not used as a strict filter. Instead, a conservation score (based on number of species in which site is conserved) and the type of seed match (7-9mer) are used to assign miRNA recognition element (MRE) scores for the predicted target site. miRNA target gene scores are also assigned by summing all MRE scores (weighted by seed sequence type) across the 3′-UTR for a given GOI.
miRanda algorithm
The miRanda algorithm, unlike the previously described algorithms, does not rely solely on strict seed sequence complementarity for initial target site prediction (Betel et al., 2008; Enright et al., 2003; John et al., 2004). The algorithm first performs a dynamic programming local alignment modified to detect sequence complementarity between the complete miRNA sequence and putative target sites. The algorithm does weight 5′ end complementarity more heavily than 3′ end, but imperfect or non-canonical seed matches can be compensated by increased 3′ end complementarity. Local alignment optimization is followed by assessment of the thermodynamic stability of the putative duplex interaction. Target sites are then filtered by setting threshold scores for alignment and free energy. Sites are further filtered by assessing cross-species conservation. A recent algorithm, mirSVR, has been paired with miRanda target site predictions to predict miRNA efficacy at a given miRanda-predicted target site (Betel et al., 2010). mirSVR is a machine learning algorithm trained empirically on a large mRNA expression data set following miRNA transfections.
PITA and rna22 algorithms
Other popular prediction algorithms include PITA, which takes into account target site secondary structure to determine accessibility of miRNAs to predicted target sites (Kertesz et al., 2007), and a pattern-based algorithm, rna22 (Miranda et al., 2006). rna22 utilizes pattern-recognition algorithms to capture sequence motifs from a large input of mature miRNA sequences. The reverse complement of these pattern motifs are then searched for in 3′-UTRs. Locations, where many motifs are concentrated are termed target islands and presumed to represent miRNA target sites. Therefore, rna22 initially identifies target sites without respect for seed sequences, cross-species conservation, or even the identity of the miRNA that binds to the target site.
Advantages and limitations of predictor algorithms
The advantage of using algorithmic predictor tools is that they are easily accessed and generally successful in identifying true interactions between miRNAs and mRNA target sites. Recent proteomic analyses of protein expression changes following transfection of synthetic miRNAs have demonstrated that bioinformatic predictions capture many experimentally confirmed regulatory relationships (Baek et al., 2008; Selbach et al., 2008). These global analyses revealed that predictors that rely on strict seed sequence complementarity (TargetScan, PicTar, DIANA-microT3.0) demonstrated the highest accuracy in predicting functional miRNA target sites. However, these same studies indicate that these tools still generate very high levels of false positives. Therefore, a set of miRNAs predicted to target a GOI can be expected to contain many physiologically irrelevant members. Further, this set of miRNAs is sure to exclude many physiologically relevant members not detected by the predictor algorithm (false negatives). This highlights the need for rapid and medium-throughput assays for functional validation of predicted miRNA-mRNA target interactions after compiling such a set of predictions. Other disadvantages underlying exclusive use of web-based predictors is that most have pre-assembled prediction sets that are limited to 3′-UTR only. New experimental approaches have identified functional interactions with target sites in the CDS and 5′-UTR of mRNAs (Chi et al., 2009; Hafner et al., 2010a; Wang et al., 2010b). Therefore, exclusive use of computational methods for target site identification is likely to miss out on some important functional interactions that occur outside the 3′-UTR. Finally, the requirement for cross-species conservation at putative target sites serves as a useful filter for enriching for functional sites but is also likely to eliminate the detection of species-specific miRNA-mRNA target interactions. Overall, given the ease of use and continuously improving nature of these predictors, they are an appropriate tool for initiating investigations of regulatory interactions in many cases.
Experimental methods for identifying miRNA target sites
Transcriptome profiling approach
Experimental methods attempt to circumvent the biases inherent in the use of computational predictions. While not free of their own biases, they are exempted from certain assumptions programmed into many of the predictor algorithms to enrich for true targets. For example, functional transcript target sites that do not exhibit optimal sequence contexts, cross-species conservation, target site accessibility or high duplex stability will not be automatically excluded from detection in experimental paradigms. Some of the earliest experimental approaches for identifying putative miRNA target sites relied on transfection of specific miRNAs into cell types followed by high-throughput mRNA expression analysis by microarray (Grimson et al., 2007; Lim et al., 2005; Linsley et al., 2007). Transcripts with reduced expression were then scanned for putative target sites by identifying sequences complementary to the miRNA seed sequence. These studies demonstrated that mRNA transcripts deregulated following miRNA transfection were significantly enriched for matches to the seed sequence of the transfected miRNA. Since delivery of supraphysiological levels of an exogenous miRNA may mediate non-specific effects, knockdown of miRNA availability using antisense oligonucleotides followed by micro-array analysis of global transcript expression has been employed (Elmén et al., 2008; Krützfeldt et al., 2005). Transcript destabilization is not the exclusive mechanism by which miRNA inhibit gene expression; translational repression is another. Therefore, similar studies have been performed utilizing proteomic approaches to detect repressed protein expression following miRNA transfection (Baek et al., 2008; Selbach et al., 2008). Similar scanning of deregulated transcripts revealed enrichment for sequences complementary to the seed sequence of the transfected miRNA. One issue of concern must be that direct interactions between the miRNA and mRNA are not detected. Therefore, these methods cannot distinguish between transcript expression deregulated in response to direct interaction with a targeting miRNA and transcript expression deregulated in response to indirect miRNA effects on other gene products with regulatory functions. Further, these global analyses following miRNA delivery are useful for determining the targets of a miRNA of interest but are not useful for identifying target sites within a GOI.
Pull-down assays with Argonaute proteins or miRNAs
Methods that can directly detect interactions between a transcript of interest and miRNA are needed when focusing on a GOI (Fig. 2). Most are based upon pull-down assays of either Argonaute proteins or miRNAs directly. Like the transcriptome profiling approaches described above, most of the pull-down methods have been designed to identify transcript targets of specific miRNAs of interest. We believe that most of these assays could be sufficiently modified to enable the detection of miRNAs that associate with a transcript of interest. This modified experimental design would involve overexpression of the transcript of interest in a relevant cell context (e.g. in neuronal cultures or in vivo brain via lentiviral delivery) followed by pull-down and global profiling of miRNA levels. Enrichment of a complement of miRNA in lysates from cells overexpressing the transcript of interest as compared to cells overexpressing a control gene product or empty expression vector would suggest that these miRNA interact with and are pulled-down along with the transcript of interest.
Fig. 2.

Methods of experimental target identification based upon AGO IP. The primary concept employed by the four illustrated methods is the pull-down of a ternary complex consisting of miRNA-mRNA-AGO. For RIP-Chip, AGO IP is followed by RNA extraction and high-content analysis of mRNA primarily by microarray. TAP-Tar differs from RIP-Chip in that it employs transfection of biotinylated miRNA mimics and a second pull-down purification with streptavidin to isolate specific miRNA complexes. Both HITS-CLIP and PAR-CLIP generate UV-induced crosslinks between RNA molecules and AGO to stabilize transient interactions. Mild RNA digestion then generates short fragment libraries enriched in miRNA target sites that are analyzed by deep sequencing. PAR-CLIP differs from HITS-CLIP in that it utilizes photoactivatable ribonucleosides to enhance crosslinking efficiency.
Tagged miRNA pull-down method
One of the more direct methods for detecting transcripts associated with a miRNA of interest is a biotinylated miRNA pull-down method (Orom and Lund, 2007). In this method, synthetic biotinylated miRNA are transfected into cells of interest and mildly lysed after incubation. Lysed samples are then exposed to streptavidin-coated beads and mRNAs associated with the transfected miRNAs can be identified by standard expression assays. An in vitro variant, termed labeled microRNA pull-down assay (LAMP), utilizes digoxigenin (DIG)-labeled pre-miRNA oligonucleotides that are mixed with cell extracts. Extracts are then immunoprecipitated with anti-DIG antibodies followed by analysis of co-precipitated mRNA (Hsu and Tsai, 2011; Hsu et al., 2009). Both of these methods could be modified to identify miRNA targeting a GOI by replacing labeled miRNA with labeled transcripts of interest. One difficulty with these approaches is interpreting whether the miRNA-mRNA interactions are physiological or only juxtapositional. Even if the interactions are functional, this method does not enable the detection of the specific target site in the targeted transcript. Newer methods incorporate technologies that allow for more confident interpretations. Nonetheless, the technique by Orom and Lund (2007) led to the discovery that miR-10a interacts with the 5′-UTR of ribosomal protein transcripts and drives enhanced translation by interacting with a 5′-UTR regulatory motif (5′TOP) common to these transcripts (Ørom et al., 2008).
RIP-Chip method
Ribonucleoprotein (RNP) immunoprecipitation (IP) followed by microarray chip analysis (RIP-Chip) is an important method that utilizes IP of RNA-binding proteins to pull down bound RNA targets. This method was adapted using specific anti-AGO antibodies (Ikeda et al., 2006; Nelson et al., 2007) to pull down RISC complexes comprising AGO effectors, miRNA guides, and mRNA targets (Beitzinger et al., 2007; Easow et al., 2007; Hendrickson et al., 2008; Karginov et al., 2007; Landthaler et al., 2008; Wang et al., 2010a). To identify targets of specific miRNA, cells are transfected with a synthetic miRNA or negative control. After incubation, cells are mildly lysed, AGO complexes immunoprecipitated, and captured mRNA quantified by micro-array analyses. mRNAs enriched following miRNA transfection are presumed to contain target sites for the transfected miRNA. Transfection with a transcript of interest followed by miRNA enrichment analysis (e.g. by microarray) could presumably be employed for studying a GOI. Given that AGO proteins are the RISC effectors, the complement of miRNA-mRNA interactions pulled down in complex with AGO should be enriched for functional interactions. However, validation is still necessary to confirm this assumption. This approach has been used to determine that AD-dysregulated miR-107 functionally interacts with and regulates granulin (Wang et al., 2010b). Importantly, this interaction was found to occur in the granulin open reading frame and was therefore not predicted by standard computational approaches. One drawback of RIP-Chip is that it detects all miRNA and mRNA bound to AGO, irrespective of whether they exist as a ternary miRNP complex.
TAP-Tar method
A modification, called tandem affinity purification of miRNA targets (TAP-Tar), addresses this deficiency by combining RIP-Chip with the labeled miRNA approach (Nonne et al., 2010). In TAP-Tar, cells stably expressing Flag-HA-AGO1/2 are transfected with biotinylated miRNA, and cell extracts are subjected to sequential purifications. First, miRNP are pulled down by anti-FLAG IP. Then miRNP are further fractionated by streptavidin pull-down. This approach enables detection of AGO-loaded mRNAs that are specifically in complex with the transfected miRNA.
A deficiency of both RIP-Chip and TAP-Tar is that neither enables the specific detection of the target sites in target transcripts. Also, the nature of the interactions inside the miRNA-mRNA-AGO complex may be transient. These techniques do not use any stabilizing methods (e.g. crosslinking) to ensure that transient interactions are not lost during the IP step (Nelson et al., 2010). As a result, analyses have shown that target site identification is biased towards those specific targets that survive the pull-down method (Hausser et al., 2009; Hong et al., 2009). Finally, a previous study identified a significant number of non-overlapping miRNA bound to AGO1 versus AGO2 following RIP-Chip, indicating that many miRNA associations may be AGO-specific (Beitzinger et al., 2007). Therefore, the choice of which AGO protein to IP is a critical consideration when performing RIP-Chip or TAP-Tar.
HITS-CLIP method
Several similar methods based on UV-induced covalent crosslinking were developed to stabilize miRNP interactions prior to AGO pull down. The goal of stabilization is to enhance the capture of more transient miRNA-mRNA interactions and reduce selection bias inherent in RIP-Chip. The first method is called high throughput sequencing of RNA isolated by crosslinking and IP (HITS-CLIP) (Chi et al., 2009). Biological samples are first irradiated with UV light, which induces the formation of covalent crosslinks between RNA and bound proteins that are in direct contact. Crosslinked samples are then immunoprecipitated with anti-AGO antibodies. Mild RNA digestion releases RNA flanking fragments not in direct contact with AGO. Following stringent washing, only those miRNA and mRNA fragments directly crosslinked to AGO in RISC are retained. These RNA tags are then released and deep-sequenced using massively parallel sequencing technology. Analysis can reveal approximate miRNA binding sites on captured mRNA fragments and have been demonstrated to be enriched in miRNA target sequences. This technique was utilized to map probable ternary complexes between AGO2, miRNA guides and mRNA target sites across the transcriptome for the top 20 most abundant miRNA in the mouse brain (Chi et al., 2009). When combined with transfection of a miRNA of interest, this technique can allow for direct detection of transcript fragments enriched in the AGO-IP fraction, with the fragment providing clues to the miRNA target site.
While the resolution for miRNA target site identification in the original HITS-CLIP data set was limited to approximately 45–62 nt (AGO footprint), a recent reanalysis has revealed single nucleotide resolution (Zhang and Darnell, 2011). Cross-linking induced mutation sites (CIMS) consisting of deletions were found in 8% of sequence reads and were presumably the result of reverse transcriptase read-through at the crosslinked site on the mRNA fragment. The investigators observed that deletions reproducibly clustered near sites containing miRNA seed sequences. Analysis of CIMS resulted in a higher rate of discovery and significantly enhanced signal-to-noise ratio for detecting miRNA seed matches in sequenced mRNA tags. Therefore, CIMS analysis can identify nearly the exact location of crosslink formation between AGO and targeted transcripts.
PAR-CLIP method
A modification of HITS-CLIP is the photo-activatable-ribonucleoside-enhanced-CLIP (PAR-CLIP), which utilizes photoactivatable ribonucleosides to enhance crosslinking efficiency and fold-recovery of RNA following wash steps (Hafner et al., 2010b). The special ribonucleosides are mostly non-toxic and readily taken up by cultured cells. The authors report that, similar to HITS-CLIP, crosslink formation between these modified ribonucleosides and RNA-binding proteins induce a high rate of modified T to C conversions during the reverse transcription process used prior to deep sequencing. Therefore, by analyzing locations of T to C conversions, the exact site of crosslinks can be determined, which greatly aid the process of identifying the miRNA target site on targeted transcripts. We hypothesize that both of these techniques could be modified such that delivery of a transcript of interest (via direct transfection of in vitro transcribed RNA or lentiviral delivery) would enable the detection of enriched AGO-bound miRNA fractions that bind to the transcript of interest at detectable miRNA target sites. We intend to investigate whether such strategies would enable detection of novel APP mRNA-miRNA interactions not predicted by the standard bioinformatic approaches.
Advantages and limitations of crosslinking approaches
In summary, crosslinking approaches represent robust methods for detecting miRNA target sites, with a much lower false positive rate than computational methods. Unfortunately, they are also the most expensive and technically challenging option for detecting target sites. Significant computational effort must also be expended for analyzing deep sequencing reads. However, unlike RIP-Chip, these crosslinking strategies provide a means to directly identify, at single-nucleotide resolution, the sites of interaction in the miRNP. However, HITS-CLIP and PAR-CLIP experiments may also reflect a selection bias for strongly interacting miRNP complexes (Zhang and Darnell, 2011). Therefore, while target sites identified by HITS-CLIP and PAR-CLIP are highly specific, they likely under-represent all miRNA target sites in a transcript of interest.
Functional validation of putative miRNA-transcript interactions
Whether miRNA target sites are identified by computational predictions or via direct experimental detection, none of these methods assess the functionality of the interaction. Therefore, after identifying possible miRNA target sites on the transcript of interest, one must validate that the miRNAs exhibit regulatory actions on the GOI. One of the most common methods for fast, medium-throughput validation is the use of reporter assays. Validated hits then are verified for regulatory effects on native mRNA and protein expression. As an example, in our efforts to study miRNA regulation of APP expression, we chose to validate computational predictions using a luciferase-based reporter assay and by examining the direct effect of the predicted miRNA on native APP expression (Long and Lahiri, 2011c).
Reporter assay validation of putative miRNA target sites
The most optimum strategy for reporter screens is to directly clone the 3′-UTR of interest into a reporter vector, of which many are commercially available. One such reporter is psiCHECK-2 (Promega), a vector that contains a Renilla luciferase gene under the transcriptional control of a SV40 promoter. The multiple cloning site is positioned just downstream of the luciferase CDS such that an insert will be positioned in the 3′-UTR. If the predicted target site is not in the 3′-UTR, the cloning strategy will have to be necessarily modified such that the sequence containing the putative target site is fused to the appropriate portion of the reporter transcript (5′-UTR or CDS). Modified reporter vectors, such as pmiRGLO (Promega) have been created specifically for investigating miRNA regulatory functions. This reporter is driven by the more moderately-expressing PGK promoter, enabling more sensitive detection of changes in reporter expression.
Synthetic miRNA mimics are commercially available from multiple sources, each with their own proprietary chemistries to enhance potency and specificity of miRNA-mediated effects. To perform the reporter screen, the reporter construct is co-transfected into a relevant, easy-to-transfect cell type along with the miRNA mimic predicted to interact with the putative target site. Reporter expression should always be analyzed in relation to transfections with a negative control mimic to ensure changes in reporter expression do not reflect a non-specific response to small RNA delivery. Dual reporter assays enable the normalization of reporter signal that may be affected by variations in cell number, transfection efficiency, and other stochastic effects. The choice of cell type must be considered carefully, as high endogenous expression of the exogenously delivered miRNA mimic may result in saturation of the regulatory effect mediated at the putative target site. If expression profiles are known (see “Measuring miRNA Expression Levels” below), it is best to choose a cell type with low to moderate expression levels of the miRNAs to be tested. If not known, a viable strategy would be to analyze reporter expression in multiple, different cell types to ensure effects on reporter expression are not cell-type specific or related to endogenous miRNA expression. An example of the results generated from this type of reporter assay is shown in Fig. 3. A series of miRNA predicted to target the APP 3′-UTR by various predictor algorithms were transfected along with the specific APP 3′-UTR reporter construct. Reporter expression was assayed 48 h post-transfection, although other time points should also be considered.
Fig. 3.
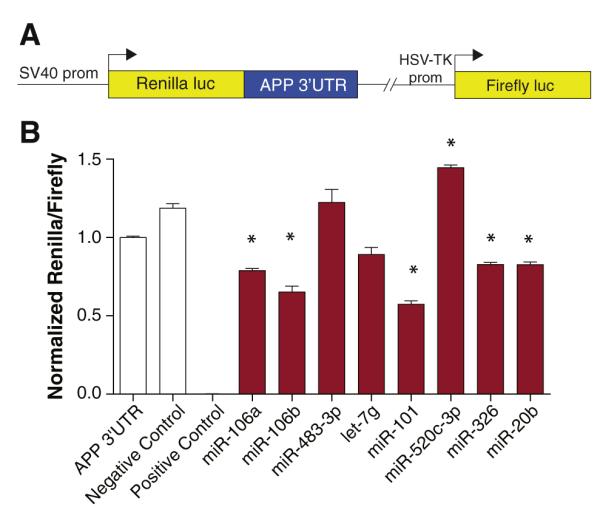
Validation of predicted miRNA targets by reporter assay. (A) An APP 3′-UTR reporter construct was generated as previously described (Long and Lahiri, 2011b). This reporter construct contains the APP 3′-UTR inserted downstream of the Renilla luciferase coding sequence and transcription driven by the SV40 promoter. Expression of firefly luciferase reporter is driven by an independent HSV-TK promoter and serves to normalize for experimental variables such as cell density, transfection efficiency, and cell toxicity. (B) HeLa cells were co-transfected with an APP 3′-UTR reporter construct and miRNAs (50 nM) predicted to target the APP 3′-UTR by various predictor algorithms. Negative control miRNA not predicted to target the APP 3′-UTR was also cotransfected. A positive control was included that consisted of co-transfection with miR-1 and a miR-1 MRE reporter. Renilla and firefly luciferase activity was assayed 48 h post-transfection using the Dual-Glo assay (Promega). Renilla luminescence values are normalized to firefly luminescence and scaled. Statistically significant differences were assessed by ANOVA followed by post-hoc Dunnett’s test. n=6; *p<0.05 relative to negative control miRNA.
Once the effect has been demonstrated at the reporter level, the exact sequence motif that enables target site binding to the miRNA can be assessed by introducing specific mutations in the predicted seed sequence through site-directed mutagenesis. If these mutations disrupt the effect on reporter expression, this location serves as the confirmed miRNA target site.
Validation of miRNA effects on native protein expression
To confirm that effects mediated by miRNAs on reporter expression translate to native protein expression, miRNA mimics are similarly transfected alone into a chosen cell type and native protein expression assayed by Western blot, ELISA or some other validated biochemical assay. Choice of cell type is more critical here, as endogenous expression of the native protein must be verified beforehand. Further, a more potent effect is likely to be observed if the cell type does not already express the exogenously supplied miRNAs at a high level. If possible, cell types relevant to the biological pathway of interest should be chosen so as to confirm that the regulatory relationship exists in a physiologically relevant context. Transfection efficiency may be an issue in difficult-to-transfect cell types (e.g. neurons), as miRNA effects are generally moderate and require high delivery efficiencies to be detected. In these cases, viral delivery of a miRNA-expressing gene cassette by either adeno-associated or lentivirus may be appropriate. These delivery systems are also particularly well-suited for in vivo delivery of miRNA. We and other investigators have employed these approaches in APP-miRNA studies to overexpress miRNA in difficult-to-transfect neuronal cell types (unpublished data, Vilardo et al., 2010). An exciting recent method for in vivo brain delivery of siRNA by systemical injection of targeted exosomes was found to be effective at reducing brain BACE1 levels and may also be suitable for systemic delivery of miRNA to the in vivo brain (Alvarez-Erviti et al., 2011).
Important caveats in functional validation studies
With standard transfection approaches in easy-to-transfect cell types, concentrations of delivered miRNA mimics might easily reach supraphysiological levels. Since high levels of miRNA may promote regulatory interactions that are not physiologically relevant, changes in reporter and native protein expression should be interpreted with caution. It is prudent to confirm changes in reporter and native protein expression following miRNA-target disruption (as discussed below) before making claims regarding the nature of the regulatory interaction.
Cell culture conditions may also dramatically alter miRNA biology, and maintaining uniform culture conditions between experiments is essential for ensuring reproducible results. In high density cultures, cell-to-cell contacts can induce global miRNA biogenesis leading to skewed expression levels (Hwang et al., 2009). Two separate reports suggest that proliferating cells and some cancer cell lines may utilize alternative polyadenylation sites that favor shorter 3′-UTRs and therefore the potential elimination of functional miRNA target sites (Mayr and Bartel, 2009; Sandberg et al., 2008). Further, cell cycle arrest can lead miRNAs to activate translation of their target transcripts rather than inhibit translation as occurs during normal cell cycle progression (Mortensen et al., 2011; Vasudevan et al., 2007). Therefore, cell culture conditions should be kept as constant as possible across all experiments.
Measuring miRNA expression levels
Subsequent to miRNA target validation, the natural question to address is whether validated target sites in the transcript of interest are physiologically regulated by the targeting endogenously expressed miRNA. The first requirement for physiological regulation is that the miRNA must be co-expressed with the GOI in relevant tissue and/or cell types. Therefore, prior to initiating experiments probing the endogenous role of miRNA in modulating GOI expression, it is appropriate to confirm that the miRNA is expressed in the tissue or cell model system. In the absence of published expression data for the targeting miRNAs in the model system being employed, it will be necessary to directly measure levels of the miRNAs, either in absolute copy numbers or relative to miRNA known to be highly and weakly expressed in the model system. When focusing on miRNA that target a GOI, generally low-throughput detection methods (e.g. Northern blot, RT-qPCR, in situ hybridization) should be sufficient since the number of functionally validated target sites is generally limited. If pursuing experimental methods for identifying miRNA target sites, as discussed above, high-throughput methods will be required (e.g. miRNA microarray, deep sequencing).
MiRNA expression analysis presents unique challenges as compared to standard mRNA expression analysis. To begin, the miRNA sequence is very short. Since most detection methods employ complementary oligonucleotide primer or probe hybridizations, this means there are limited choices for primer/probe sequence optimization. GC content and, therefore, melting temperatures (Tm) vary dramatically between miRNA sequences providing challenges in optimizing hybridization protocols. As a result, specificity may be compromised if no adjustments are applied. Additionally, high sequence similarity between miRNA paralogs provides additional challenges in designing primer/probes with sufficient sequence discrimination. Finally, the functional, mature miRNA sequence is also present in pri-miRNA and pre-miRNA molecules. Primer/probe design must allow for discrimination between these miRNA species. We will now briefly discuss the methods used in miRNA detection and how they have been adapted to circumvent these challenges.
Northern blot analysis
Northern blot miRNA analysis is performed using high percentage polyacrylamide gel electrophoresis (PAGE) of microgram quantities of total RNA followed by transfer to membrane, stabilization of small RNA on membrane by cross-linking or baking, hybridization of a labeled complementary probe to membrane, washing, and finally, detection of labeled probe by autoradiography or other detection method. While Northern blot is easy to implement and quite specific, its sensitivity is rather low relative to other detection techniques, requiring large quantities of input RNA. The assay also has a limited dynamic range. However, unlike other techniques, one can visually distinguish mature miRNA from pre-miRNA as a consequence of PAGE size fractionation. In fact, the original studies identifying the first miRNA utilized Northern analysis to identify short ~21-nt mature miRNA and longer ~61-nt pre-miRNA bands that would guide the detection of hairpin precursors (Lagos-Quintana et al., 2001; Lau et al., 2001; Lee and Ambros, 2001; Lee et al., 1993; Reinhart et al., 2000).
The same challenges described above in regard to probe design apply to Northern blotting. One modified probe design selectively incorporates locked nucleic acids (LNA) in place of standard nucleotides (Válóczi et al., 2004; Várallyay et al., 2007). LNA are nucleotides with modified ribose moieties containing a 2′-O,4′-C methylene bridge that locks the nucleic acid in a specific conformation. Oligonucleotides containing LNA have significantly enhanced Tm that increases with each subsequent LNA addition (Kurreck et al., 2002). This allows probes to be designed with optimal Tm by selective incorporation of LNA. This probe design results in higher sensitivity for detecting low abundance miRNAs and enables greater mismatch discrimination needed to distinguish between miRNA paralogs. Other modifications for enhancing Northern sensitivity have been described that target probe labeling. Standard probe end-labeling utilizes terminal deoxynucleotide transferase or polynucleotide kinase along with γ-32P-dATP to label the 5′- or 3′-terminal nucleotide with a radioactive phosphate. In the StarFire method (Behlke et al., 2000), the probe is instead appended with a 32P-labeled poly(A) tail. This is achieved by incorporating into the probe a 3′ hexamer extension that is recognized by a template oligonucleotide containing a 5′ stretch of poly(T) and a 3′ end complementary to the hexamer extension. Klenow polymerase and α-32P-dATP are then used to extend the 3′end of the probe with the poly(A) tail containing radiolabeled adenine nucleotides. This probe has much higher specific activity per molecule since it contains multiple radioactive phosphates and can be used to sensitively detect miRNAs (Sempere et al., 2004). Non-radioactive probe labeling can also be achieved by end-labeling with tags, such as digoxygenin (DIG) or biotin. Hybridized probe can then be detected using anti-DIG antibodies or streptavidin-conjugated enzymes compatible with standard chemiluminescent detection methods. This modification enables highly sensitive miRNA detection and significantly reduces exposure time for detecting signal as compared to radiolabeled probes (Kim et al., 2010).
Reverse transcription quantitative real-time polymerase chain reaction (RT-qPCR)
RT-qPCR is one of the most commonly used methods for detecting miRNA and considered a “gold standard” by many given its exquisite sensitivity as compared to other methods. RT-qPCR also has a much wider dynamic range (up to seven logs) than Northern. However, unlike Northern analysis, visualization of size-fractionated species cannot be used to distinguish mature miRNA from pre-miRNA amplicons. Like Northern analysis, the short length and varying GC content of miRNA presents a challenge for designing reverse transcription (RT) and qPCR primers and probes with necessary Tm and sequence specificity, especially for discriminating between highly similar paralogs. Many modifications have been developed for RT-qPCR detection of miRNA to compensate for these challenges.
The general RT-qPCR protocol begins by reverse transcribing total RNA (including miRNA) from cell or tissue extracts into cDNA followed by qPCR analysis of cDNA. Most protocols use special strategies during RT to ensure mature miRNA, and not pre-miRNA, are amplified during subsequent qPCR. Most also append universal sequences to cDNA by either miRNA ligation, miRNA tailing or as RT primer 5′ extensions to compensate for limited sequence options when designing qPCR primers and probes. During qPCR, both SYBR Green (Simpson et al., 2000) and hydrolysis probe (Heid et al., 1996) detection chemistries may be utilized to detect real-time amplicon accumulation. Hydrolysis probes add specificity to amplicon detection but require unique probe design for each target. SYBR Green is easier to implement but requires additional validation to ensure specific amplification, such as melting curve or gel analysis.
One of the most commonly used miRNA RT-qPCR methods employs miRNA-specific RT primers with complementarity to several ribonucleotides on the 3′ end of the targeted miRNA along with a stem-loop structure on the primer 5′ end (Chen et al., 2005). Since pre-miRNA species have additional nucleotides downstream of the mature sequence, steric hindrance between the RT primer secondary structure and pre-miRNA sequence impedes unwanted pre-miRNA cDNA synthesis. A miRNA-specific forward primer and reverse primer targeting a universal sequence located within the loop of the RT primer are used for qPCR amplification. The assay uses a hydrolysis probe for added specificity. Stem-loop RT-qPCR has been modified for use with SYBR Green by employing two miRNA-specific qPCR primers in a hemi-nested PCR design (Wan et al., 2010).
A second approach employs poly(A) polymerase (PAP) to append a poly(A) tail to miRNA 3′ termini (Shi and Chiang, 2005). cDNA is then synthesized with a RT primer containing a complementary 3′ oligo(dT) sequence and 5′ universal sequence. miRNA-specific forward and universal reverse qPCR primers are used with SYBR Green detection. A modification of this method uses the same RT strategy but uses two miRNA-specific qPCR primers that are spiked with LNA to enhance Tm and enable shorter primer design (Andreasen et al., 2010). The reverse qPCR primer is complementary to cDNA portions of both the mature miRNA and poly(A) tail, meaning that pre-miRNA sequences should not be amplified by this method. Recent reports claim that LNA-spiked primers may reduce PCR efficiency and that standard DNA primers with 5′ extensions to enhance Tm are equally successful for this method of miRNA RT-qPCR (Balcells et al., 2011).
A third strategy for RT-qPCR amplification uses T4 RNA ligase to attach a universal adapter sequence to miRNA 3′ termini prior to RT (Benes and Castoldi, 2010). RT proceeds using a primer against the universal sequence. Forward and reverse miRNA-specific qPCR primers bind to cDNA, with the reverse primer preventing pre-miRNA amplification by binding to cDNA portions of both the miRNA and 3′ adapter. The advantage of poly(A) tailing and adapter ligation methods is that there is global miRNA cDNA during RT. This means a universal cDNA pool can be used for qPCR analysis of any miRNA. In contrast, the stem-loop RT-qPCR method requires a separate RT reaction for each miRNA analysis.
When performing relative quantification, proper normalization of miRNA RT-qPCR data is a critical consideration. Small RNA with stable expression (such as other miRNA or small nuclear RNA) should be used as reference controls. Multiple reference gene normalization is the best approach for single miRNA analysis (Vandesompele et al., 2002). Other aspects of experimental set-up, validation, controls and normalization in RT-qPCR are beyond the scope of this review and discussed elsewhere (Bustin et al., 2009, 2010).
In situ hybridization (ISH)
ISH is likely not the first choice for surveying miRNA expression in a model system or across a panel of samples. The method is more technically challenging than Northern or RT-qPCR analysis and has low sensitivity and limited dynamic range. However, ISH is the only technique that provides miRNA expression data at cellular- and subcellular-level resolutions. Other detection methods measure miRNA levels averaged across all cell types in a heterogeneous sample. Therefore, in the CNS, where cell type diversity is high, ISH provides complementary information that may be crucial for proper data interpretation (Nelson and Wilfred, 2009). As with other methods discussed, the short length and sequence similarity of miRNAs provide the greatest challenge for designing ISH probes that specifically and sensitively detect endogenous miRNA. Technically, ISH probes may detect both mature miRNA and pre-miRNA, but pre-miRNA expression is often below the detection limit of ISH (Nuovo, 2008).
The general ISH protocol consists of tissue or cell fixation and sectioning followed by protease digestion to increase sample permeability and eliminate protein-RNA cross-links that may prevent probe hybridization. Samples are then incubated with labeled ISH probes (generally DIG or other hapten) against a specific miRNA species under high stringency conditions (high temperature and stringent hybridization buffer) and then washed with high stringency buffers. Probe is detected microscopically based on the type of label used (often by using an enzymatically-labeled primary antibody against probe hapten). High stringency incubation and washes are needed in ISH to prevent binding of probes with non-specific targets. But this need for high stringency is in direct conflict with the design constraints imposed by short miRNA length and low probe Tm. Though some protocols have been published using standard RNA oligonucleotide probes (Deo et al., 2006; Thompson et al., 2007), most protocols compensate for short probe length by using LNA-modified probes. These probes have elevated Tm, higher sensitivity for miRNA detection and enhanced single base mismatch discrimination for paralogs (Kloosterman et al., 2006). Notably, these probes have also been successfully used in formalin fixed and paraffin embedded (FFPE) specimens and cryosections (Nelson et al., 2006; Obernosterer et al., 2007). This approach along with RT in situ PCR and immunohistochemistry can also be used to simultaneously detect mature miRNA, pre-miRNA and miRNA protein targets within the same or serial sections (Nuovo, 2008; Nuovo et al., 2009). Signal amplification strategies used with LNA-fluorescence ISH (LNA-FISH) greatly enhance ISH sensitivity. Examples include tyramide amplification (Silahtaroglu et al., 2007) and enzyme linked fluorescence (ELF). In ELF, alkaline phosphatase converts reagent substrate to a photostable, highly fluorescent precipitate. The method is sensitive enough to detect single miRNA molecules in situ (Lu and Tsourkas, 2009). A separate strategy for enhancing ISH sensitivity is to increase cross-linking efficiency. Formaldehyde cross-links are reversible and may cause some miRNA release during processing. Dual fixation with formaldehyde and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) leads to increased miRNA retention and enhanced sensitivity (Pena et al., 2009), though not all studies have replicated this effect (Jørgensen et al., 2010; Lu and Tsourkas, 2009; Søe et al., 2011).
High-throughput methods for miRNA detection
High-throughput methods are generally not needed by investigators studying miRNA regulation of a GOI, so a light treatment is provided here. The exception would be if experimental techniques for identifying miRNA targets in a transcript are employed. In such cases, microarray analysis of total RNA, deep sequencing of small RNA libraries, or high-throughput RT-qPCR assays are the typical choices for high content analysis. These experiments can be technically challenging and contain certain pitfalls of which investigators should be cognizant (Nelson et al., 2008).
Microarray analysis is the most widely used method for high-throughput analysis and is cheaper and less technically challenging than deep sequencing. Early variants included a membrane-based, manually-spotted dot blot (Krichevsky et al., 2003) along with numerous custom-printed, slide-based microarrays. As with the other methods described, LNA have been incorporated into microarray design by using Tm-normalized LNA capture probes (Castoldi et al., 2006) with enhanced sensitivity over standard probes. Published methods also vary in the mechanism of small RNA labeling, including radioactive (Krichevsky et al., 2003), biotin (Sun et al., 2004) and fluorophore end-labeling (Babak et al., 2004), along with strategies to label small RNA during first strand cDNA synthesis (Liu et al., 2004; Zhao et al., 2006), by PCR amplification with labeled primers (Baskerville and Bartel, 2005; Miska et al., 2004), and by poly(A) tailing using PAP (Shingara et al., 2005). Commercially available microarrays from Agilent (Wang et al., 2007), Exiqon (Castoldi et al., 2006), and Affymetrix, among others, are now commonly employed and have been analyzed for cross-platform concordance (Git et al., 2010; Pradervand et al., 2010).
Deep sequencing is more technically challenging and expensive than microarray analysis and requires significant bioinformatic analysis for data interpretation. However, unlike microarray analysis, deep sequencing is unbiased in that it is not limited by current miRNA database annotations. This means deep sequencing can be used for simultaneous novel miRNA discovery and digital expression analysis and can detect miRNA terminal modifications that might interfere with microarray detection (Newman et al., 2011). Commercial deep sequencing platforms used for miRNA analysis include the 454 (Ruby et al., 2006), Illumina (Morin et al., 2008; Shao et al., 2010), and SOLiD (Hafner et al., 2008) sequencing systems. Despite being relatively unbiased relative to microarray analyses, recent studies demonstrate that deep sequencing does exhibit some sequence biases related to adapter ligations during cDNA library preparation (Hafner et al., 2011; Linsen et al., 2009). miRNA sequence and secondary structure apparently underlie these biases. However, since these sequence biases are highly reproducible, relative quantification between samples should be unaffected.
Finally, high-throughput RT-qPCR arrays are available and offer the high sensitivity and large dynamic range of qPCR analysis on a high-throughput scale (Mestdagh et al., 2008). However, similar to microarray analysis, these assays only profile a subset of miRNAs present in annotated databases. Distinct normalization strategies are recommended when performing global miRNA RT-qPCR analysis as compared to single miRNA analysis (Mestdagh et al., 2009).
Disrupting endogenous miRNA function to confirm physiological regulation
After confirming that a miRNA modulates both reporter and native protein expression and is expressed in the relevant tissue or cell model system, a remaining question to address is whether the miRNA can be shown to participate in endogenous regulation of the transcript of interest. Certain tools are available to disrupt the interaction between endogenous miRNAs and endogenous mRNA target sites.
Antisense inhibition of miRNA function
The first molecular tool is the miRNA antisense inhibitor (antimiR). These are RNA oligonucleotides that exhibit perfect complementarity to the targeted miRNA and inhibit miRNA function through an antisense mechanism. These inhibitors generally incorporate chemical modifications to enhance stability and hybridization strength in the form of 2′-O-methylation (Hutvágner et al., 2004), LNA (Ørom et al., 2006), phosphorothioate backbone modifications (Esau et al., 2006; Lennox and Behlke, 2010) or various design implementations shown to enhance potency, such as hairpin secondary structure (Vermeulen et al., 2007). Inhibitor designs incorporating cholesterol conjugates, termed antagomirs, enhance in vivo delivery (Krützfeldt et al., 2005). Short, fully LNA- and phosphorothioate-modified oligos termed tiny LNAs inhibit whole miRNA families by binding to seed sequences and are taken up in vivo in unconjugated form (Obad et al., 2011). Lentiviral-based delivery of miRNA inhibitors driven from a human H1 promoter (Scherr et al., 2007) may be useful when transfection efficiency is problematic or in vivo inhibition is needed.
Genetic approaches for disrupting miRNA function
Genetic manipulation in mouse models is another strategy for eliminating miRNA function. One transgenic strategy employs miRNA sponges (Ebert and Sharp, 2010; Ebert et al., 2007) that work in a similar manner to antisense oligonucleotide inhibitors. These transgenes express transcripts containing tandem repeats of complementary sequences targeted against a specific miRNA. Other genetic approaches employ gene knockouts. One strategy has been to globally reduce miRNA production by conditional deletion of Dicer in the CNS (Davis et al., 2008; Hébert et al., 2010; Konopka et al., 2010; Schaefer et al., 2007). Driving postnatal loss of Dicer in Purkinje cells using a Pcp2-Cre system (Schaefer et al., 2007), in forebrain using an α-CaMKII-Cre system (Davis et al., 2008; Hébert et al., 2010), or even in astroglia using a Gfap-Cre system results in a slowly developing neurodegenerative phenotype, though early learning and cognition may actually be enhanced in certain models (Konopka et al., 2010). An issue with this strategy is that its global approach does not allow one to dissect the contribution of single miRNA to the phenotype. Also, non-canonical processing of some miRNAs persists in the absence of Dicer or Dgcr8 (Babiarz et al., 2011). Instead, single miRNA knockouts can be utilized (Magill et al., 2010; Zhao et al., 2007). However, miRNA genomic architecture may make this difficult or impossible in many cases. Due to gene duplication events, many miRNA are expressed from multiple loci, requiring sequential deletion events for complete knockout. Further, many miRNA are clustered in the genome and expressed as polycistronic transcripts. Therefore, designing a knockout strategy that only eliminates expression of a single miRNA in the cluster is challenging. As with any knockout strategy, functional redundancy may compensate for loss of miRNA function over time, complicating interpretation of animal phenotypes.
Genetic deletion and miRNA inhibitors block the functionality of the miRNA with all of its target transcripts. Therefore, it is difficult to confirm whether the response to miRNA disruption is due to blockade of the interaction between the miRNA and the transcript of interest or due to indirect effects mediated by blockade with other target transcripts.
Target site interference of miRNA function
A more targeted approach is to block the interaction of the miRNA with a specific target site in a transcript of interest. Target protectors are modified synthetic RNA or morpholino oligonucleotides (Choi et al., 2007) or transgenically overexpressed RNA species (Gehrke et al., 2010) that are complementary to a specific target site and block the hybridization of targeting miRNAs. Due to chemical modifications, synthetic target protectors do not load into RISC and therefore do not mediate siRNA-like effects. Target protectors are not just for experimental manipulation. An endogenous long, non-coding RNA, which targets BACE1 transcript and is dysregulated in AD, acts like a target protector by blocking interaction between the transcript and miR-485-5p (Faghihi et al., 2010).
Delivery of miRNA modulators
Experiments utilizing either inhibitors or target protectors simply require they be transfected or otherwise delivered into cell culture or other model systems followed by the measurement of gene expression. In vivo delivery of miRNA modulators to the CNS is still difficult but direct cortical or intraventricular injection of modified synthetic molecules (Krützfeldt et al., 2007) or transgene-expressing viral particles is an option. The exosome strategy previously mentioned may also represent a facile method for systemic administration of miRNA modulators to the CNS and warrants further testing (Alvarez-Erviti et al., 2011). Finally, it is important to be aware that many regulatory networks have built-in redundancy and robust compensatory feedback loops to maintain homeostatic levels of critical proteins. Therefore, even subtle changes in expression following manipulation by miRNA inhibitors or target protectors may be physiologically meaningful in this context.
Conclusions
The search for miRNAs that regulate gene products implicated in CNS disorders is likely to reveal novel drug targets. However, the appropriate tools are needed to make such discoveries. This review has outlined many of those tools available to researchers pursuing this endeavor. The experimental workflow and methodologies discussed herein are summarized in Fig. 4. A specific example of how this workflow was implemented in Long and Lahiri (2011c) is also illustrated. Each method has important limitations that have been discussed and should be thoughtfully considered prior to incorporation in an experimental paradigm. Through careful implementation of the appropriate methodologies, we are optimistic that important discoveries related to miRNA function in CNS disorders are on the horizon. Hopefully breakthrough discoveries on this front will lead to the development of novel therapeutic strategies that alleviate human suffering.
Fig. 4.

Summary of methodological workflow for studying miRNA regulation of GOI. (A) The workflow described in text begins with a specific predetermined GOI. Putative target sites in the transcript of the GOI are identified by either bioinformatic or experimental approaches. Target sites are then validated by both 3′-UTR reporter assays and direct analysis of native protein levels following transfection of putative targeting miRNA. If not already known, miRNA levels are then measured in the experimental model of choice to confirm relevant expression levels. Endogenous miRNA function is then disrupted to assess whether endogenous miRNAs physiologically regulate expression of the GOI. (B) A specific example of the experimental workflow employed by Long and Lahiri (2011c) is presented. Predictor algorithms predicted two putative miR-101 target sites in the APP 3′-UTR. The APP 3′-UTR was cloned into a dual luciferase reporter construct. The 3′ -UTR reporter was co-transfected into cultured human cells along with miR-101 mimics. Luciferase expression was assessed using the dual luciferase assay. Following site validation, the effect of miR-101 on native APP mRNA and protein expression was assessed by transfection of miR-101 followed by RT-qPCR and Western blot analysis of cell extracts. Endogenous miRNA regulation was also assessed in separate experiments by transfecting a miR-101 inhibitor and target protector.
Acknowledgments
Funding was provided by grants from Alzheimer’s Association (Zenith award) and the NIH (AG18379 and AG18884) to DKL.
Abbreviations
- CNS
central nervous system
- GOI
gene of interest
- mRNA
messenger RNA
- siRNA
short interfering RNA
- miRNA
microRNA
- pri-miRNA
primary microRNA
- pre-miRNA
microRNA precursor
- AD
Alzheimer disease
- Aβ
amyloid-β
- APP
amyloid-β precursor protein
- UTR
untranslated region
- RNP
ribonucleoprotein
- miRNP
microribonucleo-protein
- RISC
RNA-induced silencing complex
- MRE
microRNA recognition element
- CDS
coding sequence
- LAMP
labeled microRNA pull down assay
- IP
immunoprecipitation
- RIP-Chip
ribonucleoprotein immunoprecipitation followed by microarray chip analysis
- TAP-Tar
tandem affinity purification of microRNA targets
- HITS-CLIP
high throughput sequencing of RNA isolated by crosslinking and immunopurification
- CIMS
crosslinking induced mutation sites
- PAR-CLIP
photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation
- PAGE
polyacrylamide gel electrophoresis
- LNA
locked nucleic acid
- Tm
melting temperature
- DIG
digoxygenin
- RT-qPCR
reverse transcription quantitative real-time polymerase chain reaction
- PAP
poly(A) polymerase
- FFPE
formalin fixed paraffin embedded
- ISH
in situ hybridization
- FISH
fluorescent in situ hybridization
- ELF
enzyme linked fluorescence
References
- Alley GM, Bailey JA, Chen D, Ray B, Puli LK, Tanila H, Banerjee PK, Lahiri DK. Memantine lowers amyloid-beta peptide levels in neuronal cultures and in APP/PS1 transgenic mice. J. Neurosci. Res. 2010;88:143–154. doi: 10.1002/jnr.22172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJA. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011;29:341–345. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- Andreasen D, Fog JU, Biggs W, Salomon J, Dahslveen IK, Baker A, Mouritzen P. Improved microRNA quantification in total RNA from clinical samples. Methods. 2010;50:S6–S9. doi: 10.1016/j.ymeth.2010.01.006. [DOI] [PubMed] [Google Scholar]
- Babak T, Zhang W, Morris Q, Blencowe BJ, Hughes TR. Probing microRNAs with microarrays: tissue specificity and functional inference. RNA. 2004;10:1813–1819. doi: 10.1261/rna.7119904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiarz JE, Hsu R, Melton C, Thomas M, Ullian EM, Blelloch R. A role for noncanonical microRNAs in the mammalian brain revealed by phenotypic differences in Dgcr8 versus Dicer1 knockouts and small RNA sequencing. RNA. 2011;17:1489–1501. doi: 10.1261/rna.2442211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Ray B, Greig NH, Lahiri DK. Rivastigmine lowers Aβ and increases sAPPα levels, which parallel elevated synaptic markers and metabolic activity in degenerating primary rat neurons. PLoS One. 2011;6:e21954. doi: 10.1371/journal.pone.0021954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balcells I, Cirera S, Busk PK. Specific and sensitive quantitative RT-PCR of miRNAs with DNA primers. BMC Biotechnol. 2011;11:70. doi: 10.1186/1472-6750-11-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Baskerville S, Bartel DP. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA. 2005;11:241–247. doi: 10.1261/rna.7240905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker RE, Greig NH. Why so few drugs for Alzheimer’s disease? Are methods failing drugs? Curr. Alzheimer Res. 2010;7:642–651. doi: 10.2174/156720510793499075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker C, Hammerle-Fickinger A, Riedmaier I, Pfaffl MW. mRNA and micro-RNA quality control for RT-qPCR analysis. Methods. 2010;50:237–243. doi: 10.1016/j.ymeth.2010.01.010. [DOI] [PubMed] [Google Scholar]
- Behlke MA, Dames SA, McDonald WH, Gould KL, Devor EJ, Walder JA. Use of high specific activity StarFire oligonucleotide probes to visualize low-abundance pre-mRNA splicing intermediates in S. pombe. Biotechniques. 2000;29:892–897. doi: 10.2144/00294pf01. [DOI] [PubMed] [Google Scholar]
- Beitzinger M, Peters L, Zhu JY, Kremmer E, Meister G. Identification of human microRNA targets from isolated argonaute protein complexes. RNA Biol. 2007;4:76–84. doi: 10.4161/rna.4.2.4640. [DOI] [PubMed] [Google Scholar]
- Benes V, Castoldi M. Expression profiling of microRNA using real-time quantitative PCR, how to use it and what is available. Methods. 2010;50:244–249. doi: 10.1016/j.ymeth.2010.01.026. [DOI] [PubMed] [Google Scholar]
- Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: back to the future. Neuron. 2010;68:270–281. doi: 10.1016/j.neuron.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–D153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betel D, Koppal A, Agius P, Sander C, Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010;11:R90. doi: 10.1186/gb-2010-11-8-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burroughs AM, Kawano M, Ando Y, Daub CO, Hayashizaki Y. pre-miRNA profiles obtained through application of locked nucleic acids and deep sequencing reveals complex 5′/3′ arm variation including concomitant cleavage and polyuridylation patterns. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr903. doi:10.1093/nar/gkr903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- Bustin SA, Beaulieu J-F, Huggett J, Jaggi R, Kibenge FSB, Olsvik PA, Penning LC, Toegel S. MIQE précis: practical implementation of minimum standard guidelines for fluorescence-based quantitative real-time PCR experiments. BMC Mol. Biol. 2010;11:74. doi: 10.1186/1471-2199-11-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoldi M, Schmidt S, Benes V, Noerholm M, Kulozik AE, Hentze MW, Muckenthaler MU. A sensitive array for microRNA expression profiling (miChip) based on locked nucleic acids (LNA) RNA. 2006;12:913–920. doi: 10.1261/rna.2332406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Grosshans H. Active turnover modulates mature microRNA activity in Caenorhabditis elegans. Nature. 2009;461:546–549. doi: 10.1038/nature08349. [DOI] [PubMed] [Google Scholar]
- Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, Shiekhattar R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436:740–744. doi: 10.1038/nature03868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W-Y, Giraldez AJ, Schier AF. Target protectors reveal dampening and balancing of Nodal agonist and antagonist by miR-430. Science. 2007;318:271–274. doi: 10.1126/science.1147535. [DOI] [PubMed] [Google Scholar]
- Cogswell JP, Ward J, Taylor IA, Waters M, Shi Y, Cannon B, Kelnar K, Kemppainen J, Brown D, Chen C, Prinjha RK, Richardson JC, Saunders AM, Roses AD, Richards CA. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J. Alzheimers Dis. 2008;14:27–41. doi: 10.3233/jad-2008-14103. [DOI] [PubMed] [Google Scholar]
- Davis TH, Cuellar TL, Koch SM, Barker AJ, Harfe BD, McManus MT, Ullian EM. Conditional loss of Dicer disrupts cellular and tissue morphogenesis in the cortex and hippocampus. J. Neurosci. 2008;28:4322–4330. doi: 10.1523/JNEUROSCI.4815-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deo M, Yu J-Y, Chung K-H, Tippens M, Turner DL. Detection of mammalian microRNA expression by in situ hybridization with RNA oligonucleotides. Dev. Dyn. 2006;235:2538–2548. doi: 10.1002/dvdy.20847. [DOI] [PubMed] [Google Scholar]
- Easow G, Teleman AA, Cohen SM. Isolation of microRNA targets by miRNP immunopurification. RNA. 2007;13:1198–1204. doi: 10.1261/rna.563707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert MS, Sharp PA. MicroRNA sponges: progress and possibilities. RNA. 2010;16:2043–2050. doi: 10.1261/rna.2414110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat. Methods. 2007;4:721–726. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmén J, Lindow M, Silahtaroglu A, Bak M, Christensen M, Lind-Thomsen A, Hedtjärn M, Hansen JB, Hansen HF, Straarup EM, McCullagh K, Kearney P, Kauppinen S. Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. 2008;36:1153–1162. doi: 10.1093/nar/gkm1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, Subramaniam A, Propp S, Lollo BA, Freier S, Bennett CF, Bhanot S, Monia BP. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Faghihi MA, Zhang M, Huang J, Modarresi F, Van der Brug MP, Nalls MA, Cookson MR, St-Laurent G, III, Wahlestedt C. Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome Biol. 2010;11:R56. doi: 10.1186/gb-2010-11-5-r56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Liu Y, Jiang J, Ma Z, Wu H, Liu T, Liu M, Li X, Tang H. miR-20a promotes proliferation and invasion by targeting APP in human ovarian cancer cells. Acta Biochim. Biophys. Sin. (Shanghai) 2010;42:318–324. doi: 10.1093/abbs/gmq026. [DOI] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev. Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantier MP, McCoy CE, Rusinova I, Saulep D, Wang D, Xu D, Irving AT, Behlke MA, Hertzog PJ, Mackay F, Williams BRG. Analysis of microRNA turnover in mammalian cells following Dicer1 ablation. Nucleic Acids Res. 2011;39:5692–5703. doi: 10.1093/nar/gkr148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y-W, Ghosh C, Song W, Maloney B, Lahiri DK. Mechanism of promoter activity of the beta-amyloid precursor protein gene in different cell lines: identification of a specific 30 bp fragment in the proximal promoter region. J. Neurochem. 2004;90:1432–1444. doi: 10.1111/j.1471-4159.2004.02608.x. [DOI] [PubMed] [Google Scholar]
- Gehrke S, Imai Y, Sokol N, Lu B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. 2010;466:637–641. doi: 10.1038/nature09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Git A, Dvinge H, Salmon-Divon M, Osborne M, Kutter C, Hadfield J, Bertone P, Caldas C. Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression. RNA. 2010;16:991–1006. doi: 10.1261/rna.1947110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Farh KK-H, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landgraf P, Ludwig J, Rice A, Ojo T, Lin C, Holoch D, Lim C, Tuschl T. Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods. 2008;44:3–12. doi: 10.1016/j.ymeth.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jr., Jungkamp A-C, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010a;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jungkamp A-C, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T. PAR-CliP—a method to identify transcriptome-wide the binding sites of RNA binding proteins. J. Vis. Exp. 2010b;41:e2034. doi: 10.3791/2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Renwick N, Brown M, Mihailović A, Holoch D, Lin C, Pena JTG, Nusbaum JD, Morozov P, Ludwig J, Ojo T, Luo S, Schroth G, Tuschl T. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA. 2011;17:1697–1712. doi: 10.1261/rna.2799511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom K-H, Kim Y-K, Jin H, Kim VN. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hausser J, Landthaler M, Jaskiewicz L, Gaidatzis D, Zavolan M. Relative contribution of sequence and structure features to the mRNA binding of Argonaute/EIF2C-miRNA complexes and the degradation of miRNA targets. Genome Res. 2009;19:2009–2020. doi: 10.1101/gr.091181.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hébert SS, Horré K, Nicolaï L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, Kauppinen S, Delacourte A, De Strooper B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. U. S. A. 2008;105:6415–6420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hébert SS, Horré K, Nicolaï L, Bergmans B, Papadopoulou AS, Delacourte A, De Strooper B. MicroRNA regulation of Alzheimer’s amyloid precursor protein expression. Neurobiol. Dis. 2009;33:422–428. doi: 10.1016/j.nbd.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Hébert SS, Papadopoulou AS, Smith P, Galas M-C, Planel E, Silahtaroglu AN, Sergeant N, Buée L, De Strooper B. Genetic ablation of Dicer in adult forebrain neurons results in abnormal tau hyperphosphorylation and neurodegeneration. Hum. Mol. Genet. 2010;19:3959–3969. doi: 10.1093/hmg/ddq311. [DOI] [PubMed] [Google Scholar]
- Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- Hendrickson DG, Hogan DJ, Herschlag D, Ferrell JE, Brown PO. Systematic identification of mRNAs recruited to argonaute 2 by specific microRNAs and corresponding changes in transcript abundance. PLoS One. 2008;3:e2126. doi: 10.1371/journal.pone.0002126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson DG, Hogan DJ, McCullough HL, Myers JW, Herschlag D, Ferrell JE, Brown PO. Concordant regulation of translation and mRNA abundance for hundreds of targets of a human microRNA. PLoS Biol. 2009;7:e1000238. doi: 10.1371/journal.pbio.1000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo I, Joo C, Cho J, Ha M, Han J, Kim VN. Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Mol. Cell. 2008;32:276–284. doi: 10.1016/j.molcel.2008.09.014. [DOI] [PubMed] [Google Scholar]
- Heo I, Joo C, Kim Y-K, Ha M, Yoon M-J, Cho J, Yeom K-H, Han J, Kim VN. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell. 2009;138:696–708. doi: 10.1016/j.cell.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Hong X, Hammell M, Ambros V, Cohen SM. Immunopurification of Ago1 miRNPs selects for a distinct class of microRNA targets. Proc. Natl. Acad. Sci. U. S. A. 2009;106:15085–15090. doi: 10.1073/pnas.0908149106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu R-J, Tsai H-J. Performing the Labeled microRNA pull-down (LAMP) assay system: an experimental approach for high-throughput identification of microRNA-target mRNAs. Methods Mol. Biol. 2011;764:241–247. doi: 10.1007/978-1-61779-188-8_16. [DOI] [PubMed] [Google Scholar]
- Hsu R-J, Yang H-J, Tsai H-J. Labeled microRNA pull-down assay system: an experimental approach for high-throughput identification of microRNA-target mRNAs. Nucleic Acids Res. 2009;37:e77. doi: 10.1093/nar/gkp274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutvágner G, McLachlan J, Pasquinelli AE, Bálint E, Tuschl T, Zamore PD. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293:834–838. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- Hutvágner G, Simard MJ, Mello CC, Zamore PD. Sequence-specific inhibition of small RNA function. PLoS Biol. 2004;2:E98. doi: 10.1371/journal.pbio.0020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang H-W, Wentzel EA, Mendell JT. Cell–cell contact globally activates microRNA biogenesis. Proc. Natl. Acad. Sci. U. S. A. 2009;106:7016–7021. doi: 10.1073/pnas.0811523106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Satoh M, Pauley KM, Fritzler MJ, Reeves WH, Chan EKL. Detection of the argonaute protein Ago2 and microRNAs in the RNA induced silencing complex (RISC) using a monoclonal antibody. J. Immunol. Methods. 2006;317:38–44. doi: 10.1016/j.jim.2006.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human Micro-RNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen S, Baker A, Møller S, Nielsen BS. Robust one-day in situ hybridization protocol for detection of microRNAs in paraffin samples using LNA probes. Methods. 2010;52:375–381. doi: 10.1016/j.ymeth.2010.07.002. [DOI] [PubMed] [Google Scholar]
- Kai ZS, Pasquinelli AE. MicroRNA assassins: factors that regulate the disappearance of miRNAs. Nat. Struct. Mol. Biol. 2010;17:5–10. doi: 10.1038/nsmb.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karginov FV, Conaco C, Xuan Z, Schmidt BH, Parker JS, Mandel G, Hannon GJ. A biochemical approach to identifying microRNA targets. Proc. Natl. Acad. Sci. U. S. A. 2007;104:19291–19296. doi: 10.1073/pnas.0709971104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh T, Sakaguchi Y, Miyauchi K, Suzuki T, Kashiwabara S-I, Baba T, Suzuki T. Selective stabilization of mammalian microRNAs by 3′ adenylation mediated by the cytoplasmic poly(A) polymerase GLD-2. Genes Dev. 2009;23:433–438. doi: 10.1101/gad.1761509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- Kim SW, Li Z, Moore PS, Monaghan AP, Chang Y, Nichols M, John B. A sensitive non-radioactive northern blot method to detect small RNAs. Nucleic Acids Res. 2010;38:e98. doi: 10.1093/nar/gkp1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiriakidou M, Nelson PT, Kouranov A, Fitziev P, Bouyioukos C, Mourelatos Z, Hatzigeorgiou A. A combined computational-experimental approach predicts human microRNA targets. Genes Dev. 2004;18:1165–1178. doi: 10.1101/gad.1184704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman WP, Wienholds E, de Bruijn E, Kauppinen S, Plasterk RHA. In situ detection of miRNAs in animal embryos using LNA-modified oligonucleotide probes. Nat. Methods. 2006;3:27–29. doi: 10.1038/nmeth843. [DOI] [PubMed] [Google Scholar]
- Konopka W, Kiryk A, Novak M, Herwerth M, Parkitna JR, Wawrzyniak M, Kowarsch A, Michaluk P, Dzwonek J, Arnsperger T, Wilczynski G, Merkenschlager M, Theis FJ, Köhr G, Kaczmarek L, Schütz G. MicroRNA loss enhances learning and memory in mice. J. Neurosci. 2010;30:14835–14842. doi: 10.1523/JNEUROSCI.3030-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause JE, Chenard BL. Opportunities and challenges in the discovery of new central nervous system drugs. Ann. N. Y. Acad. Sci. 2008;1144:243–250. doi: 10.1196/annals.1418.024. [DOI] [PubMed] [Google Scholar]
- Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. Combinatorial microRNA target predictions. Nat. Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- Krichevsky AM, King KS, Donahue CP, Khrapko K, Kosik KS. A microRNA array reveals extensive regulation of microRNAs during brain development. RNA. 2003;9:1274–1281. doi: 10.1261/rna.5980303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol J, Busskamp V, Markiewicz I, Stadler MB, Ribi S, Richter J, Duebel J, Bicker S, Fehling HJ, Schübeler D, Oertner TG, Schratt G, Bibel M, Roska B, Filipowicz W. Characterizing light-regulated retinal microRNAs reveals rapid turnover as a common property of neuronal microRNAs. Cell. 2010;141:618–631. doi: 10.1016/j.cell.2010.03.039. [DOI] [PubMed] [Google Scholar]
- Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with “antagomirs”. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- Krützfeldt J, Kuwajima S, Braich R, Rajeev KG, Pena J, Tuschl T, Manoharan M, Stoffel M. Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res. 2007;35:2885–2892. doi: 10.1093/nar/gkm024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurreck J, Wyszko E, Gillen C, Erdmann VA. Design of antisense oligonucleotides stabilized by locked nucleic acids. Nucleic Acids Res. 2002;30:1911–1918. doi: 10.1093/nar/30.9.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- Lahiri DK. Application of a multidisciplinary approach to Alzheimer’s disease to develop novel drug targets by integrating biomarkers, imaging, genetic and molecular strategies. Curr. Alzheimer Res. 2011;8:1–3. doi: 10.2174/156720511794604534. [DOI] [PubMed] [Google Scholar]
- Lahiri DK, Nall C. Promoter activity of the gene encoding the beta-amyloid precursor protein is up-regulated by growth factors, phorbol ester, retinoic acid and interleukin-1. Brain Res. Mol. Brain Res. 1995;32:233–240. doi: 10.1016/0169-328x(95)00078-7. [DOI] [PubMed] [Google Scholar]
- Lahiri DK, Robakis NK. The promoter activity of the gene encoding Alzheimer beta-amyloid precursor protein (APP) is regulated by two blocks of upstream sequences. Brain Res. Mol. Brain Res. 1991;9:253–257. doi: 10.1016/0169-328x(91)90009-m. [DOI] [PubMed] [Google Scholar]
- Lahiri DK, Farlow MR, Sambamurti K, Greig NH, Giacobini E, Schneider LS. A critical analysis of new molecular targets and strategies for drug developments in Alzheimer’s disease. Curr. Drug Targets. 2003;4:97–112. doi: 10.2174/1389450033346957. [DOI] [PubMed] [Google Scholar]
- Lahiri DK, Ge Y-W, Maloney B. Characterization of the APP proximal promoter and 5′-untranslated regions: identification of cell type-specific domains and implications in APP gene expression and Alzheimer’s disease. FASEB J. 2005;19:653–655. doi: 10.1096/fj.04-2900fje. [DOI] [PubMed] [Google Scholar]
- Lahiri DK, Maloney B, Zawia NH. The LEARn model: an epigenetic explanation for idiopathic neurobiological diseases. Mol. Psychiatry. 2009;14:992–1003. doi: 10.1038/mp.2009.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam LT, Lu X, Zhang H, Lesniewski R, Rosenberg S, Semizarov D. A micro-RNA screen to identify modulators of sensitivity to BCL2 inhibitor ABT-263 (navitoclax) Mol. Cancer Ther. 2010;9:2943–2950. doi: 10.1158/1535-7163.MCT-10-0427. [DOI] [PubMed] [Google Scholar]
- Landthaler M, Gaidatzis D, Rothballer A, Chen PY, Soll SJ, Dinic L, Ojo T, Hafner M, Zavolan M, Tuschl T. Molecular characterization of human Argonaute-containing ribonucleoprotein complexes and their bound target mRNAs. RNA. 2008;14:2580–2596. doi: 10.1261/rna.1351608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson E, Sander C, Marks D. mRNA turnover rate limits siRNA and microRNA efficacy. Mol. Syst. Biol. 2010;6:433. doi: 10.1038/msb.2010.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- Lee Y, Kim M, Han J, Yeom K-H, Lee S, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennox KA, Behlke MA. A direct comparison of anti-microRNA oligonucleotide potency. Pharm. Res. 2010;27:1788–1799. doi: 10.1007/s11095-010-0156-0. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Shih I.-hung, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- Linsen SEV, de Wit E, Janssens G, Heater S, Chapman L, Parkin RK, Fritz B, Wyman SK, de Bruijn E, Voest EE, Kuersten S, Tewari M, Cuppen E. Limitations and possibilities of small RNA digital gene expression profiling. Nat. Methods. 2009;6:474–476. doi: 10.1038/nmeth0709-474. [DOI] [PubMed] [Google Scholar]
- Linsley PS, Schelter J, Burchard J, Kibukawa M, Martin MM, Bartz SR, Johnson JM, Cummins JM, Raymond CK, Dai H, Chau N, Cleary M, Jackson AL, Carleton M, Lim L. Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Mol. Cell. Biol. 2007;27:2240–2252. doi: 10.1128/MCB.02005-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C-G, Calin GA, Meloon B, Gamliel N, Sevignani C, Ferracin M, Dumitru CD, Shimizu M, Zupo S, Dono M, Alder H, Bullrich F, Negrini M, Croce CM. An oligonucleotide microchip for genome-wide microRNA profiling in human and mouse tissues. Proc. Natl. Acad. Sci. U. S. A. 2004;101:9740–9744. doi: 10.1073/pnas.0403293101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Liu C, Zhu J, Shu P, Yin B, Gong Y, Qiang B, Yuan J, Peng X. MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer’s-associated pathogenesis in SAMP8 mice. Neurobiol. Aging. 2010 doi: 10.1016/j.neurobiolaging.2010.04.034. doi:10.1016/j.neurobiolaging.2010.04.034. [DOI] [PubMed] [Google Scholar]
- Long JM, Lahiri DK. A novel microRNA interaction relevant to Alzheimer’s disease pathogenesis and drug target discovery: miR-346 induces expression of amyloid precursor protein. Alzheimers Dement. 2011a;7:S566–S567. [Google Scholar]
- Long JM, Lahiri DK. Current drug targets for modulating Alzheimer’s amyloid precursor protein: role of specific micro-RNA species. Curr. Med. Chem. 2011b;18:3314–3321. doi: 10.2174/092986711796504592. [DOI] [PubMed] [Google Scholar]
- Long JM, Lahiri DK. MicroRNA-101 downregulates Alzheimer’s amyloid-β precursor protein levels in human cell cultures and is differentially expressed. Biochem. Biophys. Res. Commun. 2011c;404:889–895. doi: 10.1016/j.bbrc.2010.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Tsourkas A. Imaging individual microRNAs in single mammalian cells in situ. Nucleic Acids Res. 2009;37:e100. doi: 10.1093/nar/gkp482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukiw WJ. Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippo-campus. Neuroreport. 2007;18:297–300. doi: 10.1097/WNR.0b013e3280148e8b. [DOI] [PubMed] [Google Scholar]
- Lund E, Güttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science. 2004;303:95–98. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- Magill ST, Cambronne XA, Luikart BW, Lioy DT, Leighton BH, Westbrook GL, Mandel G, Goodman RH. microRNA-132 regulates dendritic growth and arborization of newborn neurons in the adult hippocampus. Proc. Natl. Acad. Sci. U. S. A. 2010;107:20382–20387. doi: 10.1073/pnas.1015691107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney B, Ge Y-W, Greig N, Lahiri DK. Presence of a “CAGA box” in the APP gene unique to amyloid plaque-forming species and absent in all APLP-1/2 genes: implications in Alzheimer’s disease. FASEB J. 2004;18:1288–1290. doi: 10.1096/fj.03-1703fje. [DOI] [PubMed] [Google Scholar]
- Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. Alzheimer’s disease: clinical trials and drug development. Lancet Neurol. 2010;9:702–716. doi: 10.1016/S1474-4422(10)70119-8. [DOI] [PubMed] [Google Scholar]
- Maragkakis M, Alexiou P, Papadopoulos GL, Reczko M, Dalamagas T, Giannopoulos G, Goumas G, Koukis E, Kourtis K, Simossis VA, Sethupathy P, Vergoulis T, Koziris N, Sellis T, Tsanakas P, Hatzigeorgiou AG. Accurate micro-RNA target prediction correlates with protein repression levels. BMC Bioinformatics. 2009a;10:295. doi: 10.1186/1471-2105-10-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maragkakis M, Reczko M, Simossis VA, Alexiou P, Papadopoulos GL, Dalamagas T, Giannopoulos G, Goumas G, Koukis E, Kourtis K, Vergoulis T, Koziris N, Sellis T, Tsanakas P, Hatzigeorgiou AG. DIANA-microT web server: elucidating microRNA functions through target prediction. Nucleic Acids Res. 2009b;37:W273–W276. doi: 10.1093/nar/gkp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673–684. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbella EG, Bertrand S, Huez G, Octave JN. A GG nucleotide sequence of the 3′ untranslated region of amyloid precursor protein mRNA plays a key role in the regulation of translation and the binding of proteins. Mol. Cell. Biol. 2000;20:4572–4579. doi: 10.1128/mcb.20.13.4572-4579.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestdagh P, Feys T, Bernard N, Guenther S, Chen C, Speleman F, Vandesompele J. High-throughput stem-loop RT-qPCR miRNA expression profiling using minute amounts of input RNA. Nucleic Acids Res. 2008;36:e143. doi: 10.1093/nar/gkn725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestdagh P, Van Vlierberghe P, De Weer A, Muth D, Westermann F, Speleman F, Vandesompele J. A novel and universal method for microRNA RT-qPCR data normalization. Genome Biol. 2009;10:R64. doi: 10.1186/gb-2009-10-6-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda KC, Huynh T, Tay Y, Ang Y-S, Tam W-L, Thomson AM, Lim B, Rigoutsos I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–1217. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- Miska EA, Alvarez-Saavedra E, Townsend M, Yoshii A, Sestan N, Rakic P, Constantine-Paton M, Horvitz HR. Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol. 2004;5:R68. doi: 10.1186/gb-2004-5-9-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin RD, O’Connor MD, Griffith M, Kuchenbauer F, Delaney A, Prabhu A-L, Zhao Y, McDonald H, Zeng T, Hirst M, Eaves CJ, Marra MA. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18:610–621. doi: 10.1101/gr.7179508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen RD, Serra M, Steitz JA, Vasudevan S. Posttranscriptional activation of gene expression in Xenopus laevis oocytes by microRNA-protein complexes (microRNPs) Proc. Natl. Acad. Sci. U. S. A. 2011;108:8281–8286. doi: 10.1073/pnas.1105401108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourelatos Z, Dostie J, Paushkin S, Sharma A, Charroux B, Abel L, Rappsilber J, Mann M, Dreyfuss G. miRNPs: a novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev. 2002;16:720–728. doi: 10.1101/gad.974702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Wang W-X. MiR-107 is reduced in Alzheimer’s disease brain neocortex: validation study. J. Alzheimers Dis. 2010;21:75–79. doi: 10.3233/JAD-2010-091603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Wilfred BR. In situ hybridization is a necessary experimental complement to microRNA (miRNA) expression profiling in the human brain. Neurosci. Lett. 2009;466:69–72. doi: 10.1016/j.neulet.2009.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Baldwin DA, Kloosterman WP, Kauppinen S, Plasterk RHA, Mourelatos Z. RAKE and LNA-ISH reveal microRNA expression and localization in archival human brain. RNA. 2006;12:187–191. doi: 10.1261/rna.2258506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, De Planell-Saguer M, Lamprinaki S, Kiriakidou M, Zhang P, O’Doherty U, Mourelatos Z. A novel monoclonal antibody against human Argonaute proteins reveals unexpected characteristics of miRNAs in human blood cells. RNA. 2007;13:1787–1792. doi: 10.1261/rna.646007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Wang W-X, Wilfred BR, Tang G. Technical variables in high-throughput miRNA expression profiling: much work remains to be done. Biochim. Biophys. Acta. 2008;1779:758–765. doi: 10.1016/j.bbagrm.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J. Neuropathol. Exp. Neurol. 2009;68:1–14. doi: 10.1097/NEN.0b013e3181919a48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Kiriakidou M, Mourelatos Z, Tan GS, Jennings MH, Xie K, Wang W-X. High-throughput experimental studies to identify miRNA targets directly, with special focus on the mammalian brain. Brain Res. 2010;1338:122–130. doi: 10.1016/j.brainres.2010.03.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman MA, Mani V, Hammond SM. Deep sequencing of microRNA precursors reveals extensive 3′ end modification. RNA. 2011;17:1795–1803. doi: 10.1261/rna.2713611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonne N, Ameyar-Zazoua M, Souidi M, Harel-Bellan A. Tandem affinity purification of miRNA target mRNAs (TAP-Tar) Nucleic Acids Res. 2010;38:e20. doi: 10.1093/nar/gkp1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez-Iglesias J, Liu CC, Morgan TE, Finch CE, Zhou XJ. Joint genome-wide profiling of miRNA and mRNA expression in Alzheimer’s disease cortex reveals altered miRNA regulation. PLoS One. 2010;5:e8898. doi: 10.1371/journal.pone.0008898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunomura A, Hofer T, Moreira PI, Castellani RJ, Smith MA, Perry G. RNA oxidation in Alzheimer disease and related neurodegenerative disorders. Acta Neuropathol. 2009;118:151–166. doi: 10.1007/s00401-009-0508-1. [DOI] [PubMed] [Google Scholar]
- Nuovo GJ. In situ detection of precursor and mature microRNAs in paraffin embedded, formalin fixed tissues and cell preparations. Methods. 2008;44:39–46. doi: 10.1016/j.ymeth.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Nuovo GJ, Elton TS, Nana-Sinkam P, Volinia S, Croce CM, Schmittgen TD. A methodology for the combined in situ analyses of the precursor and mature forms of microRNAs and correlation with their putative targets. Nat. Protoc. 2009;4:107–115. doi: 10.1038/nprot.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obad S, dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C, Fu C, Lindow M, Stenvang J, Straarup EM, Hansen HF, Koch T, Pappin D, Hannon GJ, Kauppinen S. Silencing of microRNA families by seed-targeting tiny LNAs. Nat. Genet. 2011;43:371–378. doi: 10.1038/ng.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obernosterer G, Martinez J, Alenius M. Locked nucleic acid-based in situ detection of microRNAs in mouse tissue sections. Nat. Protoc. 2007;2:1508–1514. doi: 10.1038/nprot.2007.153. [DOI] [PubMed] [Google Scholar]
- Orom UA, Lund AH. Isolation of microRNA targets using biotinylated synthetic microRNAs. Methods. 2007;43:162–165. doi: 10.1016/j.ymeth.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Ørom UA, Kauppinen S, Lund AH. LNA-modified oligonucleotides mediate specific inhibition of microRNA function. Gene. 2006;372:137–141. doi: 10.1016/j.gene.2005.12.031. [DOI] [PubMed] [Google Scholar]
- Ørom UA, Nielsen FC, Lund AH. MicroRNA-10a binds the 5′UTR of ribosomal protein mRNAs and enhances their translation. Mol. Cell. 2008;30:460–471. doi: 10.1016/j.molcel.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Pena JTG, Sohn-Lee C, Rouhanifard SH, Ludwig J, Hafner M, Mihailovic A, Lim C, Holoch D, Berninger P, Zavolan M, Tuschl T. miRNA in situ hybridization in formaldehyde and EDC-fixed tissues. Nat. Methods. 2009;6:139–141. doi: 10.1038/nmeth.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters L, Meister G. Argonaute proteins: mediators of RNA silencing. Mol. Cell. 2007;26:611–623. doi: 10.1016/j.molcel.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Pollwein P, Masters CL, Beyreuther K. The expression of the amyloid precursor protein (APP) is regulated by two GC-elements in the promoter. Nucleic Acids Res. 1992;20:63–68. doi: 10.1093/nar/20.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradervand S, Weber J, Lemoine F, Consales F, Paillusson A, Dupasquier M, Thomas J, Richter H, Kaessmann H, Beaudoing E, Hagenbüchle O, Harshman K. Concordance among digital gene expression, microarrays, and qPCR when measuring differential expression of microRNAs. Biotechniques. 2010;48:219–222. doi: 10.2144/000113367. [DOI] [PubMed] [Google Scholar]
- Pratt AJ, MacRae IJ. The RNA-induced silencing complex: a versatile gene-silencing machine. J. Biol. Chem. 2009;284:17897–17901. doi: 10.1074/jbc.R900012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quitschke WW, Goldgaber D. The amyloid beta-protein precursor promoter. A region essential for transcriptional activity contains a nuclear factor binding domain. J. Biol. Chem. 1992;267:17362–17368. [PubMed] [Google Scholar]
- Raina P, Santaguida P, Ismaila A, Patterson C, Cowan D, Levine M, Booker L, Oremus M. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: evidence review for a clinical practice guideline. Ann. Intern. Med. 2008;148:379–397. doi: 10.7326/0003-4819-148-5-200803040-00009. [DOI] [PubMed] [Google Scholar]
- Ray B, Chauhan NB, Lahiri DK. Oxidative insults to neurons and synapse are prevented by aged garlic extract and S-allyl-L-cysteine treatment in the neuronal culture and APP-Tg mouse model. J. Neurochem. 2011a;117:388–402. doi: 10.1111/j.1471-4159.2010.07145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray B, Long JM, Sokol DK, Lahiri DK. Increased secreted amyloid precursor protein-α (sAPPα) in severe autism: proposal of a specific, anabolic pathway and putative biomarker. PLoS One. 2011b;6:e20405. doi: 10.1371/journal.pone.0020405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- Rogers JT, Leiter LM, McPhee J, Cahill CM, Zhan SS, Potter H, Nilsson LN. Translation of the Alzheimer amyloid precursor protein mRNA is up-regulated by interleukin-1 through 5′-untranslated region sequences. J. Biol. Chem. 1999;274:6421–6431. doi: 10.1074/jbc.274.10.6421. [DOI] [PubMed] [Google Scholar]
- Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, Leiter L, McPhee J, Sarang SS, Utsuki T, Greig NH, Lahiri DK, Tanzi RE, Bush AI, Giordano T, Gullans SR. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 2002;277:45518–45528. doi: 10.1074/jbc.M207435200. [DOI] [PubMed] [Google Scholar]
- Ruby JG, Jan C, Player C, Axtell MJ, Lee W, Nusbaum C, Ge H, Bartel DP. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell. 2006;127:1193–1207. doi: 10.1016/j.cell.2006.10.040. [DOI] [PubMed] [Google Scholar]
- Sambamurti K, Greig NH, Utsuki T, Barnwell EL, Sharma E, Mazell C, Bhat NR, Kindy MS, Lahiri DK, Pappolla MA. Targets for AD treatment: conflicting messages from γ-secretase inhibitors. J. Neurochem. 2011;117:359–374. doi: 10.1111/j.1471-4159.2011.07213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science. 2008;320:1643–1647. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer A, O’Carroll D, Tan CL, Hillman D, Sugimori M, Llinas R, Greengard P. Cerebellar neurodegeneration in the absence of microRNAs. J. Exp. Med. 2007;204:1553–1558. doi: 10.1084/jem.20070823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherr M, Venturini L, Battmer K, Schaller-Schoenitz M, Schaefer D, Dallmann I, Ganser A, Eder M. Lentivirus-mediated antagomir expression for specific inhibition of miRNA function. Nucleic Acids Res. 2007;35:e149. doi: 10.1093/nar/gkm971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipper HM, Maes OC, Chertkow HM, Wang E. MicroRNA expression in Alzheimer blood mononuclear cells. Gene Reg. Syst. Biol. 2007;1:263–274. doi: 10.4137/grsb.s361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider LS, Lahiri DK. The perils of Alzheimer’s drug development. Curr. Alzheimer Res. 2009;6:77–78. doi: 10.2174/156720509787313871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider LS, Insel PS, Weiner MW. Treatment with cholinesterase inhibitors and memantine of patients in the Alzheimer’s Disease Neuroimaging Initiative. Arch. Neurol. 2011;68:58–66. doi: 10.1001/archneurol.2010.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M, Lightfoot S, Menzel W, Granzow M, Ragg T. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006;7:3. doi: 10.1186/1471-2199-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004;5:R13. doi: 10.1186/gb-2004-5-3-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi P, Lukiw WJ. Micro-RNA abundance and stability in human brain: specific alterations in Alzheimer’s disease temporal lobe neocortex. Neurosci. Lett. 2009;459:100–104. doi: 10.1016/j.neulet.2009.04.052. [DOI] [PubMed] [Google Scholar]
- Shao N-Y, Hu HY, Yan Z, Xu Y, Hu H, Menzel C, Li N, Chen W, Khaitovich P. Comprehensive survey of human brain microRNA by deep sequencing. BMC Genomics. 2010;11:409. doi: 10.1186/1471-2164-11-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi R, Chiang VL. Facile means for quantifying microRNA expression by real-time PCR. Biotechniques. 2005;39:519–525. doi: 10.2144/000112010. [DOI] [PubMed] [Google Scholar]
- Shingara J, Keiger K, Shelton J, Laosinchai-Wolf W, Powers P, Conrad R, Brown D, Labourier E. An optimized isolation and labeling platform for accurate microRNA expression profiling. RNA. 2005;11:1461–1470. doi: 10.1261/rna.2610405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silahtaroglu AN, Nolting D, Dyrskjøt L, Berezikov E, Møller M, Tommerup N, Kauppinen S. Detection of microRNAs in frozen tissue sections by fluorescence in situ hybridization using locked nucleic acid probes and tyramide signal amplification. Nat. Protoc. 2007;2:2520–2528. doi: 10.1038/nprot.2007.313. [DOI] [PubMed] [Google Scholar]
- Simpson DA, Feeney S, Boyle C, Stitt AW. Retinal VEGF mRNA measured by SYBR green I fluorescence: a versatile approach to quantitative PCR. Mol. Vis. 2000;6:178–183. [PubMed] [Google Scholar]
- Siomi H, Siomi MC. Posttranscriptional regulation of microRNA biogenesis in animals. Mol. Cell. 2010;38:323–332. doi: 10.1016/j.molcel.2010.03.013. [DOI] [PubMed] [Google Scholar]
- Søe MJ, Møller T, Dufva M, Holmstrøm K. A sensitive alternative for micro-RNA in situ hybridizations using probes of 2′-O-methyl RNA + LNA. J. Histochem. Cytochem. 2011;59:661–672. doi: 10.1369/0022155411409411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol DK, Maloney B, Long JM, Ray B, Lahiri DK. Autism, Alzheimer disease, and fragile X: APP, FMRP, and mGluR5 are molecular links. Neurology. 2011;76:1344–1352. doi: 10.1212/WNL.0b013e3182166dc7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Lahiri DK. Functional identification of the promoter of the gene encoding the Rhesus monkey beta-amyloid precursor protein. Gene. 1998;217:165–176. doi: 10.1016/s0378-1119(98)00340-0. [DOI] [PubMed] [Google Scholar]
- Sperling RA, Dickerson BC, Pihlajamaki M, Vannini P, LaViolette PS, Vitolo OV, Hedden T, Becker JA, Rentz DM, Selkoe DJ, Johnson KA. Functional alterations in memory networks in early Alzheimer’s disease. Neuromolecular Med. 2010;12:27–43. doi: 10.1007/s12017-009-8109-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Koo S, White N, Peralta E, Esau C, Dean NM, Perera RJ. Development of a micro-array to detect human and mouse microRNAs and characterization of expression in human organs. Nucleic Acids Res. 2004;32:e188. doi: 10.1093/nar/gnh186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- The Lancet Why are drug trials in Alzheimer’s disease failing? Lancet. 2010;376:658. doi: 10.1016/S0140-6736(10)61316-5. [DOI] [PubMed] [Google Scholar]
- Thompson RC, Deo M, Turner DL. Analysis of microRNA expression by in situ hybridization with RNA oligonucleotide probes. Methods. 2007;43:153–161. doi: 10.1016/j.ymeth.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Válóczi A, Hornyik C, Varga N, Burgyán J, Kauppinen S, Havelda Z. Sensitive and specific detection of microRNAs by northern blot analysis using LNA-modified oligonucleotide probes. Nucleic Acids Res. 2004;32:e175. doi: 10.1093/nar/gnh171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij E. The art of microRNA research. Circ. Res. 2011;108:219–234. doi: 10.1161/CIRCRESAHA.110.227496. [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:1–12. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Várallyay E, Burgyán J, Havelda Z. Detection of microRNAs by Northern blot analyses using LNA probes. Methods. 2007;43:140–145. doi: 10.1016/j.ymeth.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up-regulate translation. Science. 2007;318:1931–1934. doi: 10.1126/science.1149460. [DOI] [PubMed] [Google Scholar]
- Vermeulen A, Robertson B, Dalby AB, Marshall WS, Karpilow J, Leake D, Khvorova A, Baskerville S. Double-stranded regions are essential design components of potent inhibitors of RISC function. RNA. 2007;13:723–730. doi: 10.1261/rna.448107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilardo E, Barbato C, Ciotti M, Cogoni C, Ruberti F. MicroRNA-101 regulates amyloid precursor protein expression in hippocampal neurons. J. Biol. Chem. 2010;285:18344–18351. doi: 10.1074/jbc.M110.112664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vostrov AA, Taheny MJ, Izkhakov N, Quitschke WW. A nuclear factor-binding domain in the 5′-untranslated region of the amyloid precursor protein promoter: implications for the regulation of gene expression. BMC Res. Notes. 2010;3:4. doi: 10.1186/1756-0500-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan G, Lim QE, Too H-P. High-performance quantification of mature micro-RNAs by real-time RT-PCR using deoxyuridine-incorporated oligonucleotides and hemi-nested primers. RNA. 2010;16:1436–1445. doi: 10.1261/rna.2001610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Ach RA, Curry B. Direct and sensitive miRNA profiling from low-input total RNA. RNA. 2007;13:151–159. doi: 10.1261/rna.234507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WX, Rajeev BW, Stromberg AJ, Ren N, Tang G, Huang Q, Rigoutsos I, Nelson PT. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 2008a;28:1213–1223. doi: 10.1523/JNEUROSCI.5065-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Sheng G, Juranek S, Tuschl T, Patel DJ. Structure of the guide-strand-containing argonaute silencing complex. Nature. 2008b;456:209–213. doi: 10.1038/nature07315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W-X, Wilfred BR, Hu Y, Stromberg AJ, Nelson PT. Anti-Argonaute RIP-Chip shows that miRNA transfections alter global patterns of mRNA recruitment to microribonucleoprotein complexes. RNA. 2010a;16:394–404. doi: 10.1261/rna.1905910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W-X, Wilfred BR, Madathil SK, Tang G, Hu Y, Dimayuga J, Stromberg AJ, Huang Q, Saatman KE, Nelson PT. miR-107 regulates granulin/progranulin with implications for traumatic brain injury and neurodegenerative disease. Am. J. Pathol. 2010b;177:334–345. doi: 10.2353/ajpath.2010.091202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- Winter J, Diederichs S. Argonaute proteins regulate microRNA stability: increased microRNA abundance by Argonaute proteins is due to microRNA stabilization. RNA Biol. 2011;8 doi: 10.4161/rna.8.6.17665. [DOI] [PubMed] [Google Scholar]
- Yeom K-H, Heo I, Lee J, Hohng S, Kim VN, Joo C. Single-molecule approach to immunoprecipitated protein complexes: insights into miRNA uridylation. EMBO Rep. 2011;12:690–696. doi: 10.1038/embor.2011.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi SH, Malter JS. Amyloid precursor protein mRNA stability is controlled by a 29-base element in the 3′-untranslated region. J. Biol. Chem. 1994;269:24007–24013. [PubMed] [Google Scholar]
- Zaidi SH, Denman R, Malter JS. Multiple proteins interact at a unique cis-element in the 3′-untranslated region of amyloid precursor protein mRNA. J. Biol. Chem. 1994;269:24000–24006. [PubMed] [Google Scholar]
- Zhang C, Darnell RB. Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat. Biotechnol. 2011;29:607–614. doi: 10.1038/nbt.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J-J, Hua Y-J, Sun D-G, Meng X-X, Xiao H-S, Ma X. Genome-wide microRNA profiling in human fetal nervous tissues by oligonucleotide microarray. Childs Nerv. Syst. 2006;22:1419–1425. doi: 10.1007/s00381-006-0173-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Dysregulation of cardio-genesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]