

Abstract
This paper reports an approach to enable rapid concentration and recovery of bacterial cells from aqueous chicken homogenates as a preanalytical step of detection. This approach includes biochemical pretreatment and prefiltration of food samples and development of an automated cell concentration instrument based on cross-flow microfiltration. A polysulfone hollow-fiber membrane module having a nominal pore size of 0.2 μm constitutes the core of the cell concentration instrument. The aqueous chicken homogenate samples were circulated within the cross-flow system achieving 500- to 1,000-fold concentration of inoculated Salmonella enterica serovar Enteritidis and naturally occurring microbiota with 70% recovery of viable cells as determined by plate counting and quantitative PCR (qPCR) within 35 to 45 min. These steps enabled 10 CFU/ml microorganisms in chicken homogenates or 102 CFU/g chicken to be quantified. Cleaning and sterilizing the instrument and membrane module by stepwise hydraulic and chemical cleaning (sodium hydroxide and ethanol) enabled reuse of the membrane 15 times before replacement. This approach begins to address the critical need for the food industry for detecting food pathogens within 6 h or less.
INTRODUCTION
The 2012 report card for food safety released by the Centers for Disease Control and Prevention (CDC) indicated a lack of recent progress in reducing food-borne infections and highlighted the need for improved prevention (1). Although many food-borne illnesses have declined in the past 15 years, the report shows that incidences of laboratory-confirmed Salmonella did not change significantly in 2012 compared with 2006 to 2008 (1).
A method for detecting food contamination ideally should be sensitive and specific to confirm the lack of contamination. The method should also be fast, requiring minimal sample preparation before analysis, and low in cost (2). Culture-based detection techniques are widely applied to detect pathogens from food and environmental samples. Cultural enrichment amplifies the target organism exponentially by as much as a millionfold so that detection is possible. However, these culture-based techniques are considered labor-intensive and time-consuming (3). Additionally, enrichment may produce false-negative results under certain circumstances, as injured cells are often not detected due to overgrowth by background microbiota during nonselective enrichment and/or inhibition of recovery of injured cells during selective enrichment (4, 5). Selective agents in enrichment media also may inhibit repair of sublethally injured pathogenic cells (4).
More than 30 years ago, when testing membrane filtration of food suspensions, Sharpe et al. (6) concluded that “food can be filtered in quantities pertinent to the maximums used in conventional plating procedures.” Microfiltration represents a conceptually simple way to reduce large samples to a small volume and effectively increase cell concentration without lengthy culturing and enrichment steps. There have been various recent reports on this approach as summarized in Table 1 (7–11). However, there were still several challenges to be overcome in order to develop a rapid filtration approach for the microorganism concentration and recovery from foods. Reports have shown that dead-end microfiltration using flat-sheet membrane is effective for concentrating microbial cells, e.g., Listeria monocytogenes and Salmonella enterica for microbiological analysis of water, dairy, and food products (12–18). We have also shown that sequential filtration through a depth filter followed by a screen filter could concentrate 100-ml samples having as few as 1.3 log CFU/ml to a volume of 50 μl with 3.3 log CFU/ml (14, 15). However, due to rapid membrane fouling in dead-end filtration, cross-flow microfiltration, in which the feed suspension flows tangentially to the membrane surface, was investigated. Cross-flow microfiltration is normally considered when the suspension to be filtered contains fine particles or microbial suspensions where the density is close to that of the suspending fluid and when the particles have the tendency to form highly resistant and compressible deposits or films formed from proteins, fats, or oils (19). Hollow-fiber membranes are characterized by high surface area-to-volume ratios and flux per unit volume of the membrane module compared to flat-sheet membranes (20), or alternately, fluid passed through the hollow fiber may wash out cells for recovery. The membrane may be back-flushed to recover concentrated cells in an aqueous buffer or flushed with an elution buffer through the fiber to recover cells from the device platforms for detection and identification (20). Hollow-fiber membranes enable pathogen concentration and recovery from water samples (21–24), but not for large volumes of aqueous food extracts due to rapid membrane plugging.
Table 1.
Review of recent reports for physical (filtration and/or centrifugation) separation and concentration of microorganisms from foods
| Target pathogen(s) | Food sample(s) | Initial vol (ml) | Final vol (ml) | Sample prepn | Detection method(s)a | Time for whole procedure (h) | Automated sample concn and recovery | Detection levelb (CFU/g) | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Salmonella enterica | Chicken | 100 | 1 | Stomaching, two filtration steps, and vortexing | qRT-PCR | 3 | No | 75 | 7 |
| S. enterica and 11 other microorganisms | Chicken and 11 other food samples | 250 | 0.1c | Stomaching, filtration, low- and high-speed centrifugation, flotation, sedimentation, and buoyant density centrifugation | RTi-qPCR | 3 | No | 101–103 | 8 |
| Escherichia coli O157:H7 | Ground beef | ≥100 | NAd | Stomaching and 3-stage filtration system | Plating | 24 | No | ≤1 | 9 |
| E. coli C3000 | Ham and lettuce | 5,000 | 200 | Cells directly inoculated in food, elution with buffer, and hollow-fiber ultrafiltration | Plating and RT-PCR | NA | No | NA | 10 |
| S. enterica, E. coli O157:H7, and Listeria monocytogenes | Deli meat, whole milk, and orange juice | 250 | NA | Stomaching, filtration and pathogen immobilization in filter, and pathogen extraction by filter incubation | qRT-PCR | ∼8 | No | ∼1 | 11 |
| S. enterica | Chicken | 250 | ∼0.5 | Stomaching, enzyme treatment, membrane prefiltration, and hollow-filter microfiltration | qPCR | ∼6 | Yes | 102 | This study |
qRT-PCR, quantitative reverse transcription-PCR; RTi-qPCR, real-time quantitative PCR.
The detection level refers the cell level that can be quantified.
It was estimated that target pathogens could theoretically be 250-fold concentrated in food samples.
NA, not available.
The goal of this study was to extend the ease of use of hollow fibers, demonstrated for water, to the microfiltration of food-derived suspensions for the purpose of concentrating and recovering viable microbial cells and quantitating low levels of food-borne pathogenic cells. This increases the number of microbial cells to a detectable level so that they may be effectively probed for the presence of pathogens.
MATERIALS AND METHODS
Preparation and biochemical pretreatment of aqueous homogenates of chicken.
Chicken legs of commercial brands were purchased locally. The aqueous chicken homogenates were made by mixing 25 g of thin-cut chicken flesh and skin with 250 ml of sterile deionized water in a sterile Filtra Bag (catalog no. 01-002-57; Fisher Scientific) and homogenizing it in a Seward Stomacher 400 circulator at 100 rpm for 30 s. The aqueous fraction was then transferred to a Falcon polypropylene graduated conical tube with cap (sterile). Protease (Protex 7L; neutral metallo endopeptidase; protein content, 1,600 azo units/g; Genencor Division of Danisco, Rochester, NY, USA) at a final loading of 0.5% (vol/vol) (equal to 0.27 mg/ml) was incubated with the crude homogenates at 200 rpm and 37°C for 1 h, which were then vacuum filtered using a glass microfiber filter (particle retention, 2.7 μm; 100% borosilicate glass; catalog no. 1823-090; Whatman). One sheet of membrane was used per aliquot of 125-ml homogenate. The prefiltration step removes crude particles and colloids that may cause clogging and/or fouling of the hollow-fiber microfiltration membrane module, while the incubation of the homogenate with protease enables microfiltration. As shown in Fig. 1, the flux of permeate that passed through the membrane during microfiltration was significantly improved by protease treatment of chicken homogenates. Prior to prefiltration, the turbidity of the homogenate was 68 Klett units, while after filtration, the filtrate was clear with a turbidity of about 0 Klett units. The prepared aqueous chicken homogenates contained 623.5 ± 155.6 μg/ml of protein and <100 μg/ml of lipids. All the above steps were performed under aseptic conditions. The spiked aqueous chicken homogenates were kept refrigerated and used within 1 h of preparation, except where otherwise stated.
Fig 1.
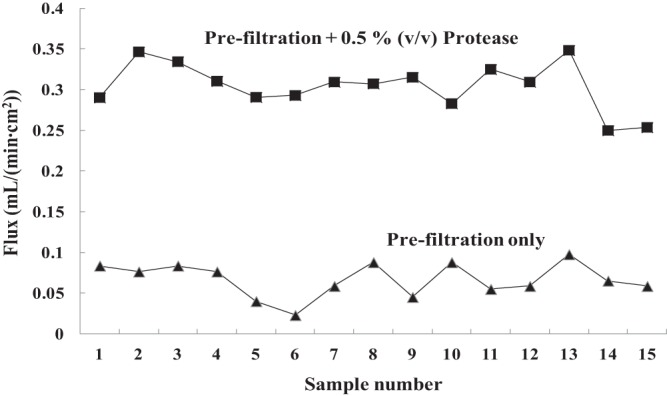
Variation of permeation flux of cross-flow microfiltration of protease-treated and untreated aqueous chicken homogenates. One hollow-fiber membrane module was used for each of the 15 samples (H. B. Vibbert, E. Ximenes, X. Li, S. Ku, X. Liu, and M. R. Ladisch, unpublished data).
Bacterial strains, culture conditions, and spiking of chicken homogenates.
Salmonella enterica serovar Enteritidis phage type (PT) 21, Salmonella Typhimurium Nos-6, Escherichia coli O157:H7 EDL 933, E. coli K-12, Pseudomonas aeruginosa, Citrobacter freundii, and Enterobacter aerogenes were obtained from Arun Bhunia's lab, Food Science Department, Purdue University. The non-Salmonella strains were used to ascertain the specificity of the primer set in the quantitative PCR (qPCR) analysis. All bacterial strains were grown overnight in BBL brain heart infusion (BHI) (BD, Sparks, MD, USA) broth at 37°C with shaking at 200 rpm. S. Enteritidis culture was serially diluted in phosphate-buffered saline (PBS) (pH 7.4) and spiked into prefiltered chicken homogenates at 1.0 to 3.0 log CFU/ml, as confirmed by viable count.
Membrane module.
The microfiltration module is a 12-fiber bundle of polysulfone hollow-fiber membranes with a nominal pore size of 0.2 μm, inner diameter (ID) of 280 μm, outer diameter (OD) of 360 μm (Minntech, Minneapolis, MN), and length of 27 cm, giving an overall effective filtration area of 2,849 mm2 and a cross-section area of 0.74 mm2. Each module (Fig. 2; see Fig. S1 in the supplemental material) is constructed with two tee junctions (1/8-in. Peek tubing [IDEX], 1/16-in. through hole), six flangeless male nuts (1/8-in. Peek tubing; IDEX), six flangeless ferrules (1/8-in. Tefzel [natural]; IDEX), two 5-cm pieces of tubing (1/8- by 0.080-in. Peek tubing; IDEX), one 15-cm piece of tubing (1/8- by 0.080-in. Peek tubing; IDEX), and approximately 0.1 g Loctite Hysol M-21HP and M-31CL epoxy; Peek and Tefzel pieces are assembled, and each hollow fiber is individually threaded through the module. Each end of the module is sealed with M-31CL epoxy to prevent leakage; M-21HP epoxy is applied as a secondary layer to the M-31CL epoxy to ensure separation of the inlet and permeate sides of the module.
Fig 2.
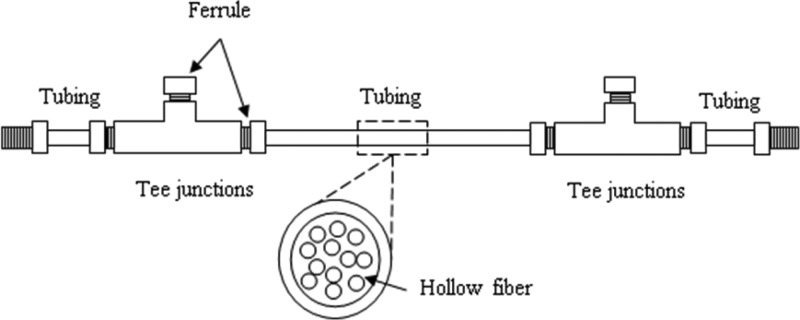
Schematic representation of the hollow-fiber membrane module.
Cell concentration instrument.
The schematic representative of the cell concentration instrument is shown in Fig. 3 (see Fig. S2 in the supplemental material). It contains four reservoirs for sample, sodium hydroxide, ethanol, and recovery elution buffer. Key microfluidic components are a 4-to-1 source select valve (medium-pressure valve; Scivex, Oak Harbor, WA), feed/retentate circulating pump (pump 1, Ismatec compact analog pump, 2 channels, 1.6 to 160 rpm, 0.004 to 50 ml/min, 115/230 VAC) (catalog no. EW-78016-10; Cole-Parmer, Vernon Hills, IL), membrane module, and a valve (3-way valve 1/16 24VDC; Cole-Parmer, Vernon Hills, IL) that controls the direction of retentate flow. A second pump (pump 2) pumps sterile deionized water into the membrane model through the permeate side to remove permeate as it is generated and to achieve uniform transmembrane pressure along the length of the hollow fiber. The cross-flow velocity through the module is controlled by adjusting the speed of pump 1. Teflon FEP Tubing (Upchurch Scientific, Oak Harbor, WA) connects the various components of the instrument.
Fig 3.

Schematic representation of the cell concentration instrument.
The flow rate of the permeate (F1), the flow rate of the sterile deionized water into the permeate side (F2), the retentate pressure (P1), and the permeate side pressure (P2) at the inlet end of the membrane module were monitored using two differential pressure flow meters (catalog no. EW-32908-43; Cole-Parmer, Vernon Hills, IL). The operational control and data acquisition occurred through a graphical interface, and the program was built with LabVIEW 2009 in a personal computer (PC) in the laboratory. The pressure and flow rate data as well as the operation mode and the status of valves are automatically recorded every second. All operations were carried out at room temperature.
Two operational modes, cell concentration and cell recovery, define the instrument's operation. Microbiota in 250 ml of prefiltered homogenate are concentrated when the initial sample is fed through the membrane module, and the microorganisms are concentrated in the retentate which is circulated to the sample reservoir using pump 1. Elution of the cells at the end of the cycle occurs when 10 ml of deionized water or PBS supplemented with 0.002% (vol/vol) Tween 20 per 250-ml initial sample is loaded. The cell concentration mode is run with the speeds of pump 1 and pump 2 adjusted to 100% and 20% of their maximum, respectively, giving a laminar cross-flow velocity of 0.375 m/s (corresponding to a Reynolds number of 117). Cell recovery occurs when the retentate is vented into a sample collection tube instead of being circulated back to the sample reservoir; only pump 1 is used, and it is set at its maximum speed.
Cleaning procedure.
Unlike water matrices, the aqueous chicken homogenates (and other types of food suspensions) are heterogeneous, consisting of naturally occurring microbiota and various components, including proteins, lipids, micron-sized particulates, and inorganic food components. Proteins and liquids are known to foul microfiltration membranes. Although tangential flow reduces the rate of the buildup of foulants on the membrane surface, regular chemical cleaning is required to ensure reproducible performance and repeated membrane use (25). A hydraulic and chemical cleaning procedure is carried out between consecutive runs of sample processing for this reason, as well as to achieve system sterilization between samples.
More specifically, sample processing is followed by an immediate system rinsing with deionized water for 5 min to remove the loosely bound sample residues in the system tubing and weaken the deposit surface layer (26, 27). Following rinsing, the system is subjected to a stepwise chemical cleaning procedure which was developed as part of this work. The steps in the chemical cleaning procedure were as follows: (i) cleaning with 0.2 M sodium hydroxide solution through the rig for 10 min, as sodium hydroxide has the ability to saponify fats and solubilize proteins to some extent; (ii) flushing with deionized water for 5 min to clean any trace of a caustic agent in the system; (iii) cleaning with 70% (vol/vol) ethanol through the rig for 10 min to return the system back to a sterile state; (iv) flushing with deionized water for 5 min to clean any trace of ethanol and rehydrate the membrane. All the steps above are carried out using flow paths for both the cell concentration and cell recovery modes, i.e., with pump 1 and pump 2 adjusted to 100% and 20% of their maximum speeds, respectively. Both permeate and retentate are disposed of in the waste tank. The polysulfone membrane and wetted components of the instrument are compatible with the reagents used.
Water flushed through the instrument after sodium hydroxide and ethanol cleaning was collected and plated. This plating examination is performed periodically (typically once per 3 runs) to ensure good sterilization of the system and enable 15 reuses of the hollow-fiber membrane.
Cell enumeration and expression of results.
The number of viable cells in samples was determined by plating and quantitative PCR. The methods of qPCR are presented in the next section. The total cells were enumerated on BBL BHI agar (BD, Sparks, MD, USA). S. Enteritidis was enumerated by plating on CHROMagar Salmonella (DGR International, Mountainside, NJ). The BHI plates were incubated at 37°C for 24 h, while the CHROMagar plates were incubated for 36 to 48 h because it may take longer for colonies to develop on selective media. Cell levels were expressed as log CFU/ml. Cell concentration and recovery efficiency were calculated using equations 1 and 2, respectively.
| (1) |
| (2) |
where cell concentrations are in CFU/milliliter and volume is in milliliters.
qPCR. (i) DNA isolation.
DNA was isolated from the different bacterial species and samples using the DNeasy minikit (Qiagen) according to the manufacturer's instructions. The DNA concentration was measured using the NanoDrop 2000 spectrophotometer (Thermo Scientific). For specificity testing, the DNA isolated from different bacterial species was diluted in double-distilled water (ddH2O) to 1 ng/μl, and qPCR was performed. The genomic DNA standard for real-time quantification was prepared by serially diluting (1:10) the DNA from pure cultures in ddH2O. The copy number of invA gene per ng was calculated based on the molecular weight of the 4,685-kbp genome of Salmonella Enteritidis (28). One nanogram of DNA corresponds to 2 × 105 genome or bacterial cell equivalents. The number of bacterial cell equivalents (BCE) in a sample was extrapolated from the qPCR signal of the standard.
(ii) SYBR green qPCR.
PCR analysis of the samples was carried out using the StepOnePlus real-time system (Applied Biosystems) using species-specific primers for the invA gene in Salmonella enterica (invAF [F stands for forward] [5′-GTGAAATAATCGCCACGTCGGCAA-3′] and invAR [R stands for reverse] [5′-TCATCGCACCGTCAAAGGAACC-3′]). The invA gene has been used as the target sequence for Salmonella species by several researchers (28, 29). Primers were synthesized using the Primer Express software (Applied Biosystems). The PCR mixture contained 10 μl of 2× SYBR green PCR master mix (Applied Biosystems), 0.3 μM of each primer, and 2 μl of DNA template and nuclease-free water to make up the volume to 20 μl. All runs included a negative control without template DNA and Salmonella Enteritidis PT21 standards to obtain the standard curve. Thermal cycling conditions were as follows: 15 min at 95°C, followed by 40 cycles, with 1 cycle consisting of 15 s at 95°C and 15 s at 68°C, and a final melt cycle of 15 s at 95°C and 60 s at 60°C with temperature increments of 0.3°C. All PCRs were performed in triplicate. Reproducibility of SYBR green qPCR was assessed by running samples independently on different days. Three replicate samples were run for each assay, and the run was repeated twice.
RESULTS
Efficiency of cell concentration and recovery of artificially spiked Salmonella Enteritidis in aqueous chicken homogenates.
Chicken homogenates spiked with three levels of S. Enteritidis, i.e., 1.0, 2.0, and 3.0 log CFU/ml, were processed and enumerated by plating (Table 2). For each starting cell level, five replicate experiments were conducted. After cell concentration, the initial sample volume (250 ml) was reduced down to 408 ± 223 μl. For total cells, the concentration factors for the three starting cell levels were 102.68, 102.71, and 102.72, respectively, on average. The corresponding recovery of total microbiota attained 75.2%, 77.5%, and 75.6%. For S. Enteritidis, the mean concentration factors for the three starting cell levels were 102.70, 102.71, and 102.65, and the recoveries were 77.8%, 74.4%, and 63.9%, respectively. The concentration and recovery processes were finished within 35 to 45 min for all runs; including the postconcentration cleaning procedure, one run could be finished within 1.5 h.
Table 2.
Concentration and recovery of total cells and artificially spiked S. enterica serovar Enteritidis in aqueous chicken homogenates (n = 15)
| Starting level of S. Enteritidis (log CFU/ml) | Concn of total cells (log CFU/ml) |
Concn of S. Enteritidis (log CFU/ml) |
Vol of concentrated sample (μl) | Concn factor |
Recovery (%) |
||||
|---|---|---|---|---|---|---|---|---|---|
| Prefiltered sample | Concentrated sample | Prefiltered sample | Concentrated sample | Total cells | S. Enteritidis | Total cells | S. Enteritidis | ||
| 3 | 3.26 | 6.28 | 3.11 | 6.20 | 200 | 3.02 | 3.09 | 84.4 | 98.5 |
| 3.34 | 6.26 | 3.30 | 6.26 | 250 | 2.91 | 2.96 | 81.8 | 90.0 | |
| 3.22 | 5.79 | 3.19 | 5.65 | 550 | 2.57 | 2.46 | 81.8 | 64.0 | |
| 3.30 | 5.84 | 3.12 | 5.76 | 400 | 2.54 | 2.64 | 55.8 | 70.4 | |
| 3.26 | 5.64 | 3.16 | 5.51 | 750 | 2.38 | 2.34 | 72.0 | 66.2 | |
| Mean ± SEM | 430 ± 225 | 2.68 ± 0.27 | 2.70 ± 0.32 | 75.2 ± 11.8 | 77.8 ± 15.5 | ||||
| 2 | 2.95 | 5.97 | 2.95 | 5.88 | 225 | 3.01 | 2.92 | 93.1 | 75.3 |
| 2.78 | 5.74 | 2.10 | 5.03 | 250 | 2.96 | 2.93 | 91.2 | 85.1 | |
| 2.48 | 5.51 | 2.33 | 5.37 | 200 | 3.03 | 3.04 | 85.7 | 87.7 | |
| 2.26 | 4.35 | 2.04 | 4.08 | 1,000 | 2.09 | 2.04 | 49.8 | 44.0 | |
| 2.46 | 4.99 | 2.30 | 4.90 | 500 | 2.53 | 2.60 | 67.7 | 80.0 | |
| Mean ± SEM | 435 ± 338 | 2.71 ± 0.41 | 2.71 ± 0.41 | 77.5 ± 18.5 | 74.4 ± 17.7 | ||||
| 1 | 1.70 | 4.56 | 1.00 | 3.80 | 300 | 2.86 | 2.80 | 86.9 | 75.7 |
| 1.78 | 4.42 | 1.30 | 3.85 | 450 | 2.64 | 2.55 | 78.6 | 63.9 | |
| 1.85 | 4.53 | 1.40 | 3.93 | 400 | 2.68 | 2.53 | 76.6 | 54.2 | |
| 1.65 | 4.32 | 1.30 | 3.96 | 350 | 2.67 | 2.66 | 65.5 | 64.0 | |
| 1.54 | 4.31 | 1.00 | 3.71 | 300 | 2.77 | 2.71 | 70.7 | 61.5 | |
| Mean ± SEM | 330 ± 65 | 2.72 ± 0.09 | 2.65 ± 0.11 | 75.6 ± 8.1 | 63.9 ± 7.7 | ||||
| Total mean ± SEM | 408 ± 223 | 2.71 ± 0.27 | 2.68 ± 0.28 | 76.1 ± 12.5 | 72.0 ± 14.6 | ||||
Efficiency of cell concentration and recovery of natural microbiota in aqueous chicken homogenates.
A total of eight runs were performed to validate the instrument in concentrating and recovering naturally occurring microbiota from aqueous chicken homogenates. The results are shown in Table 3. The initial cell levels were estimated by plating to be in the range between 1 and 3.17 log CFU/ml with variance due to the heterogeneity of the chicken samples. Cell concentration resulted in cell levels of 3.48 to 6.12 log CFU/ml (or 3,000 to 1.3 million). Correspondingly, the concentration was increased by 102.48- to 103.01-fold. With an initial volume of 250 ml, the mean volume of the concentrated sample was 295 μl with a standard deviation (SD) of 137 μl. Accordingly, the recovery of total cells varied from 47.1 to 102.8 (mean recovery equals 76.2 with an SD of 18.8). The lowest detectable amount of Salmonella was 1.3 log CFU/ml of Salmonella equivalent to −1.80 log CFU/ml in prefiltered sample or 0.16 CFU/g chicken meat. The time needed for cell concentration and recovery from 250 ml of enzyme-conditioned, prefiltered homogenate was 45 min. Again, one cycle of sample processing was completed within 1.5 h, including the cleaning cycle.
Table 3.
Concentration and recovery of natural microbiota in aqueous chicken homogenates (n = 8)
| Concn of natural microbiota (log CFU/ml) |
Vol of concentrated sample (μl) | Concn factor | Recovery (%) | Concn of S. Enteritidis (log CFU/ml) in concentrated sample | |
|---|---|---|---|---|---|
| Prefiltered sample | Concentrated sample | ||||
| 1.00 | 3.72 | 280 | 2.72 | 58.8 | |
| 1.00 | 4.01 | 250 | 3.01 | 102.8 | |
| 1.78 | 4.67 | 150 | 2.89 | 47.1 | |
| 1.78 | 4.63 | 350 | 2.85 | 99.2 | |
| 1.00 | 3.48 | 600 | 2.48 | 72.0 | |
| 1.00 | 3.83 | 280 | 2.83 | 74.9 | |
| 2.00 | 4.92 | 250 | 2.92 | 83.5 | |
| 3.17 | 6.12 | 200 | 2.93 | 71.4 | 1.3 |
| 295 ± 137 | 2.83 ± 0.17 | 76.2 ± 18.8 | |||
Quantitative PCR.
qPCR analysis of artificially inoculated Salmonella in chicken homogenate and PBS samples was carried out. The results are shown in Table 4. The sensitivity of the qPCR assay without enrichment was approximately 2.0 log CFU/ml in PBS and chicken wash. The detection of S. Enteritidis was linear for DNA isolated from samples containing 1.0 to 7.0 log CFU/ml in PBS and chicken wash with high correlation efficiencies (r2) of 0.98 and 0.99, respectively. The linear relationship between the number of cells in the pure DNA samples and the threshold cycle (CT) number was used to generate a standard curve for the quantification of S. Enteritidis in the PBS and chicken wash samples (Fig. 4). Using the standard curve as a guideline, the BCE in each sample was extrapolated based on the corresponding CT value and the DNA concentration. The PCR result was consistent with bacterial counts from plating.
Table 4.
Plate counts and quantitative PCR analysis of S. enterica serovar Enteritidis PT21 in artificially contaminated PBS and chicken homogenate samplesa
| Sample type | Sample | Plate count (log CFU/ml) (mean ± SEM) | qPCR (log GCE/mlb) (mean ± SE) |
|---|---|---|---|
| PBS (n = 4) | Unspiked sample | UDc | UD |
| Artificially spiked sample | 2.68 ± 0.09 | 2.53 ± 0.08 | |
| Concentrated sample | 4.92 ± 0.15 | 5.55 ± 0.06 | |
| Chicken homogenates (n = 8) | Unspiked sample | UD | UD |
| Artificially spiked sample | 3.26 ± 0.30 | 2.57 ± 0.13 | |
| Concentrated sample | 4.95 ± 0.54 | 5.57 ± 0.09 |
Replicate samples were artificially contaminated, and the experiment was repeated four times.
GCE, genomic cell equivalents.
UD, undetermined.
Fig 4.

Quantification of bacterial number based on qPCR of Salmonella standards. The detection of S. Enteritidis was linear for DNA isolated from samples containing 1.0 to 7.0 log CFU. Bacterial cell equivalents for samples were extrapolated from the standard curve based on CT value and the corresponding log ratio of genome cell equivalents (GCE).
DISCUSSION
Cross-flow ultra/microfiltration has been studied extensively for rapid concentration and recovery of microorganisms from a variety of water matrices. The configuration of cross-flow filtration greatly facilitates continuous operation and enables direct recovery of cells. Although cross-flow filtration can potentially separate and concentrate bacterial pathogens from food matrices to detectable levels if large sample volumes are processed, one challenge remains due to the high solid content and viscosity of typical food samples (1).
This work demonstrated that microbiota can be concentrated and efficiently recovered from aqueous chicken homogenates by cross-flow microfiltration using a hollow-fiber membrane. For both naturally occurring microbiota and inoculated S. Enteritidis, the cell levels in the concentrates were about 3 orders of magnitude greater than the initial levels, thus enabling detection of 10 CFU/ml of cells in the original samples by plating and qPCR. In this work, after cell concentration, cells were eluted in a cross-flow mode with the surfactant Tween 20 given its compatibility with polysulfone membrane and the microorganisms. The surfactant lowers the interfacial tension between the membrane surface and the attached cells and hence enhances their recovery. The possibility that microfiltration may damage cells due to stress induced by the laminar shear field or by deformation of the cell in filter pores under the transmembrane pressure was also examined in our work (30). The extent of shear damage depends on the intensity, duration, and type of shear force and also on the cell type and growth stage (31). While the intensity of stress and cell viability were not determined directly, cells were enumerated by spread plating, and the time required for the colonies to develop was not substantially different for samples before and after being microfiltered in our instrument. In addition, this work addressed the reuse of filtration membranes for 15 times before replacement by applying a hydraulic and chemical cleaning procedure following the cell concentration and recovery cycle.
In qPCR analysis, the specificity of the primer set for the detection of Salmonella sp. was ascertained by testing with other bacterial species. When the qPCR was performed using known DNA concentrations of Salmonella (S. Enteritidis and S. Typhimurium Nos-6) and non-Salmonella strains (Escherichia coli O157:H7 EDL 933, E. coli K-12, Pseudomonas aeruginosa, Citrobacter freundii, and Enterobacter aerogenes), positive amplification was obtained only with the Salmonella DNA samples, thus confirming the specificity of the primer set for detection of Salmonella species only (data not shown). The results obtained from qPCR analysis were found to be in accordance with the viable-cell counts by plating, suggesting that replacing the traditional culture method with qPCR for quantification of bacterial load is applicable. This will help significantly reduce the time required to obtain cell counts. We have shown that the pathogenic target in chicken homogenates could be quantified within 6 h. It is noted that the detection time could be even shorter, with the cleanup cycle for the cell concentration instrument being run concurrently with a qPCR.
In previous studies, other types of separation and concentration techniques have been reported in recovering pathogenic cells from food homogenates. Some recent works are summarized in Table 1. For example, Wolffs et al. used a two-step filtration protocol followed by a real-time PCR assay, allowing quantitation of 75 CFU/g of Salmonella and identification of as low as 0.22 CFU/g in chicken rinse within 3 h (7); Fukushima et al. combined filtration, centrifugation, and buoyant density gradient centrifugation, concentrating cells by 250-fold, and enabling detection of 101 to 103 CFU/g of Salmonella assay in naturally contaminated chicken using real-time qPCR (RTi-qPCR) (8). Here, when processing higher-volume samples (250 ml) using an automated instrument, significant numbers of microorganisms are recovered from chicken natural microbiota (103 to 106 CFU/ml) with a mean recovery of 76.2% of viable cells. In the natural microbiota of chicken homogenates, the lowest detectable amount of Salmonella was 1.3 log CFU/ml of Salmonella equivalent to −1.80 log CFU/ml in prefiltered sample or 0.16 CFU/g chicken meat. This illustrates the utility of recovery and concentration of targeted pathogens from food samples.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by U.S. Department of Agriculture (USDA) cooperative agreement OSQR (935-42000-049-00D), Purdue University Agricultural Research Programs, and the Department of Agricultural and Biological Engineering at Purdue University.
We also thank Youngmi Kim for helpful comments on the manuscript.
Footnotes
Published ahead of print 6 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02587-13.
REFERENCES
- 1.Centers for Disease Control and Prevention 18 April 2013. Trends in foodborne illness in the United States, 2012. Centers for Disease Control and Prevention, Atlanta, GA: http://www.cdc.gov/features/dsfoodnet2012/reportcard.html [Google Scholar]
- 2.Kennedy S. 2008. Why can't we test our way to absolute food safety? Science 322:1641–1643 [DOI] [PubMed] [Google Scholar]
- 3.Dwivedi HP, Jaykus LA. 2011. Detection of pathogens in foods: the current state-of-the-art and future directions. Crit. Rev. Microbiol. 37:40–63 [DOI] [PubMed] [Google Scholar]
- 4.Bull MK, Hayman MM, Stewart CM, Szabo EA, Knabel SJ. 2005. Effect of prior growth temperature, type of enrichment medium, and temperature and time of storage on recovery of Listeria monocytogenes following high pressure processing of milk. Int. J. Food Microbiol. 101:53–61 [DOI] [PubMed] [Google Scholar]
- 5.Brewster JD. 2003. Isolation and concentration of Salmonellae with an immunoaffinity column. J. Microbiol. Methods 55:287–293 [DOI] [PubMed] [Google Scholar]
- 6.Sharpe AN, Peterkin PI, Dudas I. 1979. Membrane filtration of food suspensions. Appl. Environ. Microbiol. 37:21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolffs PFG, Glencross K, Thibaudeau R, Griffiths MW. 2006. Direct quantitation and detection of Salmonella in biological samples without enrichment, using two-step filtration and real-time PCR. Appl. Environ. Microbiol. 72:3896–3900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fukushima H, Katsube K, Hata Y, Kishi R, Fujiwara S. 2007. Rapid separation and concentration of foodborne pathogens in food samples prior to quantification by viable-cell counting and real-time PCR. Appl. Environ. Microbiol. 73:92–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brewster JD. 2009. Large-volume filtration for recovery and concentration of Escherichia coli O157:H7 from ground beef. J. Rapid Methods Autom. Microbiol. 17:242–256 [Google Scholar]
- 10.Kim H-Y, Park H-J, Ko G. 2009. Hollow fiber ultrafiltration for the concentration and simultaneous recovery of multiple pathogens in contaminated foods. J. Food Prot. 72:2547–2552 [DOI] [PubMed] [Google Scholar]
- 11.Murakami T. 2012. Filter-based pathogen enrichment technology for detection of multiple viable foodborne pathogens in 1 day. J. Food Prot. 75:1603–1610 [DOI] [PubMed] [Google Scholar]
- 12.Besse NG, Lafarge V. 2001. Development of a membrane filtration method for enumeration of Listeria monocytogenes from soft cheese. Food Microbiol. 18:669–676 [Google Scholar]
- 13.Carroll SA, Carr LE, Mallinson ET, Lamichanne C, Rice BE, Rollins DM, Joseph SW. 2000. Development and evaluation of a 24-hour method for the detection and quantification of Listeria monocytogenes in meat products. J. Food Prot. 63:347–353 [DOI] [PubMed] [Google Scholar]
- 14.Chen WT, Hendrickson RL, Huang CP, Sherman D, Geng T, Bhunia AK, Ladisch MR. 2005. Mechanistic study of membrane concentration and recovery of Listeria monocytogenes. Biotechnol. Bioeng. 89:263–273 [DOI] [PubMed] [Google Scholar]
- 15.Chen WT, Ladisch MR, Geng T, Bhunia AK. 2005. Membrane for selective capture of the microbial pathogen Listeria monocytogenes. AIChE J. 51:3305–3308 [Google Scholar]
- 16.Entis P, Lerner I. 2000. Twenty-four-hour direct presumptive enumeration of Listeria monocytogenes in food and environmental samples using the ISO-GRID method with LM-137 agar. J. Food Prot. 63:354–363 [DOI] [PubMed] [Google Scholar]
- 17.Ladisch MR. 2001. Bioseparations engineering: principles, practice, and economics, p 17–19, 36–47, 53–106 Wiley-Interscience, New York, NY [Google Scholar]
- 18.Peterkin PI, Sharpe AN. 1980. Membrane filtration of dairy products for microbiological analysis. Appl. Environ. Microbiol. 39:1138–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foley G. 27 November 2008. Crossflow microfiltration. SciTopics. http://www.scitopics.com/Crossflow_Microfiltration.html.
- 20.Baker RW. 2004. Membrane technology and applications, 2nd ed, p 89–155 J. Wiley, New York, NY [Google Scholar]
- 21.Hill VR, Polaczyk AL, Hahn D, Narayanan J, Cromeans TL, Roberts JM, Amburgey JE. 2005. Development of a rapid method for simultaneous recovery of diverse microbes in drinking water by ultrafiltration with sodium polyphosphate and surfactants. Appl. Environ. Microbiol. 71:6878–6884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu P, Hill VR, Hahn D, Johnson TB, Pan Y, Jothikumar N, Moe CL. 2012. Hollow-fiber ultrafiltration for simultaneous recovery of viruses, bacteria and parasites from reclaimed water. J. Microbiol. Methods 88:155–161 [DOI] [PubMed] [Google Scholar]
- 23.Morales-Morales HA, Vidal G, Olszewski J, Rock CM, Dasgupta D, Oshima KH, Smith GB. 2003. Optimization of a reusable hollow-fiber ultrafilter for simultaneous concentration of enteric bacteria, protozoa, and viruses from water. Appl. Environ. Microbiol. 69:4098–4102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith CM, Hill VR. 2009. Dead-end hollow-fiber ultrafiltration for recovery of diverse microbes from water. Appl. Environ. Microbiol. 75:5284–5289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukhopadhyay S, Tomasula PM, Van Hekken DL, Luchansky JB, Call JE, Porto-Fett AC. 2009. Effectiveness of cross-flow microfiltration for removal of microorganisms associated with unpasteurized liquid egg white from process plant. J. Food Sci. 74:319–327 [DOI] [PubMed] [Google Scholar]
- 26.Blanpain-Avet P, Faille C, Bénézech T. 2009. Cleaning kinetics and related mechanisms of Bacillus cereus spore removal during an alkaline cleaning of a tubular ceramic microfiltration membrane. Desalin. Water Treat. 5:235–251 [Google Scholar]
- 27.Mukhopadhyay S, Tomasula PM, Luchansky JB, Porto-Fett A, Call JE. 2010. Removal of Salmonella Enteritidis from commercial unpasteurized liquid egg white using pilot scale cross flow tangential microfiltration. Int. J. Food Microbiol. 142:309–317 [DOI] [PubMed] [Google Scholar]
- 28.Hein I, Flekna G, Krassnig M, Wagner M. 2006. Real-time PCR for the detection of Salmonella spp. in food: an alternative approach to a conventional PCR system suggested by the FOOD-PCR project. J. Microbiol. Methods 66:538–547 [DOI] [PubMed] [Google Scholar]
- 29.Malorny B, Hoorfar J, Bunge C, Helmuth R. 2003. Multicenter validation of the analytical accuracy of Salmonella PCR: towards an international standard. Appl. Environ. Microbiol. 69:290–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maiorella B, Dorin G, Carion A, Haran D. 1991. Crossflow microfiltration of animal cells. Biotechnol. Bioeng. 37:121–126 [DOI] [PubMed] [Google Scholar]
- 31.Kang ST, Subramani A, Hoek EM, Deshusses MA, Matsumoto MR. 2004. Direct observation of biofouling in cross-flow microfiltration: mechanisms of deposition and release. J. Membr. Sci. 244:151–165 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.