

Abstract
The proteobacterium Pantoea stewartii subsp. stewartii causes Stewart's wilt disease in maize when it colonizes the xylem and secretes large amounts of stewartan, an exopolysaccharide. The success of disease pathogenesis lies in the timing of bacterial virulence factor expression through the different stages of infection. Regulation is achieved through a quorum-sensing (QS) system consisting of the acyl-homoserine lactone (AHL) synthase, EsaI, and the transcription regulator EsaR. At low cell densities, EsaR represses transcription of itself and of rcsA, an activator of the stewartan biosynthesis operon; it also activates esaS, which encodes a small RNA (sRNA). Repression or activation ceases at high cell densities when EsaI synthesizes sufficient levels of the AHL ligand N-3-oxo-hexanoyl-l-homoserine lactone to bind and inactivate EsaR. This study aims to identify other genes activated or repressed by EsaR during the QS response. Proteomic analysis identified a QS regulon of more than 30 proteins. Electrophoretic mobility shift assays of promoters of genes encoding differentially expressed proteins distinguished direct targets of EsaR from indirect targets. Additional quantitative reverse transcription-PCR (qRT-PCR) and DNA footprinting analysis established that EsaR directly regulates the promoters of dkgA, glpF, and lrhA. The proteins encoded by dkgA, glpF, and lrhA are a 2,5-diketogluconate reductase, glycerol facilitator, and transcriptional regulator of chemotaxis and motility, respectively, indicating a more global QS response in P. stewartii than previously recognized.
INTRODUCTION
Quorum sensing (QS) is a cell density-dependent method of coordinating gene expression employed by bacteria to regulate various cellular processes. Proteobacteria commonly detect and respond to the concentration of a chemical signal, an acylated homoserine lactone (AHL), produced by an AHL synthase homologous to LuxI from Vibrio fischeri. This signal is sensed by an AHL-responsive transcription regulator homologous to LuxR (1). Thus, gene expression is altered in relation to cell density such that physiological processes, such as bioluminescence, virulence, and pigment or capsule production are affected (2, 3). QS is a central component in the regulatory networks of proteobacteria, and along with other bacterial regulatory systems, it allows the bacteria to colonize and grow efficiently in a particular environment or host-associated niche (4).
The proteobacterium Pantoea stewartii subsp. stewartii is a phytopathogen known to utilize QS to regulate virulence. P. stewartii is the causative agent of Stewart's wilt that affects maize cultivars, particularly sweet corn. It is found in the midgut of Chaetocnema pulicaria, the corn flea beetle where it overwinters. Within the plant, the bacterium colonizes the xylem after migrating there from the sites of insect feeding on the leaves and stalks of maize (5). Once in the xylem, P. stewartii forms a biofilm and overproduces an exopolysaccharide, stewartan, that obstructs the xylem and likely causes the vascular wilting associated with Stewart's wilt by blocking water transport (6, 7). P. stewartii also possesses a hrp and wts gene cluster encoding a Hrp type III secretion system and effector proteins (8). Affected seedlings wither (wilt phase), and affected mature plants develop necrotic lesions (leaf blight phase), leading to losses in the crop yield (9). Other pathogenic Pantoea species include Pantoea ananatis, which causes soft rot of onions (10), and Pantoea citrea, which causes pink disease of pineapple (11). Pantoea species are also found symbiotically associated with leaf-cutter ants (12).
The expression of stewartan, a key virulence factor in P. stewartii, is controlled by the cell density-dependent QS system, consisting of EsaI/EsaR, a signaling molecule synthase and the cognate transcription regulator homologous to LuxI/LuxR (6). EsaI constitutively synthesizes a 3-oxo-hexanoyl-homoserine lactone (AHL) signal that accumulates at high cell densities. EsaR is a transcription factor that activates or represses expression of certain genes at low cell densities. Binding of the AHL ligand at high cell densities inactivates it. EsaR regulates virulence by repressing rcsA, an activator of the stewartan biosynthesis operon. RcsA in P. stewartii is functionally interchangeable with the RcsA regulator in Escherichia coli that controls capsular polysaccharide synthesis (13). Inactivation of EsaR at high cell densities by the AHL ligand permits the RcsAB heterodimer to activate stewartan synthesis (14). At low cell densities, EsaR also autorepresses itself and activates the expression of a small RNA of unknown function (15, 16). Mutants lacking esaR constitutively overproduce stewartan and are less virulent than the wild type, indicating that it is the temporal regulation of virulence factors during the stages of infection that lead to success in establishing disease in the host (14). However, the full extent of EsaR regulation and QS-controlled genes in P. stewartii is not known.
In this study, we have further defined the QS regulon using a proteomic approach. Two-dimensional (2D) SDS-PAGE experiments revealed more than 30 proteins that are differentially expressed in the presence of EsaR and, thus, are regulated by QS. Electrophoretic mobility shift assays (EMSAs) revealed several direct targets of EsaR in the regulon. Regulation of three of the promoters at the level of transcription was confirmed through quantitative reverse transcription-PCR (qRT-PCR), and the binding sites of EsaR have been defined through DNase I footprinting and additional EMSA analysis. Identification of these regulated targets provides a more robust understanding of the QS regulon in P. stewartii.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
A list of bacterial strains and plasmids utilized is provided in Table 1. Both Pantoea stewartii and Escherichia coli strains were grown in Luria-Bertani (LB) broth supplemented with ampicillin (100 μg/ml) or tetracycline (10 μg/ml) as required. Pantoea strains were grown at 30°C, and E. coli strains were grown at 37°C. Conjugation was performed using E. coli strain CC118λpir carrying the conjugative helper plasmid pEVS104 to facilitate transfer of pSVB60 or pBBR1MCS-3 into P. stewartii ESΔIR (17) to generate an EsaR-complemented strain and the control with the same selectable marker.
Table 1.
Plasmids and strains used in this study
| Bacterial strain or plasmid | Relevant informationa | Reference |
|---|---|---|
| P. stewartii strains | ||
| DC283 | Wild-type SS104; Nalr | 42 |
| ESΔIR | DC283 ΔesaR ΔesaI mutant | 7 |
| ESΔIR(pBBR1MCS-3) | DC283 ΔesaR ΔesaI mutant with vector control | This study |
| ESΔIR(pSVB60) | DC283 ΔesaR ΔesaI mutant with esaR expressed from native promoter | 14 |
| ESN51 | DC283, AHL deficient; esaI::Tn5seqN51 | 6 |
| E. coli strains | ||
| Top10 | F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80dlacZΔM15 ΔlacX74 deoR recAI araD139 Δ(ara-leu)7697 galU galK rpsL (Strr) endA1 nupG | 43 |
| CC118λpir | Δ(ara-leu) araD ΔlacX74 galE galK phoA20 thi-1 rpsE rpoB argE(Am) recA1 λpir | 44 |
| Plasmids | ||
| pBBR1MCS-3 | Broad-host-range vector; Tetr | 45 |
| pEVS104 | Conjugative helper plasmid; tra trb; Knr | 46 |
| pSVB-60 | pBBR1MCS-3 with esaR controlled by native PesaR; Tetr | 15 |
| pHMGE | Plac promoter-controlled HisMBP-GLY5-EsaR; Apr | 16 |
Apr, ampicillin resistance; Nalr, naldixic acid resistance; Tetr, tetracycline resistance; Knr, kanamacyin resistance.
Two-dimensional SDS-PAGE.
To prepare cell extracts, P. stewartii strain ESN51 grown to stationary phase was used to inoculate 1 liter of LB broth alone or supplemented with 10 μM AHL [N-(β-ketocaproyl)-l-homoserine lactone (3-oxo-C6-HSL)] (Sigma, St. Louis, MO) to an optical density (OD) at 600 nm of 0.05 and allowed to grow at 30°C with shaking to an OD at 600 nm of 0.5. The cells were harvested by centrifugation and stored at −80°C. The cell pellet was resuspended in 15 ml of cold 8 M urea plus 1% Triton buffer and lysed by passage through a French press twice at a cell pressure of 18,000 lb/in2. The lysate was then centrifuged for 10 min at 5,000 × g at 4°C before being subjected to ultracentrifugation using a Ti70 rotor at 40,000 rpm at 4°C for 1 h to clarify the cell extract. Protein samples were similarly extracted from P. stewartii strains ESΔIR(pBBR1MCS-3) and ESΔIR(pSVB60).
2D gel electrophoresis was performed by the Virginia Bioinformatics Institute (VBI) Core Laboratory (Blacksburg, VA). Briefly, samples containing 150 μg of protein were focused in the first dimension using 17-cm, pH 3 to 10 NL immobilized pH gradient (IPG) strips (Bio-Rad, Hercules, CA) and then loaded along with Precision plus protein standard plugs (Bio-Rad) on 20-cm precast 12% SDS-polyacrylamide gels (Jule Inc., Milford, CT). The postelectrophoresis gels were stained with Coomassie staining solution (2.5 g Coomassie brilliant blue R250, 450 ml methanol, and 100 ml acetic acid per liter) and stored in 0.1% sodium azide. The gels were scanned on a GS-800 calibrated densitometer (Bio-Rad) to analyze the differentially expressed protein spots.
Mass spectrometry and protein identification.
Differentially expressed proteins were identified by excising the spots of interest from the 2D gels, and their sequence was determined via matrix-assisted laser desorption/ionization tandem time of flight mass spectrometry (MALDI-TOF MS-MS) (Virginia Tech Mass Spectrometry Incubator, Blacksburg, VA) as described previously (18). Protein identifications were made by searching the peptide sequences obtained from mass spectrometry analysis against the supplied P. stewartii genome v5 from the ASAP database (19).
Purification of EsaR fusion protein.
A His-tagged maltose-binding protein (MBP) fused to a glycine linker-tagged EsaR protein (HMGE) was purified as previously described (20) with the following modifications. To ensure that only DNA-binding competent protein was used in the EMSAs, the purified HMGE was passed through a HiTrap heparin HP column (GE Healthcare Life Sciences, Piscataway, NJ) and eluted using a gradient of 800 mM NaCl. HMGE was then further purified in a HiPrep 26/60 Sephacryl 200-S (GE Healthcare Life Sciences) gel filtration column using the AKTAPrime fast-performance liquid chromatography (FPLC) system, equilibrated with 20 mM HEPES, 1 mM EDTA, 30 mM KCl, and 10% glycerol (pH 7.4).
EMSA analysis.
Phusion DNA polymerase (Thermo Fisher Scientific Inc., Waltham, MA) was used to amplify upstream promoter regions from genes of interest with specific forward and reverse primers (IDT-DNA Inc., Coralville, IA) (see Table S1 in the supplemental material). The resulting PCR fragments were subsequently purified using the QIAquick gel extraction kit (Qiagen, Valencia, CA). DNA fragments (2 to 10 pmol) were individually end labeled using [γ-32P]ATP (PerkinElmer, Waltham, MA) and T4 polynucleotide kinase (New England BioLabs, Ipswich, MA) and resuspended in distilled H2O (dH2O) to a final concentration of 20 nM. HMGE (0 to 100 nM) and 1 nM promoter DNA were incubated for 30 min in 20-μl reaction mixtures with EMSA binding buffer at a final concentration in all samples of 20 mM HEPES, 1 mM EDTA, 30 mM KCl, 0.2% Tween 20, 10 mM (NH4)2SO4, 50 ng/μl poly(dI-dC), 150 μg/ml acetylated bovine serum albumin (BSA), and 10% glycerol (pH 7.4). The constituents of the EMSA reaction were based on previous experiments (14). The postincubation reactions were separated on 6% Tris-glycine-EDTA native PAGE minigels (Bio-Rad) at 80 V for 2 h. The entire apparatus was placed in an ice-packed container to maintain low temperatures. After electrophoresis, the gels were dried and exposed to a Fisher Biotech autoradiography cassette (Fisher Scientific, Pittsburgh, PA). The screen was then visualized using a Typhoon Trio variable-mode imager (GE Healthcare, Life Sciences).
Additional EMSAs performed to determine the precise bases essential for binding were performed in a similar fashion as described above except 10 nM 6-carboxyfluorescein (FAM)-labeled DNA fragments were used instead of radiolabeled fragments, but visualization was still achieved through a Typhoon Trio variable-mode imager.
qRT-PCR.
P. stewartii ESΔIR(pBBR1MCS) and ESΔIR(pSVB60) grown to stationary phase were used to inoculate 5 ml LB supplemented with tetracycline (10 μg/ml) at an OD at 600 nm of 0.05 and allowed to grow to a final OD at 600 nm of 0.5. RNAprotect bacteria reagent (10 ml) (Qiagen) was used per the manufacturer's instructions to stabilize the RNA. The cell pellets were stored at −80°C prior to total RNA extraction using the RNeasy minikit (Qiagen) with an additional on-column DNase I digestion step using the RNase-free DNase set (Qiagen). RNA was quantified using a NanoPhotometer (Implen, Westlake Village, CA) and submitted to the VBI Core Laboratory Facility for analysis by an Agilent Bioanalyzer 2100 to check for quality. All RNA integrity number (RIN) values were above 9.7. The extracted RNA was converted to cDNA using the high-capacity cDNA reverse transcription kit (Life Technologies, Grand Island, NY) per the manufacturer's instructions. The cDNA was quantified using a NanoPhotometer and checked for purity by measuring absorbance ratios at 260/280 nm and 260/230 nm. The cDNA samples were used as the templates in a 7300 real-time PCR system (Applied Biosystems, now Life Technologies).
Primers were designed to enable amplification of coding regions of the EsaR targets dkgA, fabF, glpF, lrhA, mglB, and rpsB (see Table S1 in the supplemental material). The amplified fragments were ligated into pGEM (Promega, Madison, WI), transformed into E. coli Top10 cells (Table 1), and sequenced. They were then used as the template to optimize qRT-PCR primers within 100% ±10% efficiency. The primer pairs (Table S1) for qRT-PCR analysis of all direct targets of EsaR determined by EMSAs were designed using Primer Express 3.0 (Life Technologies). The parameters for primer design were set as follows: 18 to 24 bp in length, melting temperature (Tm) of 60°C, and amplicon length of 80 to 120 bp. Template DNA (either plasmid or cDNA) was used at concentrations ranging from 10 ng to 0.001 ng per 25-μl reaction mixture containing 300 nM (each) of the specific forward and reverse primer pair and 2× SYBR green PCR master mix (Life Technologies) diluted to a 1× concentration with dH2O. Reactions were carried out in triplicate in a MicroAmp optical 96-well reaction plate (Life Technologies). The thermal cycler settings were programmed for 10 min at 95°C followed by 45 cycles consisting of 15 s at 95°C and 1 min at 60°C, which was also set as the data collection point. A dissociation stage was added at the end of the PCR run to confirm specific product amplification. The data obtained were analyzed through a 7300 system SDS RQ software v1.4 (Life Technologies) using an automated cycle threshold, and the relative expression was calculated using the PFAFFL method (21). In total, triplicate samples from two replicate experiments were analyzed.
DNase I footprinting analysis.
A modified DNase I footprinting approach (8) involved the use of FAM-labeled and nonlabeled primer pairs (see Table S1 in the supplemental material) annealing to the promoter regions of dkgA, glpF, and lrhA to amplify the regions of interest. Fluorescently labeled DNA (50 ng) was incubated with 100 nM HMGE in 1× EMSA binding buffer (see above) supplemented with 5 mM MgCl2 and 2 mM CaCl2. The DNA was digested using 50 ng of DNase I (Worthington Biochemicals, Lakewood, NJ) for 5 min at room temperature. The reaction was stopped by the addition of 125 mM EDTA and cleaned using the QIAquick gel extraction kit (Qiagen). A 10-μl sample of DNA eluted in dH2O was processed at VBI on the 3730 DNA analyzer (Life Technologies), with a G5 dye set, running an altered default genotyping module with increased injection time and injection voltage (8). The electropherograms of the probes generated from digestion in the presence of BSA and in the presence of HMGE were overlapped using Peak Scanner v1.0 (Life Technologies) to reveal bases that are protected from cleavage by DNase I. Simultaneous manual sequencing of the labeled probe was performed using the Thermo Sequenase cycle sequencing kit (Affymetrix, Santa Clara, CA) using specific FAM-labeled primers to generate 4 electropherograms for each dideoxynucleotide, which were then overlapped to yield sequencing electropherograms that could be compared to the DNase I-digested fragments.
RESULTS
Identification of QS-regulated proteins.
To identify proteins regulated by QS in P. stewartii, 2D gel electrophoresis was first performed using cell extracts from P. stewartii ESN51 (esaI mutant) with and without exogenously added AHL (see Fig. S1 in the supplemental material). Several differentially expressed proteins were identified from this preliminary experiment. In order to identify additional proteins of the QS regulon, 2D gel electrophoresis was repeated twice using two other P. stewartii strains that exhibit an enhanced physiological response to QS, P. stewartii ESΔIR carrying pBBR1MCS-3 or pSVB60. ESΔIR is a DC283 ΔesaR ΔesaI strain that lacks the transcription regulator EsaR and the AHL synthase EsaI, making it completely deficient in QS. This QS defect is partially complemented by pSVB60, which carries the gene coding for EsaR with its expression driven from the native promoter. The vector pBBR1MCS-3 serves as a negative control. In the absence of the AHL synthase, EsaR is constitutively active, allowing for identification of proteins whose expression is activated by EsaR, as demonstrated by qualitatively higher protein levels in strain ESΔIR(pSVB60) versus ESΔIR(pBBR1MCS-3), while proteins that are repressed by EsaR are more highly expressed in ESΔIR(pBBR1MCS-3), since EsaR is completely absent (Fig. S1). When plated on medium enhancing capsule production, the ΔesaR ΔesaI mutant strain ESΔIR(pBBR1MCS-3) exhibits a hypermucoid appearance due to overproduction of capsule, whereas strain ESΔIR(pSVB60) appears dry and streaky due to constitutive repression of capsule production compared to the wild-type control (20). Over the 3 cumulative 2D SDS-PAGE trials (Fig. S1), 34 proteins were observed twice or more to be differentially expressed, 22 EsaR-activated proteins and 12 EsaR-repressed proteins (Table 2). Interestingly, RcsA, a known repressed target of EsaR was not observed, highlighting an inherent drawback due to the limited sensitivity of the proteomic approach.
Table 2.
The QS regulon of Pantoea stewartii regulated by EsaR
| Category | Genea | Proteinb |
|---|---|---|
| EsaR-activated genes/proteins | CKS-1750 | N-Acetylmuramoyl-l-alanine amidase |
| CKS-5296 | Lipoprotein, putative | |
| accA* | Acetyl-CoA carboxylase, carboxytransferase, alpha subunit | |
| aceF* | Pyruvate dehydrogenase, dihydrolipoyltransacetylase component E2 | |
| ahpF | Alkyl hydroperoxide reductase, F52a subunit, FAD/NAD(P) binding | |
| degP | Serine endoprotease (protease Do), membrane associated | |
| fabF | 3-Oxoacyl-(acyl carrier protein) synthase | |
| fabI | Enoyl-(acyl carrier protein) reductase, NADH dependent | |
| fkpA | FKBP-type peptidyl-prolyl cis-trans isomerase (rotamase) | |
| fusA | Protein chain elongation factor EF-G, GTP binding | |
| glmU | Fused N-acetylglucosamine-phosphate uridyltransferase acetyltransferase | |
| glpK | Glycerol kinase | |
| hslU* | Molecular chaperone of HslUV protease | |
| lrhA | DNA-binding transcriptional repressor of flagellar, motility, and chemotaxis genes | |
| mglB | Methyl-galactoside transporter subunit periplasmic binding component of ABC superfamily | |
| nemA | N-Ethylmaleimide reductase, FMN linked | |
| ompA | Predicted outer membrane lipoprotein | |
| pheT | Phenylalanine tRNA synthetase, β-subunit | |
| rpsB | 30S ribosomal subunit protein S2 | |
| rplA* | 50S ribosomal subunit protein L1 | |
| rplC* | 50S ribosomal subunit protein L3 | |
| rplF* | 50S ribosomal subunit protein L6 | |
| EsaR-repressed genes/proteins | CKS-2738 | UDP-glucose dehydrogenase |
| accC | Acetyl-CoA carboxylase, biotin carboxylase subunit | |
| dkgA | 2,5-Diketo-d-gluconate reductase A | |
| galE | UDP-galactose-4-epimerase | |
| galF | Predicted regulatory subunit of GalU | |
| galU | Glucose-1-phosphate uridylyltransferase | |
| htpG | Chaperone protein HtpG | |
| osmC* | Osmotically inducible, stress-inducible membrane protein | |
| osmY | Periplasmic protein | |
| pgm | Phosphoglucomutase | |
| tetR | Predicted TetR family transcriptional regulator | |
| atpA* | F1 sector of membrane-bound ATP synthase |
Genes not analyzed in EMSA analysis are indicated by an asterisk.
CoA, coenzyme A; FKBP, FK506-binding protein; FMN, flavin mononucleotide.
Identification of direct targets of EsaR.
In an attempt to ascertain indirect versus direct targets among the 34 proteins identified from the 2D SDS-PAGE experiments (Table 2), the EsaR-regulated genes with 24 promoters putatively controlling 26 of these proteins were tested for direct regulation by EsaR via EMSAs utilizing 200- to 400-bp upstream promoter regions and the EsaR fusion protein HMGE (His-MBP-glycine linker-EsaR) (20). The eight proteins whose promoters were not subjected to EMSA analysis were encoded by genes located in the middle of operons such that no promoter regions were observed in proximity to support EsaR binding, and thus they were not analyzed by EMSA. EsaR directly binds to six promoters (Fig. 1), while the rest were established as indirect targets (see Fig. S2 in the supplemental material). Specific binding was demonstrated when labeled promoter probes were bound by HMGE and the shift was successfully competed using nonlabeled promoter probes. The genes with the bound promoters are as follows: dkgA, encoding 2,5-diketo-d-gluconate reductase A; fabF, encoding 3-oxoacyl-(acyl carrier protein) synthase I; glpF, encoding glycerol facilitator (first in an operon consisting of glpK [glycerol kinase] and glpX [fructose 1,6-bisphosphatase II]); lrhA, encoding DNA-binding transcriptional repressor of flagellar, motility, and chemotaxis genes; mglB, encoding periplasmic binding component of a methyl-galactoside transporter (first in an operon with mglA [ATP-binding component] and mglC [membrane component] of the methyl-galactoside transporter); and rpsB, encoding 30S ribosomal subunit protein (Fig. 1). A noticeable difference was observed, though not quantified, in the intensity of the bound probe of PdkgA and PlrhA compared to the other probes, with PdkgA and PlrhA showing stronger, more-complete shifts. Furthermore, in initial EMSA trials, three additional promoters, for the genes CKS-5296, encoding putative lipoprotein, galU, encoding glucose-1-phosphate uridylyltransferase, and fusA, encoding GTP-binding protein chain elongation factor EF-G, were also seen to be weakly bound by HMGE but were unable to successfully compete with the same unlabeled probe (Fig. S2) and hence they were not pursued further.
Fig 1.
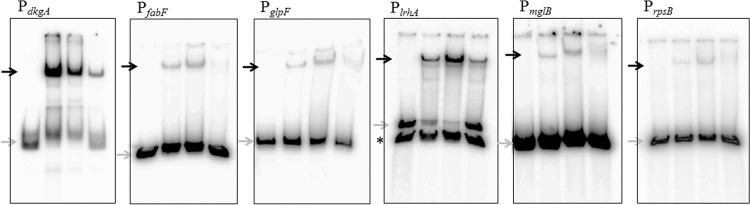
Electrophoretic mobility shift assays of putative QS regulon promoters with the His-MBP-glycine linker-EsaR fusion protein, HMGE (20). Gray arrows indicate free, unbound probe; black arrows indicate probe bound by HMGE. The black asterisk to the left of the PlrhA gel indicates a DNA fragment not bound by EsaR that was produced during PCR amplification of PlrhA. The concentration of DNA probe in all lanes is 1 nM. The lanes within each panel consist of the following: from left to right, DNA probe, DNA probe with 50 nM HMGE, DNA probe with 100 nM HMGE, and DNA probe with 100 nM HMGE plus 100 nM unlabeled DNA probe. From left to right, the gene promoters analyzed are PdkgA (dkgA encodes 2,5-diketo-d-gluconate reductase A), PfabF (fabF encodes 3-oxoacyl-[acyl carrier protein] synthase I), PglpF (glpF encodes glycerol facilitator), PlrhA (lrhA encodes a DNA-binding transcriptional repressor of flagellar, motility, and chemotaxis genes), PmglB (mglB encodes a periplasmic binding component of ABC superfamily methyl-galactoside transporter), and PrpsB (rpsB encodes 30S ribosomal subunit protein).
Verification of transcriptional control of dkgA, glpF, and lrhA.
To quantify the fold change in expression of the six genes with promoters bound by EsaR, qRT-PCR was performed on RNA extracted from P. stewartii strain ESΔIR(pBBR1MCS-3) and ESΔIR(pSVB60) (Fig. 2). Relative mRNA expression of glpF and lrhA was increased nearly 4-fold in the presence of EsaR when normalized to the expression of a 16S rRNA gene. Expression of dkgA was repressed more than 5-fold in the presence of EsaR. Thus, the 2D SDS-PAGE, EMSA, and qRT-PCR results are consistent for these three direct targets. Interestingly, the relative expression of fabF, mglB, and rpsB was not significantly altered in the two RNA samples. Although EsaR binds the promoters of these genes as demonstrated by EMSA and the protein encoded by the genes was differentially expressed in previous 2D SDS-PAGE experiments, the mRNA levels remain unchanged in the presence of EsaR.
Fig 2.

Relative change in mRNA expression in P. stewartii ESΔIR(pSVB60) versus ESΔIR(pBBR1MCS-3). Genes activated (A) or repressed (B) by EsaR. Error bars were calculated from triplicate samples obtained from two replicate experiments. All values are normalized with respect to the change in mRNA expression of 16S rRNA which was set at 1.
Determination of the EsaR binding site at the dkgA, glpF, and lrhA promoters.
A modified DNase I footprinting technique using fluorescently labeled probes and an automated capillary DNA fragment analysis instrument (8) was optimized for the 181-bp PdkgA DNA fragment, 226-bp PglpF DNA fragment, and the 284-bp PlrhA DNA fragment (Fig. 3). Overlapping electropherograms generated from digestion in the presence of BSA and in the presence of HMGE revealed bases that are protected from cleavage by DNase I only in the presence of HMGE.
Fig 3.

DNase I footprinting assay of EsaR binding sites in the noncoding strand of PdkgA, coding strand of PglpF, and noncoding strand of PlrhA. (A to C) Capillary electrophoresis of FAM-labeled DNA fragments PdkgA (A), PglpF (B), and PlrhA (C) from DNase I protection assays in the presence (black) and absence (gray) of HMGE, demonstrating that HMGE binds to specific sequences in the three promoter regions and protects against DNase I digestion. The rounded rectangles highlight the binding regions. The protected sequences are shown at the bottom of the rounded rectangles, and the bases believed to be the 20-bp binding site determined through EMSAs are indicated by underlining. The x axis denotes size in base pairs, and the y axis denotes relative fluorescence units.
The binding site on PdkgA is 48 bases upstream of the predicted ATG start codon. This binding site was further delineated by using EMSA analysis on probes that deleted two base pairs at a time until a loss of binding was observed (Fig. 4). This allowed us to narrow down the 20 bp that are essential for binding by considering similarity to previously known esa boxes (Fig. 5) (14, 16). The newly discovered EsaR binding site at dkgA is conserved in 11 of 20 bases of the previously known esa box in the esaR promoter (Fig. 5). The EsaR binding site is centered around −22 bp from the initiation site of transcription (based on unpublished RNA sequencing [RNA-Seq] data in A. M. Stevens' lab) and overlaps a putative −10 RNA polymerase binding site, consistent with the repressed nature of this promoter in the presence of EsaR.
Fig 4.

Nested deletion EMSA analysis of EsaR direct targets. (A to C) The region protected by DNase I digestion in PdkgA (A), PglpF (B), and PlrhA (C) is the gray-shaded sequence (5′ to 3′), and the underlined bases are the 20-bp EsaR binding sites. Sequences above and below the bold sequence are forward and reverse primers used to identify the required bases, and the bold letters in the primer sequence indicate the 5′ start base for the primer. EMSAs in boxes denote the point where binding by EsaR is lost upon removing flanking bases of the EsaR binding site. The concentration of DNA probe in all lanes is 10 nM. Lanes within each panel consist of the following (from left to right): DNA probe, DNA probe with 50 nM HMGE, DNA probe with 100 nM HMGE, and DNA probe with 100 nM HMGE plus 100 nM unlabeled DNA probe.
Fig 5.

Alignment of esa boxes recognized by EsaR in P. stewartii. The distances of the esa box elements from the translational start sites of the downstream open reading frames are indicated. EsaR binds to esa boxes on the esaR and rcsA promoter and to the lux box on the luxI promoter in in vitro assays (14, 16). The five newly discovered esa boxes were added to the alignment to generate a consensus sequence. In the consensus sequence, n denotes either one of four nucleotides. The sequence logo was constructed using Weblogo 3.0 (41).
A DNase I footprinting analysis of the EsaR-activated glpF promoter revealed two 20-bp protected regions ∼275 bases upstream of the predicted ATG start site separated by 11 bases. The 51-bp region was protected by cleavage by DNase I in the presence of EsaR (Fig. 3). The binding sites were further delineated by using EMSA analysis on probes that deleted three base pairs at a time to identify bases essential for binding (Fig. 4). The 20-bp sites sharing the most homology with the esa box, with 8 and 9 out of 20 bases conserved with the esa box were identified (Fig. 5).
Similar analysis of the EsaR-activated lrhA promoter using a fluorescently labeled 284-bp upstream promoter DNA identified two distinct 20-bp binding sites separated by 2 bases (Fig. 3). EsaR binding at these two sites was confirmed using EMSA analysis of short probes that deleted three base pairs at a time until a loss of binding was observed (Fig. 4). The newly discovered EsaR binding sites, with the highest similarity to previously known esaR boxes, are conserved in 10 and 11 of 20 bases present in the esa box in the esaR promoter (Fig. 5). These sites are ∼475 bp upstream of the predicted ATG start codon and centered at −170 bp from a predicted transcriptional start site (based on unpublished RNA-Seq data in A. M. Stevens' lab). The relative positions of the esa boxes in the promoters of EsaR direct targets are shown in Fig. S3 in the supplemental material.
Analysis of EsaR binding sites.
Previously, only two native binding sites for EsaR had been identified; the esa box in the promoter of esaR and the esa box in the promoter of rcsA (15). EsaR has also been shown to bind to the lux box in a recombinant E. coli harboring a V. fischeri luxI promoter in an in vivo experiment (16). Using these three known binding sites and the five newly identified 20-bp EsaR binding sites identified via DNase I footprinting and EMSAs, an alignment was generated to identify a possible consensus sequence for EsaR binding, ACCTGTACTNNAGTACAGNT (where N is any nucleotide) (Fig. 5). This sequence is highly palindromic, similar to the conserved palindromes seen in other lux box-like sequences (14). Five bases, C3, T4, G5, C16, and A17, are similar to five of the six essential bases of LuxR-DNA interaction identified in the lux box of V. fischeri (22). The sixth residue at position 18 however, was not conserved, as both A and G residues were observed with similar frequencies. In addition, residues A1, A12, G13, T14, and T20 are also more conserved in the esa boxes and could be critical for EsaR-DNA interaction, distinguishing the consensus from the LuxR-binding consensus.
DISCUSSION
In this study, we have completed a proteomic approach identifying and characterizing genes involved in the QS regulon of P. stewartii. Comparison of the differentially expressed proteins in the strains proficient and deficient in QS allowed the identification of over 30 proteins that were previously not known to be regulated by EsaR (Table 2). These proteins were observed to play possible roles in capsular synthesis, fatty acid metabolism, and virulence, indicating a more-global cell density-dependent regulation of cellular functions in P. stewartii than previously known. While establishing the physiological function of the newly discovered QS-regulated proteins and definitively making connections to virulence in P. stewartii is beyond the scope of this study, plausible roles for some of the EsaR-regulated proteins are discussed below.
Two proteins essential in capsule production and previously shown to be not regulated by rcsA have been shown to be repressed by EsaR. The genes encoding these two proteins, GalE (UDP-galactose-4-epimerase) and GalF (a putative regulatory subunit for GalU), are found in an operon downstream of the wceI operon needed for stewartan production (23). Another repressed protein, GalU (glucose-1-phosphate uridylyltransferase), sometimes found in a complex with GalF (24), is a limiting factor in the production of exopolysaccharide. Thus, by repressing expression of these three proteins, EsaR further inhibits steps in the biosynthesis of stewartan at low cell densities. Interestingly, another previously identified repressed target of EsaR, RcsA, which activates the capsule operon (14), was not observed via 2D SDS-PAGE analysis. This may be due to the specific growth conditions used here or the fact it was simply below the limits of detection. However, OsmC, an osmotically induced membrane protein known to be activated by the Rcs phosphorelay in E. coli, was observed to be repressed by EsaR (25). Thus, the proteomic approach has revealed additional possible connections to the regulation of capsule production but has likely still not provided a complete list of the involved factors.
In this study, six promoters that are directly regulated by EsaR were identified and the EsaR binding sites in three of the promoters of the dkgA, glpF, and lrhA genes (Fig. 3 and 4) were fully characterized. DkgA belongs to the aldo-keto reductase (AKR) family that constitutes NADPH-dependent oxidoreductases with broad substrate ranges. DkgA is hypothesized to catalyze the conversion of 2,5-diketogluconate to 2-keto-l-gluconate in Erwinia species (26), and it has been linked to furfural tolerance and ascorbic acid biosynthesis in E. coli (27, 28). In Tatumella citrea, formerly Pantoea citrea (29), the production of 2,5-diketogluconate, is directly responsible for the intense coloration characteristic of pink disease of pineapple (11), presumably due to pathways containing DkgA-like enzymes. While it is unknown whether T. citrea possesses a dkgA gene and a QS system similar to those of P. stewartii and whether a similar regulation is observed, it is interesting to speculate that DkgA may play a role in causing Stewart's wilt similar to its importance in causing the pink disease of pineapple.
The glpF gene codes for a glycerol facilitator in the inner membrane and facilitates transport of glycerol. It is the first gene in a tricistronic operon consisting of glpK and glpX, which code for a glycerol kinase and a fructose biphosphatase, respectively. Glycerol is found in the midguts of beetles in winter, so activating the glycerol utilization operon at low cell densities may be relevant, as a response to P. stewartii overwintering in hibernating beetles (30). The glpFKX operon is regulated by other transcriptional regulators in E. coli, GlpR and the cyclic AMP (cAMP)-cAMP receptor protein (CRP) regulator (31). Potential GlpR and CRP binding sites were observed 87 and 406 bases upstream of the ATG start site, respectively, suggesting similar repressor and activator functions by the two proteins in P. stewartii.
LrhA is an indirect repressor of the chemotaxis, motility, and flagellar biosynthesis operon and also represses the expression of genes involved in the synthesis of type 1 fimbriae and the expression of the master regulator FlhDC, through which it indirectly regulates many other genes in E. coli (32, 33). In E. coli, the expression of lrhA is activated by itself and indirectly repressed by the RcsCDB phosphorelay system and FtsK, the DNA motor protein (34). LrhA in P. stewartii shares 69% identity with HexA from Pectobacterium carotovora, where it is known to negatively regulate the small RNA rsmB and RpoS levels (35). In P. stewartii, motility at low cell density from the initial site of plant inoculation to the xylem would logically seem to be necessary. However, the swarming motility exhibited by P. stewartii is more critical during biofilm formation within the xylem (36). Swarming motility requires synthesis of capsule, which occurs at high cell density once EsaR is inactive, and when LrhA would also be deactivated. In addition, it is also possible that LrhA-mediated repression of fimbrial expression prevents attachment at low cell density. It is interesting to note that the identified EsaR binding site in PlrhA was more than 475 bases upstream of the ATG start site. This is not unusual however, as the known esa box documented on the promoter of rcsA is more than 400 bases upstream of the ATG start site. The presence of two sites so far upstream of the start site for translation could be due to the presence of a long 5′ untranslated region, indicating the possibility of further regulatory elements such as small noncoding RNAs.
Three additional possible direct targets of EsaR were also found in the EMSA analysis but not by qRT-PCR. The apparent absence of transcriptional control of EsaR of these three targets is inconsistent with the proteomic and EMSA results and could be due to secondary methods of regulation, such as posttranscriptional control by small noncoding RNAs. It is also possible that transcriptional control occurred at an earlier stage of growth with differential protein expression being maintained over a longer period of time. The fabF gene, at the end of a fatty acid biosynthesis operon, codes for a 3-oxoacyl-(acyl carrier protein) synthase. Acyl carrier proteins are acyl donors for AHLs and the sugars involved in capsule synthesis. FabF also regulates fatty acid composition of the membrane phospholipids in response to cold in E. coli (37). The mglB gene is part of a tricistronic operon coding for components of methyl-galactoside transporter and functions as a galactose chemoreceptor and the periplasmic binding component for the transporter in E. coli (38). Galactose in an important component of stewartan (39). The rpsB gene codes for a 30S ribosomal subunit protein S2p. There is one report describing elevated levels of RpsB in biofilms of Listeria monocytogenes, wherein it was hypothesized to serve as a sensor of physical and chemical changes in the surrounding environment (40). RpsB may serve a similar role in P. stewartii.
Complete analysis of the QS regulon and the interactions of various other pathways with QS could illustrate how the cumulative effect of subtle changes in gene expression confers physiological benefits to the bacterium. Therefore, a transcriptomic approach would aid in recognizing the complete picture of additional indirect and direct targets of EsaR. The detailed analysis of the EsaR binding site in the promoters of dkgA, lrhA, and glpF has allowed the development of a more robust consensus sequence for EsaR binding that may prove useful to identify other EsaR-regulated genes using bioinformatic approaches. Deletion analysis of each EsaR direct target individually would aid in understanding the precise roles of these genes in plant pathogenesis and the necessity of regulation by QS in P. stewartii. It is probable that the temporal regulation afforded by QS helps the bacterium in adapting to different physiological conditions during colonization and infection.
Supplementary Material
ACKNOWLEDGMENTS
We thank the laboratory of S. von Bodman (University of Connecticut) for providing us with plasmids and strains and sharing unpublished results and W. K. Ray and R. Helm at the Virginia Tech (VT) Mass Spectrometry Incubator for MALDI-TOF MS-MS analysis.
This work is supported by the National Science Foundation grant MCB-0919984, Virginia Tech GSA GRDP award cycle II in 2012 and 2013, and the 2012-2013 Biological Sciences GSDA fellowship.
Footnotes
Published ahead of print 2 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01744-13.
REFERENCES
- 1.Fuqua C, Winans SC, Greenberg EP. 1996. Census and consensus in bacterial ecosystems: the LuxR-LuxI family of quorum-sensing transcriptional regulators. Annu. Rev. Microbiol. 50:727–751 [DOI] [PubMed] [Google Scholar]
- 2.Waters CM, Bassler BL. 2005. Quorum sensing: cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol. 21:319–346 [DOI] [PubMed] [Google Scholar]
- 3.Stevens AM, Schuster M, Rumbaugh KP. 2012. Working together for the common good: cell-cell communication in bacteria. J. Bacteriol. 194:2131–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Withers H, Swift S, Williams P. 2001. Quorum sensing as an integral component of gene regulatory networks in Gram-negative bacteria. Curr. Opin. Microbiol. 4:186–193 [DOI] [PubMed] [Google Scholar]
- 5.Pataky JK. 2003. Stewart's wilt of corn. APSnet Features 0703. 10.1094/APSnetFeature-2003-0703 [DOI] [Google Scholar]
- 6.Beck von Bodman S, Farrand SK. 1995. Capsular polysaccharide biosynthesis and pathogenicity in Erwinia stewartii require induction by an N-acylhomoserine lactone autoinducer. J. Bacteriol. 177:5000–5008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.von Bodman SB, Majerczak DR, Coplin DL. 1998. A negative regulator mediates quorum-sensing control of exopolysaccharide production in Pantoea stewartii subsp. stewartii. Proc. Natl. Acad. Sci. U. S. A. 95:7687–7692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merighi M, Majerczak DR, Zianni M, Tessanne K, Coplin DL. 2006. Molecular characterization of Pantoea stewartii subsp. stewartii HrpY, a conserved response regulator of the Hrp type III secretion system, and its interaction with the hrpS promoter. J. Bacteriol. 188:5089–5100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roper MC. 2011. Pantoea stewartii subsp. stewartii: lessons learned from a xylem-dwelling pathogen of sweet corn. Mol. Plant Pathol. 12:628–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coutinho TA, Venter SN. 2009. Pantoea ananatis: an unconventional plant pathogen. Mol. Plant Pathol. 10:325–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pujol CJ, Kado CI. 2000. Genetic and biochemical characterization of the pathway in Pantoea citrea leading to pink disease of pineapple. J. Bacteriol. 182:2230–2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aylward FO, Currie CR, Suen G. 2012. The evolutionary innovation of nutritional symbioses in leaf-cutter ants. Insects 3:41–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Torres-Cabassa A, Gottesman S, Frederick RD, Dolph PJ, Coplin DL. 1987. Control of extracellular polysaccharide synthesis in Erwinia stewartii and Escherichia coli K-12: a common regulatory function. J. Bacteriol. 169:4525–4531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minogue TD, Carlier AL, Koutsoudis MD, von Bodman SB. 2005. The cell density-dependent expression of stewartan exopolysaccharide in Pantoea stewartii ssp. stewartii is a function of EsaR-mediated repression of the rcsA gene. Mol. Microbiol. 56:189–203 [DOI] [PubMed] [Google Scholar]
- 15.Minogue TD, Wehland-von Trebra M, Bernhard F, von Bodman SB. 2002. The autoregulatory role of EsaR, a quorum-sensing regulator in Pantoea stewartii ssp. stewartii: evidence for a repressor function. Mol. Microbiol. 44:1625–1635 [DOI] [PubMed] [Google Scholar]
- 16.Schu DJ, Carlier AL, Jamison KP, von Bodman SB, Stevens AM. 2009. Structure/function analysis of the Pantoea stewartii quorum-sensing regulator EsaR as an activator of transcription. J. Bacteriol. 191:7402–7409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koutsoudis MD, Tsaltas D, Minogue TD, von Bodman SB. 2006. Quorum-sensing regulation governs bacterial adhesion, biofilm development, and host colonization in Pantoea stewartii subspecies stewartii. Proc. Natl. Acad. Sci. U. S. A. 103:5983–5988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilbert ER, Williams PM, Ray WK, Li H, Emmerson DA, Wong EA, Webb KE. 2010. Proteomic evaluation of chicken brush-border membrane during the early posthatch period. J. Proteome Res. 9:4628–4639 [DOI] [PubMed] [Google Scholar]
- 19.Glasner JD. 2003. ASAP, a systematic annotation package for community analysis of genomes. Nucleic Acids Res. 31:147–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schu DJ, Ramachandran R, Geissinger JS, Stevens AM. 2011. Probing the impact of ligand binding on the acyl-homoserine lactone-hindered transcription factor EsaR of Pantoea stewartii subsp. stewartii. J. Bacteriol. 193:6315–6322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Antunes LCM, Ferreira RBR, Lostroh CP, Greenberg EP. 2008. A mutational analysis defines Vibrio fischeri LuxR binding sites. J. Bacteriol. 190:4392–4397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carlier A, Burbank L, von Bodman SB. 2009. Identification and characterization of three novel EsaI/EsaR quorum-sensing controlled stewartan exopolysaccharide biosynthetic genes in Pantoea stewartii ssp. stewartii. Mol. Microbiol. 74:903–913 [DOI] [PubMed] [Google Scholar]
- 24.Marolda CL, Valvano MA. 1996. The GalF protein of Escherichia coli is not a UDP-glucose pyrophosphorylase but interacts with the GalU protein possibly to regulate cellular levels of UDP-glucose. Mol. Microbiol. 22:827–840 [DOI] [PubMed] [Google Scholar]
- 25.Sturny R, Cam K, Gutierrez C, Conter A. 2003. NhaR and RcsB independently regulate the osmCp1 promoter of Escherichia coli at overlapping regulatory sites. J. Bacteriol. 185:4298–4304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yum DY, Lee BY, Pan JG. 1999. Identification of the yqhE and yafB genes encoding two 2,5-diketo-d-gluconate reductases in Escherichia coli. Appl. Environ. Microbiol. 65:3341–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turner PC, Miller EN, Jarboe LR, Baggett CL, Shanmugam KT, Ingram LO. 2011. YqhC regulates transcription of the adjacent Escherichia coli genes yqhD and dkgA that are involved in furfural tolerance. J. Ind. Microbiol. Biotechnol. 38:431–439 [DOI] [PubMed] [Google Scholar]
- 28.Jeudy S, Monchois V, Maza C, Claverie J-M, Abergel C. 2006. Crystal structure of Escherichia coli DkgA, a broad-specificity aldo-keto reductase. Proteins 62:302–307 [DOI] [PubMed] [Google Scholar]
- 29.Brady CL, Venter SN, Cleenwerck I, Vandemeulebroecke K, De Vos P, Coutinho T. 2010. Transfer of Pantoea citrea, Pantoea punctata and Pantoea terrea to the genus Tatumella emend. as Tatumella citrea comb. nov., Tatumella punctata comb. nov. and Tatumella terrea comb. nov. and description of Tatumella morbirosei sp. nov. Int. J. Syst. Evol. Microbiol. 60:484–494 [DOI] [PubMed] [Google Scholar]
- 30.Cohen E. 2012. Roles of aquaporins in osmoregulation, desiccation and cold hardiness in insects. Entomol. Ornithol. Herpetol. S1:001. 10.4172/2161-0983.S1-001 [DOI] [Google Scholar]
- 31.Weissenborn DL, Wittekindt N, Larson TJ. 1992. Structure and regulation of the glpFK operon encoding glycerol diffusion facilitator and glycerol kinase of Escherichia coli K-12. J. Biol. Chem. 267:6122–6131 [PubMed] [Google Scholar]
- 32.Blumer C, Kleefeld A, Lehnen D, Heintz M, Dobrindt U, Nagy G, Michaelis K, Emödy L, Polen T, Rachel R, Wendisch VF, Unden G. 2005. Regulation of type 1 fimbriae synthesis and biofilm formation by the transcriptional regulator LrhA of Escherichia coli. Microbiology 151:3287–3298 [DOI] [PubMed] [Google Scholar]
- 33.Lehnen D, Blumer C, Polen T, Wackwitz B, Wendisch VF, Unden G. 2002. LrhA as a new transcriptional key regulator of flagella, motility and chemotaxis genes in Escherichia coli. Mol. Microbiol. 45:521–532 [DOI] [PubMed] [Google Scholar]
- 34.Peterson CN, Carabetta VJ, Chowdhury T, Silhavy TJ. 2006. LrhA regulates rpoS translation in response to the Rcs phosphorelay system in Escherichia coli. J. Bacteriol. 188:3175–3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mukherjee A, Cui Y, Ma W, Liu Y, Chatterjee AK. 2000. HexA of Erwinia carotovora ssp. carotovora strain Ecc71 negatively regulates production of RpoS and rsmB RNA, a global regulator of extracellular proteins, plant virulence and the quorum-sensing signal, N-(3-oxohexanoyl)-l-homoserine. Environ. Microbiol. 2:203–215 [DOI] [PubMed] [Google Scholar]
- 36.Herrera CM, Koutsoudis MD, Wang X, von Bodman SB. 2008. Pantoea stewartii subsp. stewartii exhibits surface motility, which is a critical aspect of Stewart's wilt disease development on maize. Mol. Plant Microbe Interact. 21:1359–1370 [DOI] [PubMed] [Google Scholar]
- 37.Garwin JL, Klages AL, Cronan JE. 1980. Beta-ketoacyl-acyl carrier protein synthase II of Escherichia coli. Evidence for function in the thermal regulation of fatty acid synthesis. J. Biol. Chem. 255:3263–3265 [PubMed] [Google Scholar]
- 38.Harayama S, Bollinger J, Iino T, Hazelbauer GL. 1983. Characterization of the mgl operon of Escherichia coli by transposon mutagenesis and molecular cloning. J. Bacteriol. 153:408–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, Yang F, von Bodman SB. 2012. The genetic and structural basis of two distinct terminal side branch residues in stewartan and amylovoran exopolysaccharides and their potential role in host adaptation. Mol. Microbiol. 83:195–207 [DOI] [PubMed] [Google Scholar]
- 40.Trémoulet F, Duché O, Namane A, Martinie B, Labadie JC. 2002. Comparison of protein patterns of Listeria monocytogenes grown in biofilm or in planktonic mode by proteomic analysis. FEMS Microbiol. Lett. 210:25–31 [DOI] [PubMed] [Google Scholar]
- 41.Crooks GE, Hon G, Chandonia J-M, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dolph PJ, Majerczak DR, Coplin DL. 1988. Characterization of a gene cluster for exopolysaccharide biosynthesis and virulence in Erwinia stewartii. J. Bacteriol. 170:865–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grant SG, Jessee J, Bloom FR, Hanahan D. 1990. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc. Natl. Acad. Sci. U. S. A. 87:4645–4649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Herrero M, De Lorenzo V, Timmis KN. 1990. Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram-negative bacteria. J. Bacteriol. 172:6557–6567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176 [DOI] [PubMed] [Google Scholar]
- 46.Stabb EV, Ruby E. 2002. RP4-based plasmids for conjugation between Escherichia coli and members of the Vibrionaceae. Methods Enzymol. 358:413–426 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.