

Abstract
Bioluminescence in Vibrio fischeri ES114 is activated by autoinducer pheromones, and this regulation serves as a model for bacterial cell-cell signaling. As in other bacteria, pheromone concentration increases with cell density; however, pheromone synthesis and perception are also modulated in response to environmental stimuli. Previous studies suggested that expression of the pheromone-dependent bioluminescence activator LuxR is regulated in response to glucose by cyclic AMP (cAMP) receptor protein (CRP) (P. V. Dunlap and E. P. Greenberg, J. Bacteriol. 164:45–50, 1985; P. V. Dunlap and E. P. Greenberg, J. Bacteriol. 170:4040–4046, 1988; P. V. Dunlap, J. Bacteriol. 171:1199–1202, 1989; and W. F. Friedrich and E. P. Greenberg, Arch. Microbiol. 134:87–91, 1983). Consistent with this model, we found that bioluminescence in V. fischeri ES114 is modulated by glucose and stimulated by cAMP. In addition, a Δcrp mutant was ∼100-fold dimmer than ES114 and did not increase luminescence in response to added cAMP, even though cells lacking crp were still metabolically capable of producing luminescence. We further discovered that CRP regulates not only luxR but also the alternative pheromone synthase gene ainS. We found that His-tagged V. fischeri CRP could bind sequences upstream of both luxR and ainS, supporting bioinformatic predictions of direct regulation at both promoters. Luminescence increased in response to cAMP if either the ainS or luxR system was under native regulation, suggesting cAMP-CRP significantly increases luminescence through both systems. Finally, using transcriptional reporters in transgenic Escherichia coli, we elucidated two additional regulatory connections. First, LuxR-independent basal transcription of the luxI promoter was enhanced by CRP. Second, the effect of CRP on the ainS promoter depended on whether the V. fischeri regulatory gene litR was also introduced. These results suggest an integral role for CRP in pheromone signaling that goes beyond sensing cell density.
INTRODUCTION
The observation that pheromones govern bioluminescence in Vibrio fischeri was a fundamental discovery that shed light on bacterial cell-cell communication. It is now appreciated that diverse bacteria similarly utilize pheromones to regulate numerous processes (1); however, the study of bioluminescence and its regulation in V. fischeri still serves as a powerful model, enabling researchers to explore the mechanisms by which pheromone signals and their regulatory circuits are used by bacteria. In particular, strain ES114 has become a widely used experimental model for V. fischeri, because this isolate's bioluminescent symbiosis with the Hawaiian bobtail squid, Euprymna scolopes, can be reconstituted in the laboratory.
The genes primarily responsible for bioluminescence in V. fischeri are separated into two divergent transcripts composed of luxR and luxICDABEG (2–4). LuxI and LuxR underpin pheromone-mediated regulation of bioluminescence. LuxI encodes a pheromone synthase that generates N-3-oxo-hexanoyl-homoserine lactone (3-oxo-C6-HSL), which is also referred to as an autoinducer (5). As the bacterial population density increases, the membrane-permeable 3-oxo-C6-HSL accumulates, and at a threshold concentration it binds to the transcriptional regulator LuxR (2, 6, 7). This LuxR-HSL complex binds the lux box located within the intergenic region between luxR and luxI to activate transcription of luxICDABEG.
Two additional autoinducer pheromones, N-octanoyl-HSL (C8-HSL), generated by AinS, and AI-2, generated by LuxS, also regulate bioluminescence (8, 9). C8-HSL and AI-2 are thought to function through distinct receptors that both act via LuxU, LuxO, Hfq, and the small RNA Qrr to increase levels of LitR, which then activates luxR expression (10–13). C8-HSL can also activate LuxR directly, although it is a weaker activator than 3-oxo-C6-HSL (11).
Although pheromone-mediated regulation such as that controlling bioluminescence is frequently framed in terms of cell density-dependent quorum sensing, environmentally responsive regulators often govern these systems. As a result, a high cell density may be necessary but not sufficient to achieve stimulatory pheromone levels. For example, symbiotic V. fischeri ES114 cells are ∼1,000-fold brighter and produce more 3-oxo-C6-HSL than cultured cells grown to similar density (14, 15), indicating that environmental conditions are important in luxICDABEG expression in the squid. Several studies have suggested a role for the environmentally responsive cyclic AMP (cAMP) receptor protein (CRP) in regulating V. fischeri bioluminescence (16–18). CRP was originally identified in Escherichia coli for its role in catabolite repression (19, 20) and has since been shown to activate transcription from numerous promoters (21–23). In E. coli, when glucose concentrations are low, adenylate cyclase (Cya) activity and intracellular cAMP levels are relatively high, and this effector molecule associates with CRP (24). Together the cAMP-CRP complex binds target DNA and promotes transcription of many genes, particularly those involved in growth on substrates other than glucose (21, 24, 25).
Although previous research supports a link between catabolite repression and pheromone-mediated regulation of luminescence, the exact relationship and mechanism have remained unclear, particularly in V. fischeri ES114. In transgenic E. coli cells carrying the lux operon from strain MJ-1, CRP and cAMP stimulate the induction of luminescence, while the addition of glucose causes a 1- to 2-h delay in the onset of luciferase activity (16). Analyses of transcriptional regulators further suggested that the cAMP-CRP complex activates transcription of luxR (16, 17), and CRP from E. coli showed binding to DNA upstream of luxR (26). Studies of V. fischeri have suggested strain-specific differences. Luminescence in strain MJ-1 is decreased in the presence of glucose (27); however, it was reported that in strain ES114 glucose had no effect (14). In both strains the addition of cAMP increased light production (14, 27). Furthermore, undefined cya- and crp-like mutants of MJ-1 produce low levels of luminescence, and in the cya-like mutant luminescence was restored by supplying cAMP exogenously (18).
In this study, we exploited genomic and genetic advances in understanding V. fischeri ES114 to examine the underpinnings of glucose- or cAMP-mediated effects on luminescence and to explore the regulatory role of CRP. Our results suggest a multilayered integration of carbon metabolism and pheromone-mediated regulation.
MATERIALS AND METHODS
Bacteria and media.
Bacterial strains used in this study are listed in Table 1. V. fischeri ES114 was the wild-type strain used throughout (14). Plasmids were transformed into E. coli strain DH5α (28) or DH5αλpir (29). E. coli was grown in LB medium (30) or brain heart infusion medium (BHI), and V. fischeri was grown in LBS medium (31) or SWTO medium (32). Solid media were prepared with 15 mg ml−1 agar for plating. For selection of E. coli, ampicillin (Amp), chloramphenicol (Cam), kanamycin (Kan), and tetracycline (Tet) were added to LB at final concentrations of 100, 20, 40, and 12 μg ml−1, respectively, and 150 μg ml−1 erythromycin (Erm) was added to BHI. For selection of V. fischeri on LBS, Cam, Erm, and Kan were used at concentrations of 2, 5, and 100 μg ml−1, respectively. Where specified, glucose, cyclic AMP (cAMP), and/or C8-HSL were added to cultures at final concentrations of 20 mM, 10 mM, and 250 nM, respectively.
Table 1.
Bacterial strains, plasmids, and oligonucleotides used in this study
| Strain, plasmid, or oligonucleotide | Relevant characteristic(s) or sequencea | Source or reference |
|---|---|---|
| E. coli | ||
| DH5α | F− φ80dlacZΔM15 Δ(lacZYA-argF)U169 deoR supE44 hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | 28 |
| DH5αλpir | DH5α lysogenized with λpir | 29 |
| M182 | Δ(lacIPOZY)X74 galK galU rpsL150 (strA) mel | 43 |
| M182 Δcrp | M182 Δcrp | 42 |
| UQ3811 | K12 Δ(lacIPOZY), crp::camR, ilv::Tn10 cya | 40 |
| V. fischeri | ||
| ES114 | Wild-type isolate from E. scolopes | 14 |
| CL53 | ES114 ΔluxR::ermR | 9 |
| DC1 | ES114 Δcrp lacIq PA1/34-luxCDABEG | This study |
| DC22 | C8-HSL bioreporter; ES114 ΔainS ΔluxR-luxI, mutant luxR (MJ1-T33A, R67M, S116A, M135I), PluxI-luxCDABEG | This study |
| JB22 | ES114 lacIq PA1/34-luxCDABEG | 38 |
| JB24 | ES114 Δcrp | 32 |
| NL60 | ES114 ΔainS | This study |
| NL62 | ES114 ΔluxR::ermR, ΔainS | This study |
| Plasmidsb | ||
| pCL149 | ΔluxR::ermR allele; ColE1, oriTRP4, camR | 9 |
| pCR-BluntII-TOPO | PCR product cloning vector; ColE1, kanR | Invitrogen |
| pDCRP | E. coli crp in pBR322 derivative; ColEI, ampR | 34 |
| pDCRP TA158 | E. coli crp variant TA158 in pBR322 derivative; ColEI, ampR | 34 |
| pDCRP HL159 | E. coli crp variant HL159 in pBR322 derivative; ColEI, ampR | 44 |
| pDCRP HY19 | E. coli crp variant HY19 in pBR322 derivative; ColEI, ampR | 45 |
| pDCRP KE101 | E. coli crp variant KE101 in pBR322 derivative; ColEI, ampR | 45 |
| pDU9 | Δcrp in pBR322 derivative; ColEI, ampR | 35 |
| pEVS122 | Suicide vector; R6Kγ, oriTRP4, ermR | 29 |
| pEXT20 | Ptac expression vector; ColEI, ampR | 39 |
| pJLB36 | PluxR-gfp reporter in pVSV33; pES213, R6Kγ, oriTRP4, kanR | 32 |
| pJLB113 | luxR in pCR-BluntII-TOPO; ColE1, kanR | This study |
| pJLB117 | V. fischeri Δcrp allele; ColE1, R6Kγ, oriTRP4, camR, kanR | 32 |
| pJLB123 | Porf1-luxR; pES213, R6Kγ, oriTRP4, kanR | 32 |
| pMTV1 | Pcon-gfp reporter in pVSV33; pES213, R6Kγ, oriTRP4, kanR | This study |
| pMTV2 | PainS-gfp reporter in pVSV33; pES213, R6Kγ, oriTRP4, kanR | This study |
| pNL12 | crp in pCR-BluntII-TOPO; ColE1, kanR | This study |
| pNL15 | Porf1-crp in pVSV105; pES213, R6Kγ, oriTRP4, camR | This study |
| pNL19 | Ptac-crp (His6 tag) in pEXT20; ColEI, ampR | This study |
| pNL31 | ainS upstream fragment in pCR-BluntII-TOPO; ColE1, kanR | 37 |
| pNL53 | ainS downstream fragment in pEVS122; R6Kγ, oriTRP4, ermR | This study |
| pNL56 | E. coli crp in pVSV105; pES213, R6Kγ, oriTRP4, camR | This study |
| pNL62 | ΔainS allele; ColEI, R6Kγ, oriV, oriTRP4, kanR, ermR | This study |
| pNL66 | 129–269 bp of luxR in pCR-BluntII-TOPO; ColE1, kanR | This study |
| pNL69 | V. fischeri crp in pDU9; ColEI, ampR | This study |
| pNL73 | PluxR-lacZ reporter in pRW50; oriV, tetR | This study |
| pNL76 | PainS-lacZ reporter in pRW50; oriV, tetR | This study |
| pNL81 | 98–260 bp of lpxA in pCR-BluntII-TOPO; ColE1, kanR | This study |
| pNL82 | PluxI-lacZ reporter in pRW50; oriV, tetR | This study |
| pNL84 | PluxI-lacZ reporter in pRW50; oriV, tetR, luxR | This study |
| pNL85 | litR in pCR-BluntII-TOPO; ColE1, kanR | This study |
| pNL86 | PainS-lacZ reporter in pRW50; oriV, tetR, litR | This study |
| pNL90 | 91–244 bp of ainS in pCR-BluntII-TOPO; ColE1, kanR | This study |
| pRW50 | Promoterless lacZYA; oriV, tetR | 41 |
| pVSV33 | Promoterless camR-gfp; pES213, R6Kγ, oriTRP4, kanR | 33 |
| pVSV105 | Shuttle vector; pES213, R6Kγ, oriTRP4, camR, lacZα | 33 |
| Oligonucleotidesc | ||
| DMC2 | GGC GGT ACC AGA ACC AAG ACC TGC TCG TGC TAA | This study |
| JBCRP4 | TGC AGG GCA ACG TTG TAC TTG TGC | 32 |
| JBCRP5 | GCA TCC TCC AGC AGC CAT TAA GAC C | 32 |
| JBLUXR1 | GAA GGA GAT ATA CAT ATG AAC ATT AAA AAT ATA AAT GC | 32 |
| JBLUXR2 | CGC CAA GAT TTT ATG GAA ATG TAT GAG | 32 |
| JBPromoter1 | CTA GTT GAC ATG ATA GAA GCA CTC TAC TAT ATT | This study |
| JBPromoter2 | CTA GAA TAT AGT AGA GTG CTT CTA TCA TGT CAA | This study |
| Lux1 | GGG GTC TAG AGC TTT AGA AAT ACT TT | 38 |
| Lux2 | GGA TCC GCT AGG GCG GCC GCC TAA CT | 38 |
| pr_MTV1 | CGC GGA ATT CAG GAA CTA TAA ACT ATG GTT CTA GGT AAA CCT CAA ACA | This study |
| pr_MTV2d | GCG CAA GCT TAT TAA TGG TGA TGG TGA TGG TGA TGA TAA CGA TGA CC GTA AAC TAC AAT | This study |
| pr_NL54 | CGC TAT TAT CTA TCC TCA CTC AAT AAT TAA ACC TGA TG | This study |
| pr_NL55 | GAA TGA TGG GAC TTA GAG TAA TCG ACT ACA GGG TC | This study |
| pr_NL58 | GGC AGG CCT CAT CAG TTG TTG AAG TAA ATT AAA ATT CTG CG | This study |
| pr_NL78.2 | GCG CCC TAG GTG ACT TTT ATA TAA ATG TTA ACT ACT TTA C | This study |
| pr_NL79 | CCT CGA CT AAT CGA ATA GAT ATA GAA CTT TTA T | This study |
| pr_NL80 | GCG CGA ATT CCA TGA TCA TAA CAA ACT GAT GCA TTA CGG | This study |
| pr_NL81 | GCG CAA GCT TGT TCA TTT TTT GTT CAC CTA GCT TAT TGT TA | This study |
| pr_NL86 | GCG CGA ATT CAG AAC CAA GAC CTG CTC GTG CTA A | This study |
| pr_NL87 | GCG CAA GCT TGA TCA GTT GTT GAA GTA AAT TAA AAT TCT GCG | This study |
| pr_NL94 | CTG GTA ATG TAA CGA TTG GCG AAG GTA CGG AAG TG | This study |
| pr_NL95 | CCG ATA ACA ACT GTT GTT GCT TCA CCA CCA TAT TTC | This study |
| pr_NL96 | GCG CGA ATT CCA CCA ATT TGG AGG TTT GGT GAT ATC GC | This study |
| pr_NL97 | GCG CAA GCT TTA TCA TTA CAG CCA TGC AAC CTC TCT TAT TT | This study |
| pr_NL100 | GCG CCC TAG GGG TTT CCC GAC TGG AAA GCG GGC | This study |
| pr_NL101 | GCG CTC TAG AGG TTT CCC GAC TGG AAA GCG GGC | This study |
| pr_NL102 | CTG TTG AAT ATA TAA GCA GCC TTA TGT AAA GTT ATT GAG | This study |
| pr_NL103 | TCA GCG GTT TTT TAT TAT TAT CAT TTC ATG AAT CCT G | This study |
| pr_NL108 | GGC GGA ACG ATT GGA AAT TTG GAA TAC TTA TTT TCA ACA TC | This study |
| pr_NL109 | CAG TAC TGC ATT TCA AAA GAC AAC CAA AAA CTT TGA TAG CC | This study |
Drug resistance gene designations: ampR, ampicillin resistance (bla); camR, chloramphenicol resistance (cat); ermR, erythromycin resistance; kanR, kanamycin resistance (aph); and tetR, tetracycline resistance (tetM).
All alleles cloned in this study are from V. fischeri strain ES114. The replication origins of each vector are listed as R6Kγ, ColE1, oriV, and/or pES213. Plasmids based on pES213 are stable and do not require antibiotic selection for maintenance (33).
All oligonucleotides are shown 5′ to 3′.
Underlined bases indicate the C-terminal His6 tag used in protein purification and a newly created stop codon.
Molecular genetic techniques and analyses.
Plasmids (Table 1) were constructed using standard molecular techniques. DNA ligase and restriction enzymes were obtained from New England BioLabs (Beverly, MA). Oligonucleotides used for PCR and cloning (Table 1) were synthesized by Integrated DNA Technologies (Coralville, IA). PCR products were generated using KOD HiFi DNA polymerase (Novagen, Madison, WI) or Phusion high-fidelity DNA polymerase (New England BioLabs, Beverly, MA) with an iCycler (Bio-Rad Laboratories, Hercules, CA). Plasmids used in cloning were purified with the GenElute plasmid miniprep kit (Sigma-Aldrich, Inc., St. Louis, MO), and DNA from PCR, digestion, and ligation reactions was purified with the DNA clean and concentrator-5 kit (Zymo Research, Orange, CA). When specified, PCR products were cloned into the pCR-BluntII-TOPO vector using a ZeroBlunt-TOPO PCR cloning kit (Invitrogen, Carlsbad, CA), and white colonies were identified on plates containing 5-bromo-4-chloro-3-indolyl-β-d-galactoside at a final concentration of 60 μg ml−1. All cloned PCR products were sequenced at the University of Michigan DNA Sequencing Core Facility, and sequences were analyzed using Sequencher 4.1.2 (Gene Codes Corp., Ann Arbor, MI).
To construct a constitutively expressed gfp gene as a control for promoter-gfp reporters, oligonucleotides JBPromoter1 and JBPromoter2 were designed to anneal together, forming an E. coli consensus promoter and single-stranded overhangs compatible with SpeI-digested DNA. The oligonucleotides were combined in a 1:1 ratio, heated to 100°C, and then annealed by allowing the mix to cool slowly at room temperature. This fragment was then ligated into SpeI-digested pVSV33 (33) to generate pMTV1. To construct the PainS-gfp reporter, the 427-bp region upstream of ainS was PCR amplified using primers DMC2 and pr_NL58. The PCR product was digested with KpnI and StuI and ligated into KpnI- and StuI-digested pVSV33 (33) to generate pMTV2. Plasmids pNL56 and pNL69 (Table 1) were generated for interspecies complementation of crp mutants between E. coli and V. fischeri. To generate pNL56, E. coli crp from SalI- and PvuI-digested pDCRP (34) was ligated into SalI- and PvuI-digested pVSV105 (33). To generate pNL69, V. fischeri ES114 crp from ClaI- and XhoI-digested pNL15 was ligated into ClaI- and SalI-digested pDU9 (35).
Mutant strain construction.
Plasmids carrying mutant alleles were transferred from E. coli into V. fischeri by triparental matings using conjugative helper strain CC118λpir pEVS104 (36). Recombinational insertion and marker exchange were identified by screening for antibiotic resistance, and putative mutants were tested by PCR. To generate a ΔainS allele, the 1,833-bp region downstream of ES114 ainS was PCR amplified using primers pr_NL78.2 and pr_NL79. This product was cloned into SmaI-digested pEVS122 (29) to generate pNL53. Plasmid pNL31, which contains the region upstream of ainS (37), and pNL53 were linearized with AvrII and ligated together to generate pNL62, which contains the upstream and downstream sequences fused at the ΔainS allele, with the ainS start and stop codons separated by the 6-bp AvrII recognition sequence. The ΔainS allele from pNL62 was crossed into ES114 and CL53 (9) to generate strains NL60 and NL62, respectively, and the deletion of ainS was confirmed by PCR. To generate DC1, the Δcrp allele on pJLB117 (32) was crossed into the genome of JB22 (38) and allelic exchange was confirmed by PCR and by isopropyl-β-d-thiogalactopyranoside (IPTG) inducibility of luminescence in the resulting strain.
Luminescence and fluorescence measurements.
Overnight cultures of V. fischeri were diluted 1:1,000 in 20 or 50 ml of SWTO in 125- or 250-ml flasks, respectively, and then incubated with shaking (200 rpm) at 24°C. At regular intervals, 500-μl samples were removed to measure optical density at 595 nm (OD595) with a BioPhotometer (Brinkman Instruments, Westbury, NY). Relative luminescence was measured with a Glomax TD-20/20 luminometer (Promega, Madison, WI) immediately following shaking to aerate the sample (38). Specific luminescence was calculated as the luminescence per OD595 unit. Fluorescence expressed from reporter plasmids carrying gfp was measured with a TD-700 fluorometer (Turner Designs, Sunnyvale, CA) using excitation and emission filters of 486 and >510 nm, respectively. Fluorescence of strains carrying the promoterless vector pVSV33 (33) was subtracted as the background. Specific fluorescence was calculated as the fluorescence per unit of OD595 from cells growing in SWTO supplemented with 20 mM glucose, and the mean specific fluorescence is reported for cultures at an OD595 of ∼2.0.
Quantitative PCR and transcript analysis.
Overnight cultures of V. fischeri ES114 or JB24 were diluted 1:1,000 in 20 ml of SWTO in 125-ml flasks and then incubated with shaking (200 rpm) at 24°C. At an OD595 of ∼0.5, 5-ml samples were removed and a 1/5 volume 5% acidic phenol and 95% ethanol (vol/vol) was added. Samples were incubated on ice for 30 min, and RNA was isolated using the Stratagene absolutely RNA miniprep kit (Agilent Technologies, La Jolla, CA). Isolated RNA was further treated with Ambion Turbo DNase (Applied Biosystems, Foster City, CA). cDNA was generated from 1 μg of DNase-treated RNA using a Superscript VILO cDNA synthesis kit (Invitrogen, Carlsbad, CA). Reverse transcription-PCRs (RT-PCRs) were completed with iQ SYBR green Supermix (Bio-Rad, Hercules, CA), and 10 ng cDNA was analyzed using the MyiQ real-time PCR detection system (Bio-Rad, Hercules, CA). The luxR cDNA was amplified using primers pr_NL54 and pr_NL55, and ainS cDNA was amplified using primers pr_NL108 and pr_NL109. The lpxA cDNA was amplified using primers pr_NL94 and pr_NL95. All RNA and cDNA concentrations were determined with a Synergy 2 plate reader and Gen5 software (BioTek, Winooski, VT).
To generate standard curves, the primer sets described above (pr_NL54 and pr_NL55, pr_NL108 and pr_NL109, and pr_NL94 and pr_NL95) were used to PCR amplify luxR, ainS, and lpxA, respectively, using ES114 DNA as a template. The blunt luxR, ainS, and lpxA PCR products were ligated into the pCR-BluntII-TOPO vector to generate pNL66, pNL90, and pNL81, respectively. These plasmids were utilized as standards in real-time PCR analyses. The efficiency of each primer set was calculated to be ∼98, ∼96, and ∼95% using the standard curves generated from serial dilutions of pNL66, pNL90, and pNL89, respectively. To ensure that chromosomal DNA contamination was not affecting the real-time analyses, control samples lacking reverse transcriptase were also examined using each primer set, and no-template controls were included in each assay.
Purification of V. fischeri CRP.
For CRP purification, V. fischeri crp was PCR amplified using primers JBCRP4 and JBCRP5, and the blunt product was cloned into pCR-BluntII-TOPO to generate pNL12. The wild-type crp allele from pNL12 was then used as a template for PCR with primers pr_MTV1 and pr_MTV2. The latter primer mutated the crp stop codon, added a C-terminal His6 tag, and created a new stop codon (Table 1). The PCR product was digested with HindIII and EcoRI and then ligated into HindIII- and EcoRI-digested pEXT20 expression vector (39) to generate pNL19.
V. fischeri crp was expressed from pNL19 in E. coli strain UQ3811 (40). Overnight cultures were diluted 1:100 and grown with shaking (200 rpm) in LB at 37°C to an OD595 of ∼0.5. At this OD595, CRP overexpression was induced by adding IPTG to a final concentration of 1 mM. Following induction, cultures were incubated for 5 h and then cells were pelleted by centrifugation. Cell pellets were resuspended in 50 mM Tris-HCl (pH 8.0), 500 mM KCl, and 1 mM EDTA and lysed using a French pressure apparatus. Cell debris was pelleted by centrifugation at 10,000 × g for 10 min. The cell lysate was loaded onto a Ni-MAC cartridge (Novagen, EMD Chemicals Inc., Gibbstown, NJ), the column was washed with 10 volumes of 50 mM Tris-HCl (pH 8.0), 500 mM KCl, and 50 mM imidazole, and then it was eluted with 50 mM Tris-HCl (pH 8.0), 500 mM KCl, and 250 mM imidazole in 1-ml fractions. To remove imidazole, elution fractions containing protein were dialyzed overnight in 50 mM Tris-HCl (pH 8.0), 100 mM KCl, and 1 mM EDTA.
Fluorescence polarization DNA binding assays.
DNA binding assays were completed using 5′-fluorescein-labeled oligonucleotides annealed to unlabeled complementary oligonucleotides, both synthesized by Sigma-Aldrich (St. Louis, MO). Oligonucleotides were resuspended to a final concentration of 1 mM and then combined in a 1:1 ratio, boiled for 5 min, and allowed to anneal by cooling slowly at room temperature. Specified concentrations of purified CRP were added to binding assays, which contained a final concentration of 50 mM Tris-HCl (pH 8.0), 100 mM KCl, 1 mM EDTA, 15 nM oligonucleotide probe, and 20 ng ml−1 salmon sperm DNA. Binding assay mixtures were incubated at room temperature for 15 min, and then samples were excited at 485 nm and emission measured at 528 nm using a Synergy 2 plate reader (BioTek) set to read the extent of fluorescence polarization. Each sample was assayed two additional times, after 25 and 35 min, and all three measurements were averaged to generate the anisotropy values reported. The data shown below reflect two biological replicates, each of which was assayed three times to determine a value.
Beta-galactosidase transcriptional reporter assays.
Beta-galactosidase transcriptional reporters were constructed using the promoterless lacZ plasmid pRW50 (41). The luxR promoter was amplified with primers pr_NL80 and pr_NL81, the ainS promoter was amplified with primers pr_NL86 and pr_NL87, and the luxI promoter was amplified with primers pr_NL96 and pr_NL97. The PCR products were digested with EcoRI and HindIII and then ligated into EcoRI- and HindIII-digested pRW50 to generate pNL73, pNL76, and pNL82, respectively.
Because these reporters were used in E. coli, which lacks specific key regulators found in V. fischeri, the complete luxR and litR genes were added to the PluxI-lacZ and PainS-lacZ reporter plasmids, respectively. To construct pNL84, luxR was amplified using primers JBLUXR1 and JBLUXR2, and then the blunt PCR product was ligated into the pCR-BluntII-TOPO vector, generating pJLB113. The vector-derived lacZ promoter and luxR insert from pJLB113 were amplified with primers pr_NL100 and pr_NL101, and the PCR product was digested with AvrII and ligated into AvrII-digested pNL76. To construct pNL86, litR was amplified using primers pr_NL102 and pr_NL103, and the blunt PCR product was ligated into the pCR-BluntII-TOPO vector, generating pNL85. Both litR and the vector-derived lacZ promoter upstream of it were amplified from pNL85 using primers pr_NL100 and pr_NL101, and the PCR product was digested with AvrII and ligated into AvrII-digested pNL76.
All reporter plasmids were cotransformed into E. coli strain Δcrp (42), which is a crp mutant of E. coli M182 (43), along with pDCRP (34), pDCRP-TA158 (34), pDCRP-HL159 (44), pDCRP-HY19 (45), pDCRP-KE101 (45), or pDU9 (35). Cotransformants were tested in beta-galactosidase assays. Overnight cultures of cotransformants were subcultured 1:100 into 20 ml of LB containing Amp and Tet in 125-ml flasks and then incubated with shaking (200 rpm) at 28°C. At an OD595 of ∼0.5 for strains with pNL73, pNL82, and pNL84 and ∼1.0 for strains with pNL76 and pNL86, 1-ml samples were removed, pelleted, and stored at −80°C. For beta-galactosidase assays, pelleted cells were resuspended in 1 ml of Z buffer and 0.5 ml of each resuspension was assayed as previously described (30).
C8-HSL bioassays.
To assess C8-HSL production by particular strains, overnight cultures were diluted 1:1,000 in 15 ml SWTO medium in 125-ml flasks and grown at 24°C with shaking (200 rpm) to an OD595 of 2.4 to 2.5. Cells were pelleted by centrifugation, and supernatants were filter sterilized before adding an equal volume of acidified ethyl acetate (1:1,000 glacial acetic acid:ethyl acetate) and incubating for 30 min on a rotary shaker at 55 rpm to extract acyl-homoserine lactones (AHLs). Three-ml samples of the organic phase were removed and added to sterile glass beakers, and the ethyl acetate was allowed to evaporate overnight. Extracts were dissolved in 3 ml SWTO, and 200-μl samples were added to alternating wells of a 96-well microplate. Wells were then inoculated with the C8-HSL bioreporter strain DC22 (Table 1), which will be described in greater detail elsewhere. It was engineered to lack the pheromone synthases encoded by luxI and ainS and to have expression of the luxCDABEG genes activated by a mutant LuxR derivative (LuxRMJ1 T33A, R67M, S116A, M135I) that is responsive to C8-HSL but not to 3-oxo-C6-HSL (46). C8-HSL standards were likewise prepared, and C8-HSL concentration was determined by measuring luminescence of DC22 using a Synergy 2 plate reader (BioTek).
Experimental repetitions and statistical analyses.
Each figure represents data from one experiment but is representative of at least three experimental repetitions. Means, standard errors, and significance in Student's t tests were calculated using Microsoft Excel.
RESULTS
Luminescence in ES114 is influenced by glucose.
To determine the effect of glucose on V. fischeri ES114 luminescence, we examined cultures grown in SWTO alone and with 20 mM glucose (Fig. 1A). We found that glucose decreased luminescence when the culture OD595 was below 2.0, with a maximum effect of ∼15-fold. Later, a brief peak of brighter luminescence was observed in the glucose-supplemented culture at an OD595 of ∼2.3. At this peak, during a brief but reproducible window in cell density, cultures grown with glucose were ∼3-fold brighter than cultures grown without glucose (Fig. 1A).
Fig 1.
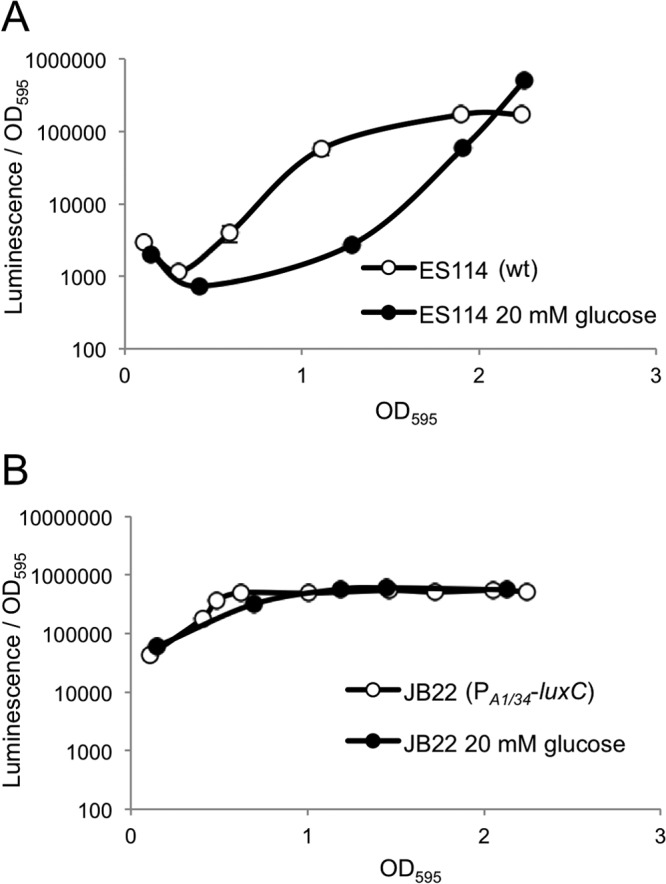
Effect of glucose on luminescence. Specific luminescence is shown for wild-type ES114 (A) or strain JB22 (lacIq-PA1/34-luxC) (B) grown in SWTO either without (open circles) or with (filled circles) 20 mM glucose added. Averages and standard errors were calculated from replicate flasks (n = 2). Many error bars are smaller than the circles used to mark the means and are difficult to visualize.
The effect of glucose on the luminescence of ES114 in Fig. 1A might be due to regulation of genes involved in light production, or growing on glucose might directly affect the luminescence output of cells, for example, simply by altering the availability of substrates for luciferase. To distinguish between these possibilities, we examined the luminescence of mutant JB22, which contains the constitutive promoter, lacIq-PA1/34, inserted between luxI and luxC, resulting in constitutive expression of luxCDABEG (38). In the lacIq-PA1/34-luxC background, glucose had no effect on luminescence below an OD595 of ∼2.5 (Fig. 1B). Because constitutive luxCDABEG expression eliminated glucose-mediated repression of luminescence, these data suggest that at these culture densities glucose affects ES114 luminescence by controlling gene expression rather than by altering the physiological capacity of cells to produce luminescence.
In both ES114 and JB22, luminescence declines rapidly at cell densities around an OD595 of 2.5 to 2.7, and the data shown in Fig. 1 precede this rapid decline in light production, which is observed roughly coinciding with a rapid drop in pH (data not shown), possibly owing to fermentation of the glucose.
Luminescence in ES114 is affected by cAMP and crp.
Previous work using transgenic E. coli carrying the lux genes from V. fischeri strain MJ-1 suggested that cAMP-CRP activated luxR transcription and luminescence (16, 17). In E. coli, cAMP levels are relatively low in the presence of glucose, but exogenous addition of 4 mM cAMP to lux-containing E. coli stimulated luminescence (16), an effect attributed to enough cAMP entering cells to activate CRP. Similarly, it was reported that addition of 10 mM cAMP stimulates luminescence in V. fischeri cultures (14, 18).
We likewise found that addition of cAMP to ES114 stimulated luminescence ∼10-fold (Fig. 2A). It was difficult to interpret the effects of glucose on luminescence in a V. fischeri Δcrp mutant, because this strain grew very poorly without glucose; however, we could test the effect of cAMP addition on both ES114 and the Δcrp mutant in media with added glucose. In glucose-containing media we found that the Δcrp mutant was both dimmer than the wild type and unresponsive to cAMP (Fig. 2B).
Fig 2.
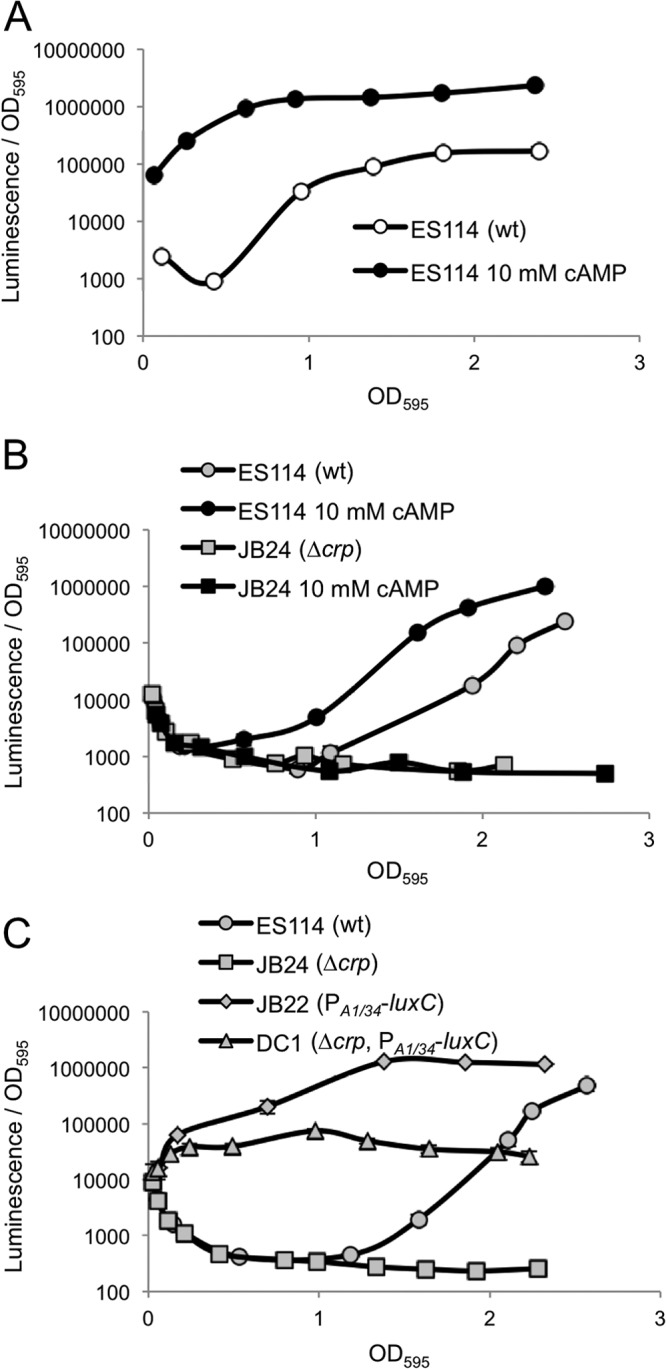
Effect of cAMP on luminescence. (A) Specific luminescence of ES114 (wild type) grown in SWTO without (open circles) or with (filled circles) 10 mM cAMP added. (B) Specific luminescence of ES114 and Δcrp mutant JB24 grown in SWTO containing 20 mM glucose without (gray circles) or with (filled circles) 10 mM cAMP. (C) Specific luminescence of ES114, JB22 (lacIq-PA1/34-luxC), Δcrp mutant JB24, and Δcrp lacIq-PA1/34-luxC mutant DC1 grown with 20 mM glucose. Averages and standard errors were calculated from replicate flasks (n = 2).
We considered the possibility that pleiotropic physiological effects associated with the Δcrp mutation underlie the low luminescence of this mutant. Therefore, we tested the effect of the Δcrp allele in the JB22 strain background with lux expressed from a constitutive nonnative PA1/34 promoter. We found that deleting crp led to an ∼15-fold decrease in luminescence in the constitutive lux background (Fig. 2C); however, this does not match the ∼1,000-fold decrease in luminescence seen when crp is deleted from ES114 (Fig. 2C). Moreover, the Δcrp constitutive lux strain DC1 is ∼100-fold brighter than the Δcrp mutant JB24. This phenotype of DC1 indicates that cells with the Δcrp mutation are metabolically capable of much brighter luminescence than that displayed by the Δcrp mutant JB24. Taken together, our data suggest that the attenuation of luminescence caused by the Δcrp allele in JB24 in large part reflects a limitation of lux gene expression with a smaller contribution arising from another, possibly metabolic, mechanism.
CRP influences luxR and ainS transcription.
To identify genes involved in luminescence that may be controlled by cAMP-CRP, we used Virtual Footprint to analyze the V. fischeri ES114 genome using that program's position weight matrix default setting for CRP, which is derived from E. coli K-12 CRP binding sites (47). As expected based on previous studies, a putative CRP binding site was identified upstream of luxR. More surprisingly, a potential CRP binding site was also identified upstream of ainS which encodes the autoinducer synthase responsible for generating C8-HSL (8). In fact, the putative CRP binding site upstream of ainS was a better match to the weighted consensus CRP binding site than the site in the lux intergenic region, as Virtual Footprint returned position weight matrix scores of 7.0 and 7.6 for putative CRP sites upstream of luxR and ainS, respectively. Both sites are similar matches to the core CRP binding consensus of 5′-TGTGA(N6)TCACA-3′ (see Fig. 5A).
Fig 5.
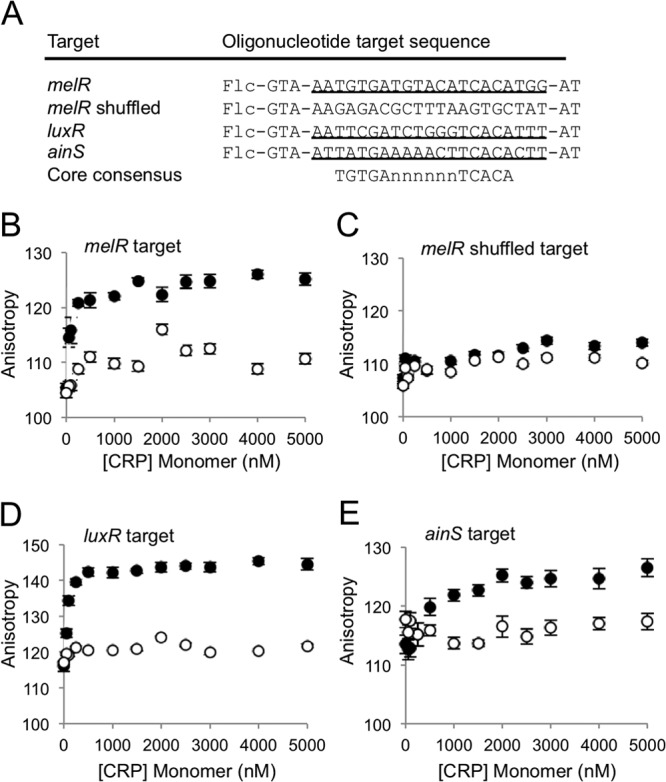
Fluorescence polarization analysis of CRP binding to the luxR and ainS promoter regions. (A) Fluorescein-labeled oligonucleotides utilized for binding analyses. Flc indicates the position of fluorescein modification at the 5′ end of the oligonucleotide target sequences. The melR sequence was previously shown to bind CRP (40) and is used in this study as a positive control. Target oligonucleotides are each 26 bp long, encompassing the putative CRP binding site, and each is flanked by 5′-GTA-target-AT-3′. The melR shuffled sequence was generated using the melR sequence and the shuffle DNA application from The Sequence Manipulation Suite (http://www.bioinformatics.org/sms/). The core, most heavily conserved portion of the CRP binding consensus, 5′-TGTGA(N6)TCACA, is shown for comparison. Fluorescence polarization curves without (open circles) or with (filled circles) 10 mM cAMP added are shown for the melR target (B), shuffled melR target (C), luxR target (D), and ainS target (E). Averages and standard errors were calculated from replicate samples (n = 2) as described in Materials and Methods.
We next examined the effect of CRP on the expression of the luxR and ainS promoters using PluxR-gfp and PainS-gfp promoter-reporters. Fluorescence expressed from each reporter was measured in the wild type and the Δcrp mutant (Fig. 3). The PluxR-gfp and PainS-gfp reporters yielded a statistically significant (P ≤ 0.05) ∼15- and ∼10-fold decrease in fluorescence in the Δcrp mutant, respectively. A constitutively active promoter-reporter with a consensus promoter driving gfp expression was also examined in the wild type and crp mutant to ensure that the Δcrp allele did not decrease fluorescence of all gfp reporters. Fluorescence of the Pcon-gfp reporter was actually significantly higher in the Δcrp mutant, suggesting that, if anything, the effects of crp on the PluxR- and PainS-gfp reporters underestimate the positive effects of crp on transcription of these genes.
Fig 3.

CRP regulation of luxR and ainS promoter activity. Specific fluorescence was generated from the reporters Pcon-gfp (pMTV1), PluxR-gfp (pJLB36), and PainS-gfp (pMTV2) carried within ES114 (gray bars) or crp mutant JB24 (black bars) grown in SWTO with 20 mM glucose. Fluorescence from each strain harboring the promoterless vector pVSV33 was subtracted as the background. Data represent the average specific fluorescence when the culture OD595 was ∼2.0. Averages and standard errors were calculated from replicate flasks (n = 2). An asterisk indicates a significant difference between fluorescence in reporters expressed from ES114 or JB24 cultures, as determined by a Student's t test (P ≤ 0.05).
To measure directly whether CRP controls the transcript levels of luxR and ainS, we utilized quantitative real-time PCR to determine the copy number of both luxR and ainS transcripts in V. fischeri ES114 and in the Δcrp mutant (Fig. 4A and B). The copy number of the luxR and ainS transcripts was decreased by ∼250- and ∼7-fold, respectively. To test whether all transcripts appear decreased in the Δcrp mutant, we also examined the lpxA transcript as a control. LpxA is involved in lipid A biosynthesis in Gram-negative bacteria and is an essential housekeeping gene (48) that is not part of the CRP regulon in E. coli (49). The transcript level of lpxA was equivalent in both the wild type and the Δcrp mutant (Fig. 4C). These results support the model that CRP regulates luxR and ainS expression.
Fig 4.
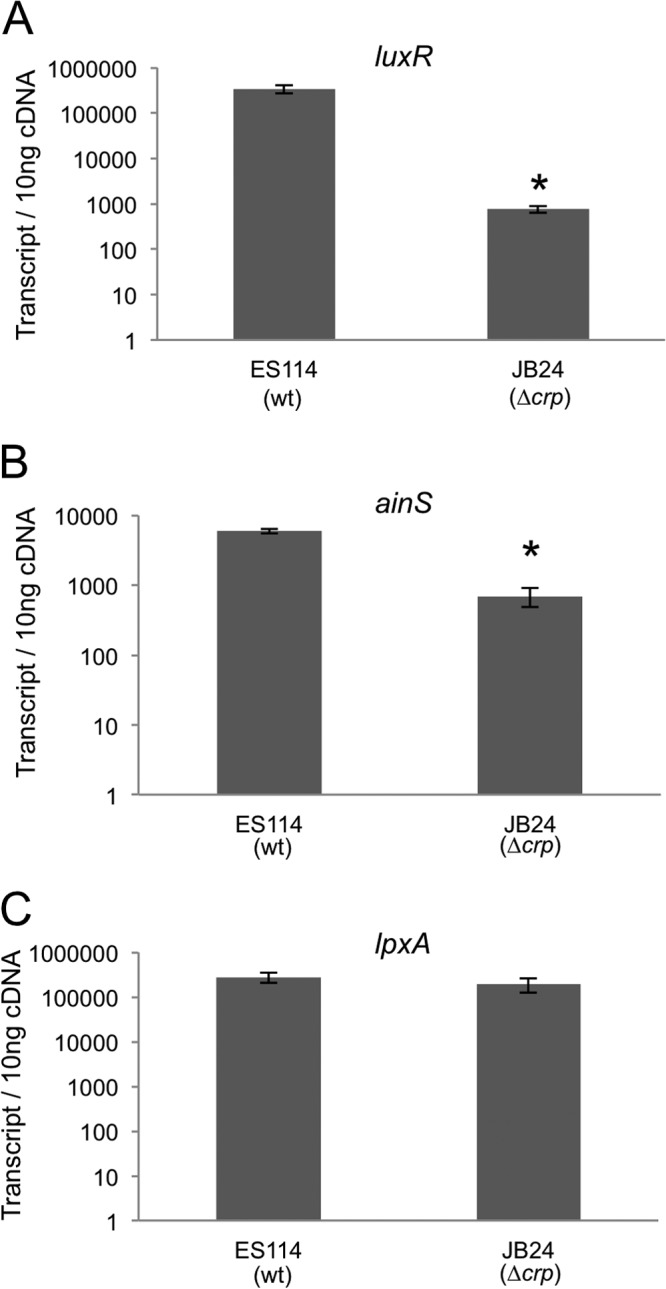
RT-PCR transcript analysis of luxR and ainS. Copy number of luxR (A), ainS (B), and lpxA (C) transcripts in 10 ng total cDNA generated from ES114 (wild type) or crp mutant JB24 RNA. Bars indicate standard errors (n = 3). An asterisk indicates a significant difference in transcript number between wild-type and crp mutant transcript copy numbers as determined by a Student's t test (P ≤ 0.0005).
CRP binds upstream of luxR and ainS.
Previous work showed that E. coli CRP can bind upstream of luxR from V. fischeri strain ATCC 7744 (26), and we sought to test whether this would also be true for CRP from V. fischeri binding the ES114 lux promoter and whether binding upstream of ainS could be detected. Using purified His-tagged V. fischeri CRP, we tested in vitro binding to 5′ fluorescein-labeled DNA fragments (Fig. 5A). The transcriptional start sites for luxR and ainS in V. fischeri remain uncertain, but the sequences upstream of these genes along with the target binding sequences are highlighted in Fig. S1 in the supplemental material. A melR promoter target region was previously shown to associate with CRP (40), and we used it in this study as a positive control (Fig. 5A). The melR shuffled target contains the same nucleotides as the melR target, but they were randomized, thereby serving as a negative control for binding. With increased anisotropy serving as a measure of target binding, we observed cAMP-dependent binding of CRP to the melR target and no binding to the melR shuffled target (Fig. 5B and C).
We next tested if CRP binds the predicted CRP target sequences upstream of luxR and ainS. As with melR, we observed a cAMP-dependent increase in anisotropy as CRP was added to both the luxR and ainS targets, indicating that CRP binds sequences found upstream of these genes (Fig. 5D and E). These results support the model that cAMP-CRP binds near the promoters of luxR and ainS.
Regulation of both luxR and ainS may contribute to cAMP activation of luminescence.
Although CRP binds upstream of both ainS and luxR, it remained possible that one or the other of these CRP sites was solely responsible for the effect of cAMP-CRP on luminescence in ES114. To explore whether regulation of the luxR promoter alone could account for the effect of cAMP-CRP on luminescence, we determined the effect of cAMP on luminescence of a luxR mutant (CL53) (9) complemented by luxR constitutively expressed in trans (pJLB123) (32) (Fig. 6). By using this construct, we eliminated expression of luxR from the CRP-regulated native promoter and maintained constitutive expression of luxR. When 10 mM cAMP was added to this strain, luminescence nonetheless increased ∼15-fold (Fig. 6). Thus, the cAMP-mediated induction of luminescence is not due solely to CRP regulation of the native luxR promoter.
Fig 6.
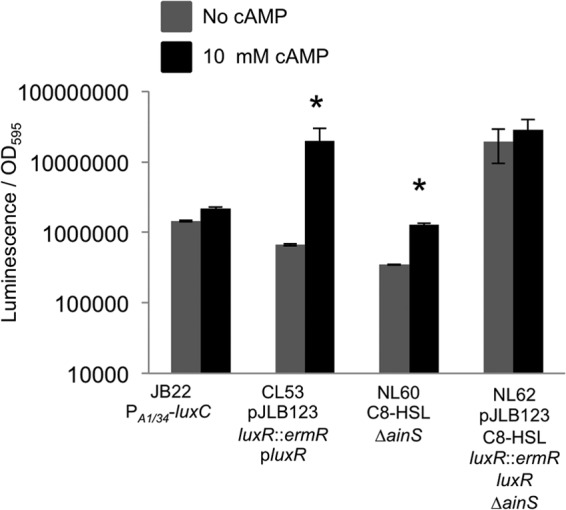
Regulation of both ainS and luxR contributes to the effect of cAMP on luminescence. Maximal specific luminescence is shown for JB22 (lacIq-PA1/34-luxC), a luxR mutant complemented with constitutive luxR in trans (CL53 pJLB123), an ainS mutant biochemically complemented with 250 mM C8-HSL (NL60 C8-HSL), and a luxR ainS double mutant complemented with luxR in trans and 250 mM C8-HSL (NL62 pJLB123 C8-HSL). Cultures were grown in SWTO alone (gray bars) or with 10 mM cAMP added (black bars). Standard errors were calculated from two replicates. An asterisk indicates significant differences between cultures grown without and with cAMP as determined by a Student's t test (P ≤ 0.05).
We also examined the luminescence of an ainS mutant supplemented with C8-HSL (NL60) alone or with 10 mM cAMP (Fig. 6). With constant C8-HSL, the ainS mutant's luminescence increased ∼5-fold when 10 mM cAMP was added (Fig. 6). Thus, as with luxR, the cAMP-mediated induction of luminescence appears to not be solely due to CRP regulation of ainS.
We then tested the effect of cAMP on a luxR ainS double mutant (NL62) complemented with pJLB123 and supplemented with C8-HSL. Using this strain under these conditions, neither C8-HSL nor LuxR levels should be subject to direct regulatory control by cAMP-CRP over the ainS or luxR promoters. In this setup, a cAMP-mediated increase in luminescence was not observed (Fig. 6). Although this strain supplemented with C8-HSL was also the brightest treatment in the experiment, it is more than an order of magnitude dimmer than the luminescence capacity of ES114 (e.g., supplemented with 3-oxo-C6-HSL), suggesting that it would be possible to see a cAMP-mediated increase in luminescence if one existed. Taken together, these results suggest that cAMP-CRP control of both ainS and luxR is significant in determining the luminescence of V. fischeri ES114.
CRP activates transcription of luxR, luxI, and ainS promoters through different mechanisms in transgenic E. coli.
CRP usually regulates transcription by recruiting RNA polymerase (RNAP) to promoters following binding to a target sequence (21). CRP-dependent promoters are grouped into three classes. Class I-type promoters require one point of contact with RNAP at activating region I (ARI). Class II-type promoters require two points of contact between CRP and RNAP at both activating region I and activating region II (ARI and ARII). Finally, class III-type promoters involve multiple regulators (i.e., more than one CRP or CRP and another regulator), and either these require ARI or ARII or, at some class III-type promoters, CRP exerts an effect on a second regulatory protein through DNA binding and protein-protein interaction, in which case ARI and ARII may not be required (21, 50–52).
V. fischeri promoters were examined using a reporter system developed in E. coli wherein known ARI and ARII crp mutations are used to classify CRP-regulated promoters (34, 35, 45). In this dual plasmid system, promoter-lacZ reporters are generated in pRW50 and crp alleles are expressed from a compatible plasmid (41). Plasmid pDCRP contains the wild-type E. coli crp and is used as a positive control, whereas plasmid pDU9 lacks crp and is used as a negative control. The crp alleles on pDCRP-TA158 and pDCRP-HL159 are mutated at ARI, and the crp alleles pDCRP-HY19 and pDCRP-KE101 are mutated at ARII. To ensure that E. coli CRP activates transcription from V. fischeri promoters, we tested whether the wild-type E. coli crp would complement the growth and luminescence defect of the V. fischeri crp mutant. When the E. coli wild-type crp allele was expressed in trans on pNL56, luminescence and growth in the absence of glucose were restored in the V. fischeri crp mutant (data not shown). We similarly observed that crp from ES114 on plasmid pNL69 could complement E. coli mutant M182Δcrp (data not shown).
Expression from the PluxR-lacZ (pNL73) promoter-reporter increased ∼100-fold in E. coli cells cotransformed with the wild-type CRP allele relative to cotransformation with the empty vector control (Fig. 7A). This increase in activity is also evident in cells cotransformed with ARII variant alleles but not in cells cotransformed with ARI variant alleles, indicating that the effect of CRP on luxR expression requires only ARI, consistent with a CRP class I-type promoter.
Fig 7.
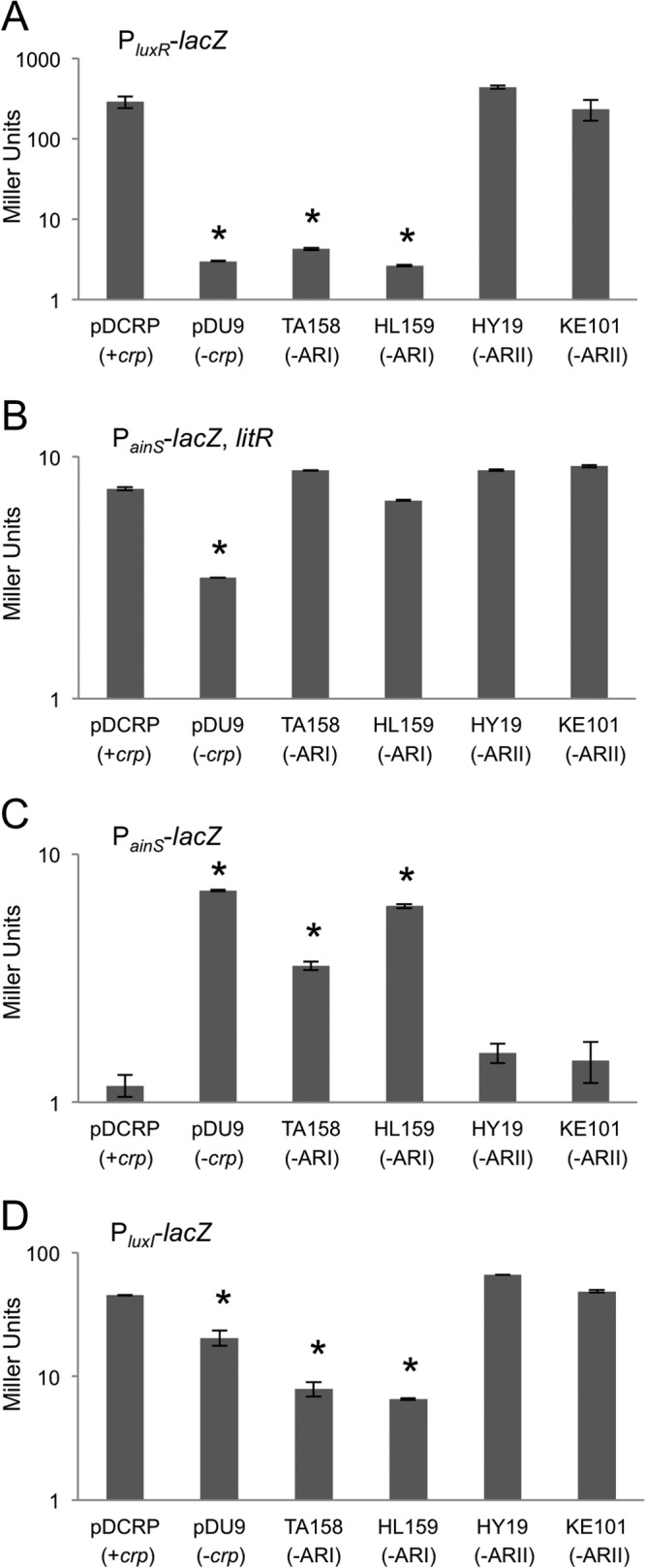
Effects of ARI and ARII crp mutations on luxR, ainS, and luxI promoter activity in E. coli. Data are given as Miller units generated from the reporters PluxR-lacZ (pNL73) (A), PainS-lacZ (pNL76) (B), PainS-lacZ, litR (pNL76) (C), and PluxI-lacZ (pNL82) (D). Bars indicate standard errors (n = 2). An asterisk indicates a significant difference from pDCRP as determined by a Student's t test (P ≤ 0.05).
We next examined the ainS promoter using a PainS-lacZ reporter. Because previous research suggested a role for LitR in ainS expression (11), litR was cloned into the PainS-lacZ reporter plasmid. In the presence of litR, the wild-type CRP allele increased beta-galactosidase activity from the PainS-lacZ reporter ∼2-fold relative to cotransformation with the empty vector control (Fig. 7B). This increase in activity was also observed in cells cotransformed with either the ARI or ARII variant allele, indicating that the activating regions that define class I and class II promoters are not required for this regulatory effect.
We also examined expression of the ainS reporter in the absence of litR (Fig. 7C). These experiments showed higher expression of PainS-lacZ when litR is present (Fig. 7B and C), consistent with the proposed role of LitR in ainS regulation. Surprisingly, when litR was absent we observed higher PainS-lacZ activity in the presence of the vector control or vectors carrying CRP with ARI mutant alleles (Fig. 7C). These results suggest an additional layer of regulation, at least in E. coli, whereby CRP mediates negative regulation of ainS.
Finally, we used this reporter system to test the effect of CRP on the luxI promoter. The CRP binding site upstream of luxR likewise is upstream of the divergently transcribed luxI. Previous work, also in transgenic E. coli, suggested that in the absence of luxR CRP has a negative effect on expression of the luxI promoter from V. fischeri strain MJ-1 (16). We found that our PluxI-lacZ reporter derived from strain ES114 increased ∼3-fold in expression in the presence of the wild-type crp or with the ARII mutants (Fig. 7D), suggesting that CRP activates LuxR-independent basal expression of luxI through a mechanism that requires ARI but not ARII. We also added luxR to the PluxI-lacZ reporter plasmid under the control of a nonnative constitutive promoter and observed the expected autoinducer-dependent increase in PluxI-lacZ expression (data not shown); however, when the 3-oxo-C6-HSL autoinducer combined with LuxR to enhance transcription, no further increase was seen when crp was present (data not shown).
C8-HSL levels affected by glucose.
Based on the data described above, we proposed that the effect of glucose on ES114 luminescence (Fig. 1) is due in part to lower C8-HSL levels in glucose-grown cells, reflecting regulation of ainS by cAMP-CRP in response to the carbon source. As predicted, we saw decreased levels of C8-HSL in ES114 cultures supplemented with glucose (Fig. 8). However, contrary to our prediction, when cultures were supplemented with glucose we also saw higher C8-HSL levels in the Δcrp mutant than in the wild type (Fig. 8), a phenomenon that will be discussed below.
Fig 8.
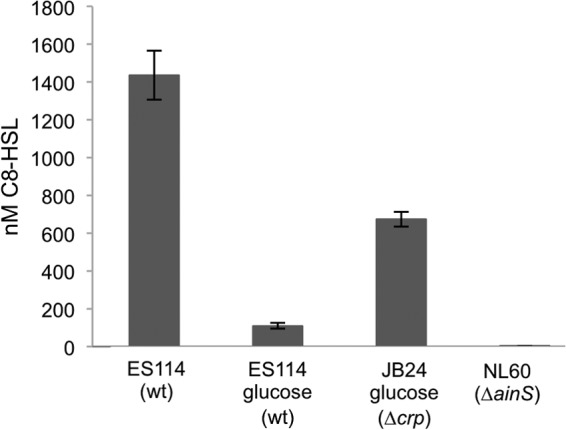
Effects of glucose and Δcrp mutation on C8-HSL accumulation. C8-HSL was extracted and estimated from cultures of ES114, the Δcrp mutant JB24, or the ainS mutant NL60. The C8-HSL synthase mutant NL60 serves as a negative control. Cultures were grown to an OD595 of 2.4 to 2.5 in SWTO either amended with 20 mM glucose or left unamended, as indicated. C8-HSL levels were determined by bioassays of ethyl acetate-extracted cultures, and bars indicate standard errors (n = 3).
DISCUSSION
V. fischeri ES114 controls luminescence using pheromone-mediated regulation, and like many other bacterial pheromone systems, this signaling is both cell density dependent and regulated in response to the environment. Although high cell density may be necessary for lux induction in ES114, it is not sufficient to achieve stimulatory pheromone levels. Some environmentally responsive regulators must also control luxICDABEG expression and are critical for symbiotic induction of luminescence (14, 15, 37). Previous reports suggested a role for glucose and CRP-mediated regulation of V. fischeri bioluminescence (14, 16–18, 26). However, the model that emerged of cAMP-CRP regulating luxR predated the development of genetic and genomic tools in V. fischeri or the discovery of key regulatory elements, such as the AinS/AinR pheromone system.
Our goal in this study was to test and define the role of glucose and CRP in lux regulation in V. fischeri isolate ES114. Recently, ES114 has become a popular experimental model, and the genetic tools developed for this strain allowed us to explore this area in ways that were not possible when the effects of glucose and CRP on lux were first examined. It seemed especially prudent not to assume that previous conclusions about CRP's role in lux regulation held for ES114, because the lux system in ES114 has diverged significantly from that in strains like MJ-1 (53). Moreover, it was reported that ES114 does not respond to glucose in the way MJ-1 does (14).
We found that CRP regulates the ainS pheromone synthase system, which is absent from studies with transgenic E. coli, underscoring the value of incorporating experiments in ES114 itself. We also generated support for the previous conclusion that CRP regulates luxR in ES114. Taken together, our data suggest that the regulatory mechanisms connecting CRP, pheromone signaling, and luminescence are more complex than previously appreciated. A new model for this CRP-mediated regulation of luminescence is presented in Fig. 9.
Fig 9.
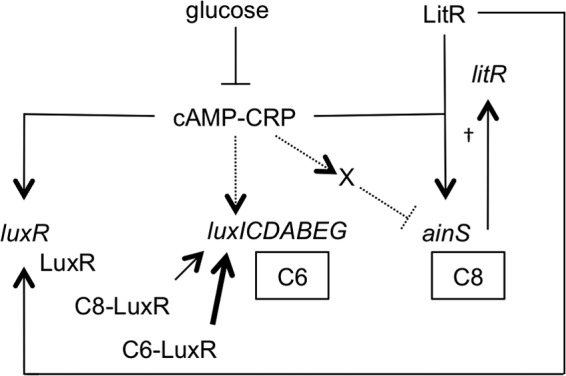
Proposed model of CRP-mediated regulation in V. fischeri luminescence. Direct activation of luxI and of a potential negative regulator of ainS (indicated by an “X”) are based on data from transgenic E. coli (Fig. 7) and are shown with dashed lines. It should be noted that this potential negative regulator need not exist if cAMP-CRP acts as a simple repressor of ainS, and other mechanisms are likewise viable. The autoinducers 3-oxo-C6-HSL and C8-HSL are abbreviated as C6 and C8, respectively. The dagger indicates that C8-HSL functions through a phosphorelay involving AinR, LuxU, LuxO, Hfq, and the small RNA qrr that increases levels of LitR to activate luxR expression (10, 13). C8-HSL can also activate LuxR directly, although it is a weaker activator than 3-oxo-C6-HSL (11), and this is represented by the relative weights of the arrows from C8-LuxR and C6-LuxR.
Regulation by CRP or similar proteins is integral to pheromone circuitry in diverse systems.
Interestingly, the role of CRP in modulating the pheromone signaling circuitry of V. fischeri apparently illustrates a widespread trend. Several bacteria with mechanistically diverse pheromone signaling systems integrate CRP, CRP-like proteins, or catabolite repression into regulation of their pheromone signaling systems. For example, Vibrio cholerae and Vibrio harveyi lack the V. fischeri LuxI/LuxR system, but the quorum-sensing master regulator HapR in V. cholerae and its homolog in V. harveyi are regulated by CRP (54–56). V. cholerae mutants defective in CRP express lower levels of hapR, resulting in increased production of cholera toxin and enhanced biofilm formation (54, 55). Similarly, CRP indirectly regulates the pheromone synthase luxS in E. coli (57), and CRP activates transcription of the acyl homoserine lactone-dependent regulators encoded by lasR and expR in Pseudomonas aeruginosa and Erwinia chrysanthemi, respectively (58, 59). Further afield taxonomically from V. fischeri, in Staphylococcus aureus the Agr pheromone signaling system regulates virulence and antibiotic resistance and is modulated by carbon-catabolite protein A (CcpA) (60).
Taken together, these studies strongly suggest that pheromone signaling in diverse bacteria communicates carbon source availability as well as cell density. The trend appears to be upregulation of pheromone synthases and/or receptors when the preferred carbon source, glucose, is absent. This phenomenon is easily rationalized in the many instances where pheromones control expression of exoenzymes that help cells scavenge for alternative growth substrates, but it is less clear why, for example, regulation of luminescence would be tied to growth in the absence of glucose. Importantly, pheromone-controlled genes are not simply coregulated by CRP; rather, pheromone signaling itself is controlled. Thus, it is clear that pheromone levels will reflect environmental context as well as cell density, and future studies should take this fact into account when building models to explain the biological role(s) of these systems.
Role of glucose in luminescence regulation.
The regulatory role of glucose in V. fischeri has been difficult to interpret due in part to potential strain-specific effects. Previous research showed that luminescence in strain MJ-1 is decreased when glucose is added (27), yet glucose was reported to have no effect in ES114 (14). We found that glucose modulates luminescence in V. fischeri ES114, and this phenomenon appears to involve gene regulation and not simply metabolic changes. Our results also show that depending on when a culture is sampled, one might see positive, negative, or no apparent effect at all of glucose on ES114 (Fig. 1). We found that C8-HSL accumulation was lower when ES114 was grown with glucose (Fig. 8), and the opposite effects of glucose on luminescence at low or high cell densities (Fig. 1) might reflect the contrary roles of C8-HSL. In ES114, at low to moderate cell densities, luminescence expression is primarily driven by LuxR combined with C8-HSL (11); however, as cell density increases, C8-HSL competes with the stronger activator 3-oxo-C6-HSL for binding with LuxR, such that C8-HSL becomes effectively an inhibitor of luminescence (11, 37), as was seen in MJ-1 (8, 61). We speculate that in ES114 grown with glucose, there is less active cAMP-CRP to promote expression of ainS, leading to less C8-HSL production and opposing effects on luminescence in the absence or presence of stimulatory 3-oxo-C6-HSL at low or high OD, respectively. Such a scenario would lead to low C8-HSL levels throughout growth with effects on luminescence that vary depending on cell density.
With respect to this model, one confounding result was the observation of higher C8-HSL levels in a Δcrp mutant than in ES114 when both were grown on glucose (Fig. 8). These data seem most directly in conflict with the results shown in Fig. 3 and 4, which indicate decreased expression of ainS in the Δcrp mutant, leading us to predict less C8-HSL in the Δcrp mutant, not more. Alternative explanations for this apparent contradiction include cAMP-CRP activating (i) a protease involved in AinS protein turnover, (ii) an unknown enzyme capable of C8-HSL degradation, and/or (iii) pathways competing with AinS for the same substrates. These three scenarios, and probably others, could result in increased AinS and/or C8-HSL accumulation in a crp mutant despite lower ainS transcription. In interpreting these results, it is important to keep in mind that while glucose modulates cAMP-CRP levels, the Δcrp mutation has more profound effects by eliminating CRP altogether. Thus, while deleting crp is useful for elucidating regulatory connections, the Δcrp mutant is probably unlike the wild type on any carbon source. We believe it remains possible that the effect of glucose on C8-HSL levels in ES114 reflects ainS regulation by cAMP-CRP; however, we cannot rule out the possibility of another glucose-responsive system being involved in AinS regulation and/or C8-HSL accumulation.
CRP and regulation of luxR.
Our data support the previous model (16, 17, 26) that cAMP-CRP directly regulates luxR (Fig. 3, 4, and 5). The intergenic lux region has diverged significantly between ES114, MJ-1, and other V. fischeri strains, and while a putative CRP binding site is evident in the same location in a variety of strains, the exact sequence of the site does vary (53). The sequence revealed in CRP footprinting analyses of V. fischeri ATCC 7744 DNA by Shadel et al. (26) aligns with the ES114 sequence used in our binding studies (Fig. 5), although there are several differences in the sequences, particularly in the variable central region of the binding site (53). In addition to demonstrating binding, using a transgenic E. coli system we found that the effect of CRP on luxR requires only the CRP-activating region I, consistent with this effect being mediated by a class I-type promoter interaction. Defining the luxR promoter(s) and transcriptional start site(s) in V. fischeri ES114 would provide a more detailed picture of the mechanism by which CRP regulates this gene.
CRP and regulation of ainS.
We were surprised to find that when luxR expression was artificially held constant, addition of cAMP still influenced luminescence (Fig. 6), leading us to investigate other luminescence regulators that might be controlled by CRP. Bioinformatic analysis revealed a potential CRP binding site upstream of ainS, and we subsequently found evidence that CRP regulates ainS. In addition, we show that LitR is important for CRP-dependent ainS expression, and this effect appears independent of CRP-activating region I or II (Fig. 7B). It is tempting to speculate that these data reflect a class III promoter interaction involving LitR, although such a mechanism remains to be tested. Moreover, at least in E. coli, an additional layer of CRP- and ARI-dependent regulation represses ainS expression (Fig. 7C). These new wrinkles in the ainS and lux regulatory circuitry are depicted in Fig. 9, although some, particularly the mechanism of CRP-dependent ainS repression, remain speculative. We hope in the future to further elucidate regulators of ainS, which may also inform our understanding of CRP's role at this promoter.
CRP and regulation of luxI.
Interestingly, we also found that CRP may directly regulate transcription of luxI. In transgenic E. coli carrying the MJ-1 lux genes, CRP appeared to inhibit luxI transcription in the absence of LuxR (17); however, in our transgenic E. coli system using an ES114-derived lux promoter, we observed the opposite effect (Fig. 7D). The spacing of the CRP binding site to the luxI promoter differs by one nucleotide between strains ES114 and MJ-1 (53) (see Fig. S1 in the supplemental material). We do not know whether direct regulation of luxI by CRP is relevant in V. fischeri, where the effects of CRP on luxR and ainS may overpower direct modulation of the luxICDABEG promoter. If cAMP-CRP has a significant and direct effect on the luxI promoter, it would probably be at very low cell densities where a lack of pheromone accumulation renders LuxR inactive and effects on ainS expression irrelevant. In the future, studies focused on low cell densities may reveal if CRP plays a role in modulating the basal LuxR-independent expression of the luxICDABEG operon.
Studying cAMP-CRP in the ES114 model.
We have found that cAMP-CRP regulates luminescence and pheromone signaling in V. fischeri ES114. It remains to be determined exactly how environmental conditions are connected to cAMP and CRP levels in this bacterium or if there are other regulatory connections between either CRP or carbon source and pheromone signaling. By laying the groundwork using strain ES114, it should be possible to explore these areas and examine the relevance of cAMP-CRP-mediated regulation in a natural symbiotic infection.
Supplementary Material
ACKNOWLEDGMENTS
We thank Anne Weeks and Alecia N. Septer for technical assistance and Stephen J. W. Busby for E. coli strains M182 and M182Δcrp, the pRW50 promoterless lacZα vector, and promoter-reporter constructs. We also thank Gary P. Roberts for the pEXT20 expression vector and E. coli strain UQ3811, utilized in the purification of CRP.
This research was supported by the National Science Foundation (NSF) under grants CAREER MCB-0347317, OCE-0929081, and IOS-1121106.
Footnotes
Published ahead of print 30 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00751-13.
REFERENCES
- 1.Miller MB, Bassler BL. 2001. Quorum sensing in bacteria. Annu. Rev. Microbiol. 55:165–169 [DOI] [PubMed] [Google Scholar]
- 2.Engebrecht J, Nealson K, Silverman M. 1983. Bacterial bioluminescence: isolation and genetic analysis of functions from Vibrio fischeri. Cell 32:773–781 [DOI] [PubMed] [Google Scholar]
- 3.Engebrecht J, Silverman M. 1984. Identification of genes and gene products necessary for bacterial bioluminescence. Proc. Natl. Acad. Sci. U. S. A. 81:4154–4158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meighen EA. 1994. Genetics of bacterial bioluminescence. Annu. Rev. Genet. 28:117–139 [DOI] [PubMed] [Google Scholar]
- 5.Eberhard A, Burlingame AL, Eberhard C, Kenyon GL, Nealson KH, Oppenheimer NJ. 1981. Structural identification of autoinducer of Photobacterium fischeri luciferase. Biochemistry 20:2444–2449 [DOI] [PubMed] [Google Scholar]
- 6.Kaplan HB, Greenberg EP. 1985. Diffusion of autoinducer is involved in regulation of the Vibrio fischeri luminescence system. J. Bacteriol. 163:1210–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Urbanowski ML, Lostroh CP, Greenberg EP. 2004. Reversible acyl-homoserine lactone binding to purified Vibrio fischeri LuxR protein. J. Bacteriol. 186:631–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuo A, Blough NV, Dunlap PV. 1994. Multiple N-acyl-L-homoserine lactone autoinducers of luminescence in the marine symbiotic bacterium Vibrio fischeri. J. Bacteriol. 176:7558–7565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lupp C, Ruby EG. 2004. Vibrio fischeri LuxS and AinS: comparative study of two signal synthases. J. Bacteriol. 186:3873–3881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fidopiastis PM, Miyamoto CM, Jobling MG, Meighen EA, Ruby EG. 2002. LitR, a new transcriptional activator in Vibrio fischeri, regulates luminescence and symbiotic light organ colonization. Mol. Microbiol. 45:131–143 [DOI] [PubMed] [Google Scholar]
- 11.Lupp C, Urbanowski M, Greenberg EP, Ruby EG. 2003. The Vibrio fischeri quorum-sensing systems ain and lux sequentially induce luminescence gene expression and are important for persistence in the squid host. Mol. Microbiol. 50:319–331 [DOI] [PubMed] [Google Scholar]
- 12.Miyamoto CM, Lin YH, Meighen EA. 2000. Control of bioluminescence in Vibrio fischeri by the LuxO signal response regulator. Mol. Microbiol. 36:594–607 [DOI] [PubMed] [Google Scholar]
- 13.Miyashiro T, Wollenberg MS, Cao X, Oehlert D, Ruby EG. 2010. A single qrr gene is necessary and sufficient for LuxO-mediated regulation in Vibrio fischeri. Mol. Microbiol. 77:1556–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boettcher KJ, Ruby EG. 1990. Depressed light emission by symbiotic Vibrio fischeri of the sepiolid squid Euprymna scolopes. J. Bacteriol. 172:3701–3706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boettcher KJ, Ruby EG. 1995. Detection and quantification of Vibrio fischeri autoinducer from symbiotic squid light organs. J. Bacteriol. 177:1053–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunlap PV, Greenberg EP. 1985. Control of Vibrio fischeri luminescence gene expression in Escherichia coli by cyclic AMP and cyclic AMP receptor protein. J. Bacteriol. 164:45–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dunlap PV, Greenberg EP. 1988. Control of Vibrio fischeri lux gene transcription by a cyclic AMP receptor protein-LuxR protein regulatory circuit. J. Bacteriol. 170:4040–4046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dunlap PV. 1989. Regulation of luminescence by cyclic AMP in cya-like and crp-like mutants of Vibrio fischeri. J. Bacteriol. 171:1199–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emmer M, de Crombrugghe B, Pastan I, Perlman R. 1970. Cyclic AMP receptor protein of E. coli: its role in the synthesis of inducible enzymes. Proc. Natl. Acad. Sci. U. S. A. 66:480–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zubay G, Schwartz D, Beckwith J. 1970. Mechanism of activation of catabolite-sensitive genes: a positive control system. Proc. Natl. Acad. Sci. U. S. A. 66:104–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Busby S, Ebright RH. 1999. Transcription activation by catabolite activator protein (CAP). J. Mol. Biol. 293:199–213 [DOI] [PubMed] [Google Scholar]
- 22.Grainger DC, Hurd D, Harrison M, Holdstock J, Busby SJ. 2005. Studies of the distribution of Escherichia coli cAMP-receptor protein and RNA polymerase along the E. coli chromosome. Proc. Natl. Acad. Sci. U. S. A. 102:17693–17698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kolb A, Busby S, Buc H, Garges S, Adhya S. 1993. Transcriptional regulation by cAMP and its receptor protein. Annu. Rev. Biochem. 62:749–795 [DOI] [PubMed] [Google Scholar]
- 24.Botsford JL, Harman JG. 1992. Cyclic AMP in prokaryotes. Microbiol. Rev. 56:100–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lawson CL, Swigon D, Murakami KS, Darst SA, Berman HM, Ebright RH. 2004. Catabolite activator protein: DNA binding and transcription activation. Curr. Opin. Struct. Biol. 14:10–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shadel GS, Devine JH, Baldwin TO. 1990. Control of the lux regulon of Vibrio fischeri. J. Biolumin. Chemilumin. 5:99–106 [DOI] [PubMed] [Google Scholar]
- 27.Friedrich WF, Greenberg EP. 1983. Glucose repression of luminescence and luciferase in Vibrio fischeri. Arch. Microbiol. 134:87–91 [Google Scholar]
- 28.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557–580 [DOI] [PubMed] [Google Scholar]
- 29.Dunn AK, Martin MO, Stabb EV. 2005. Characterization of pES213, a small mobilizable plasmid from Vibrio fischeri. Plasmid 54:114–134 [DOI] [PubMed] [Google Scholar]
- 30.Miller JH. 1992. A short course in bacterial genetics. Cold Spring Harbor Laboratory Press, New York, NY [Google Scholar]
- 31.Stabb EV, Reich KA, Ruby EG. 2001. Vibrio fischeri genes hvnA and hvnB encode secreted NAD+-glycohydrolases. J. Bacteriol. 183:309–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bose JL, Kim U, Bartkowski W, Gunsalus RP, Overley AM, Lyell NL, Visick KL, Stabb EV. 2007. Bioluminescence in Vibrio fischeri is controlled by the redox-responsive regulator ArcA. Mol. Microbiol. 65:538–553 [DOI] [PubMed] [Google Scholar]
- 33.Dunn AK, Millikan DS, Adin DM, Bose JL, Stabb EV. 2006. New rfp- and pES213-derived tools for analyzing symbiotic Vibrio fischeri reveal patterns of infection and lux expression in situ. Appl. Environ. Microbiol. 72:802–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.West D, Williams R, Rhodius V, Bell A, Sharma N, Zou C, Fujita N, Ishihama A, Busby S. 1993. Interactions between the Escherichia coli cyclic AMP receptor protein and RNA polymerase at class II promoters. Mol. Microbiol. 10:789–797 [DOI] [PubMed] [Google Scholar]
- 35.Bell A, Gaston K, Williams R, Chapman K, Kolb A, Buc H, Minchin S, Williams J, Busby S. 1990. Mutations that alter the ability of the Escherichia coli cyclic AMP receptor protein to activate transcription. Nucleic Acids Res. 18:7243–7250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stabb EV, Ruby EG. 2002. RP4-based plasmids for conjugation between Escherichia coli and members of the Vibrionaceae. Methods Enzymol. 358:413–426 [DOI] [PubMed] [Google Scholar]
- 37.Lyell NL, Dunn AK, Bose JL, Stabb EV. 2010. Bright mutants of Vibrio fischeri ES114 reveal conditions and regulators that control bioluminescence and expression of the lux operon. J. Bacteriol. 192:5103–5114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bose JL, Rosenberg CS, Stabb EV. 2008. Effects of luxCDABEG induction in Vibrio fischeri: enhancement of symbiotic colonization and conditional attenuation of growth in culture. Arch. Microbiol. 190:169–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dykxhoorn DM, St Pierre R, Linn T. 1996. A set of compatible tac promoter expression vectors. Gene 177:133–136 [DOI] [PubMed] [Google Scholar]
- 40.Youn H, Kerby RL, Conrad M, Roberts GP. 2006. Study of highly constitutively active mutants suggests how cAMP activates cAMP receptor protein. J. Biol. Chem. 281:1119–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lodge J, Fear J, Busby S, Gunasekaran P, Kamini NR. 1992. Broad host range plasmids carrying the Escherichia coli lactose and galactose operons. FEMS Microbiol. Lett. 74:271–276 [DOI] [PubMed] [Google Scholar]
- 42.Busby S, Kotlarz D, Buc H. 1983. Deletion mutagenesis of the Escherichia coli galactose operon promoter region. J. Mol. Biol. 167:259–274 [DOI] [PubMed] [Google Scholar]
- 43.Casadaban MJ, Cohen SN. 1980. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J. Mol. Biol. 138:179–207 [DOI] [PubMed] [Google Scholar]
- 44.Steed PM, Wanner BL. 1993. Use of the rep technique for allele replacement to construct mutants with deletions of the pstSCAB-phoU operon: evidence of a new role for the PhoU protein in the phosphate regulon. J. Bacteriol. 175:6797–6809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rhodius VA, West DM, Webster CL, Busby SJ, Savery NJ. 1997. Transcription activation at class II CRP-dependent promoters: the role of different activating regions. Nucleic Acids Res. 25:326–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collins CH, Leadbetter JR, Arnold FH. 2006. Dual selection enhances the signaling specificity of a variant of the quorum-sensing transcriptional activator LuxR. Nat. Biotechnol. 24:708–712 [DOI] [PubMed] [Google Scholar]
- 47.Munch R, Hiller K, Grote A, Scheer M, Klein J, Schobert M, Jahn D. 2005. Virtual Footprint and PRODORIC: an integrative framework for regulon prediction in prokaryotes. Bioinformatics 21:4187–4189 [DOI] [PubMed] [Google Scholar]
- 48.Crowell DN, Anderson MS, Raetz CR. 1986. Molecular cloning of the genes for lipid A disaccharide synthase and UDP-N-acetylglucosamine acyltransferase in Escherichia coli. J. Bacteriol. 168:152–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng D, Constantinidou C, Hobman JL, Minchin SD. 2004. Identification of the CRP regulon using in vitro and in vivo transcriptional profiling. Nucleic Acids Res. 32:5874–5893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ebright RH. 1993. Transcription activation at class I CAP-dependent promoters. Mol. Microbiol. 8:797–802 [DOI] [PubMed] [Google Scholar]
- 51.Zhou Y, Zhang X, Ebright RH. 1993. Identification of the activating region of catabolite gene activator protein (CAP): isolation and characterization of mutants of CAP specifically defective in transcription activation. Proc. Natl. Acad. Sci. U. S. A. 90:6081–6085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Niu W, Kim Y, Tau G, Heyduk T, Ebright RH. 1996. Transcription activation at class II CAP-dependent promoters: two interactions between CAP and RNA polymerase. Cell 87:1123–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bose JL, Wollenberg MS, Colton DM, Mandel MJ, Septer AN, Dunn AK, Stabb EV. 2011. Contribution of rapid evolution of the luxR-luxI intergenic region to the diverse bioluminescence outputs of Vibrio fischeri strains isolated from different environments. Appl. Environ. Microbiol. 77:2445–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liang W, Pascual-Montano A, Silva AJ, Benitez JA. 2007. The cyclic AMP receptor protein modulates quorum sensing, motility and multiple genes that affect intestinal colonization in Vibrio cholerae. Microbiology 153:2964–2975 [DOI] [PubMed] [Google Scholar]
- 55.Silva AJ, Benitez JA. 2004. Transcriptional regulation of Vibrio cholerae hemagglutinin/protease by the cyclic AMP receptor protein and RpoS. J. Bacteriol. 186:6374–6382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chatterjee J, Miyamoto CM, Zouzoulas A, Lang BF, Skouris N, Meighen EA. 2002. MetR and CRP bind to the Vibrio harveyi lux promoters and regulate luminescence. Mol. Microbiol. 46:101–111 [DOI] [PubMed] [Google Scholar]
- 57.De Lay N, Gottesman S. 2009. The Crp-activated small noncoding regulatory RNA CyaR (RyeE) links nutritional status to group behavior. J. Bacteriol. 191:461–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Albus AM, Pesci EC, Runyen-Janecky LJ, West SE, Iglewski BH. 1997. Vfr controls quorum sensing in Pseudomonas aeruginosa. J. Bacteriol. 179:3928–3935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reverchon S, Bouillant ML, Salmond G, Nasser W. 1998. Integration of the quorum-sensing system in the regulatory networks controlling virulence factor synthesis in Erwinia chrysanthemi. Mol. Microbiol. 29:1407–1418 [DOI] [PubMed] [Google Scholar]
- 60.Seidl K, Stucki M, Ruegg M, Goerke C, Wolz C, Harris L, Berger-Bachi B, Bischoff M. 2006. Staphylococcus aureus CcpA affects virulence determinant production and antibiotic resistance. Antimicrob. Agents Chemother. 50:1183–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kuo A, Callahan SM, Dunlap PV. 1996. Modulation of luminescence operon expression by N-octanoyl-L-homoserine lactone in ainS mutants of Vibrio fischeri. J. Bacteriol. 178:971–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.