

Abstract
Soil fungi play a major role in terrestrial ecosystem functioning through interactions with soil structure, plants, micro- and mesofauna, and nutrient cycling through predation, pathogenesis, mutualistic, and saprotrophic roles. The diversity of soil fungi was assessed by sequencing their 28S rRNA gene in Alaskan permafrost and Oklahoma tallgrass prairie soils at experimental sites where the effect of climate warming is under investigation. A total of 226,695 reads were classified into 1,063 genera, covering 62% of the reference data set. Using the Bayesian Classifier offered by the Ribosomal Database Project (RDP) with 50% bootstrapping classification confidence, approximately 70% of sequences were returned as “unclassified” at the genus level, although the majority (∼65%) were classified at the class level, which provided insight into these lesser-known fungal lineages. Those unclassified at the genus level were subjected to BLAST analysis against the ARB-SILVA database, where ∼50% most closely matched nonfungal taxa. Compared to the more abundant sequences, a higher proportion of rare operational taxonomic units (OTU) were successfully classified to genera at 50% bootstrap confidence, indicating that the fungal rare biosphere in these sites is not composed of sequencing artifacts. There was no significant effect after 1 year of warming on the fungal community structure at both sites, except perhaps for a few minor members, but there was a significant effect of sample depth in the permafrost soils. Despite overall significant community structure differences driven by variations in OTU dominance, the prairie and permafrost soils shared 90% and 63% of all fungal sequences, respectively, indicating a fungal “seed bank” common between both sites.
INTRODUCTION
Fungi represent a large, still incompletely discovered, repository of biological diversity (1) whose role is crucial in ecosystem functioning. By directly influencing the food web as plant pathogens, predators of micro- and mesofauna, mutualists, and decomposers, fungi play a key role in soil function (1). As one of the most diverse groups of the Eukarya, only a small fraction of the estimated total fungal diversity (2) has been assessed to date. Fungal communities are influenced by spatiotemporal as well as biotic and abiotic factors. For example, edaphic characteristics such as soil type (3), substrate composition and pH (4, 5, 6), as well as seasonality (7, 8, 9), have been found to shape community composition. Plant community composition may also play an important role (8, 10) through a direct influence on endophytes, pathogens, and mycorrhizal fungi or on saprotrophs through differences in litter quality and source.
The diversity of the fungal component in microbial communities has become more accessible through high-throughput marker gene sequencing. Among the primer sets proposed for the amplification of the 28S gene, the LR3/LR0R primer combination that spans D1/D2 is suitable for both classification accuracy and resolution to the genus level (11). While 28S gene sequence assignment has typically been performed using iterations of BLAST (12, 13), a fungal 28S naive Bayesian classifier (NBC) was recently made available at the Ribosomal Database Project (RDP) that allows for direct, rapid sequence classification without clustering. The classifier has been independently validated (14) and is based on a manually archived large subunit (LSU) gene training set (11). An advantage of this fungal 28S classifier is the ability to select bootstrap values from 0% to 95%, which indicates the number of times a genus is selected out of 100 bootstrap trials using a subset of one-eighth of the eight-character subsequences (words) comprising each query sequence. Hence, higher bootstrap values result in a larger proportion of sequences binned as “unclassified.” These unclassified bins occur at various taxonomic ranks; an unclassified sequence at the genus level may remain classified at coarser taxonomic levels. This group can be further investigated by examining the composition at different taxonomic levels to give more insight into the lesser-known fungal lineages, including their distribution relative to environmental attributes.
With the exception of a few deep sequencing studies (7, 10, 15, 16), most have not addressed the extent or composition of the unclassified members other than establishing them as nonfungal reads. The definition of unclassified reads is largely dependent on the classification method. In most internal transcribed spacer (ITS)-based studies where BLAST or phylogenetic relationships inferred through trees based on alignments are used, an unclassified bin or cluster would result from a closest sequence match to an unclassified or uncultured sequence or no matches above a predetermined similarity or E value. The abundance of fungal unclassified reads, even at higher subphylum or class levels (7), illustrates the dearth of sequences available from classified fungi. As such, the composition of any fungal database precludes the identification of the majority of sequences (15, 17, 18). As Lentendu et al. (15) pointed out, the effect of the definition “unclassified” on fungal diversity estimates has been largely ignored.
In this study, we used 28S gene-based pyrosequencing coupled with direct classification using the RDP-naive Bayesian fungal classifier of fungal communities from Alaskan permafrost samples (AK samples) and Oklahoma tallgrass prairie samples (OK samples), both of which have plots at natural temperatures and warmed 1.5 to 2°C above the ambient temperature to simulate global warming effects. Our objectives were (i) to gain more insight into the difficult to classify taxa by using the classifier's ability to resolve these sequences at different taxonomic levels, (ii) to determine if short-term warming resulted in a shift in community structure, (iii) to assess if depth in the Alaska soil (through the active layer and into permafrost) affected community structure, and (iv) to establish what proportion of the known fungal taxa resided in these two very different ecosystem types and which taxa they had in common.
MATERIALS AND METHODS
Site descriptions.
A permafrost warming experiment, the Carbon in Permafrost Experimental Heating Research project (CiPEHR), is located in the northern foothills of the Alaska Range near Denali National Park and Preserve in the region of Eight Mile Lake, Alaska (63°52′59″N, 149°13′32″W). Established in September 2008, the goal of the study site is to determine the effects of soil, air, and permafrost warming on tundra ecosystems (19). The site lies within a region of discontinuous permafrost where thawing and thermokarst (areas with standing water due to permafrost thaw and resulting soil depression) have been occurring over several decades. Winter soil temperatures were passively increased by accumulation of snow with snow fences in the winter and removal of snow in the spring. Warming has increased the soil temperature by 1.5°C (integrated over a depth of 5 to 40 cm) (19) and melted the upper permafrost layers.
Soils are classified in the soil order Gelisol, and vegetation is dominated by the deciduous shrub Vaccinium uliginosum and the tussock-forming sedge Eriophorum vaginatum. Other common vascular plants include Betula nana and Carex bigelowii, with the nonvascular Sphagnum spp., feather moss, and several lichen species also present (19, 20, 21, 22). Three replicate soil cores, up to 65-cm depth, were taken from winter warming (WW) and control (C) plots after 1 year of warming treatment in May 2009 and, while the active layer remained frozen, sliced into 10-cm-depth increments through the entire soil core. The depth to the permanently frozen layer (permafrost) was determined in the field for each core.
The Oklahoma samples originated from the unclipped (no biomass harvest) plots at the Great Plain Apiaries site (34°58′54″N, 97°31′14″W). The study site was implemented in November 1999 to characterize soil responses to experimental warming (22, 23, 24, 25, 26, 27). Soils are classified as silt loams, and the grassland is dominated by the C4 grasses Schizachyrium scoparium, Sorghastrum nutans, and Eragrostis curvula and the C3 forbs Ambrosia psilostachyia and Xanthocephalum texanum. Plots were warmed with an infrared heater, while the control plot contained a “dummy” heater to simulate the shading effect. Experimental warming increased mean soil temperatures by 1.8 to 2.7°C. Soil cores were collected in October 2010 from the topsoil (0 to 15 cm) of six replicates in both warming and control plots.
All samples from both sites were kept frozen until DNA extraction. Plant roots were not removed. DNA was extracted and purified using the freeze-grinding mechanical lysis method (28).
454 pyrosequencing.
Amplification was performed using fungal 28S gene primers LR3/LR0R (http://www.biology.duke.edu/fungi/mycolab/primers.htm), which results in 625-bp fragments (11). Bar codes were included on both the forward and reverse primers to facilitate sample discrimination. Quadruplicate 20-μl PCRs were performed with the following materials: 4 μl Promega GoTaq buffer, 0.5 μl GoTaq DNA polymerase, 1.5 μl Roche 25 mM MgCl2, 1 μl Invitrogen 10 mM deoxynucleoside triphosphate (dNTP) mix, 1 μl each primer (10 pmol μl−1), 0.2 μl New England BioLabs 10 mg ml−1 bovine serum albumin (BSA), 1 μl (10 ng μl−1) template, and 9.8 μl H2O. The cycling conditions were an initial denaturation of 94°C for 3 min followed by 30 cycles of 94°C for 1 min, 51°C for 40 s, and 72°C for 1 min, followed by an extension at 72°C for 10 min. Replicates were pooled and gel purified using the Qiagen gel purification kit following band excision. Products were further purified using the Qiagen PCR purification kit. Following adapter ligation, amplicons were sequenced by Macrogen (Seoul, South Korea) using Lib-L kits and processed using the shotgun protocol.
Raw sequences read from both the A and B adapters were quality processed and sorted through the RDP pyrosequencing pipeline (http://pyro.cme.msu.edu). The following filter parameters were used: 0 mismatches to forward primer, 400-bp-length filter, ambiguous base N count = 0, and quality score (Q) ≥ 20. The sequences that fully matched the bar codes were selected and then subjected to the RDP fungal LSU classifier (http://rdp.cme.msu.edu/classifier) using training set 1 at both 0% (best match) and 50% bootstrap confidence levels. In both cases, nonfungal and Eukaryota incertae sedis reads were removed for downstream statistical analyses. For the 50% bootstrap confidence classification, taxa were identified as “unclassified” at a particular taxonomic level when the classification confidence was <50%. This confidence level was chosen because it preceded large incremental increases in the percentage of unclassified taxa at higher confidence levels. The abundances of sequences belonging to taxa classified at the phylum, class, order, family, and genus levels were treated as bins for downstream statistical analysis. Approximately 1.7% of all sequences were identified as chimeras based on UCHIME (29). Putative chimeras were not removed, as this frequency falls within the false-positive detection rate (30).
Data analysis.
Raw abundances were normalized by Hellinger transformation (square root of relative abundance), and a Bray-Curtis dissimilarity matrix was constructed for nonmetric multidimensional scaling (NMDS). Significant community differences were tested using analysis of similarity (ANOSIM) (31) and permutational multivariate analysis of variance (PERMANOVA) (32). Cluster analyses were performed with the similarity profile analysis (SIMPROF) test (33), and similarity percentages (SIMPER) analysis (34) was used to identify the operational taxonomic unit (OTU) that contributed to the discrimination among treatments. Sample dispersion was tested by permutational analysis of multivariate dispersions (PERMDISP) (35) using PRIMER-E (36). Significant differences in species richness (Margalef; d), Shannon diversity (H′), and Pielou's evenness (J′) were assessed with one-way analysis of variance (ANOVA) using StatPlus 5.8.2.0 (AnalystSoft, Inc.).
Random resampling was performed to 5,180 Oklahoma and 3,522 Alaska sequences per sample for downstream analyses. For direct comparison between sites, 3,522 sequences were randomly resampled from both data sets.
Nucleotide sequence accession numbers.
Nucleotide sequences have been deposited in the European Nucleotide Archive (http://www.ebi.ac.uk/ena/) under study no. PRJEB3968 with accession no. ERS250779 to ER250807.
RESULTS
Oklahoma site.
From a total of 147,434 sequences subjected to filtering, the Oklahoma (OK) samples yielded a total of 96,669 best-match (0% bootstrap confidence) classified sequences containing 936 genera in 276 families, 96 orders, 31 classes, and 7 phyla. A total of 130 genus bins were singletons. Approximately 95% of the reads were successfully classified as Fungi or Eukaryota incertae sedis. Rarefaction curves were not saturated (see Fig. S1A in the supplemental material). Communities were dominated by the fungal classes Agaricomycetes, Sordariomycetes, Lecanoromycetes, and Glomeromycetes. The top 10 and 20 genera accounted for 36.3% and 46.9% of all sequences, respectively, as evidenced by the species accumulation curve (Fig. 1A). The OK fungal communities were dominated by the Agaricales (Moniliophthora, Leucopaxillus, Camarophyllus, and Camarophyllopsis), the Hysteriales (Hysteropatella), Chaetothyriales (Sorocybe), Onygenales (Arachnomyces), Ascomycota incertae sedis (Everhartia), Lecanorales (Boreoplaca), Pezizales (Phialea), Schizosaccharomycetales (Schizosaccharomyces), and the Sordiales (Cercophora) (Fig. 2), while two predominant soil molds of warm prairie soils, Penicillum and Aspergillus, constituted <0.1% of all sequences. There was no consistent difference among genus-level fungal communities between the control and warming treatments according to NMDS ordination (see Fig. S2 in the supplemental material) and as tested through ANOSIM (P = 0.64) and PERMANOVA (P = 0.66). This was consistent among all taxonomic levels. Only 10 mostly lower-abundance genera changed significantly with treatment (t test, P < 0.05).
Fig 1.
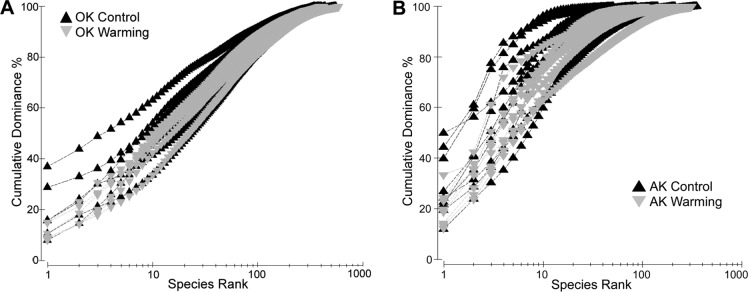
Species accumulation curves for (A) Oklahoma (OK) and (B) Alaska CiPEHR (AK) samples.
Fig 2.

Composition of the top 10 taxa at the (A) class, (B) order, and (C) family levels and the top 20 taxa at the (D) genus level for Oklahoma samples. The total percentages of all sequences are (A) 97%, (B) 75.1%, (C) 49.3%, and (D) 46.9%.
Alaska site.
Of a total of 153,403 sequences, the Alaska (AK) samples generated a total of 130,026 classified sequences at best match, containing 743 genera in 276 families, 101 orders, 33 classes, and 9 phyla. Rarefaction curves showed higher coverage than OK samples, with saturation approached in many of the deeper soil samples (see Fig. S1B in the supplemental material). A total of 157 genera were singletons. The distribution of genera revealed a dominant community, with the top 10 and 20 genera accounting for 54.7% and 69.9% of all sequences, respectively (Fig. 1B). Fungal communities were dominated by the Helotiales genera Godronia, Chloroscypha, Hyaloscypha, Hyalodendriella, Sclerotinia, Lachnum, Crinula, Vibrissea, Ascocoryne, and Fulvoflamma, as well as Chaetothyriales (Sorocybe), Saccharomycetales (Dipodascus), Russulales (Russula), Polyporales (Abortiporus), Chytridiales (Karlingiomyces), and Zoopagales (Kuzuhaea), which comprised an average of 62.2% of all sequences.
There was no significant effect of warming (PERMANOVA and ANOSIM), with only seven significantly different genera (t test, P < 0.05) that comprised 5.9 and 14.9% of all sequences at the control and warming sites. However, there was a significant difference in fungal community structure with depth by parsing of samples between 0 and −20 cm (shallow) from the −20- to −50-cm (deep) samples (ANOSIM, global R = 0.42, P < 0.01; PERMANOVA, F = 4.53, P < 0.01) (see Fig. S2B in the supplemental material). Differences in fungal community structures between the shallow and deep samples were present from the class to genus taxonomic levels in both relative abundance and presence or absence (see Fig. S3 and S4 in the supplemental material). While both depth increments were dominated by the fungal class Leotiomycetes, the identity and abundance of the lesser classes changed with depth. SIMPER analysis for this comparison was weighted heavily toward a few genera, with the top 10 comprising 52.7% of the dissimilarity and 53.1% of all sequences (Fig. 3). In all, 18 genera were significantly different (t test, P < 0.05) between the shallow and deeper layers, generally corresponding to the more abundant taxa in each layer.
Fig 3.

SIMPER analysis showing the top contributing genera to the difference between the 0- to −20-cm and −20- to −50-cm soil layers in the Alaska samples. Bars indicate average percent relative abundances in the resampled data set. Percentages to the right indicate the percent dissimilarity contribution of each genus.
Composition of the unclassified taxa.
Classification was reperformed for the entire data set at a 50% bootstrap confidence level to gain insight into the composition of the underrepresented taxa within the current database. For the OK samples, at this confidence, “unclassified” taxa ranged from 8.4% (kingdom) to 67.1% (genus) of all sequences, depending on the taxonomic level and were contained within 268 family-level bins (Fig. 4A), leaving 390 classified genera, a reduction of the original genus-level diversity by 68.3%. At the phylum level, an average of 32.9% of sequences were unclassified, with the majority (25.1%) most closely assigned to the Ascomycota. At the order level, 47.2% were unclassified, with the majority belonging to the Sordariomycetes (6.0%), Dothideomycetes (7.4%), Eurotiomycetes (6.2%), and Pezizomycetes (4.9%). At the genus level, unclassified sequences were heavily weighted toward relatively few taxa (see Fig. S5 in the supplemental material). Both NMDS ordination and ANOSIM results were largely unaffected when unclassified bins were included for the Oklahoma data (data not shown). Overall, the abundances of the unclassified genera between the AK shallow, AK deep, and OK samples at all taxonomic levels were significantly different (two-way ANOVA, P < 0.05).
Fig 4.
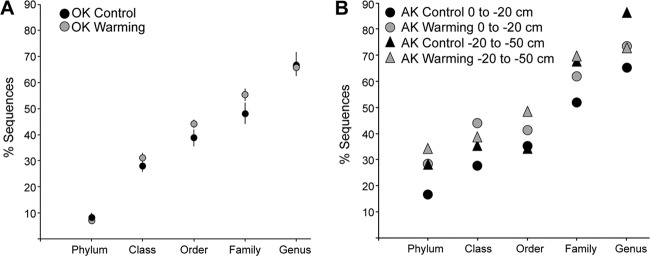
Percent unclassified as determined by classification at 50% bootstrap confidence using the RDP fungal LSU classifier at each taxonomic level in the (A) Oklahoma and (B) Alaska samples.
In the Alaska data set, unclassified taxa ranged from an average of 36.8% to 88.1% of sequences from the phylum to genus level (Fig. 4B). At the order level, 38.9% were unclassified—the majority in the Chytridiomycetes (19.5%), Fungi incertae sedis (16.9%), Leotiomycetes (9.4%), and Sordariomycetes (8.5%). A total of 241 genera were placed into unclassified bins at the corresponding family level, which were again heavily weighted toward a few taxa (see Fig. S6 in the supplemental material). This resulted in a 67.6% reduction of classified genera compared to the original data set. Of the top 30 unclassified family bins, only 7 were shared between OK and AK samples. The majority of the top 20 genera obtained by best match remained prevalent, although at lower abundances due to binning into members of associated unclassified families. A higher proportion of unclassified Fungi at the family level were associated with the deeper permafrost samples (27.8% versus 16.6%).
Community comparison between OK and AK samples.
Not surprisingly, fungal communities were fundamentally different between the Alaska permafrost and Oklahoma grassland samples. The OK samples exhibited significantly higher diversity and evenness than the AK shallow (0- to −20-cm) and deep (20- to −65-cm) samples (ANOVA, P < 0.01) (Fig. 5). Significant differences in abundance between the sites were most pronounced at the higher taxonomic levels (t test, P < 0.05) (see Tables S1 and S2 in the supplemental material) when all depths were included. Of the top 50 most abundant genera at 0% bootstrap confidence from both sites at all depths comprising 56% (AK) and 76% (OK) of all sequences, 11 genera were shared. A total of 105 genera (∼10% of sequences, 49 singletons) were unique to the OK samples, while 503 genera (∼37% of sequences, 320 singletons) were unique to AK. Both NMDS for shallow sample comparison (Fig. 6) and cluster analysis for all depths (see Fig. S7 in the supplemental material) showed a clear delineation between AK and OK samples (PERMANOVA, P = 0.001; ANOSIM, P = 0.001). As evidenced from the NMDS, sample dispersion was significantly different between the shallow AK and OK samples (PERMDISP, F = 25.26, P = 0.001). The top 29 genera reported from SIMPER analysis contributed to 21.2% of the dissimilarity (Fig. 7). The Leotiomycetes and Chytridiomycetes were most associated with the Alaska samples, while the Agaricomycetes, Eurotiomycetes, Sordariomycetes, and Lecanoromycetes dominated at the Oklahoma site.
Fig 5.
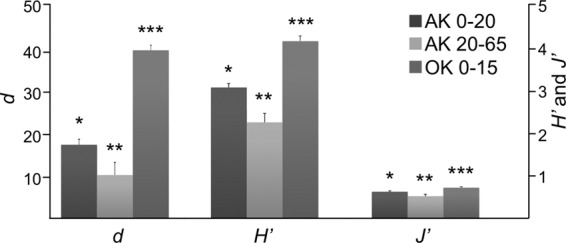
Measures of species richness (Margalef; d), Shannon diversity (H′), and evenness (Pielou; J′) for the Oklahoma (OK) (0- to −15-cm depth) and Alaska (AK) samples, parsed into shallow (0- to −20-cm) and deep (−20- to −65-cm) samples with standard errors. Asterisks indicate grouping based on ANOVA (P < 0.01).
Fig 6.
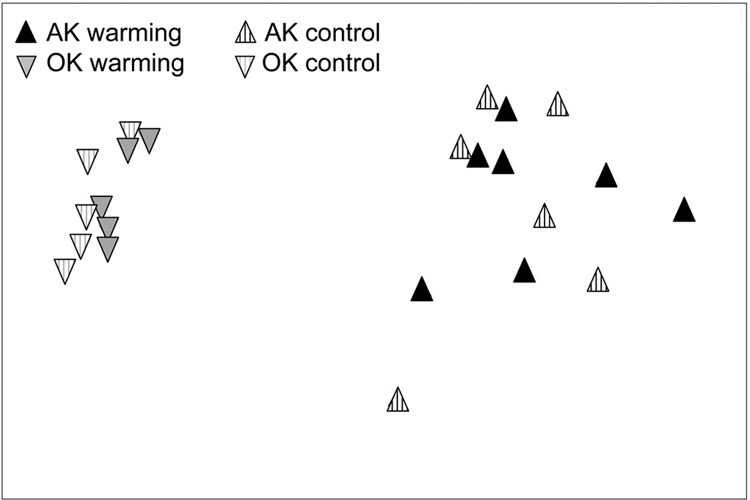
NMDS ordinations based on Bray-Curtis dissimilarity matrices for the comparison between Oklahoma (OK) (0- to −15-cm) and Alaska (AK) (0- to −20-cm) samples with control and warming treatments indicated (2-dimensional stress = 0.07).
Fig 7.

Similarity percentage (SIMPER) analysis differentiating Alaska (AK, 0- to −20-cm) and Oklahoma (OK, 0 to −15 cm) samples. Bars indicate percentages of sequence relative abundance within each genus-level bin with standard errors. Percentages to the right indicate the percent dissimilarity contributed by each bin. The line graph indicates the cumulative percent dissimilarity (Cum. % Diss.) between sites.
ARB-SILVA LSU reference database comparison.
To investigate more deeply the composition of the abundant unclassified genus-level bins and any bias associated with the RDP fungal classifier, we subjected these 158,860 “unclassified” sequences to BLAST against the ARB-SILVA LSU reference database (release 111; www.arb-silva.de). This resulted in hits to 917 taxa, of which 364 were classified as Fungi. These 80,312 sequences classified as Fungi comprised significantly different (ANOVA, P < 0.05) relative abundances of the previously unclassified taxa, with 32%, 48%, and 62% of the AK deep, AK shallow, and OK totals, respectively. The majority of new fungal hits were to relatively few taxa (see Table S3 in the supplemental material). The remaining RDP unclassified hits were mostly to Metazoa (especially Gomphiocephalus hodgsoni; 7.9% of all “unclassified” reads), cyanobacteria, and green algae (Viridiplantae) sequences. The deep AK samples were dominated by hits to the yeast Cyberlindnera meyerae (80% of sequences, 88.1% average identity), while the shallow AK and OK samples each contained ∼25% of hits to the brown rot fungus Serpula lacrymans and 21% and 12% of hits, respectively, to Phaeosphaeria avenaria. Between sites, AK samples shared 47.5% of all SILVA previously unclassified best matches, while OK samples shared 67.4%.
To verify that the inclusion of these SILVA fungal hits would not alter our overall results, we repeated our analyses with these data in combination with the 50% RDP classifier data with the “unclassified” bins removed to create a new RDP_SILVA best-match data set. Differences between the shallow AK versus OK samples remained significant (ANOSIM, P < 0.05; PERMANOVA, P < 0.01), as did the comparisons between the shallow and deep AK samples (ANOSIM, P < 0.05; PERMANOVA, P < 0.01). Warming treatment effects remained nonsignificant. The addition of the SILVA hits did not influence the significant variation previously found with species richness, evenness, and Shannon diversity between the AK shallow, AK deep, and OK samples. However, overall richness and Shannon diversity decreased in the AK sites and increased in the OK site.
DISCUSSION
Diversity profiling of grassland and permafrost soil fungi.
Direct classification of 226,695 filtered 28S sequences yielded 1,063 genera from the two ecosystems, covering 62% of the reference data set's genera. The number of genera recovered per sample ranged from 351 to 594 in the OK grassland samples to 87 to 351 in AK permafrost samples. After normalization for reads per sample, our genus-level diversity was higher than other 97 to 98% cluster-based estimates of species richness from a variety of environments determined by cloning and sequencing (8, 37), ITS pyrosequencing (10, 38, 39), and 28S pyrosequencing but with 95% clustering (40). However, our results agreed with the ITS “species”-level OTU in recent studies from various Mediterranean land use categories (41) and neotropical rain forests (42) but were lower than that found in alpine tundra habitats (15). Such comparisons are difficult to interpret, as the classification resolution differs between the ITS and 28S genes.
Rarefaction curves were nearly saturated in the permafrost samples, especially in the deeper samples, where 4,000 to 8,000 reads was sufficient to cover the majority of fungal diversity present. Comparatively, the sequencing depth was not sufficient to cover the majority of the higher alpha diversity in the Oklahoma prairie samples. As illustrated by lower Pielou's evenness, relatively few taxa dominated the permafrost soils. Together these results suggest that fungal diversity is constrained in permafrost samples, as observed in another study from high alpine soils (43) and especially in our deep soils, where plant influences are limited. Despite the significant differences in overall community structure, the sharing of genera encompassing 90% (OK) and 63% (AK) of all fungal sequences (at 0% bootstrap) between the two sites indicated the possible presence of a fungal “seed bank” analogous to that identified with bacteria in a marine system (44).
Prairie fungal communities.
The dominance of the orders Agaricales, Pleosporales, and Sordariales and the most abundant genus Moniliophthora (4.5% of all sequences, 84.1% ± 0.3% average classification confidence) in the Oklahoma prairie samples is in agreement with findings from healthy grass root zones in semiarid (45) and desert (46) grassland. Moniliophthora is related to the causative agents of witches' broom and frosty pod rot in cacao (47). As noted by Khidir et al. (45), this may be due to an ecological shift from pathogen to endophyte that is not yet understood. We also identified Cercophora sp. (2.3%), an abundant endophyte found colonizing Bouteloua gracilis (46) and Phialea sp. (4.2%), the causative agent of blind-seed disease of ryegrass (48). As a possible artifact of seasonal arbuscular mycorrhizal (AM) fungi, we detected very little Glomus sp. (0.06%), an AM genus identified as being dominant in grasslands (49) and found to be widespread among different global ecosystems (50). Primer coverage of the Glomeromycota was verified with the NCBI primer tool. Matches were identified to 2,103 of the 9,441 Glomeromycota 28S sequences in NCBI. Approximately 8,600 of the deposited sequences are of partial length and therefore may or may not contain both primer binding regions. Little is known about the abundant Arachnomyces (6.5%) species, other than they have been found associated with dung and decaying wood (51) as well as implicated in human nail infections (52). The most abundant basidiomycete was Leucopaxillus (3.2%), described as nonectomycorrhizal but saprotrophic in grasslands and forests (53) and which includes L. giganteus, which produces grassland “fairy rings.”
Permafrost fungal communities.
As found elsewhere (3, 7) in tundra soils, Ascomycota (63.5% of reads) dominated over Basidiomycota (11.5%) in our samples. However, this did not agree with the findings of Wallenstein et al. (8), who found higher levels of Basidiomycota in Arctic tundra tussock soils. This may be largely dependent on the composition of the plant community. The dominance of the order Helotiales (average abundance, 43.3%), which is also abundant in other Arctic studies (3, 54), points to its broad niche range (55) and likely reflects substrate heterogeneity within the deep soil profiles. The mostly aquatic Chytridiomycetes, which constitute 8.5% of all sequences, were found at higher dominance in high-elevation sites in Nepal and Colorado (56) and were easily isolated from other periglacial soils (57).
According to the RDP classifier results, among the most abundant genera, which comprised 41% of all sequences, were the Kuzuhaea (11.2%), belonging to the amoeba predatory order Zoopagales. The amoebae, which comprised the majority of nonfungal sequences, with the genera Amoebidium and Nuclearia accounting for 3.1% of all sequences in the raw data set, are known to be an important component of the C cycle at high latitudes (58). The Karlingiomycetes (7.3%), comprised of chitinophilic and cellulostic species (59), and Godronia (7.2%), which is known to infect blueberry and has also been found on lingonberry (Vaccinium vitus-idaea) branches (60), which populate this site (19), were also abundant. Following in abundance are Chloroscypha (5.7%), characterized by endophytic habits (61), the dematiaceous hypohomycete Sorocybe (resin fungus) (5.6%), the wood endophyte Hyalodendriella (4.6%), and the putative biotrophic parasite or saprobic Hyaloscypha (4.0%) (62), as well as the boreal ectomycorrhizal genus Russula (1.8%) (63), which were abundant in Arctic tundra associated with Betula nana, which was also abundant at our site (54).
Significant differences in fungal community compositions between the shallow and deep soil samples were likely attributed to influences of the mineral versus organic soil horizons (3), substrate composition, and pH (4, 5, 6). These factors serve to shape the niche partitioning between fungi and bacteria, resulting in competitive exclusion and thus influencing community structure. Fungal community partitioning with depth was observed elsewhere with fungi (64, 65), as well as with Actinobacteria (66). Within our AK site, shallower samples are mostly contained within the permafrost thaw depth, where soil is saturated, temperatures are warmer, and decomposition processes are more rapid (19).
Warming effects.
Although warming was not a significant factor in shaping the permafrost or prairie fungal community as a whole, we did identify a few relationships of interest within the permafrost samples. In a fungal clone study, Arctic tundra warming was also shown to influence individual fungal groups but not overall community structure (3). While our observed reduction in Helotiacea abundances did not agree with a previous Arctic warming study (3), the mycorrhizal fungi Russula, Lactarius, and Cortinarius increased with warming, in agreement with previous studies that found increases in mycorrhizal abundance or diversity with warming (54, 67, 68). Although warming has resulted in enhanced graminoid growth (20), other factors may play a combined role in shaping fungal communities in the permafrost. In addition, random sample noise related to the reproducibility of amplicon pyrosequencing may have negatively affected the discrimination between treatments (69).
For the Oklahoma prairie site, previous studies have found an increase in the ratio of fungi to bacteria and a change in the percentage of signature fungal phospholipid-derived fatty acids (PLFAs) (25) and increased CO2 efflux (26) with warming but little change in fungal enzyme abundances (27). Although warming at this site has resulted in increases in C4 but not C3 biomass (25, 70), which has increased the C/N ratio of litter inputs, there were only 10 low-abundance fungal genera that significantly changed with treatment. Previously, the influences of warming treatment on bacterial community structure at this site have been heavily influenced by precipitation, with the largest changes associated with wet months (71).
Regarding the lack of significant overall warming effects, the Alaska CiPEHR samples only represent 1 year of warming, a short time frame to expect microbial community shifts. However, the Oklahoma samples represent over a decade of warming. This lack of warming influences is likely due to the overwhelming influence of plant communities, as other studies have found that shading, precipitation, and plant composition (8, 72, 73, 74, 75), as well as seasonality (7, 8, 9), are the main drivers of fungal communities, which may overwhelm the marginal (∼2 to 3°C) warming effects. Although we did not find significant changes in community structure with warming, it should also be noted that this was based on genus-level resolution, and there are considerable genetic differences that remain at the species level that are not addressed in this study.
Unclassified fungi.
Most fungal sequencing studies have not addressed the abundance or composition of the unclassified taxa because the use of BLAST or hidden Markov model algorithms (Fungal ITS Pipeline) (18) for closest-match taxonomic assignment provides little information about the unclassified sequences at different taxonomic levels. Lentendu et al. (15) expanded on this method to allow fungus-targeted sequence assignments at the coarse phylum, kingdom, and domain levels. When an unfiltered reference data set was used to assign ITS reads, Buée et al. (10) found that 71.5% of sequences lacked an explicit taxonomic annotation, on par with our estimations at a finer taxonomic resolution. After database filtering of unknown fungal species, their unclassified proportion dropped to 11% of the OTU by forcing assignment to a classified nearest neighbor. As such, taxonomic assignment is dependent on the composition of the reference database, which is further complicated by the high fungal species diversity in soil (10, 15), estimated to be 7.12 × 105 at the lower boundary (76).
“Unclassified” sequences, defined here as those with <50% classification confidence using the RDP classifier, are a consequence of either (i) high sequence dissimilarity to a nearest match or (ii) equal classification probability to multiple genera. In both cases, BLAST retrieves a best-match hit used for taxonomic assignment without confidence estimates. By invoking a confidence cutoff, we reduced the likelihood of erroneous taxonomic assignment, although there are cases where a high confidence may also be associated with an incorrect call. For example, a query in which the closest genus is not present in the database may still be assigned to the genus with the higher rank-order likelihood score while supported by bootstrap replication (14).
Likely a consequence of the composition of the reference database, the more thoroughly characterized Ascomycota and Basidiomycota contained the smallest proportion of sequences classified at less than 50% confidence. The reference database Fungal_LSU_train_1400bp_8506_mod used for the RDP classifier and tested by Liu et al. (11) contains 8,506 unique sequences distributed into 1,702 genera, 412 families, 124 orders, 43 classes, and 9 phyla, with the majority of sequences belonging to the Basidiomycota (70.8%) and Ascomycota (28.2%). In contrast, of the approximately 100,000 accepted, described species of fungi (77), approximately 64,000 belong to the Ascomycota and 32,000 to Basidiomycota. Although these numbers probably greatly underrepresent the true diversity of these fungi, they are likely a reasonable approximation of the relative diversity of these two phyla (78). As in other ITS databases, several phyla, such as the Glomeromycota, were underrepresented (10, 16, 79, 80) leading to abundant “unclassified” bins. Of the abundant unclassified family bins (see Fig. S5 and S6 in the supplemental material), the unclassified Tricholomataceae within the prairie site (5.4% of all reads) are unique. This family is among the most highly represented in the database, with 8.3% of all sequences in 121 genera. Two types of “unclassified” sequences are likely represented here due to the underrepresentation of many genera of Tricholomataceae in the RDP database and the polyphyletic nature of the genera (81) within the family. Conversely, the unclassified Helotiaceae contain 20.1% of all reads in permafrost but are only represented by 19 sequences covering 13 genera in the database.
The results from subjecting our “unclassified” sequences to BLAST against the ARB-SILVA LSU reference database showed that 50.6% of those sequences had best matches to fungi, while the remaining binned to other Eukarya. The wide range of BLAST percent identities to fungi (see Table S3 in the supplemental material) illustrate potential novelty on the low end (e.g., 78.0% to Synchytrium decipiens) and cases in which low RDP confidence was likely based on two or more closely matching sequences on the high end (e.g., 96.6% identity to Chytridium sp. strain JEL341). Reads binned into Eukaryota incertae sedis and Fungi incertae sedis in the original data sets comprised an average of 30.7% of all the unclassified sequences at the genus-level bootstrap, compared to 60% binning into nonfungi after BLAST analyses of the same data set. This increase is most likely due to the composition of the SILVA database; 94% of all reference sequences are nonfungal (1,802 sequences are fungal). Thus, sequences that RDP binned into unclassified fungi due to high divergence likely have a higher probability of a closest match to the abundant nonfungal SILVA sequences. However, the inclusion of additional nonfungal sequences into the RDP database will likely change the binning results in both the best-match and 50% bootstrap databases, as divergent sequences alter the statistical underpinning of the naive Bayesian classifier model.
Rare biosphere.
Since a significant proportion of rare OTU were successfully assigned to a genus at 50% bootstrap confidence (68.7% of singletons, 59.7% of doubletons, and 70.3% of tripletons), the assignment of singletons as sequencing artifacts is not justified. This is supported by Lentendu et al. (15), who found that 53.3% of rare ITS molecular OTU (MOTUs0.98) could be successfully assigned to Fungi. Our singletons contained 24.1% of all genera but accounted for less than 0.01% of all sequences. This is lower than the singleton abundances of 38% (15) and 60% (10) reported using ITS cluster-based methods. Differences in the bioinformatic approach, such as clustering, rather than sequencing depth, are likely responsible for a large portion of the differences in singleton abundances among these studies as the addition of singletons decreases proportionally with increased sequencing coverage, as reflected by the flattening of rarefaction curves.
In summary, while there was no effect of artificial warming on fungal communities in AK and OK soils after 1 year of warming, we did observe significant changes between sites and with depth at the AK sites. The ability to directly classify 28S sequences at different confidence levels without the limits imposed by clustering and representative sequence generation allowed us to explore in greater depth the composition of “unclassified” sequences. The majority of “unclassified” reads were assigned to the more abundant families in each site, whereas most of the rare biosphere could be successfully assigned to the genus level. This illustrated the current dearth of fungal sequences from classified taxa, likely a consequence of high community heterogeneity among environments. The implementation of BLAST with the ARB-SILVA database showed instances where low NBC classification confidence could be due to either a large distance to the nearest relative or the presence of an equal probability of classification to two or more taxa. This direct comparison between methods showed limitations regarding the choice of taxonomic assignment method and the coverage of current fungal databases. While a majority of fungal sequencing studies have targeted subpopulations, such as the AM fungi, to address plant-fungus relationships, current and developing sequencing technology continues to reduce the cost, allowing higher coverage, such that whole-community sequencing will increase database coverage while applying environmental relationships to taxa, which is essential in understanding global fungal distribution.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported by the Department of Energy, Biological Systems Research on the Role of Microbial Communities in Carbon Cycling Program (DE-SC0004601).
We thank Amanda Pham, Christina Hazekamp, and Travis Baes for laboratory assistance, the staff of the Ribosomal Database Project at Michigan State University for implementation of the Bayesian Fungal Classifier, especially Benli Chai and Jordan Fish, and Greg Thorn for providing suggestions on fungal taxonomy.
Footnotes
Published ahead of print 6 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01702-13.
REFERENCES
- 1.Mueller GM, Schmit JP. 2007. Fungal biodiversity. What do we know? What can we predict? Biodivers. Conserv. 16:1–5 [Google Scholar]
- 2.Hibbett DS, Ohman A, Glotzer D, Nuhn M, Kirk P, Nilsson RH. 2011. Progress in molecular and morphological taxon discovery in fungi and options for formal classification of environmental sequences. Fungal Biol. Rev. 25:38–47 [Google Scholar]
- 3.Deslippe JR, Hartmann M, Simard SW, Mohn WW. 2012. Long-term warming alters the composition of Arctic soil microbial communities. FEMS Microbiol. Ecol. 1:1–13 [DOI] [PubMed] [Google Scholar]
- 4.Bååth E, Anderson AH. 2003. Comparison of soil fungal/bacterial ratios in a pH gradient using physiological and PLF-based techniques. Soil Biol. Biochem. 35:955–963 [Google Scholar]
- 5.Zinger L, Shahnavaz B, Baptis F, Geremia RA, Choler P. 2009. Microbial diversity in alpine tundra soils correlates with snow cover dynamics. ISME J. 3:850–859 [DOI] [PubMed] [Google Scholar]
- 6.Rousk J, Bååth E, Brookes PC, Lauber CL, Lozupone C, Caporaso JG, Knight R, Fierer N. 2010. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 4:1340–1351 [DOI] [PubMed] [Google Scholar]
- 7.Schadt CW, Martin AP, Lipson DA, Schmidt SK. 2003. Seasonal dynamics of previously unknown fungal lineages in tundra soils. Science 301:1359–1361 [DOI] [PubMed] [Google Scholar]
- 8.Wallenstein MD, McMahon S, Schimel J. 2007. Bacterial and fungal community structure in Arctic tundra tussock and shrub soils. FEMS Microbiol. Ecol. 59:428–435 [DOI] [PubMed] [Google Scholar]
- 9.Toberman H, Freeman C, Evans C, Fenner N, Artz RRE. 2008. Summer drought decreases soil fungal diversity and associated phenol oxidase activity in upland Calluna heathland soil. FEMS Microbiol. Ecol. 66:426–436 [DOI] [PubMed] [Google Scholar]
- 10.Buée M, Reich M, Murat C, Morin E, Nilsson RH, Uroz S, Martin F. 2009. 454 pyrosequencing analyses of forest soils reveals unexpectedly high fungal diversity. New Phytol. 184:449–456 [DOI] [PubMed] [Google Scholar]
- 11.Liu K-L, Porras-Alfaro A, Kuske CR, Elchorst SA, Xie G. 2012. Accurate, rapid taxonomic classification of fungal large-subunit rRNA genes. Appl. Environ. Microbiol. 78:1523–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eichorst SA, Kuske CR. 2012. Identification of cellulose-responsive bacterial and fungal communities in geographically and edaphically different soils by using stable isotope probing. Appl. Environ. Microbiol. 78:2316–2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krüger D, Kapturska D, Fischer C, Daniel R, Wubet T. 2012. Diversity measures in environmental sequences are highly dependent on alignment quality-data from old and new LSU primers targeting Basidiomycetes. PLoS One 7:e32139. 10.1371/journal.pone.0032139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porter TM, Golding GB. 2012. Factors that affect large subunit ribosomal amplicon sequencing studies of fungal communities: classification method, primer choice, and error. PLoS One 7:e35749. 10.1371/journal.pone.0035749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lentendu G, Zinger L, Manel S, Coissac E, Choler P, Geremia RA, Melodelima C. 2011. Assessment of soil fungal diversity in different alpine tundra habitats by means of pyrosequencing. Fungal Divers. 49:113–123 [Google Scholar]
- 16.Ryberg M, Kristiansson E, Sjökvist E, Nilsson RH. 2009. An outlook on the fungal internal transcribed spacer sequences in GenBank and the introduction of a web-based tool for the exploration of fungal diversity. New Phytol. 181:471–477 [DOI] [PubMed] [Google Scholar]
- 17.Nilsson RH, Ryberg M, Kristiansson E, Abarenkov K, Larsson KH, Koljalg U. 2006. Taxonomic reliability of DNA sequences in public sequence databases: a fungal perspective. PLoS One 1:e59. 10.1371/journal.pone.0000059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nilsson RH, Bok G, Ryberg M, Kristiansson E, Hallenberg N. 2009. A software pipeline for processing and identification of fungal ITS sequences. Source Code Biol. Med. 4:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Natali SM, Schuur EAG, Trucco C, Pries CEH, Crummer KG, Lopez AFB. 2011. Effects of permafrost warming of air, soil and permafrost on carbon balance in Alaskan tundra. Global Change Biol. 17:1394–1407 [Google Scholar]
- 20.Schuur EAG, Crummer KG, Vogel JG, Mack MC. 2007. Plant species composition and productivity following permafrost thaw and thermokarst in Alaskan tundra. Ecosystems 10:280–292 [Google Scholar]
- 21.Schuur EAG, Vogel JG, Crummer KG, Lee H, Sickman JO, Osterkamp TE. 2009. The effect of permafrost thaw on old carbon release and net carbon exchange from tundra. Nature 459:556–559 [DOI] [PubMed] [Google Scholar]
- 22.Luo T, Wan S, Hui D, Wallace LL. 2001. Acclimatization of soil respiration to warming in tallgrass prairie. Nature 413:622–625 [DOI] [PubMed] [Google Scholar]
- 23.Wan S, Luo Y, Wallace LL. 2002. Change in microclimate induced by experimental warming and clipping in tallgrass prairie. Glob. Change Biol. 8:754–768 [Google Scholar]
- 24.Wan S, Yuan T, Bowdish S, Wallace L, Russell SD, Luo Y. 2002. Response of an allergenic species, Ambrosia psilostachya (Asteraceae), to experimental warming and clipping: implications for public health. Am. J. Bot. 89:1843–1846 [DOI] [PubMed] [Google Scholar]
- 25.Zhang W, Parker KM, Luo Y, Wan S, Wallace LL, Hu S. 2005. Soil microbial responses to experiment warming and clipping in a tallgrass prairie. Glob. Change Biol. 11:266–277 [Google Scholar]
- 26.Zhou J, Wan S, Luo Y. 2007. Source components and interannual variability of soil CO2 efflux under experimental warming and clipping in a grassland ecosystem. Glob. Change Biol. 13:761–775 [Google Scholar]
- 27.Zhou J, Xue K, Xie J, Deng Y, Wu L, Cheng X, Fei S, Deng S, He Z, Van Nostrand JD, Luo Y. 2012. Microbial mediation of carbon-cycle feedbacks to climate warming. Nat. Climate Change 2:106–110 [Google Scholar]
- 28.Zhou JZ, Bruns MA, Tiedje JM. 1996. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 62:316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wright ES, Yilmaz LS, Noguera DR. 2012. DECIPHER, a search-based approach to chimera identification for 16S rRNA sequences. Appl. Environ. Microbiol. 78:717–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clarke KR. 1993. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 18:117–143 [Google Scholar]
- 32.Anderson MJ. 2001. A new method for non-parametric multivariate analysis of variance. Aust. Ecol. 26:32–46 [Google Scholar]
- 33.Clarke RK, Somerfield PJ, Gorley RN. 2008. Testing of null hypotheses in exploratory community analyses: similarity profiles and biota-environment linkage. J. Exp. Mar. Biol. Ecol. 366:57–69 [Google Scholar]
- 34.Warwick RM, Platt HM, Clarke KR, Agard J, Gobin J. 1990. Analysis of macrobenthic and meiobenthic community structure in relationship to pollution and disturbance in Hamilton Harbour, Bermuda. J. Exp. Mar. Biol. Ecol. 138:119–142 [Google Scholar]
- 35.Anderson MJ, Ellingsen KE, McArdle BH. 2006. Multivariate dispersion as a measure of beta diversity. Ecol. Lett. 9:683–693 [DOI] [PubMed] [Google Scholar]
- 36.Clarke KR, Gorley RN. 2006. PRIMER v6: user manual/tutorial. PRIMER-E, Plymouth, United Kingdom [Google Scholar]
- 37.O'Brien HE, Parrent JL, Jackson JA, Moncalvo JM, Vilfalys R. 2005. Fungal community analysis by large-scale sequencing of environmental samples. Appl. Environ. Microbiol. 71:5544–5550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu L, Ravnskov S, Larson J, Nicolaisen M. 2012. Linking fungal communities in roots, rhizosphere, and soil to the health status of Pisum sativum. FEMS Microbiol. Ecol. 82:736–745 [DOI] [PubMed] [Google Scholar]
- 39.Xu L, Ravnskov S, Larson J, Nicolaisen M. 2012. Soil fungal community structure along a soil health gradient in pea fields examined using deep amplicon sequencing. Soil Biol. Biochem. 46:26–32 [Google Scholar]
- 40.Gottel NR, Castro HF, Kerley M, Yang Z, Pelletier DA, Podar M, Karpinets T, Uberbacher E, Tuskan GA, Vilgalys R, Doktycz MJ, Schadt CW. 2011. Distinct microbial communities within the endosphere and rhizosphere of Populus deltoids roots across contrasting soil types. Appl. Environ. Microbiol. 77:5934–5944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orgiazzi A, Lumini E, Nilsson RH, Girlanda M, Vizzini A, Bonfante P, Bianciotto V. 2012. Unravelling soil fungal communities from different Mediterranean land-use backgrounds. PLoS One 7:e34847. 10.1371/journal.pone.0034847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGuire KL, Fierer N, Bateman C, Treseder KK, Turner BL. 2012. Fungal community composition in neotropical rain forests: the influence of tree diversity and precipitation. Microb. Ecol. 63:804–812 [DOI] [PubMed] [Google Scholar]
- 43.Schmidt SK, Naff CS, Lynch RC. 2012. Fungal communities at the edge: ecological lessons from high alpine fungi. Fungal Ecol. 5:443–452 [Google Scholar]
- 44.Gibbons SM, Caporaso JG, Pirrung M, Field D, Knight R, Gilbert JA. 2013. Evidence for a persistent microbial seed bank throughout the global ocean. Proc. Natl. Acad. Sci. U. S. A. 110:4651–4655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khidir HH, Eudy DM, Porras-Alfaro A, Herrea J, Natvig DO, Sinsabaugh RL. 2010. A general suite of fungal endophytes dominate the roots of two dominant grasses in a semiarid grassland. J. Arid Environ. 74:35–42 [Google Scholar]
- 46.Porras-Alfaro A, Herrera J, Sinsabaugh RL, Odenback KJ, Lowrey T, Natvig DO. 2008. Novel root fungal consortium associated with a dominant desert grass. Environ. Microbiol. 74:2805–2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aime MC, Phillips-Mora W. 2005. The causal agents of witches' broom and frosty pod rot of cacao (chocolate, Theobroma cacao) form a new lineage of Marasmiaceae. Mycologia 97:1012–1022 [DOI] [PubMed] [Google Scholar]
- 48.Calvert EL, Muskett AE. 1944. Blind-seed disease of rye-grass. Nature 153:287–288 [Google Scholar]
- 49.Santos-González JC, Finlay RD, Tehler A. 2007. Seasonal dynamics of arbuscular mycorrhizal fungal communities in roots in a seminatural grassland. Appl. Environ. Microbiol. 73:5613–5623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Öpik M, Moora Liira M, Zobel JM. 2006. Composition of root-colonizing arbuscular mycorrhizal fungal communities in different ecosystems around the globe. J. Ecol. 94:778–790 [Google Scholar]
- 51.Malloch D, Cain RF. 1970. The genus Arachnomyces. Can. J. Bot. 48:839–845 [Google Scholar]
- 52.Gibas CF, Sigler L, Summerbell RC, Hofstader SL, Gupta AK. 2002. Arachnomyces kanei (anamorph Onychocola kanei) sp. nov., from human nails. Med. Mycol. 40:573–580 [DOI] [PubMed] [Google Scholar]
- 53.Tedersoo L, May TW, Smite ME. 2010. Ectomycorrhizal lifestyle in fungi: global diversity, distribution, and evolution of phylogenetic lineages. Mycorrhiza 20:217–263 [DOI] [PubMed] [Google Scholar]
- 54.Deslippe JR, Hartmann M, Mohn WW, Simard SW. 2011. Long-term experimental manipulation of climate alters the ectomycorrhizal community of Betula nana in Arctic tundra. Glob. Change Biol. 17:1625–1636 [Google Scholar]
- 55.Wang Z, Binder M, Schoch GL, Johnston PR, Spatafora JW, Hibbett DS. 2006. Evolution of helotialean fungi (Leotiomycetes, Pezizomycotina): a nuclear rDNA phylogeny. Mol. Phylogenet. Evol. 41:295–312 [DOI] [PubMed] [Google Scholar]
- 56.Freeman KR, Martin AP, Karki D, Lynch RC, Mitter MS, Meyer AF, Longcore JE, Simmons DR, Schmidt SK. 2009. Evidence that chytrids dominate fungal communities in high-elevation soils. Proc. Natl. Acad. Sci. U. S. A. 106:18315–18320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Simmons DR, James TY, Meyer AF, Longcore JE. 2009. Lobulomycetales, a new order in the Chytridiomycota. Mycol. Res. 113:450–460 [DOI] [PubMed] [Google Scholar]
- 58.Anderson OR. 2012. The role of bacterial-based protist communities in aquatic and soil ecosystems and the carbon biogeochemical cycle, with emphasis on naked amoebae. Acta Protozool. 51:209–221 [Google Scholar]
- 59.Blackwell WH, Letcher PM, Powell MJ. 2004. Synopsis and systematic reconsideration of Karlingiomyces (Chytridiomycota). Mycotaxon 89:259–276 [Google Scholar]
- 60.Massee G. 1895. British fungus—flora, vol IV William Clowes and Sons, New York, NY [Google Scholar]
- 61.Wang Z, Johnston PR, Takamatsu S, Spatafora JW, Hibbett DS. 2006. Towards a phylogenetic classification of the Leotiomycetes based on rDNA data. Mycologia 98:1065–1075 [DOI] [PubMed] [Google Scholar]
- 62.Baral H-O, de Sloover JR, Huhtinen S, Laukka T, Stenroos S. 2009. An emendation of the genus Hyaloscypha to include Fuscoscypha (Hyaloscyphaceae, Helotiales, Ascomycotina). Karstenia 49:1–17 [Google Scholar]
- 63.Geml J, Laursen GA, Herriott IC, McFarland JM, Booth MG, Lennon N, Nusbaum HC, Taylor DL. 2010. Phylogenetic and ecological analyses of soil and sporocarp DNA sequences reveal high diversity and strong habitat partitioning in the boreal ectomycorrhizal genus Russula (Russulales; Basidiomycota). New Phytol. 187:494–507 [DOI] [PubMed] [Google Scholar]
- 64.Clemmensen KE, Bahr A, Ovaskainen O, Dahlberg A, Ekblad A, Wallander H, Stenlid J, Finlay RD, Wardle DA, Lindahl BD. 2013. Roots and associated fungi drive carbon sequestration in boreal forest. Science 339:1615–1618 [DOI] [PubMed] [Google Scholar]
- 65.Weber CF, Vilgalys R, Kuske CR. 2013. Changes in fungal community composition in response to elevated atmospheric CO2 and nitrogen fertilization varies with soil horizon. Front. Microbiol. 4:78. 10.3389/fmicb.2013.00078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kotiaho M, Fritze H, Merilä P, Tuomivirta T, Väliranta M, Korhola A, Karofeld E, Tuittila E-S. 2012. Actinobacteria community structure in the peat profile of boreal bogs follows a variation in the microtopographical gradient similar to vegetation. Plant Soil 10.1007/s11104-012-1546-3. [DOI] [Google Scholar]
- 67.Staddon PL, Thompson K, Jakobsen I, Grime JP, Askew AP, Fitter AH. 2003. Mycorrhizal fungal abundance is affected by long-term climatic manipulations in the field. Glob. Change Biol. 9:186–194 [Google Scholar]
- 68.Clemmensen KE, Michelsen A, Jonasson S, Shaver GR. 2006. Increased ectomycorrhizal fungal abundance after long-term fertilization and warming of two Arctic tundra ecosystems. New Phytol. 171:391–404 [DOI] [PubMed] [Google Scholar]
- 69.Zhou JZ, Wu LY, Deng Y, Zhi XY, Jiang YH, Tu QC, Xie JP, Van Nostrand JD, He ZH, Yang YF. 2011. Reproducibility and quantitation of amplicon sequencing-based detection. ISME J. 5:1303–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sherry RA, Weng E, Arnone JA, III, Johnson DW, Schimel DS, Verburg PS, Wallace LL, Luo Y. 2008. Lagged effects of experimental warming and doubled precipitation on annual and seasonal aboveground biomass production in a tallgrass prairie. Glob. Change Biol. 14:2923–2936 [Google Scholar]
- 71.Sheik CS, Beasley WH, Elshahed MS, Zhou X, Luo Y, Krunholz LR. 2011. Effect of warming and drought on grassland microbial communities. ISME J. 5:1692–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Walker JF, Miller OK, Jr, Horton JL. 2008. Seasonal dynamics of ectomycorrhizal fungus assemblages on oak seedlings in the southeastern Appalachian Mountains. Mycorrhiza 18:123–132 [DOI] [PubMed] [Google Scholar]
- 73.Jumpponen A, Jones KL, Mattox JD, Yaege C. 2010. Massively parallel 454-sequencing of fungal communities in Quercus spp. ectomycorrhizas indicates seasonal dynamics in urban and rural sites. Mol. Ecol. 19:S41–S53 [DOI] [PubMed] [Google Scholar]
- 74.Dumbrell AJ, Ashton PD, Aziz N, Feng G, Nelson M, Dytham C, Fitter AH, Helgason T. 2011. Distinct seasonal assemblages of arbuscular mycorrhizal fungi revealed by massively parallel pyrosequencing. New Phytol. 190:794–804 [DOI] [PubMed] [Google Scholar]
- 75.Davey ML, Heegaard E, Halvorsen R, Ohlson M, Kauserud H. 2012. Seasonal trends in the biomass and structure of bryophyte associated fungal communities explored by 454 pyrosequencing. New Phytol. 195:844–856 [DOI] [PubMed] [Google Scholar]
- 76.Schmit JP, Mueller GM. 2007. An estimate of the lower limit of global fungal diversity. Biodivers. Conserv. 16:99–111 [Google Scholar]
- 77.Kirk PM, Cannon PF, Minter DW, Stalpers JA. 2008. Dictionary of the fungi, 10th ed. CAB International, Wallingford, United Kingdom [Google Scholar]
- 78.Hawksworth DL. 2001. The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycol. Res. 105:1422–1432 [Google Scholar]
- 79.Vilgays R. 2003. Taxonomic misidentification in public DNA databases. New Phytol. 160:4–5 [DOI] [PubMed] [Google Scholar]
- 80.Nilsson RH, Kristiansson E, Ryberg M, Hallenberg N, Larsson KH. 2008. Intraspecific ITS variability in the kingdom Fungi as expressed in the international sequence database and its implications for molecular species identification. Evol. Bioinform. 4:193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hibbett DS, Thorn RG. 2001. Basidiomycota: Homobasidiomycetes, p 121–128 In McLaughlin DJ, McLaughlin EG, Lemke PA. (ed), The Mycota: systematics and evolution, vol 7 Springer-Verlag, Berlin, Germany [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.