

Abstract
Streptococcus agalactiae (group B Streptococcus [GBS]) is a Gram-positive bacterium that colonizes the cervicovaginal tract in approximately 25% of healthy women. Although colonization is asymptomatic, GBS can be vertically transmitted to newborns peripartum, causing severe disease such as pneumonia and meningitis. Current prophylaxis, consisting of late gestation screening and intrapartum antibiotics, has failed to completely prevent transmission, and GBS remains a leading cause of neonatal sepsis and meningitis in the United States. Lack of an effective vaccine and emerging antibiotic resistance necessitate exploring novel therapeutic strategies. We have employed a host-directed immunomodulatory therapy using a novel peptide, known as EP67, derived from the C-terminal region of human complement component C5a. Previously, we have demonstrated in vivo that EP67 engagement of the C5a receptor (CD88) effectively limits staphylococcal infection by promoting cytokine release and neutrophil infiltration. Here, using our established mouse model of GBS vaginal colonization, we observed that EP67 treatment results in rapid clearance of GBS from the murine vagina. However, this was not dependent on functional neutrophil recruitment or CD88 signaling, as EP67 treatment reduced the vaginal bacterial load in mice lacking CD88 or the major neutrophil receptor CXCr2. Interestingly, we found that EP67 inhibits GBS growth in vitro and in vivo and that antibacterial activity was specific to Streptococcus species. Our work establishes that EP67-mediated clearance of GBS is likely due to direct bacterial killing rather than to enhanced immune stimulation. We conclude that EP67 may have potential as a therapeutic to control GBS vaginal colonization.
INTRODUCTION
Streptococcus agalactiae (group B Streptococcus [GBS]) is currently a leading pathogen responsible for early-onset neonatal sepsis in the United States (1). However, GBS predominantly exists as a commensal colonizer of the lower gastrointestinal and vaginal tract of approximately 25% of healthy adults (2). Neonatal exposure to this pathobiont occurs in utero or peripartum through contact with vaginal fluids (3). More recently, several clinical cases have suggested that transmission also occurs postpartum through consumption of breast milk (4). Early-onset septicemia typically results in pneumonia and respiratory failure, and the late-onset form, occurring up to 7 months after birth, presents with bacteremia and meningitis (3). Current guidelines by the Centers for Disease Control include intrapartum antibiotic prophylaxis (IAP) administered to mothers who are GBS positive based on rectovaginal cultures at 35 to 37 weeks gestation. While IAP has reduced the incidence of early-onset infections by 80%, the late-onset incidence has remained unaffected (1) and adult GBS infections are on the rise (5). Lack of an effective vaccine for GBS and emerging antibiotic resistance impel development of novel treatment strategies to control GBS vaginal colonization and abrogate transmission to the vulnerable newborn.
One promising strategy is to exploit or modulate the innate immune system to enhance the host's ability to combat microbial infection while limiting inflammation-induced tissue injury. Accordingly, we have engineered a conformationally biased, response-selective analogue of the biologically active C-terminal region of the human complement component C5a65–74 (6). This analogue, termed EP67, has the sequence Tyr-Ser-Phe-Lys-Asp-Met-Pro-(N-methylLeu)-d-Ala-Arg, or YSFKDMP(MeL)aR, and was designed to bind C5a receptors (CD88) on antigen-presenting cells (APCs) such as macrophages but to limit inflammatory properties by not engaging C5aRs on polymorphonuclear cells (PMNs) (6, 7). EP67 has shown adjuvant potential in vaccine designs against ovalbumin (6) and Coccidioides (8) by enhancing activation of a TH1 response via engagement of C5aR-bearing APCs. Additionally, our previous work has demonstrated that prophylactic EP67 treatment limited infection with community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA) in a mouse model of dermonecrosis where we observed that EP67 treatment reduced skin lesion size, an effect dependent on both PMN recruitment and signaling through CD88 (9). EP67 has also been shown to lessen MRSA biofilm infection (10).
Considering these previous results, we hypothesized that EP67 may similarly exhibit immunostimulatory properties through enhancing innate immune signaling pathways to prevent GBS colonization. In this study, we demonstrated that EP67 treatment promotes rapid clearance of GBS using a murine model of vaginal colonization. In contrast to our studies with staphylococcal infections, however, this effect is largely independent of the presence of neutrophil receptor CXCr2 and CD88 signaling. Interestingly, in vitro assays reveal that EP67 possesses bactericidal activity against GBS and other Streptococcus species. Our results indicate that EP67 exhibits protective activity against GBS infection and colonization primarily through targeted bacterial killing rather than through immune stimulation.
MATERIALS AND METHODS
Peptide synthesis and preparation.
EP67 [YSFKDMP(MeL)aR] and an inactive scrambled sequence, sEP67 [(MeL)RMYKPaFDS], were generated by solid-phase methods using the Fmoc method of orthogonal synthesis, purified by analytical and preparative reverse-phase high-performance liquid chromatography (HPLC), and characterized by electrospray mass spectrometry according to previously published methods (11). For in vitro and in vivo assays, peptides were suspended in 1× phosphate-buffered saline (PBS) (CellGrow) at given concentrations.
Bacterial strains and growth conditions.
Streptococcus agalactiae clinical isolates A909 (serotype Ia) (12), 515 (serotype Ia) (13), NCTC 10/84 (1169-NT1; ATCC 49447) (serotype V) (14), and COH1 (serotype III) (15) and Streptococcus pyogenes (GAS) serotype M1T1 clinical isolate 5448 (16), Streptococcus equi (ATCC 6580), Streptococcus salivarius (ATCC 13419), and Streptococcus gordonii strain M99 (17) were grown aerobically in Todd-Hewitt broth (THB) (Hardy Diagnostics) at 37°C. Streptococcus pneumoniae serotype 2 strain D39 (NCTC 7466) (18) was grown in THB supplemented with 1.5% yeast extract (THY) at 37°C with 5% CO2. Staphylococcus aureus USA300 isolate (TCH1516-HOU-MR; ATCC accession number BAA-1717) (19) was grown aerobically in tryptone soy broth (TS) (Oxoid) at 37°C. Salmonella enterica serovar Typhimurium 14028S 1/9 (20) was grown aerobically in Luria broth (LB) (Criterion) at 37°C. Proteus mirabilis, Ruminococcus albus, and Enterococcus faecalis (murine vaginal isolates from our laboratory and verified with 16S sequencing at Eton Biosciences Inc.) were grown in LB at 37°C.
Murine strains.
All mouse work was approved by the Office of Lab Animal Care at San Diego State University and conducted under accepted veterinary standards. Female CD1 and BALB/c mice (8 to 20 weeks old) were obtained from Charles River Laboratories. Breeding pairs of CXCr2 (CXCL2 receptor) knockout (KO) mice (formerly IL8r KO mice) were originally purchased [C.129S2(B6)-Cxcr2tm1Mwm/J; Jackson Laboratories], and a homozygous × homozygous breeding colony was established at the University of California San Diego Veterans Affairs (UCSD VA) Hospital. Mice were maintained on water containing cotrimoxazole (200 μg/ml sulfamethoxazole and 40 μg/ml trimethoprim), and for the 17-week-old females used in this study, antimicrobial treatment was stopped 48 h prior to inoculation with GBS. Female C5aR1 (CD88) KO mice, 8 weeks of age, were purchased from Jackson Laboratories [C.129S4(B6)-C5ar1tm1Cge/J].
In vivo model of GBS vaginal colonization.
Female CD1, BALB/c, CXCr2 KO, and CD88 KO mice were used for colonization assays adapted from previous work (21, 22). To synchronize estrus and promote bacterial colonization (23, 24), mice were injected intraperitoneally (i.p.) with 0.5 mg 17β-estradiol suspended in sesame oil (Sigma) 24 to 72 h prior to inoculation. Mice were inoculated with 1 × 107 CFU (in 10 μl PBS) GBS A909 in the vaginal lumen. Immediately prior to inoculation, vaginal lavage was performed by pipetting 20 μl of PBS several times in the vaginal lumen to collect cells and cytokines as described elsewhere (25, 26). Bacterial load was determined by swabbing the vaginal lumen with ultrafine calcium alginate-tipped swabs and serial dilution plating of swab samples. GBS and native Enterococcus strains were identified by the presence of mauve- and blue-pigmented colonies, respectively, on CHROMagar Strep B agar (DRG International Inc.) (27). GBS was allowed to colonize for at least 24 h prior to peptide dosage. After colonization was established, 250 μg (in 12.5 μl PBS) of EP67 or sEP67 was administered into the vaginal lumen every 24 h after first collecting lavage fluid and swab samples. Enzyme-linked immunosorbent assays (ELISAs) were performed on vaginal lavage fluid for MIP-2 and KC (R&D Systems) as described by the manufacturer.
In vivo model of GBS peritoneal infection.
Female BALB/c, CXCr2 KO, and CD88 KO mice were used for peritoneal infection assays adapted from previous work (28). Mice were injected i.p. with 1 × 107 CFU GBS A909 suspended in 100 μl PBS or with 100 μl PBS only for uninfected controls. Immediately following infection, 750 μg of EP67 or sEP67, or an equivalent volume of PBS (100 μl) for uninfected controls, was injected i.p. After a 2-h incubation, all the mice were euthanized by CO2 asphyxiation and peritoneal cavities subjected to lavage with 5 ml PBS. To quantity the percentage of the original inoculum recovered, lavage fluid was serially diluted and plated on THB agar to enumerate viable bacteria. Additionally, total leukocytes present in lavage fluid were quantified using a hemocytometer loaded with 10 μl of unstained lavage fluid.
Flow cytometry.
Vaginal lavage fluid from BALB/c and CD88 KO mice and peritoneal lavage fluid from BALB/c mice were subjected to flow cytometry to identify specific immune cell populations present. Cells from lavage fluid were incubated for 30 min in RPMI medium containing 30% serum (mouse and fetal bovine serum [FBS]) to block nonspecific binding. Samples were stained with fluorochrome-conjugated antibodies specific for CD11b-AlexaFluor647, CD11c-phycoerithrin (PE) Cy7, and NK1.1-PE (eBioscience) as well as antibodies specific for Ly6G-fluorescein isothiocyanate (FITC) and B220-PE (BD Biosciences). Neutrophils were identified as small cells, by forward scatter analysis, and were Ly6G+ CD11b+. Macrophages were identified as large cells that were CD11c+ CD11b+ and Ly6G− B220−. Dendritic cells were determined by being highly CD11c+ and B220−. Natural killer cells were determined to be small cells that were Ly6G− and NK1.1+ CD11c+. Samples were analyzed with an Accuri C6 cytometer (BD Biosciences), with 20,000 events recorded per sample. Due to the presence of cell debris and large cells in vaginal samples, immune cell populations were determined from a population gated for cell size. Cell populations were assessed for percentages of fluorescent staining and staining brightness using Accuri analysis software.
Antimicrobial assays.
MICs were determined during aerobic growth in a 96-well plate format as described previously (29) with several modifications. Briefly, bacterial strains were grown to mid-exponential phase (approximately 1 × 108 CFU/ml). The cultures were diluted 1:100, and 140 μl was added to each well. A 10-μl volume of EP67 or sEP67 was added to each well in 2-fold increment concentrations of from 0 to 403.2 μM (500 μg/ml). Bacterial growth was measured by optical density at 600 nm (OD600) after a 24-h incubation at 37°C. The MIC was expressed as the lowest concentration of peptide that inhibited microbial growth completely. Assays were performed in triplicate and were repeated at least 3 times. Minimum bactericidal concentrations (MBC) were determined by subculturing bacteria from wells onto an agar plate using a 48-prong replicator. MBC was determined by the lowest peptide concentration exhibiting no visible growth on the agar. Kinetic killing assays were performed as described above for MIC assays with the following modifications: mid-exponential-phase cultures were diluted 1:100, and 1.4 ml was combined with 100 μl of 201.6 μM (250 μg/ml) EP67 or sEP67 and incubated at 37°C. Samples were collected at the indicated time points and plated on THB agar to determine viable CFU.
Live/Dead bacterial viability stain.
GBS viability during EP67 treatment was determined using a Live/Dead Bac Light bacterial viability kit (Molecular Probes, Inc.) per the manufacturer's directions with several modifications. Briefly, mid-exponential-phase GBS cultures were diluted 1:100, and 1.4 ml was combined with 250 μg EP67 or sEP67 or an equivalent volume of PBS (100 μl) and incubated for 2, 5, or 24 h at 37°C. Bacteria were pelleted and resuspended in 100 μl of diluted THB (equal parts THB and 0.85% NaCl), 0.25 μl of Syto 9, and 0.25 μl of propidium iodide. Suspensions were incubated in the dark at room temperature for 15 min, and 10 μl was wet mounted onto slides. Fluorescent images were collected at ×1,000 magnification (Zeiss Axio Observer D1 with an attached Zeiss MRc camera). Green and red channel images were merged using AxioVision software (Zeiss).
Statistical analyses.
GraphPad Prism version 5.0f was used for statistical analyses. Differences in in vivo peritoneal assays, including determinations of recovered CFU, total leukocyte counts, and flow cytometry cell populations, were calculated using unpaired Student's t test analysis or one-way analysis of variance (ANOVA) with Tukey's multiple-comparisons test. Differences in bacterial persistence in mouse vaginal experiments were calculated using log-rank (Mantel-Cox) or Mann-Whitney tests. Differences in cytokine levels from vaginal lavage fluid were calculated using two-way ANOVA with a Bonferroni posttest. Statistical significance was determined at a P < 0.05.
RESULTS
EP67 treatment promotes rapid GBS clearance from the murine vaginal tract.
Previous work has demonstrated that EP67 is protective in reducing CA-MRSA dermal infection (9) and promoting survival of lethal infection with influenza A virus (30). To determine if EP67 has similar protective activity during GBS colonization, we used our established GBS vaginal colonization mouse model (22). Briefly, we treated 8-week-old CD1 mice with 17β-estradiol 1 day prior to bacterial inoculation to promote successful colonization. We inserted 1 × 107 CFU GBS into the vagina and, 24 h postinoculation, began administering EP67 or the inactive scrambled control, sEP67, into the vaginal lumen. On successive days, the vaginal lumen was first swabbed to determine changes in bacterial load over time and then dosed with either EP67 or sEP67. Consistent with other infection models, EP67 treatment significantly decreased GBS persistence over the 5-day regimen compared to treatment with sEP67 (P = 0.0124) (Fig. 1). Moreover, we observed this same significant effect with another GBS clinical isolate, COH1 (data not shown).
Fig 1.
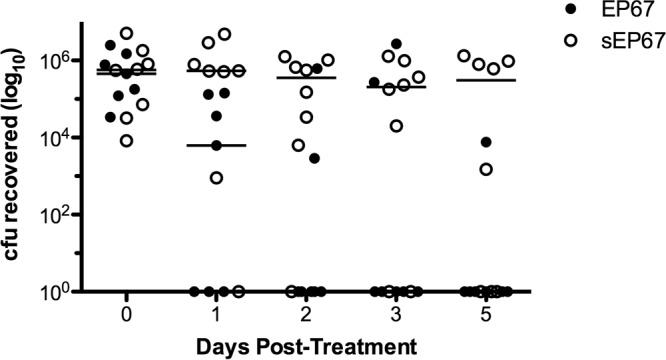
EP67 treatment reduces GBS vaginal colonization. GBS, 1 × 107 CFU, was inoculated into the vaginal lumen of 8-week-old CD1 mice (n = 7 to 8 per group). GBS was allowed to establish colonization for 24 h prior to initial treatment with EP67 or sEP67. GBS persistence was measured by swabbing the vagina and enumerating recovered bacteria. Lines represent median CFU for each treatment group (the median was 0 CFU for the EP67 group on days 2, 3, and 5). In vivo experiments were conducted independently at least three times, and data from one representative experiment are shown. EP67 significantly decreased GBS persistence compared to sEP67 (P = 0.0124) as calculated using a log rank test.
EP67-mediated reduction of vaginal GBS does not require neutrophil recruitment or C5a signaling.
In work using EP67 to control CA-MRSA, neutrophils were found to be the key responders limiting dermonecrosis (9). We have also shown that neutrophil antimicrobial activity corresponds with GBS clearance from the murine vaginal tract early on during colonization (22). Therefore, we examined the requirement for neutrophil signaling and recruitment during EP67-mediated clearance of GBS in the vaginal tract. Murine chemokine MIP-2 (CXCL2) is a homologue of human CXCL8 (interleukin-8 [IL-8]) and engages CXCr2 (IL-8 receptor homologue) on neutrophils, recruiting them to the site of chemokine production (31). Prior work has shown limited neutrophil recruitment in CXCr2 KO mice during several states of pathogenesis, including Escherichia coli urinary tract infection (32), pneumonic plague (33), and Streptococcus pneumoniae-induced pulmonary infection (34). Consequently, we examined the effect of impaired neutrophil recruitment on EP67 activity during GBS vaginal colonization using CXCr2 KO mice. As seen in CD1 mice (Fig. 1), treatment with EP67 enhanced GBS vaginal clearance in BALB/c mice compared to treatment with sEP67 (Fig. 2A and C). Surprisingly, we also observed that EP67 significantly accelerated GBS clearance in CXCr2 KO mice (P = 0.0033) (Fig. 2B and C). Furthermore, at 9 days postinoculation, although MIP-2 levels were fluctuating as expected during the estrous cycle (25) in both BALB/c and CXCr2 KO mice, there were no significant differences observed between groups (Fig. 2D). These results suggest that EP67-mediated GBS reduction in the vagina is occurring independently of neutrophil recruitment through the CXCL2 receptor.
Fig 2.

GBS vaginal clearance by EP67 does not require signaling through the CXCL2 receptor. (A and B) Approximately 1 × 107 CFU GBS was inoculated into the vaginal lumen of 16-week-old BALB/c mice (n = 5 to 10 per group) (A) or CXCr2 KO mice (n = 3 to 9 per group) (B). GBS was allowed to colonize for 9 days to ensure colonization following antibiotic removal (see maintenance of CXCr2 KO mice in Materials and Methods) prior to initial treatment with EP67 or sEP67. GBS persistence was measured by swabbing the vagina and enumerating recovered bacteria; horizontal lines represent median CFU recovered (A and B). (C) The percentages of mice that were colonized over time were calculated following EP67 or sEP67 treatment. (D) Chemokine MIP-2 levels were quantified by ELISA in vaginal lavage fluid collected from mice at the indicated time points.
EP67 interacts with C5aR (CD88), resulting in proinflammatory signaling cascades from cells such as splenic antigen-presenting cells (6) with subsequent recruitment of neutrophils, natural killer (NK) cells, and dendritic cells (DCs) (9, 30). To investigate if CD88 signaling contributes to the EP67-mediated GBS vaginal reduction in vivo, we utilized CD88 KO mice. As seen previously (Fig. 2), EP67 treatment significantly accelerated GBS vaginal clearance in BALB/c mice by day 3 posttreatment compared to the sEP67 control (P = 0.0404) (Fig. 3A and C). We also observed reduced GBS vaginal persistence in CD88 KO mice treated with EP67 compared to sEP67, but this difference was not significant (P = 0.1163) (Fig. 3B and C). This suggests that CD88 signaling may contribute in part to EP67 action. However, analysis of vaginal lavage fluid from both wild-type (WT) and CD88 KO mice colonized with GBS and treated with either EP67 or sEP67 revealed no differences in Ly6G+ CD11b+ cell populations (Fig. 3D) or total CD11b+ leukocyte populations (data not shown). These data are consistent with our results described above demonstrating that increased immune signaling is likely not responsible for the observed reduction of bacterial colonization during EP67 treatment.
Fig 3.

Increased GBS vaginal clearance during EP67 treatment does not require signaling through the CD88 receptor. (A and B) Approximately 1 × 107 CFU GBS was inoculated into the vaginal lumen of 8-week-old BALB/c mice (n = 7 per group) (A) or CD88 KO mice (n = 10 per group) (B). GBS was allowed to colonize for 2 days prior to initial treatment with EP67 or sEP67. GBS persistence was measured by swabbing the vagina and enumerating recovered bacteria. Lines represent median CFU recovered (A and B). (C) The percentages of mice that were colonized over time were calculated following EP67 or sEP67 treatment. (D) Neutrophils (Ly6G+ CD11b+) present in vaginal lavage fluid collected from mice at the indicated time points were quantified by flow cytometry as described in Materials and Methods.
EP67 exhibits direct antibacterial activity.
Since our results thus far suggested that certain immune signaling pathways are not critical for EP67 action, we sought to determine if EP67 exhibited direct antimicrobial activity against GBS. We assessed bacterial growth in the presence of EP67 and observed dramatic growth inhibition of GBS strain A909 (MIC > 100 μM), while treatment with sEP67 resulted in no inhibition (Fig. 4A). We also observed that EP67 was bactericidal at concentrations greater than 100 μM, whereas sEP67 was not bactericidal at any concentration tested, as determined by MBC assays (data not shown). Other GBS clinical isolates (COH1, NCTC 10/84, and 515) were similarly inhibited (Table 1). Additionally, we tested other Streptococcus species and observed growth inhibition in the presence of EP67 (Table 1). However, EP67 did not inhibit growth of other Gram-positive bacteria, including Staphylococcus, Ruminococcus, and Enterococcus, or of Gram-negative bacteria such as Salmonella and Proteus (Table 1).
Fig 4.

EP67 exhibits antibacterial activity against GBS. (A) GBS strain A909 was grown to the logarithmic phase and subjected to increasing levels of EP67 or sEP67. After 24 h, GBS growth was measured by absorbance (OD600). (B and C) Killing kinetics of WT GBS strains A909 and COH1 upon exposure to 200 μM EP67 or sEP67 control as measured by CFU (B) or Live-Dead staining as described in Materials and Methods (C). GBS with intact membrane fluoresces green, whereas GBS with damaged membranes fluoresces red. Representative images are shown. Magnification, ×1,000. All experiments were conducted independently at least twice, and representative data from one experiment are shown.
Table 1.
EP67 Minimal inhibitory concentrations
| Species; strain | EP67 concn [μM] |
|---|---|
| Streptococci | |
| S. agalactiae; A909 | 100.8 |
| S. agalactiae; COH1 | 100.8 |
| S. agalactiae; NCTC 10/84 | 100.8 |
| S. agalactiae; 515 | 100.8 |
| S. pyogenes; 5448 | 50.4 |
| S. pneumoniae; D39 | 12.6 |
| S. gordonii; M99 | 100.8 |
| S. equi; ATCC 6580 | 100.8 |
| S. salivarius; ATCC 13419 | 100.8 |
| Murine vaginal isolates | |
| Proteus mirabilis | >404.2 |
| Ruminococcus albus | >404.2 |
| Enterococcus faecalis | >404.2 |
| Additional human pathogens | |
| Staphylococcus aureus; USA300 | >404.2 |
| Salmonella enterica; serovar Typhimurium 14028S 1/9 | >404.2 |
To evaluate the killing kinetics of EP67 activity against GBS, bacterial survival was assessed over time in the presence of EP67 or sEP67. Our results demonstrated that approximately 95% of both the A909 and COH1 strains were killed by EP67 within 3 h, whereas sEP67 did not inhibit growth of either GBS strain (Fig. 4B). To visually confirm the ability of EP67 to directly kill GBS, we performed a two-color fluorescence assay of bacterial viability. This assay labels all bacteria with a green fluorescent nucleic acid stain (Syto 9) and selectively stains bacteria with permeable membranes using a red fluorescent nucleic acid (propidium iodide). Previous work has established that combining these two dyes at equal ratios can detect membrane damage, as demonstrated by decreased Syto 9 fluorescence and increased propidium iodide uptake in permeable cells (35, 36). We incubated GBS with EP67, sEP67, or PBS and at multiple time points conducted fluorescent staining. Microscopic analysis revealed that by 2 h post-EP67 exposure, the majority of bacteria were red, indicating membrane permeability (Fig. 4C). As expected, minimal numbers of cells showed membrane permeability with sEP67 or PBS treatment (Fig. 4C). These results further confirm that EP67 treatment results in compromised membrane integrity and corresponding cell death.
EP67 treatment reduces GBS bacterial survival in vivo.
Because EP67 promoted GBS clearance in the vaginal tract amid native flora and estrous cycle influences and in vitro assays displayed direct killing capability, we sought to examine this peptide-microbe interaction in a more controlled in vivo environment. We selected a mouse model of intraperitoneal (i.p.) infection adapted from previous work (28). Immediately following GBS A909 peritoneal infection, BALB/c mice were injected i.p. with EP67, sEP67, or PBS for uninfected controls. After 2 h of incubation, mice were sacrificed, the peritoneal cavity was subjected to lavage, and bacterial survival was assessed via enumeration of recovered GBS CFU. Viable GBS load was significantly reduced in animals treated with EP67 compared to sEP67 (P = 0.0003) (Fig. 5A). We quantified and identified innate immune cells in peritoneal lavage fluid of EP67 and sEP67 2 h postinfection with GBS using flow cytometry and antibodies to distinguish neutrophils, macrophages, dendritic cells (DC), and natural killer (NK) cells. Macrophages, DCs, and NK cells were rare and did not differ in numbers between treatment groups (Fig. 5B and data not shown). Neutrophils were the predominant innate immune cell present, with infected mice having significantly more neutrophils than uninfected controls (P = 0.0043) (Fig. 5B). Interestingly, neutrophil levels in the peritoneal cavity did not differ between EP67- and sEP67-treated mice at this early 2-h time point (Fig. 5B). Thus, these results that suggest EP67-mediated GBS reduction may be due to direct antibacterial killing instead of immune activation.
Fig 5.
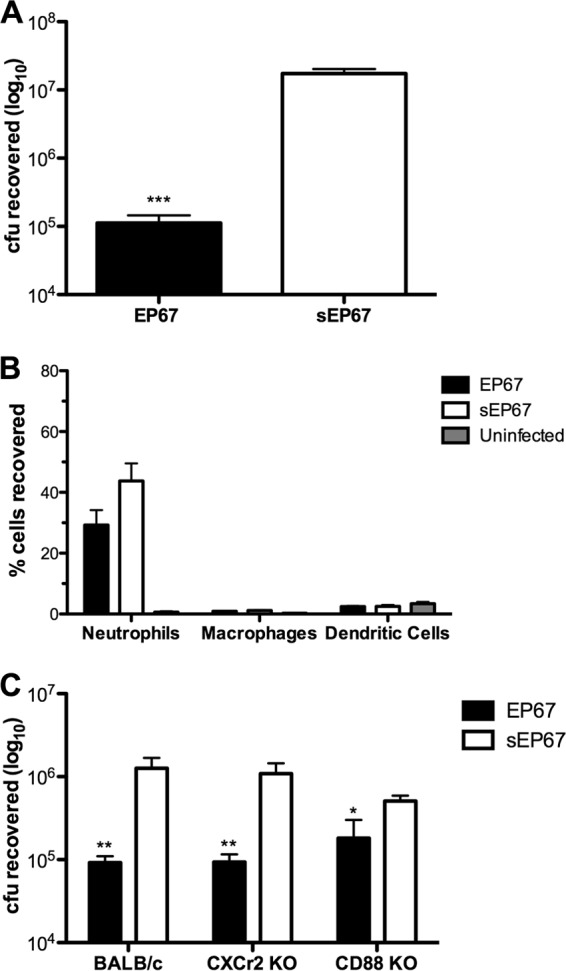
EP67 treatment inhibits bacterial load in vivo but not immune cell influx and acts independently of CXCr2 and CD88 signaling pathways. (A) Approximately 1 × 107 CFU GBS was injected into the peritoneal cavity of 20-week-old BALB/c mice (n = 5 per group). Immediately following GBS injection, mice were treated with EP67 or sEP67 in the peritoneal cavity. Peritoneal lavage fluid was conducted 2 h posttreatment, and GBS survival was quantified. Peritoneal infection was conducted independently at least twice, and data from one representative experiment are shown. (B) Lavage fluid samples from the mice described in the panel A legend were analyzed for neutrophil (Ly6G+ CD11b+), macrophage (B220− CD11c+), and dendritic cell (B220− CD11c+, B220− CD11c+ CD11b+) infiltration by flow cytometry. Data are expressed as percentages of 20,000 cells analyzed per sample. (C) Peritoneal infection and peptide treatment were performed as in described in the panel A legend using 8- to 16-week-old BALB/c (n = 5 to 8 per group), CXCr2 KO (n = 4 to 8 per group), and CD88 KO (n = 7 per group) mice. Bacterial CFU were recovered from peritoneal lavage fluid 2 h posttreatment. Data were analyzed by the use of an unpaired Student's t test. *, P < 0.05; **, P < 0.005; ***, P < 0.001.
We further performed peritoneal infection in both CXCr2 and CD88 KO mice. Interestingly, we found that EP67 treatment significantly reduced bacterial load compared to sEP67 treatment in the peritoneal cavity of BALB/c (P = 0.0039), CXCr2 KO (P = 0.0023), and CD88 KO (P = 0.0440) mice (Fig. 5C). Additionally, we quantified total leukocytes present in the peritoneal cavity and observed no differences between treatment groups or mouse strains (data not shown). These data align with results seen in our vaginal model (Fig. 2 and 3) and suggest that EP67 activity reduces GBS bacterial load independent of immune activation.
DISCUSSION
GBS is a leading cause in neonatal bacterial meningitis and sepsis, and current prophylaxis, although dramatically reducing early-onset incidence rates, is insufficient for controlling GBS transmission and infection. Despite such intervention, early-onset GBS infection in the United States remains at 1 in 3,000 live births, corresponding to approximately 1,200 infected infants per year (1). Thus, the development of novel therapeutics and improvement of risk factor assessments and treatment strategies are needed. Through our in vivo mouse vaginal colonization model and subsequent in vitro assays, we have identified a response-selective, conformationally biased C5a agonist, EP67, as a potential candidate for controlling GBS vaginal colonization. The effect of EP67 was independent of anticipated immune signaling and indicated direct killing as the main mechanism of action.
Using our murine model, we have observed GBS persistence in CD1, C57BL/6, and FVB mice for several weeks and in BALB/c mice for several months (22) (K. A. Patras and K. S. Doran, unpublished data). Factors contributing to GBS vaginal persistence include estrous stage and continuous treatment with β-estradiol (Patras and Doran, unpublished), as seen in other murine vaginal colonization/infection models (37–39). Within the mouse vaginal tract, innate immune cell populations are tightly regulated by the estrous cycle, and steroid hormones directly or indirectly steer immune cell recruitment and activation (40). CXCr2 functions as the predominant receptor for both KC (CXCL1) and MIP-2 (CXCL2), and previous work has shown that MIP-2 binds to CXCr2 with higher affinity than KC (41). In our murine vaginal model, we observed similar levels of MIP-2 and KC in wild-type and CXCr2 KO strains, suggesting that neither chemokine production nor neutrophil recruitment accounts for GBS reduction mediated by EP67. Although CXCr2 deficiency delays neutrophil recruitment, there are alternative pathways to recruit neutrophils, including CC chemokines and complement component C5a, and these pathways are independent of CXCr2 (41). Even so, we did not observe differences in vaginal leukocyte populations between EP67 and sEP67 groups.
Because previous work has shown that EP67 activates both neutrophil recruitment (9) and TH1 immune activation (6) through C5aR (CD88), we investigated the role of CD88 signaling during EP67 treatment. Human vaginal epithelial cells (HVECs) possess C5aRs, as measured by microarray; however, in vitro exposure of HVECs to GBS minimally alters C5aR expression, as measured by microarray (22). Moreover, the murine C5aR1 (NM_007577; probe identification [ID] no. 101728) has been determined to be present in vaginal tissue (42). Given the presence of this receptor, it is possible that EP67 engages C5aR on the epithelium, and yet we still observed reduction of GBS in CD88 KO mice treated with EP67. Of note, GBS possesses a C5a peptidase (SCPB) which cleaves human C5a or C5adesarg between the His and the Lys at amino acid positions 67 and 68 very near the C terminus (43). However, SCPB has been shown to have little protease activity with rodent C5a, even though there is high homology (43). EP67 is a peptide with only 10 residues that lacks this His-Lys bond, making sensitivity to the GBS SCPB highly unlikely. Further, using subtractive electrospray (ES) mass spectrometry, we observed no proteolytic cleavage of EP67 following exposure to GBS (unpublished data).
Because EP67-mediated clearance of GBS from the vaginal tract did not appear to require immunostimulatory properties, we investigated if EP67 was alternatively acting as an antimicrobial peptide (AMP) due to its similarity to members of the complement system. Aside from the well-known complement cascade involving proteins C1 to C9, C5a functions as a powerful anaphylatoxin and inflammatory mediator, causing leukocyte chemotaxis, production of cytokines, and vascular permeability (44). Furthermore, recent work demonstrated that another complement component, C3a, exhibits direct antimicrobial activity by inducing breaks in bacterial membranes (45). Many AMPs are cationic, which permits binding to negatively charged microbial surfaces (2). EP67 is also slightly positively charged (+1), which may promote initial contact with GBS cell wall and/or membrane surfaces.
Our in vitro assays indicate that EP67 is bactericidal to GBS and other Streptococcus species. Although not as potent as endogenous C3a and C3a-derived peptides, which kill E. faecalis and S. pyogenes at concentrations of <10 μM (45), or as the AMP LL-37 that kills GBS at concentrations of <16 μM (46), EP67 differentially targets GBS in vivo without inhibiting or altering native murine vaginal flora (Table 1; see also Fig. S1 in the supplemental material). AMPs that directly target bacterial membranes, such as LL-37 or its derivatives, can permeate Gram-positive and Gram-negative membranes within a matter of minutes (47). In contrast, EP67 initiates a microbicidal effect that leads to GBS cell death and membrane damage over a longer time period, suggesting the possibility of its interaction with an internal target(s) to exert its antimicrobial effect. Combining our in vivo and in vitro work, we proposed EP67-mediated direct killing of GBS in a clinically relevant model of GBS vaginal colonization. Although future work must continue to characterize the mechanism of action, here we reveal that EP67 contains unique properties as a potential therapeutic to control GBS colonization and infection.
Supplementary Material
ACKNOWLEDGMENTS
CXCr2 KO mice were a generous gift from Joshua Fierer, UCSD School of Medicine. We thank Mansi Garg for collecting and identifying native murine vaginal isolates.
This work was supported by grants from the California State University Program for Education and Research in Biotechnology (CSUPERB) and NS051247 from the NIH to K.S.D.
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 26 August 2013
This article is dedicated to our beloved colleague and coauthor Ed Morgan (1952-2012).
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01590-13.
REFERENCES
- 1.Verani JR, Schrag SJ. 2010. Group B streptococcal disease in infants: progress in prevention and continued challenges. Clin. Perinatol. 37:375–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maisey HC, Doran KS, Nizet V. 2008. Recent advances in understanding the molecular basis of group B Streptococcus virulence. Expert Rev. Mol. Med. 10:e27. 10.1017/S1462399408000811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doran KS, Nizet V. 2004. Molecular pathogenesis of neonatal group B streptococcal infection: no longer in its infancy. Mol. Microbiol. 54:23–31 [DOI] [PubMed] [Google Scholar]
- 4.Lombard F, Marchandin H, Jacquot A, Cambonie G, Rodiere M, Filleron A. 2012. Streptococcus agalactiae late-onset neonatal infections: should breast milk be more systematically tested for bacterial contamination? Acta Pædiatr. 101:e529–e530 [DOI] [PubMed] [Google Scholar]
- 5.Phares CR, Lynfield R, Farley MM, Mohle-Boetani J, Harrison LH, Petit S, Craig AS, Schaffner W, Zansky SM, Gershman K, Stefonek KR, Albanese BA, Zell ER, Schuchat A, Schrag SJ. 2008. Epidemiology of invasive group B streptococcal disease in the United States, 1999–2005. JAMA 299:2056–2065 [DOI] [PubMed] [Google Scholar]
- 6.Morgan EL, Morgan BN, Stein EA, Vitrs EL, Thoman ML, Sanderson SD, Phillips JA. 2009. Enhancement of in vivo and in vitro immune functions by a conformationally biased, response-selective agonist of human C5a: implications for a novel adjuvant in vaccine design. Vaccine 28:463–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vogen SM, Paczkowski NJ, Kirnarsky L, Short A, Whitmore JB, Sherman SA, Taylor SM, Sanderson SD. 2001. Differential activities of decapeptide agonists of human C5a: the conformational effects of backbone N-methylation. Int. Immunopharmacol. 1:2151–2162 [DOI] [PubMed] [Google Scholar]
- 8.Hung CY, Hurtgen BJ, Bellecourt M, Sanderson SD, Morgan EL, Cole GT. 2012. An agonist of human complement fragment C5a enhances vaccine immunity against Coccidioides infection. Vaccine 30:4681–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheen TR, Cavaco CK, Ebrahimi CM, Thoman ML, Sanderson SD, Morgan EL, Doran KS. 2011. Control of methicillin resistant Staphylococcus aureus infection utilizing a novel immunostimulatory peptide. Vaccine 30:9–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanke ML, Heim CE, Angle A, Sanderson SD, Kielian T. 2013. Targeting macrophage activation for the prevention and treatment of Staphylococcus aureus biofilm infections. J. Immunol. 190:2159–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phillips JA, Morgan EL, Dong Y, Cole GT, McMahan C, Hung CY, Sanderson SD. 2009. Single-step conjugation of bioactive peptides to proteins via a self-contained succinimidyl bis-arylhydrazone. Bioconjug. Chem. 20:1950–1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Madoff LC, Michel JL, Kasper DL. 1991. A monoclonal antibody identifies a protective C-protein alpha-antigen epitope in group B streptococci. Infect. Immun. 59:204–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wessels MR, Paoletti LC, Rodewald AK, Michon F, DiFabio J, Jennings HJ, Kasper DL. 1993. Stimulation of protective antibodies against type Ia and Ib group B streptococci by a type Ia polysaccharide-tetanus toxoid conjugate vaccine. Infect. Immun. 61:4760–4766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilkinson HW. 1977. Nontypable group B streptococci isolated from human sources. J. Clin. Microbiol. 6:183–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson CB, Weaver WM. 1985. Comparative susceptibility of group B streptococci and Staphylococcus aureus to killing by oxygen metabolites. J. Infect. Dis. 152:323–329 [DOI] [PubMed] [Google Scholar]
- 16.Cole JN, Pence MA, von Köckritz-Blickwede M, Hollands A, Gallo RL, Walker MJ, Nizet V. 2010. M protein and hyaluronic acid capsule are essential for in vivo selection of covRS mutations characteristic of invasive serotype M1T1 group A Streptococcus. mBio 1:e00191-10. 10.1128/mBio.00191-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sullam PM, Valone FH, Mills J. 1987. Mechanisms of platelet aggregation by viridans group streptococci. Infect. Immun. 55:1743–1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berry AM, Yother J, Briles DE, Hansman D, Paton JC. 1989. Reduced virulence of a defined pneumolysin-negative mutant of Streptococcus pneumoniae. Infect. Immun. 57:2037–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Highlander SK, Hulten KG, Qin X, Jiang H, Yerrapragada S, Mason EO, Jr, Shang Y, Williams TM, Fortunov RM, Liu Y, Igboeli O, Petrosino J, Tirumalai M, Uzman A, Fox GE, Cardenas AM, Muzny DM, Hemphill L, Ding Y, Dugan S, Blyth PR, Buhay CJ, Dinh HH, Hawes AC, Holder M, Kovar CL, Lee SL, Liu W, Nazareth LV, Wang Q, Zhou J, Kaplan SL, Weinstock GM. 2007. Subtle genetic changes enhance virulence of methicillin resistant and sensitive Staphylococcus aureus. BMC Microbiol. 7:99. 10.1186/1471-2180-7-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Sorge NM, Zialcita PA, Browne SH, Quach D, Guiney DG, Doran KS. 2011. Penetration and activation of brain endothelium by Salmonella enterica serovar Typhimurium. J. Infect. Dis. 203:401–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheen TR, Jimenez A, Wang NY, Banerjee A, van Sorge NM, Doran KS. 2011. Serine-rich repeat proteins and pili promote Streptococcus agalactiae colonization of the vaginal tract. J. Bacteriol. 193:6834–6842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patras KA, Wang NY, Fletcher EM, Cavaco CK, Jimenez A, Garg M, Fierer J, Sheen TR, Rajagopal L, Doran KS. 30 January 2013. Group B Streptococcus CovR regulation modulates host immune signalling pathways to promote vaginal colonization. Cell. Microbiol. [Epub ahead of print.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Furr PM, Hetherington CM, Taylor-Robinson D. 1989. The susceptibility of germ-free, oestradiol-treated, mice to Mycoplasma hominis. J. Med. Microbiol. 30:233–236 [DOI] [PubMed] [Google Scholar]
- 24.Cheng Q, Nelson D, Zhu S, Fischetti VA. 2005. Removal of group B streptococci colonizing the vagina and oropharynx of mice with a bacteriophage lytic enzyme. Antimicrob. Agents Chemother. 49:111–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sonoda Y, Mukaida N, Wang JB, Shimada-Hiratsuka M, Naito M, Kasahara T, Harada A, Inoue M, Matsushima K. 1998. Physiologic regulation of postovulatory neutrophil migration into vagina in mice by a C-X-C chemokine(s). J. Immunol. 160:6159–6165 [PubMed] [Google Scholar]
- 26.Caligioni CS. 2009. Assessing reproductive status/stages in mice. Curr. Protoc. Neurosci. 48:A.4I.1–A.4I.8. 10.1002/0471142301.nsa04is48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poisson DM, Evrard ML, Freneaux C, Vives MI, Mesnard L. 2011. Evaluation of CHROMagar StrepB agar, an aerobic chromogenic medium for prepartum vaginal/rectal group B Streptococcus screening. J. Microbiol. Methods 84:490–491 [DOI] [PubMed] [Google Scholar]
- 28.Nijnik A, Madera L, Ma S, Waldbrook M, Elliott MR, Easton DM, Mayer ML, Mullaly SC, Kindrachuk J, Jenssen H, Hancock RE. 2010. Synthetic cationic peptide IDR-1002 provides protection against bacterial infections through chemokine induction and enhanced leukocyte recruitment. J. Immunol. 184:2539–2550 [DOI] [PubMed] [Google Scholar]
- 29.Maisey HC, Quach D, Hensler ME, Liu GY, Gallo RL, Nizet V, Doran KS. 2008. A group B streptococcal pilus protein promotes phagocyte resistance and systemic virulence. FASEB J. 22:1715–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanderson SD, Thoman ML, Kis K, Virts EL, Herrera EB, Widmann S, Sepulveda H, Phillips JA. 2012. Innate immune induction and influenza protection elicited by a response-selective agonist of human C5a. PLoS One 7:e40303. 10.1371/journal.pone.0040303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olson TS, Ley K. 2002. Chemokines and chemokine receptors in leukocyte trafficking. Am. J. Physiol. Regul. Integr. Comp. Physiol. 283:R7–R28 [DOI] [PubMed] [Google Scholar]
- 32.Svensson M, Yadav M, Holmqvist B, Lutay N, Svanborg C, Godaly G. 2011. Acute pyelonephritis and renal scarring are caused by dysfunctional innate immunity in mCxcr2 heterozygous mice. Kidney Int. 80:1064–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eisele NA, Lee-Lewis H, Besch-Williford C, Brown CR, Anderson DM. 2011. Chemokine receptor CXCR2 mediates bacterial clearance rather than neutrophil recruitment in a murine model of pneumonic plague. Am. J. Pathol. 178:1190–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herbold W, Maus R, Hahn I, Ding N, Srivastava M, Christman JW, Mack M, Reutershan J, Briles DE, Paton JC, Winter C, Welte T, Maus UA. 2010. Importance of CXC chemokine receptor 2 in alveolar neutrophil and exudate macrophage recruitment in response to pneumococcal lung infection. Infect. Immun. 78:2620–2630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Virta M, Lineri S, Kankaanpaa P, Karp M, Peltonen K, Nuutila J, Lilius EM. 1998. Determination of complement-mediated killing of bacteria by viability staining and bioluminescence. Appl. Environ. Microbiol. 64:515–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boulos L, Prevost M, Barbeau B, Coallier J, Desjardins R. 1999. LIVE/DEAD BacLight: application of a new rapid staining method for direct enumeration of viable and total bacteria in drinking water. J. Microbiol. Methods 37:77–86 [DOI] [PubMed] [Google Scholar]
- 37.Furr PM, Taylor-Robinson D. 1989. Oestradiol-induced infection of the genital tract of female mice by Mycoplasma hominis. J. Gen. Microbiol. 135:2743–2749 [DOI] [PubMed] [Google Scholar]
- 38.Packiam M, Wu H, Veit SJ, Mavrogiorgos N, Jerse AE, Ingalls RR. 2012. Protective role of Toll-like receptor 4 in experimental gonococcal infection of female mice. Mucosal Immunol. 5:19–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gilbert NM, Lewis WG, Lewis AL. 2013. Clinical features of bacterial vaginosis in a murine model of vaginal infection with Gardnerella vaginalis. PLoS One 8:e59539. 10.1371/journal.pone.0059539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hickey DK, Patel MV, Fahey JV, Wira CR. 2011. Innate and adaptive immunity at mucosal surfaces of the female reproductive tract: stratification and integration of immune protection against the transmission of sexually transmitted infections. J. Reprod. Immunol. 88:185–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee J, Cacalano G, Camerato T, Toy K, Moore MW, Wood WI. 1995. Chemokine binding and activities mediated by the mouse IL-8 receptor. J. Immunol. 155:2158–2164 [PubMed] [Google Scholar]
- 42.Suzuki A, Urushitani H, Watanabe H, Sato T, Iguchi T, Kobayashi T, Ohta Y. 2007. Comparison of estrogen responsive genes in the mouse uterus, vagina and mammary gland. J. Vet. Med. Sci. 69:725–731 [DOI] [PubMed] [Google Scholar]
- 43.Bohnsack JF, Chang JK, Hill HR. 1993. Restricted ability of group B streptococcal C5a-ase to inactivate C5a prepared from different animal species. Infect. Immun. 61:1421–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor SM, Sherman SA, Kirnarsky L, Sanderson SD. 2001. Development of response-selective agonists of human C5a anaphylatoxin: conformational, biological, and therapeutic considerations. Curr. Med. Chem. 8:675–684 [DOI] [PubMed] [Google Scholar]
- 45.Nordahl EA, Rydengard V, Nyberg P, Nitsche DP, Morgelin M, Malmsten M, Bjorck L, Schmidtchen A. 2004. Activation of the complement system generates antibacterial peptides. Proc. Natl. Acad. Sci. U. S. A. 101:16879–16884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dorschner RA, Lin KH, Murakami M, Gallo RL. 2003. Neonatal skin in mice and humans expresses increased levels of antimicrobial peptides: innate immunity during development of the adaptive response. Pediatr. Res. 53:566–572 [DOI] [PubMed] [Google Scholar]
- 47.Wang G, Epand RF, Mishra B, Lushnikova T, Thomas VC, Bayles KW, Epand RM. 2012. Decoding the functional roles of cationic side chains of the major antimicrobial region of human cathelicidin LL-37. Antimicrob. Agents Chemother. 56:845–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.