

Abstract
The arginine deiminase system (ADS) is associated with arginine catabolism and plays a role in virulence of several pathogenic bacteria. In Streptococcus pneumoniae, the ADS genes exist as a locus consisting of arcABCDT. A recent genome-wide mutagenesis approach revealed that both arcD and arcT are potentially essential in a chinchilla otitis media (OM) model. In the present study, we generated ΔarcD, ΔarcT, and ΔarcDT mutants by homologous recombination and evaluated their infectivity. Our results showed that only arcD, and not arcT, of an OM isolate is required during chinchilla middle ear infection. Additionally, D39 ΔarcD exhibited enhanced nasopharyngeal colonization and was attenuated in both mouse pneumonia and bacteremia models. In vitro, D39 ΔarcD displayed enhanced adherence to A549 epithelial cells and increased phagocytosis by J774A.1 macrophages compared to those with the parental strain. This mutant also exhibited an impaired capsule, as detected using electron microscopy, immunofluorescence, and a capsule assay. We demonstrated that the capsule defect in the D39 ΔarcD mutant may not be associated with a deficiency in arginine but rather is likely caused by a loss of interaction between the capsule and the transmembrane protein ArcD.
INTRODUCTION
Streptococcus pneumoniae is a commensal bacterium found in the human respiratory tract. However, it can still cause a variety of infections mostly in infants and in immunocompromised elderly people, such as pneumonia, meningitis, otitis media (OM), and bacteremia (1, 2). This pathogen remains a major cause of morbidity and mortality worldwide (3). The capsular polysaccharides of S. pneumoniae are essential virulence factors and protect the bacterium against opsonophagocytosis (4, 5). Currently, more than 90 distinct capsular serotypes have been identified, and at least half of them have been structurally characterized (6).
Initially, pneumococcal vaccine efforts were directed toward capsular polysaccharides because antibodies to the polysaccharides are highly protective against invasive infections (7). However, a 23-valent pneumococcal vaccine exhibited less than 60% efficacy in preventing invasive pneumococcal infections caused by various vaccinated serotypes (8) and is less effective in preventing pneumonia and other localized respiratory infections (9). In addition, a recent 11-valent pneumococcal conjugate vaccine has been found to be highly effective in children under 5 years of age (10), but it has not been approved for use in adults (11). Due to the limitations of the current pneumococcal vaccines, more effective vaccines are urgently needed. Furthermore, the emergence of pneumococcal strains that are resistant to multiple classes of antibiotics also drives the development of new therapeutic approaches against pneumococcal diseases (12). Therefore, a better understanding of how the bacterium causes infection is a key to achieve these goals. Although a plethora of virulence determinants have been recognized in this pathogen, how metabolic pathways enable the pathogen to acquire nutrition and/or cause diseases remains largely unknown.
The enzymes in the arginine deiminase system (ADS) convert arginine to ammonia and carbon dioxide with the concomitant release of ATP. These enzymes include (i) arginine deiminase (ADI), which catalyzes the conversion of arginine to citrulline; (ii) ornithine carbamoyltransferase (OTC), which catalyzes the phosphorolysis of citrulline, yielding ornithine and carbamoyl phosphate; and (iii) carbamate kinase (CK), which catalyzes the transfer of phosphate from carbamoyl phosphate to ADP, generating ATP, ammonia, and carbon dioxide. The three enzymes are encoded by arcA, arcB, and arcC, respectively, which tend to exist as an operon (13) (see Fig. S1A in the supplemental material). The ADS has been described as an energy-generating system in several microorganisms, including both Gram-positive and Gram-negative bacteria (14). These ADS enzymes have been characterized in several species of the genera Pseudomonas (15, 16), Clostridium (17, 18), Bacillus (19), and Streptococcus (20). In addition, these enzymes also provide protection to pathogens from acid stress by producing ammonia (21, 22).
Apart from the arcABC genes, an arginine/ornithine antiporter (arcD) and a dipeptidase (arcT) are also associated with the arc operon in several pathogens (23, 24). All these genes are typically located in the same locus (25). In Streptococcus lactis, the arginine-ornithine exchange coordinates with the ADS by modulating the intracellular levels of ornithine and arginine (26–28). Pseudomonas aeruginosa ArcD possesses a 43% sequence identity with Corynebacterium glutamicum LysI (29), which transports arginine, ornithine, and lysine in an energy-independent manner (30). Despite the potential importance of these genes, the functions of arcD and arcT in S. pneumoniae remain unknown. The putative activities of S. pneumoniae arcABCD genes are summarized in Fig. S1B in the supplemental material.
Several genome-wide signature-tagged mutagenesis (STM) studies have revealed that numerous attenuated mutants are associated with nutrient transport and utilization (31–33). By using a human OM isolate, ST556, a recent study mapped 169 pneumococcal genes, including the arcD and arcT genes in the arc locus, that were essential in a chinchilla OM model (34). In the present study, we showed that S. pneumoniae ArcD is an arginine transporter, and deletion of arcD in D39 affects the capsule, which does not correlate with the bacterial arginine levels. This is the first report deciphering the arc locus in S. pneumoniae.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All of the S. pneumoniae strains used in the present study are listed in Table 1. D39 (serotype 2; ATCC) and ST606, a streptomycin-resistant derivative of clinical OM isolate ST556 (serotype 19F, originally provided by Michael Jacobs) (34, 35), were used as wild type (WT) strains, except as specified. Pneumococci were grown in Todd-Hewitt broth containing 0.5% yeast extract (THY) (BD Biosciences) or in a chemically defined medium (CDM) (purchased from JRH Bioscience, except as specified) supplemented with 0.1% choline, 0.25% sodium bicarbonate, and 0.073% cysteine (36). Competence THY medium was prepared by adding 0.2% glucose, 0.2% CaCl2, and 0.02% bovine serum albumin (BSA) to THY medium, pH 7.2 to 7.4, which was used for transformation of pneumococcal strains (37). Transformants were grown on tryptic soy agar (TSA) plates containing 3% sheep blood. If necessary, kanamycin (200 μg/ml), erythromycin (2 μg/ml), or gentamicin (2.5 μg/ml) was used for selection. Escherichia coli DH5α was routinely used for cloning and was grown in Luria-Bertani (LB) broth or on LB agar plates. If necessary, erythromycin (200 μg/ml) was used for selection. All pneumococcal strains were grown at 37°C with 5% CO2, and E. coli strains were grown at 37°C with aeration.
Table 1.
Pneumococcal strains and plasmids used in the present study
| Strain or plasmid | Descriptiona | Reference(s) or source |
|---|---|---|
| Strains | ||
| D39 | Serotype 2, encapsulated | 63 |
| R6 | D39 derivative, unencapsulated | 64 |
| ST556 | Serotype 19F, OM clinical isolate | 34, 35 |
| ST606 | ST556 derivative with streptomycin resistance; Strepr | 34 |
| ST1733 | ST606 ΔarcDT::kan; Kanr | This study |
| ST1808 | ST606 ΔarcD::kan; Kanr | This study |
| ST1809 | ST606 ΔarcT::kan; Kanr | This study |
| ST1815 | R6 ΔarcD::kan; Kanr | This study |
| ST1898 | D39 ΔarcD::kan; Kanr | This study |
| ST1899 | D39 ΔarcT::kan; Kanr | This study |
| ST1900 | D39 ΔarcDT::kan; Kanr | This study |
| ST2611 | ST1898 (D39 ΔarcD) transformed with pST2605; Kanr Ermr | This study |
| ST2705 | ST1898 (D39 ΔarcD) transformed with pST2586; Kanr Ermr | This study |
| Plasmids | ||
| pVA838 | E. coli-S. pneumoniae shuttle vector; Ermr | 65 |
| pST2586 | gyrA promoter cloned between the BamHI and SalI sites of pVA838; Ermr | This study |
| pST2605 | D39 arcD ORF cloned at the BamHI site of pST2586; Ermr | This study |
Strepr, streptomycin resistance; Kanr, kanamycin resistance; Ermr, erythromycin resistance.
Generation of pneumococcal mutants and plasmids for complementation.
Null mutants of arcD, arcT, and arcDT in both D39 and ST606 were constructed by homologous recombination as described previously (34, 38). Briefly, an upstream fragment and a downstream fragment for each target gene were PCR amplified from D39 and ST556, respectively. The primers were designed at the conserved regions between the two strains, based on the complete sequences of D39 (GenBank accession no. CP000410) and ST556 (GenBank accession no. NC_017769). All of the primers used in the present study are listed in Table S1 in the supplemental material. The PCR products were digested and ligated with a Janus cassette, which consists of a kanamycin resistance gene and a dominant WT rpsL+ allele (39). The Janus cassette was amplified from ST588 (38) and digested with the same enzymes. The ligated fragments were used to transform D39 and ST606. Isogenic mutants that are resistant to kanamycin but sensitive to streptomycin were selected from plates containing kanamycin and were verified by PCR.
For complementation of the D39 ΔarcD mutant, the gyrA promoter was amplified from D39 with primers Pr2774 and Pr2775. Primer Pr2774 contains a SalI site and primer Pr2775 contains BamHI-SphI-KpnI sites for subsequent cloning. The PCR product was digested with SalI and BamHI and ligated with pVA838 between the SalI-BamHI sites to generate pST2586. The D39 arcD open reading frame (ORF) was subsequently amplified using primers Pr2704 and Pr1673 and was digested with BamHI. This fragment was cloned into pST2586 at the BamHI site to generate pST2605 and was then transformed into the D39 ΔarcD mutant.
Phagocytosis assay.
Murine macrophage cell line J774A.1 (ATCC) was maintained in Dulbecco's modified Eagle's medium (DMEM) (HyClone) supplemented with 10% fetal calf serum. Cells were seeded in 24-well plates at 1 × 105 cells/well and were incubated overnight at 37°C. Bacteria were grown to an optical density at 620 nm (OD620) of 0.4 and were diluted to 1 × 108 CFU/ml in phosphate-buffered saline (PBS). The bacterial suspensions were then diluted with DMEM to ∼1 × 107 CFU/ml, and 100 μl of the bacteria was further mixed with 0.9 ml of medium to inoculate in duplicate wells at a multiplicity of infection (MOI) of 5:1. Inoculated CFU were determined by plate counts. Macrophages were then incubated for 1 h at 37°C. After incubation, wells were gently rinsed twice with PBS and incubated for 1 h at 37°C using 1 ml of DMEM containing gentamicin (500 μg/ml) and vancomycin (5 μg/ml) to kill any extracellular bacteria. The wells were then rinsed twice with PBS for complete removal of antibiotics. The integrity of macrophages was checked each time after washing with PBS. Macrophages were lysed with 1 ml of cold water for 15 min at 4°C, and 100-μl volumes of lysates were spread onto TSA blood plates in duplicate for CFU enumeration.
Bacterial adhesion assay.
Human lung epithelial cell line A549 was maintained in RPMI medium (Gibco) supplemented with 10% fetal calf serum. Approximately 1 × 105 cells/well were seeded in 24-well plates and incubated overnight at 37°C. Bacteria were grown and treated as described for phagocytosis experiments except that the infection was carried out at an MOI of 50:1. Infected cells were incubated for 2 h at 37°C, followed by rinsing for five times with 500 μl/well of PBS. To detach cells, 0.05% trypsin was added at 100 μl/well and incubated for 10 min at 37°C. Cells were then diluted with sterile PBS, and 100 μl of the diluents were spread onto TSA blood plates for CFU enumeration.
Capsule assays.
Cell-associated capsule production of R6, D39, the D39 ΔarcD mutant, and the complemented mutant was analyzed using a quantitative assay of uronic acids as previously described (40, 41). Briefly, bacteria were grown in 10 ml of THY broth to an OD620 of 0.5 and were harvested by centrifugation. Each bacterial pellet was washed once with 150 mM Tris-HCl (pH 7.0) and then resuspended into 0.5 ml of the same buffer. For polysaccharide analysis, 0.2 ml of the bacterial suspension was mixed with 1.2 ml of 0.0125 M tetraborate in concentrated sulfate and was then heated for 5 min at 100°C. The sample was subsequently mixed with 20 μl of 0.15% m-hydroxydiphenyl in 0.5% NaOH, followed by measurement of the absorbance at OD520. For subtraction of the blank, the bacterial suspension was treated identically except using 20 μl of 0.5% NaOH but without m-hydroxydiphenyl.
It is known that uronic acid is absent in the capsule of serotype 19F (42); thus, the capsular polysaccharides of ST556 and its ΔarcD mutant were determined based on previous reports, with slight modification (43, 44). Briefly, bacteria were grown to an OD620 of 0.6 and were harvested by centrifugation. After being washed twice with PBS, bacteria were resuspended in 0.5 ml of water. Bacterial polysaccharides were then determined by measuring the absorbance at OD640 after the addition of a 2-ml solution containing a mixture of 20 mg of 1-ethyl-2[3-(1-ethylnaphtho[1,2-d]thiazolin-2-ylidene)-2-methylpropenyl]naphtho[1,2-d]thiazolium bromide (Stains-All reagent; Sigma) and 60 μl of glacial acetic acid dissolved in 100 ml of 50% formamide. As a control, 2 ml of Stains-All reagent was added to 0.5 ml of water and mixed thoroughly. The OD640 values were normalized by subtraction to the water control.
Immunofluorescence microscopy.
Capsular polysaccharides of the D39 WT and the ΔarcD mutant were detected by immunofluorescence microscopy following the method described earlier (38), except using different primary antibodies. In the present study, rabbit antiserum against pneumococcal type 2 capsule polysaccharide (Miravista Diagnostics) was used. Finally, images were obtained using the Openlab 4.0.4 software program at a magnification of ×63 with a Zeiss Axiovert 200 M inverted microscope and a Q Imaging or Hamamatsu Orca ER camera.
Transmission electron microscopy (TEM).
To determine the capsule morphology, bacteria were grown to an OD620 of 0.4 in THY and harvested by centrifugation. The bacterial pellets were fixed using a lysine-acetate-based formaldehyde-glutaraldehyde ruthenium red-osmium fixation procedure (LRR fixation), serially diluted, and embedded into LR white resin (Ted Pella Inc.) as described previously (21). All reagents were EM grade and were purchased from Electron Microscopy Sciences. Ultrathin sections (80 to 100 nm) were cut with a diamond knife, and sections were picked up using Formvar-coated slot copper grids (300 mesh). Sections were counterstained with 2% aqueous uranyl acetate for 10 min, washed with water three times, and then air dried. After complete drying, sections were examined using a Zeiss EM 910 transmission electron microscope at an acceleration voltage of 80 kV.
In vivo animal studies.
In vivo animal studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Research Council (45). The protocol was approved by the Institutional Animal Care and Use Committee. All intranasal infection studies were performed with 6- to 8-week-old female BALB/c mice (Taconic). The WT and the ΔarcD mutant of D39 and ST556 were used for a nasopharyngeal colonization model and a pneumonia model of infection. Bacteria were grown to an OD620 of 0.4 in THY, diluted using PBS to 1 × 108 CFU/ml, and then inoculated at ∼2 × 106 CFU into each mouse intranasally in a volume of 20 μl. The final inocula were examined with serial dilution and plate counting. Nasopharyngeal colonization was determined at 5 days postinfection. The nasopharyngeal lavage was collected using a modified protocol described previously (46). Briefly, the trachea of the anesthetized mouse was cut at the top of the larynx, and 1 ml of PBS was injected through polyethylene tubing with an internal diameter of 0.58 mm (Becton, Dickinson). The nasopharyngeal lavage fluid was washed out through the nares, serially diluted, and then plated onto TSA blood plates containing gentamicin for CFU enumeration.
For a pneumonia model of infection, bacteria were grown similarly as described above and were resuspended in PBS to 108 CFU/ml, and mice were then inoculated with 50 μl (∼ 5 × 106 CFU) of bacteria intranasally as described earlier (46). Mice were subsequently monitored for 9 days.
For a bacteremia model of infection, 100-μl volumes of bacteria thawed from enumerated frozen stocks were administered intravenously into 6- to 8-week-old inbred CBA/N mice. The CFU/mouse were 1.76 × 105 and 2.7 × 105 for D39 and its ΔarcD mutant, respectively. Mice were subsequently monitored for 7 days.
Coinfection of chinchilla middle ears was carried out as described previously (34, 47). Three 1-year-old female chinchillas (Ryerson Chinchilla Ranch) were used in each group. Each mutant was mixed with a WT (ST606) in PBS at a 1:1 ratio of CFU. Aliquots of 100-μl mixtures were used to infect the middle ears of chinchillas (∼1 × 104 CFU/ear; n = 6). Bacteria were collected 3 days postinfection and plated on plates with streptomycin for the WT and with kanamycin for the mutants. The level of virulence attenuation was expressed as the competitive index (CI), which is defined as the output CFU ratio (mutant/WT) divided by the input CFU ratio (mutant/WT).
Arginine transport assay.
S. pneumoniae D39 and its derivatives were grown to an OD620 of 0.6 in THY broth at 37°C. Aliquots of 1 ml of culture were harvested by centrifugation and washed once with 100 μl of PBS. Bacterial pellets were then resuspended in 100 μl of assay solution containing 0.1 mM l-arginine and 0.1 μCi of l-[3H]arginine (PerkinElmer) in PBS. Samples were incubated at room temperature for various periods, and bacteria were immediately washed twice with 100 μl of PBS. Finally, the bacterial pellets were resuspended into scintillation solution, and the radioactivity was measured in a Tri-Carb 2900TR liquid scintillation analyzer (Packard) for 1 min. The results were expressed as counts per minute and normalized by the total bacterial numbers.
Growth of bacteria with formulated CDM.
S. pneumoniae strains were initially grown in the commercial CDM (JRH Bioscience) at 37°C to an OD620 of 0.5, harvested by centrifugation, washed twice with PBS, and then frozen in aliquots at −80°C using CDM with 25% glycerol. The CFU of the frozen stocks were detected by serial dilution and plate counting prior to use. A formulated CDM was also prepared as reported previously (36) but lacking arginine, and this arginine dropout CDM was then supplemented with various concentrations of arginine as specified below. Bacterial growth in the arginine-supplemented media was monitored hourly at OD620.
Statistical analysis.
All pairwise comparisons of groups were analyzed using a two-tailed t test. Difference in the bacterial colonization was analyzed using a two-tailed Mann-Whitney U test. Kaplan-Meier survival curves were compared using the Gehan-Breslow-Wilcoxon test. All the analyses were performed using Prism 5 (GraphPad Software), and P values of <0.05 were considered to be statistically significant.
RESULTS
Deletion of arcD in D39 enhances nasopharyngeal colonization and leads to virulence attenuation in mouse pneumonia and bacteremia models.
A recent study using the STM approach revealed that both arcD and arcT were potentially essential in a chinchilla OM infection model (34). In the present study, we generated ΔarcD, ΔarcT, and ΔarcDT null mutants in ST606 and coinfected chinchilla middle ears with each mutant and the parental strain. In this particular experiment, the streptomycin-resistant ST556 derivative, ST606, was used as the WT to distinguish from the kanamycin-resistant mutants for evaluation of the competition. To our surprise, the virulence of only the ΔarcD and ΔarcDT mutants, and not the ΔarcT mutant, was substantially attenuated (Fig. 1). This result indicates that ArcD, but not ArcT, plays a role during pneumococcal ear infection. Subsequently, we used the S. pneumoniae D39 strain as a typical model organism to study the role of ArcD in pneumococcal pathogenesis. We also included the clinical OM isolate ST556 for in vivo and in vitro experiments as specified below. We first detected the nasopharyngeal colonization by D39, ST556, and their ΔarcD mutants at 5 days postinfection. We found that the D39 ΔarcD mutant exhibited approximately 20-fold-more CFU than the WT as detected in the nasopharyngeal lavage fluid (Fig. 2A). In contrast, the colonization of ST556 was not altered by deletion of arcD, and both the ST556 WT and its ΔarcD mutant exhibited higher levels of colonization than did the D39 strains (Fig. 2A).
Fig 1.
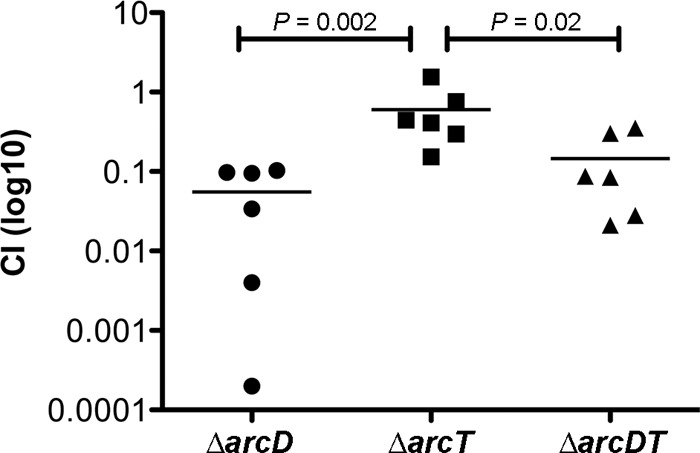
Coinfection of chinchilla middle ears (n = 6). Each ear was inoculated with a 1:1 mixture of WT (ST606) and its ΔarcD, ΔarcT, and ΔarcDT mutants. Bacteria were washed out 3 days postinfection, and the CFU were enumerated to calculate the CI. Bars indicate the mean values.
Fig 2.
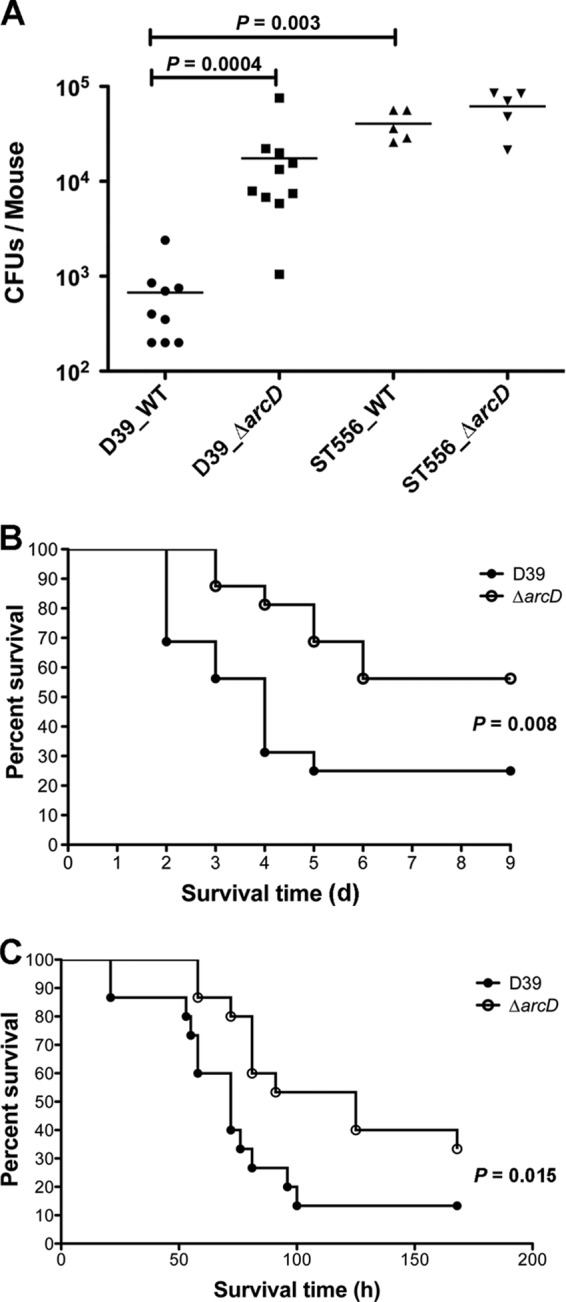
Role of ArcD in the virulence of S. pneumoniae D39. (A) Nasopharyngeal colonization of BALB/c mice infected with the WT and the ΔarcD mutant of D39 (n = 10) and ST556 (n = 5). Bacteria (∼2 × 106 CFU) in 20 μl of PBS were inoculated intranasally. Colonized bacteria were collected from the nasopharyngeal lavage fluid 5 days postinfection and were enumerated by plating serial dilutions. One mouse in the D39 WT group succumbed shortly after inoculation. Bars indicate the mean values. (B) Survival of mice during pneumococcal lung infection. BALB/c mice (n = 16) were challenged with the D39 WT and its ΔarcD mutant intranasally using ∼5 × 106 CFU of bacteria. The mice were monitored for up to 9 days (d) postinoculation. (C) Survival of mice during pneumococcal bacteremia. CBA/N mice (n = 15) were administered the D39 WT and its ΔarcD mutant intravenously using ∼2 × 105 CFU of bacteria. The mice were monitored for up to 7 days postinfection.
The virulence of the D39 ΔarcD mutant was also examined in mouse pneumonia and bacteremia models. In the pneumonia infection model, mice were inoculated intranasally and were monitored for their survival time. Only 25% of the D39 WT-infected mice (4/16) survived until the end of the experiment. However, 56% of the ΔarcD mutant-infected mice (9/16) survived for the duration of the experiment. Thus, the virulence of the D39 ΔarcD mutant was attenuated compared to that of the WT (Fig. 2B). Although this difference appears to be moderate, it is statistically significant (P < 0.01). A similar moderate attenuation by arcD deletion in D39 was also observed in the mouse bacteremia model (P < 0.05) (Fig. 2C). Taken together, these results suggest that ArcD affects the pathogenicity of D39 during in vivo infection.
Elevated phagocytosis and adherence by the ΔarcD mutant.
To further understand the role of ArcD in pneumococcal infection, we compared the D39 ΔarcD, ΔarcT, and ΔarcDT mutants with the WT during phagocytosis by macrophages and adherence to epithelial cells in vitro. Our results showed that both the ΔarcD and ΔarcDT mutants displayed increased (∼10-fold) phagocytosis by macrophages, whereas the ΔarcT mutant exhibited no difference compared to the WT (Fig. 3A). We also evaluated the adherence of D39 and its derivatives to A549 epithelial cells. In this assay, we found that about 0.01% of the inoculated WT bacteria were associated with A549 cells, which is consistent with an earlier report (48). Interestingly, both the ΔarcD and ΔarcDT mutants exhibited (∼1,000-fold) higher adherence to A549 cells, while the ΔarcT mutant was indistinguishable from the WT (Fig. 3B). The enhanced adherence to A549 cells by the ΔarcD and ΔarcDT mutants was also obvious using Gram staining (see Fig. S2 in the supplemental material). Invisibility of other infected strains using Gram staining is likely due to their weak adherence to A549 cells. These results indicate that the elevated phagocytosis and adherence exhibited by the ΔarcD and ΔarcDT mutants of D39 is associated with the deletion of arcD rather than arcT.
Fig 3.
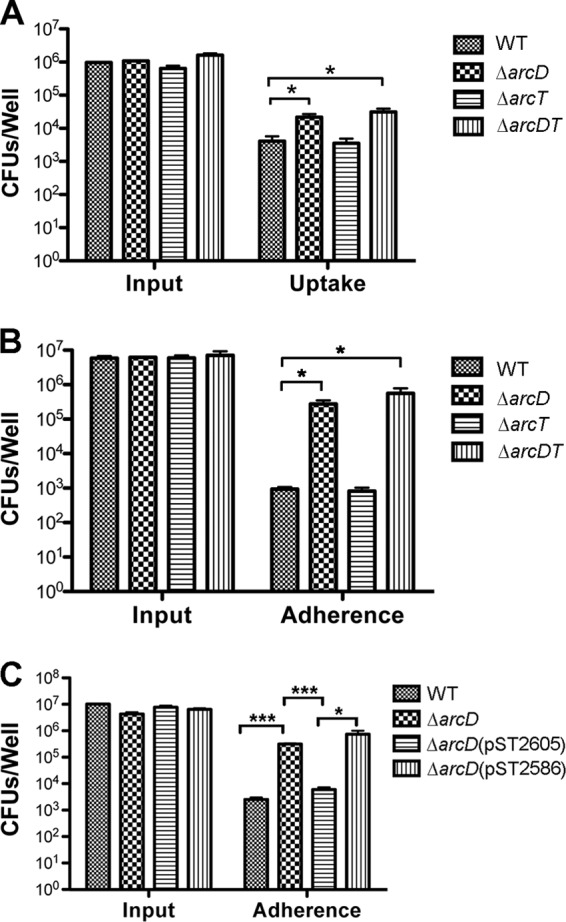
Infection of macrophages and epithelial cells. (A) Infection of J774A.1 macrophages with the D39 WT and its ΔarcD, ΔarcT, and ΔarcDT mutants. Macrophages were infected at an MOI of 5:1. The CFU of the input and the intracellular (uptake) bacteria were enumerated by plating from serial dilutions. (B) Bacterial adherence to A549 epithelial cells. A549 epithelial cells were infected at an MOI of 50:1 with the D39 WT and its ΔarcD, ΔarcT, and ΔarcDT mutants. Input and cell-associated (adherence) bacteria were enumerated by plating from serial dilutions. (C) Complementation of the D39 ΔarcD mutant. Infection of A549 cells at an MOI of 50:1 with the D39 WT, the ΔarcD and ΔarcD (pST2605) mutants, and the empty vector control strain ΔarcD (pST2586). All the data shown are the means of three independent experiments. Error bars denote SEMs. *, P < 0.05; ***, P < 0.001.
We also expressed arcD with the plasmid pST2605 controlled by a pneumococcal gyrA promoter. This construct, but not the vector control (pST2586), restored the levels in adherence to A549 cells by the ΔarcD mutant to that of the WT (Fig. 3C). This complementation result clearly indicates that the arcD gene contributes to the phenotypes that we have demonstrated with the ΔarcD mutant.
Deletion of D39 arcD leads to capsule impairment.
The capsule plays an essential role in adherence and phagocytosis of pneumococci (49). Based on the phenotypes we observed from the D39 ΔarcD mutant in vivo and in vitro, we hypothesized that deletion of arcD probably resulted in an impaired capsule in this serotype, which then enhanced the adherence to A549 cells and increased uptake by macrophages in vitro, and attenuated the virulence in mice. To further test our hypothesis, we generated a ΔarcD mutant of strain R6 (a nonencapsulated derivative of D39) and examined its adhesion to A549 cells. We found that deletion of arcD in R6 did not show any substantial increase in adherence over that of the R6 WT. Additionally, the R6 WT exhibited even higher levels of adherence to A549 cells than the D39 ΔarcD mutant (Fig. 4A), indicating that the absence of capsule dramatically enhances the bacterial adherence. These data imply that deletion of arcD in D39 affects the capsule. Therefore, we compared the capsules between the D39 WT and its ΔarcD mutant using multiple approaches.
Fig 4.

Capsule analyses. (A) Bacterial adherence to A549 epithelial cells. A549 epithelial cells were infected at an MOI of 50:1 with the WT and the ΔarcD mutant of strains D39 and R6. Input and cell-associated (adherence) bacteria were enumerated by plating from serial dilutions. Data shown are the means of three independent experiments. Error bars denote SEMs. *, P < 0.05. (B) Capsule assay of the R6 WT, the D39 WT, and its derivatives using a quantitative determination of uronic acids. Data shown are the means of three independent experiments. Error bars denote SEMs. *, P < 0.05. (C) Capsule assay of ST556 and its ΔarcD mutant using Stains-All reagent. Note that the different wavelengths indicated in panels B and C are due to different experimental procedures as described in Materials and Methods. (D) Capsule staining using immunofluorescence. The D39 WT and its ΔarcD mutant were detected using antiserum against pneumococcal type 2 capsule polysaccharides (indicated with arrows). Data are representative of two independent experiments. (E) Detection of capsule by LRR staining and transmission electron microscopy. Capsules are indicated with asterisks. Note that the magnification bars for the D39 and ST556 strains are in different scales to show the entire capsules.
First, the amount of capsular polysaccharides of D39 and its derivatives was quantified using an assay to determine uronic acids. The ΔarcD mutant was shown to have significantly fewer polysaccharides than its parental strain (Fig. 4B). This difference was fully restored by complementation with pST2605. As a control, uronic acids were undetectable in unencapsulated R6 using this assay (Fig. 4B). In contrast, the amount of capsular polysaccharides of ST556 was not altered by deletion of arcD, as detected using Stains-All (Fig. 4C). The capsule of the D39 ΔarcD mutant was also analyzed with specific anticapsular antibodies using immunofluorescence microscopy. The D39 WT showed a smooth layer of capsule, while its ΔarcD mutant exhibited interrupted fluorescent signals, suggesting capsular disruption in this mutant (Fig. 4D). These results indicate that the deletion of arcD alters the capsule in D39.
To further confirm our observations, we analyzed the morphology of the bacterial capsules using transmission electron microscopy (TEM). The D39 WT showed a condensed layer of capsule, while the capsule of its ΔarcD mutant was impaired (Fig. 4E). The complemented ΔarcD strain showed a capsule equivalent to that of its parental strain (data not shown). In contrast, the capsule of ST556 was not altered by deletion of arcD (Fig. 4E). Taken together, all these data indicate that deletion of arcD affects the capsule of D39.
We also hypothesized that the capsule defect in the D39 ΔarcD mutant may be caused by the dysregulation of the cps operon. We examined the expression of cps2A and cps2B, the first two genes of the cps operon, in the ΔarcD mutant and compared it to the WT. The quantitative RT-PCR analysis revealed that the transcriptional levels of these two genes were unaffected by arcD deletion (see Fig. S3 in the supplemental material). Thus, deletion of arcD does not regulate the expression of the cps operon.
S. pneumoniae ArcD is an arginine transporter.
Our data clearly indicate that D39 ArcD affects the capsule, but the function of this protein in pneumococci has not been experimentally established. S. pneumoniae arcD is located in a putative operon of ADS genes (see Fig. S1 in the supplemental material). In several bacteria, ArcD is a transmembrane protein that acts as an antiporter of arginine and ornithine (30, 50). Secondary-structure prediction analysis of pneumococcal ArcD revealed it to be a transmembrane protein (see Fig. S4 in the supplemental material) containing 12 helices that span the cytoplasmic membrane. Based on these analyses, we hypothesized that S. pneumoniae ArcD is an arginine transporter and tested this hypothesis by an uptake assay using 3H-labeled arginine. Our result revealed that the D39 WT obtained exogenous arginine rapidly (Fig. 5A), which is consistent with the reports that ArcD transports arginine within few minutes (25, 26). However, the arginine uptake by the ΔarcD mutant was reduced approximately 10- to 20-fold compared to that of the WT at all the tested time points (Fig. 5A). The level of labeled arginine in the mutant was restored by expression of arcD with pST2605 but not by the empty vector control alone (Fig. 5B). Similar results were observed with ST556 and its derivatives (data not shown). These results indicate that S. pneumoniae ArcD is an arginine transporter.
Fig 5.
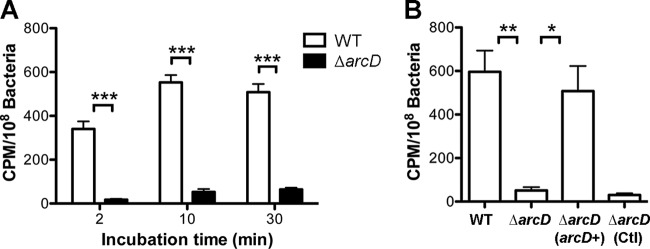
Uptake of arginine by the D39 WT and its derivatives. (A) Uptake of arginine at selected time points. Bacterial pellets were resuspended with buffer containing l-[3H]arginine as described in Materials and Methods for 0, 10, and 30 min. However, the actual incubation time of “0 min” needs approximately 2 min for the process. Therefore, it is noted as “2 min.” The counts per minute were normalized with bacterial CFU. Data shown are the means of three repeated experiments. Error bars denote SEMs. ***, P < 0.001. (B) Complementation of the ΔarcD mutant in arginine transport assay. Bacterial pellets were mixed with buffer containing l-[3H]arginine and were incubated for 10 min. Data shown are the means of three repeated experiments. Error bars denote SEMs. *, P < 0.05; **, P < 0.01.
Arginine levels are unlikely to affect the capsule of D39.
How an arginine transporter affects the D39 capsule is not clearly understood. Since we showed that ArcD was functional as an arginine transporter, it led us to postulate that the impaired capsule of the D39 ΔarcD mutant is probably due to limited intracellular arginine levels and that an “arginine-starved” D39 WT might mimic the ΔarcD mutant's phenotype. To examine this hypothesis, we prepared an arginine dropout CDM based on a published formula (51) and then supplemented this medium with various concentrations of arginine. An arginine titration curve was generated to determine the growth of the D39 WT and its ΔarcD mutant in the presence of serial dilutions of arginine (Fig. 6A). The D39 WT was unable to grow in the arginine dropout medium (Fig. 6A), suggesting that exogenous arginine is essential for pneumococcal growth. We then determined the adherence to A549 cells by D39 and its ΔarcD mutant grown in CDM with various concentrations of arginine (100, 25, 6.25, and 1.56 mg/liter, respectively). Infection was carried out in DMEM lacking arginine. To our surprise, we did not find any significant difference in adherence by the D39 WT grown in the tested CDM media (Fig. 6B), suggesting that arginine levels in the D39 WT may not affect bacterial adherence to A549 cells. Thus, it is unlikely that the intracellular arginine levels contribute directly toward the impaired capsule. The mechanism contributing to the capsule defect seen in the D39 ΔarcD mutant still remains to be investigated.
Fig 6.
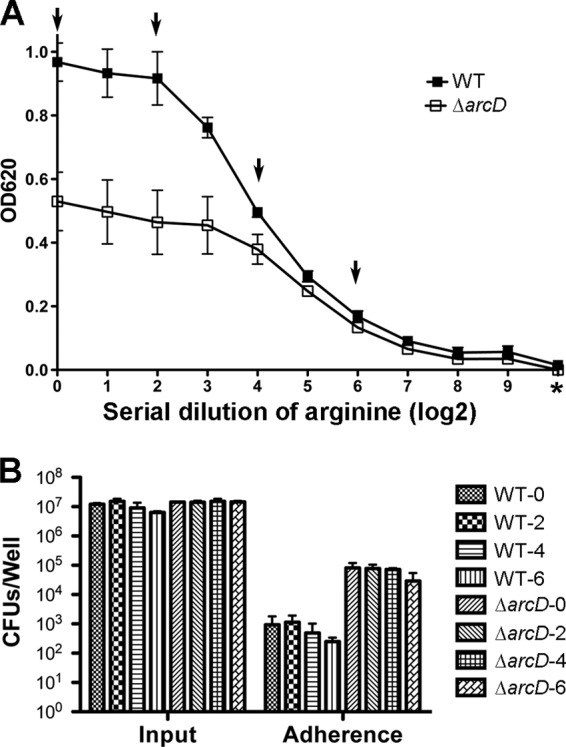
Effect of arginine on bacterial growth and adherence to A549 cells. (A) Titration of arginine concentrations in CDM. An arginine dropout CDM broth was prepared using the ingredients reported previously (36). For complete CDM, 100 mg/liter of arginine was added and was serially diluted using arginine dropout CDM, and these media were then used to grow the D39 WT and its ΔarcD mutant for 8 h. “*” denotes arginine dropout medium, and “0” indicates complete CDM. Arrows indicate various arginine dilutions in CDM that were used to grow bacteria for infection of A549 cells shown in panel B. (B) Adhesion of the D39 WT and its ΔarcD mutant grown in CDM with various amounts of arginine. Bacteria were grown for 8 h in the selected CDM as indicated in panel A. In this particular experiment, an arginine dropout DMEM was prepared using a DMEM that lacks arginine, lysine, glutamine, and sodium pyruvate (Invitrogen) and by supplementing these components, except arginine, back. This medium was then used to resuspend bacteria as inoculation medium. A549 cells were infected at an MOI of 50:1 with the D39 WT and its ΔarcD mutant. Input and cell-associated bacteria were enumerated by plating with serial dilutions. Data shown in both panels A and B are the means of three independent experiments. Error bars denote SEMs.
DISCUSSION
ArcD has been characterized as an arginine transporter in several bacteria (25, 26, 30, 50). However, little is known about the role of ArcD in bacterial virulence and pathogenicity. It has been reported that inactivation of arcD in Staphylococcus aureus leads to complete inhibition of arginine transport and also causes reduced accumulation of polysaccharide intercellular adhesin (52). In the present study, we demonstrated that S. pneumoniae ArcD functions as an arginine transporter. Surprisingly, the capsule of the D39 ΔarcD mutant was significantly impaired. This impairment is probably independent of its defect in arginine transport. In an arginine dropout CDM broth, bacteria failed to grow in the absence of exogenous arginine (Fig. 6A), suggesting that this pneumococcus is auxotrophic for arginine, which is consistent with a recent report (53) and similar to observations with other bacteria (54). In addition, the ΔarcD mutant of D39 was able to grow in a complete CDM, which contains 100 mg/liter of arginine, although its growth rate was slightly lower than that of the parental strain (see Fig. S5 in the supplemental material). These data suggest that S. pneumoniae may utilize an ArcD-independent mechanism to take up arginine from the environment, which is in accordance to the results of the arginine transport assays. The D39 WT grown in media with extremely low levels of arginine, which were minimal to maintain the bacterial growth, was unable to mimic the ΔarcD mutant in adherence to A549 cells, implying that arginine deprivation may not affect bacterial adherence. Thus, the bacterial arginine levels do not seem to influence the capsule of D39.
The impairment of the capsule in the D39 ΔarcD mutant is unlikely due to a change of glycosyl composition in the capsule. In the present study, we also purified the capsular materials from the D39 WT and the ΔarcD mutant and analyzed the glycosyl composition and linkage. Our result showed that there is no significant change in the ΔarcD mutant compared to the WT (data not shown). In S. pneumoniae, the majority of the capsular polysaccharides remain associated with bacteria via covalent linkages either to the peptidoglycan or to the membrane components (6). Based on our observations, we hypothesize that D39 ArcD plays a role in anchoring of the capsular material either directly or indirectly. Thus, deletion of arcD somehow reduces the amount of capsular materials at the bacterial surface. Surprisingly, deletion of arcD in ST556 did not affect the capsule (Fig. 4), suggesting that ArcD may play diverse roles in pathogenicity of different pneumococcal serotypes. We also demonstrated that deletion of arcD in D39 does not regulate the expression of the cps operon, but this observation does not rule out the role of ArcD in regulating capsule biosynthesis. Since D39 and ST556 possess biochemically distinct capsules (42), it is also possible that ArcD controls the synthesis of a certain component that is present in the capsule of D39 but not ST556. We are currently investigating such possibilities.
The D39 ΔarcD mutant had increased adherence in vitro and enhanced nasopharyngeal colonization in mice compared to those of the WT. Noticeably, ST556 and its ΔarcD mutant both exhibited even greater colonization than the D39 ΔarcD mutant. ST556 belongs to serotype 19F, and bacteria of this serotype are commonly isolated as carriage strains (55). The increased adherence of the D39 ΔarcD mutant can be explained by its impaired capsule, which is consistent with the observations by comparison between fully encapsulated and nonencapsulated pneumococcal isolates (48, 49, 51, 56–58). It has also been reported that during in vivo infection of S. pneumoniae, the capsule is required for the colonization to overcome the clearance by the innate defense, and several nonencapsulated derivatives are unable to colonize mice (43, 59, 60). However, substantially reduced levels of capsule may be sufficient for murine carriage, as observed with a mutant that produces capsule at only ∼20% of the parental levels (43). Therefore, the enhanced colonization of the D39 ΔarcD mutant may not directly relate to less polysaccharide material but rather is caused by a better exposure of the bacterial surface proteins that interact with host cells (59, 61, 62).
In conclusion, we show that S. pneumoniae D39 ArcD affects the capsule, which results in significantly enhanced phagocytosis by macrophages and attenuation in bacterial virulence. However, the impaired capsule of the ΔarcD mutant may not result from the lack of arginine transport but is likely caused by a loss of interaction between the capsular material and this transmembrane protein, either directly or indirectly. The molecular basis of how ArcD affects the capsule will be further investigated in a future study.
Supplementary Material
ACKNOWLEDGMENTS
We thank Michael Jacobs for providing S. pneumoniae isolates from OM patients, Paul Kolenbrander for providing the pVA838 plasmid, and Adam Underwood for technical assistance and critical reading of the manuscript. We are grateful for the support of the UCSD Glycotechnology Core Facility for analyses of sugar composition and linkage and the Wadsworth Center Electron Microscopy and Light Microscopy Core Facilities.
This study was supported by National Institutes of Health grant DC006917 to D.W.M.
Footnotes
Published ahead of print 5 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00778-13.
REFERENCES
- 1.Kim KS. 2010. Acute bacterial meningitis in infants and children. Lancet Infect. Dis. 10:32–42 [DOI] [PubMed] [Google Scholar]
- 2.van der Poll T, Opal SM. 2008. Host-pathogen interactions in sepsis. Lancet Infect. Dis. 8:32–43 [DOI] [PubMed] [Google Scholar]
- 3.Mehr S, Wood N. 2012. Streptococcus pneumoniae—a review of carriage, infection, serotype replacement and vaccination. Paediatr. Respir. Rev. 13:258–264 [DOI] [PubMed] [Google Scholar]
- 4.Hyams C, Camberlein E, Cohen JM, Bax K, Brown JS. 2010. The Streptococcus pneumoniae capsule inhibits complement activity and neutrophil phagocytosis by multiple mechanisms. Infect. Immun. 78:704–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hyams C, Yuste J, Bax K, Camberlein E, Weiser JN, Brown JS. 2010. Streptococcus pneumoniae resistance to complement-mediated immunity is dependent on the capsular serotype. Infect. Immun. 78:716–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yother J. 2011. Capsules of Streptococcus pneumoniae and other bacteria: paradigms for polysaccharide biosynthesis and regulation. Annu. Rev. Microbiol. 65:563–581 [DOI] [PubMed] [Google Scholar]
- 7.Briles DE, Tart RC, Swiatlo E, Dillard JP, Smith P, Benton KA, Ralph BA, Brooks-Walter A, Crain MJ, Hollingshead SK, McDaniel LS. 1998. Pneumococcal diversity: considerations for new vaccine strategies with emphasis on pneumococcal surface protein A (PspA). Clin. Microbiol. Rev. 11:645–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shapiro ED, Berg AT, Austrian R, Schroeder D, Parcells V, Margolis A, Adair RK, Clemens JD. 1991. The protective efficacy of polyvalent pneumococcal polysaccharide vaccine. N. Engl. J. Med. 325:1453–1460 [DOI] [PubMed] [Google Scholar]
- 9.Swiatlo E, Ware D. 2003. Novel vaccine strategies with protein antigens of Streptococcus pneumoniae. FEMS Immunol. Med. Microbiol. 38:1–7 [DOI] [PubMed] [Google Scholar]
- 10.Hansen J, Black S, Shinefield H, Cherian T, Benson J, Fireman B, Lewis E, Ray P, Lee J. 2006. Effectiveness of heptavalent pneumococcal conjugate vaccine in children younger than 5 years of age for prevention of pneumonia: updated analysis using World Health Organization standardized interpretation of chest radiographs. Pediatr. Infect. Dis. J. 25:779–781 [DOI] [PubMed] [Google Scholar]
- 11.Obaro SK. 2002. The new pneumococcal vaccine. Clin. Microbiol. Infect. 8:623–633 [DOI] [PubMed] [Google Scholar]
- 12.Doern GV, Heilmann KP, Huynh HK, Rhomberg PR, Coffman SL, Brueggemann AB. 2001. Antimicrobial resistance among clinical isolates of Streptococcus pneumoniae in the United States during 1999–2000, including a comparison of resistance rates since 1994–1995. Antimicrob. Agents Chemother. 45:1721–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griswold A, Chen YY, Snyder JA, Burne RA. 2004. Characterization of the arginine deiminase operon of Streptococcus rattus FA-1. Appl. Environ. Microbiol. 70:1321–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Venugopal V, Nadkarni GB. 1977. Regulation of the arginine dihydrolase pathway in Clostridium sporogenes. J. Bacteriol. 131:693–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramos F, Stalon V, Pierard A, Wiame JM. 1967. The specialization of the two ornithine carbamoyltransferases of Pseudomonas. Biochim. Biophys. Acta 139:98–106 [DOI] [PubMed] [Google Scholar]
- 16.Stalon V, Ramos F, Pierard A, Wiame JM. 1972. Regulation of the catabolic ornithine carbamoyltransferase of Pseudomonas fluorescens. A comparison with the anabolic transferase and with a mutationally modified catabolic transferase. Eur. J. Biochem. 29:25–35 [DOI] [PubMed] [Google Scholar]
- 17.Mitruka BM, Costilow RN. 1967. Arginine and ornithine catabolism by Clostridium botulinum. J. Bacteriol. 93:295–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt GC, Logan MA, Tytell AA. 1952. The degradation of arginine by Clostridium perfringens (BP6K). J. Biol. Chem. 198:771–783 [PubMed] [Google Scholar]
- 19.Ottow JCG. 1974. Arginine dihydrolase activity in species of the genus Bacillus revealed by thin-layer chromatography. J. Gen. Microbiol. 84:209–213 [DOI] [PubMed] [Google Scholar]
- 20.Slade HD. 1953. Hydrolysis of arginine by soluble enzymes of Streptococcus faecalis. Arch. Biochem. Biophys. 42:204–211 [DOI] [PubMed] [Google Scholar]
- 21.Benga L, Goethe R, Rohde M, Valentin-Weigand P. 2004. Non-encapsulated strains reveal novel insights in invasion and survival of Streptococcus suis in epithelial cells. Cell. Microbiol. 6:867–881 [DOI] [PubMed] [Google Scholar]
- 22.Marquis RE, Bender GR, Murray DR, Wong A. 1987. Arginine deiminase system and bacterial adaptation to acid environments. Appl. Environ. Microbiol. 53:198–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong Y, Chen YY, Snyder JA, Burne RA. 2002. Isolation and molecular analysis of the gene cluster for the arginine deiminase system from Streptococcus gordonii DL1. Appl. Environ. Microbiol. 68:5549–5553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zúñiga M, Champomier-Verges M, Zagorec M, Perez-Martinez G. 1998. Structural and functional analysis of the gene cluster encoding the enzymes of the arginine deiminase pathway of Lactobacillus sake. J. Bacteriol. 180:4154–4159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verhoogt HJ, Smit H, Abee T, Gamper M, Driessen AJ, Haas D, Konings WN. 1992. arcD, the first gene of the arc operon for anaerobic arginine catabolism in Pseudomonas aeruginosa, encodes an arginine-ornithine exchanger. J. Bacteriol. 174:1568–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Driessen AJ, Poolman B, Kiewiet R, Konings W. 1987. Arginine transport in Streptococcus lactis is catalyzed by a cationic exchanger. Proc. Natl. Acad. Sci. U. S. A. 84:6093–6097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poolman B. 1990. Precursor/product antiport in bacteria. Mol. Microbiol. 4:1629–1636 [DOI] [PubMed] [Google Scholar]
- 28.Poolman B, Driessen AJ, Konings WN. 1987. Regulation of arginine-ornithine exchange and the arginine deiminase pathway in Streptococcus lactis. J. Bacteriol. 169:5597–5604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seep-Feldhaus AH, Kalinowski J, Puhler A. 1991. Molecular analysis of the Corynebacterium glutamicum gene involved in lysine uptake. Mol. Microbiol. 5:2995–3005 [DOI] [PubMed] [Google Scholar]
- 30.Bourdineaud JP, Heierli D, Gamper M, Verhoogt HJ, Driessen AJ, Konings WN, Lazdunski C, Haas D. 1993. Characterization of the arcD arginine:ornithine exchanger of Pseudomonas aeruginosa. Localization in the cytoplasmic membrane and a topological model. J. Biol. Chem. 268:5417–5424 [PubMed] [Google Scholar]
- 31.Hava DL, LeMieux J, Camilli A. 2003. From nose to lung: the regulation behind Streptococcus pneumoniae virulence factors. Mol. Microbiol. 50:1103–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lau GW, Haataja S, Lonetto M, Kensit SE, Marra A, Bryant AP, McDevitt D, Morrison DA, Holden DW. 2001. A functional genomic analysis of type 3 Streptococcus pneumoniae virulence. Mol. Microbiol. 40:555–571 [DOI] [PubMed] [Google Scholar]
- 33.Polissi A, Pontiggia A, Feger G, Altieri M, Mottl H, Ferrari L, Simon D. 1998. Large-scale identification of virulence genes from Streptococcus pneumoniae. Infect. Immun. 66:5620–5629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen H, Ma Y, Yang J, O'Brien CJ, Lee SL, Mazurkiewicz JE, Haataja S, Yan JH, Gao GF, Zhang JR. 2008. Genetic requirement for pneumococcal ear infection. PLoS One 3:e2950. 10.1371/journal.pone.0002950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joloba ML, Windau A, Bajaksouzian S, Appelbaum PC, Hausdorff WP, Jacobs MR. 2001. Pneumococcal conjugate vaccine serotypes of Streptococcus pneumoniae isolates and the antimicrobial susceptibility of such isolates in children with otitis media. Clin. Infect. Dis. 33:1489–1494 [DOI] [PubMed] [Google Scholar]
- 36.van de Rijn I, Kessler RE. 1980. Growth characteristics of group A streptococci in a new chemically defined medium. Infect. Immun. 27:444–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pozzi G, Masala L, Iannelli F, Manganelli R, Havarstein LS, Piccoli L, Simon D, Morrison DA. 1996. Competence for genetic transformation in encapsulated strains of Streptococcus pneumoniae: two allelic variants of the peptide pheromone. J. Bacteriol. 178:6087–6090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu L, Ma Y, Zhang J-R. 2006. Streptococcus pneumoniae recruits complement factor H through the amino terminus of CbpA. J. Biol. Chem. 281:15464–15474 [DOI] [PubMed] [Google Scholar]
- 39.Sung CK, Li H, Claverys JP, Morrison DA. 2001. An rpsL cassette, Janus, for gene replacement through negative selection in Streptococcus pneumoniae. Appl. Environ. Microbiol. 67:5190–5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blumenkrantz N, Asboe-Hansen G. 1973. New method for quantitative determination of uronic acids. Anal. Biochem. 54:484–489 [DOI] [PubMed] [Google Scholar]
- 41.Morona JK, Morona R, Paton JC. 2006. Attachment of capsular polysaccharide to the cell wall of Streptococcus pneumoniae type 2 is required for invasive disease. Proc. Natl. Acad. Sci. U. S. A. 103:8505–8510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Dam J, Fleer A, Snippe H. 1990. Immunogenicity and immunochemistry of Streptococcus pneumoniae capsular polysaccharides. Antonie Van Leeuwenhoek 58:1–47 [DOI] [PubMed] [Google Scholar]
- 43.Magee AD, Yother J. 2001. Requirement for capsule in colonization by Streptococcus pneumoniae. Infect. Immun. 69:3755–3761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schrager HM, Rheinwald JG, Wessels MR. 1996. Hyaluronic acid capsule and the role of streptococcal entry into keratinocytes in invasive skin infection. J. Clin. Invest. 98:1954–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.National Research Council 1996. Guide for the care and use of laboratory animals. National Academies Press, Washington, DC [Google Scholar]
- 46.Wu HY, Virolainen A, Mathews B, King J, Russell MW, Briles DE. 1997. Establishment of a Streptococcus pneumoniae nasopharyngeal colonization model in adult mice. Microb. Pathog. 23:127–137 [DOI] [PubMed] [Google Scholar]
- 47.El Qaidi S, Yang J, Zhang JR, Metzger DW, Bai G. 2013. The vitamin B(6) biosynthesis pathway in Streptococcus pneumoniae is controlled by pyridoxal 5′-phosphate and the transcription factor PdxR and has an impact on ear infection. J. Bacteriol. 195:2187–2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adamou JE, Wizemann TM, Barren P, Langermann S. 1998. Adherence of Streptococcus pneumoniae to human bronchial epithelial cells (BEAS-2B). Infect. Immun. 66:820–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hammerschmidt S, Wolff S, Hocke A, Rosseau S, Muller E, Rohde M. 2005. Illustration of pneumococcal polysaccharide capsule during adherence and invasion of epithelial cells. Infect. Immun. 73:4653–4667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wimmer F, Oberwinkler T, Bisle B, Tittor J, Oesterhelt D. 2008. Identification of the arginine/ornithine antiporter ArcD from Halobacterium salinarum. FEBS Lett. 582:3771–3775 [DOI] [PubMed] [Google Scholar]
- 51.Talbot UM, Paton AW, Paton JC. 1996. Uptake of Streptococcus pneumoniae by respiratory epithelial cells. Infect. Immun. 64:3772–3777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu Y, Weiss EC, Otto M, Fey PD, Smeltzer MS, Somerville GA. 2007. Staphylococcus aureus biofilm metabolism and the influence of arginine on polysaccharide intercellular adhesin synthesis, biofilm formation, and pathogenesis. Infect. Immun. 75:4219–4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kloosterman TG, Kuipers OP. 2011. Regulation of arginine acquisition and virulence gene expression in the human pathogen Streptococcus pneumoniae by transcription regulators ArgR1 and AhrC. J. Biol. Chem. 286:44594–44605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ledesma OV, De Ruiz Holgado AP, Oliver G, De Giori GS, Raibaud P, Galpin JV. 1977. A synthetic medium for comparative nutritional studies of lactobacilli. J. Appl. Bacteriol. 42:123–133 [DOI] [PubMed] [Google Scholar]
- 55.Syrogiannopoulos GA, Grivea IN, Katopodis GD, Geslin P, Jacobs MR, Beratis NG. 2000. Carriage of antibiotic-resistant Streptococcus pneumoniae in Greek infants and toddlers. Eur. J. Clin. Microbiol. Infect. Dis. 19:288–293 [DOI] [PubMed] [Google Scholar]
- 56.Cundell DR, Weiser JN, Shen J, Young A, Tuomanen EI. 1995. Relationship between colonial morphology and adherence of Streptococcus pneumoniae. Infect. Immun. 63:757–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim JO, Weiser JN. 1998. Association of intrastrain phase variation in quantity of capsular polysaccharide and teichoic acid with the virulence of Streptococcus pneumoniae. J. Infect. Dis. 177:368–377 [DOI] [PubMed] [Google Scholar]
- 58.Selinger DS, Reed WP. 1979. Pneumococcal adherence to human epithelial cells. Infect. Immun. 23:545–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bogaert D, van Belkum A, Sluijter M, Luijendijk A, de Groot R, Rumke HC, Verbrugh HA, Hermans PW. 2004. Colonisation by Streptococcus pneumoniae and Staphylococcus aureus in healthy children. Lancet 363:1871–1872 [DOI] [PubMed] [Google Scholar]
- 60.Nelson AL, Roche AM, Gould JM, Chim K, Ratner AJ, Weiser JN. 2007. Capsule enhances pneumococcal colonization by limiting mucus-mediated clearance. Infect. Immun. 75:83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hammerschmidt S. 2009. Surface-exposed adherence molecules of Streptococcus pneumoniae. Methods Mol. Biol. 470:29–45 [DOI] [PubMed] [Google Scholar]
- 62.Swiatlo E, Champlin FR, Holman SC, Wilson WW, Watt JM. 2002. Contribution of choline-binding proteins to cell surface properties of Streptococcus pneumoniae. Infect. Immun. 70:412–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Avery OT, Macleod CM, McCarty M. 1944. Studies on the chemical nature of the substance inducing transformation of pneumococcal types: induction of transformation by a desoxyribonucleic acid fraction isolated from Pneumococcus type III. J. Exp. Med. 79:137–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tiraby JG, Fox MS. 1973. Marker discrimination in transformation and mutation of pneumococcus. Proc. Natl. Acad. Sci. U. S. A. 70:3541–3545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Macrina FL, Evans RP, Tobian JA, Hartley DL, Clewell DB, Jones KR. 1983. Novel shuttle plasmid vehicles for Escherichia-Streptococcus transgeneric cloning. Gene 25:145–150 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.