

Abstract
Biofilm formation and persistence are essential components for the continued survival of pathogens inside the host and constitute a major contributor to the development of chronic wounds with resistance to antimicrobial compounds. Understanding these processes is crucial for control of biofilm-mediated disease. Though chronic wound infections are often polymicrobial in nature, much of the research on chronic wound-related microbes has focused on single-species models. Klebsiella pneumoniae and Pseudomonas aeruginosa are microbes that are often found together in wound isolates and are able to form stable in vitro biofilms, despite the antagonistic nature of P. aeruginosa with other organisms. Mutants of the K. pneumoniae strain IA565 lacking the plasmid-borne mrkD1P gene were less competitive than the wild type in an in vitro dual-species biofilm model with P. aeruginosa (PAO1). PAO1 spent medium inhibited the formation of biofilm of mrkD1P-deficient mutants and disrupted preestablished biofilms, with no effect on IA565 and no effect on the growth of the wild type or mutants. A screen using a two-allele PAO1 transposon library identified the LasB elastase as the secreted effector involved in biofilm disruption, and a purified version of the protein produced results similar to those with PAO1 spent medium. Various other proteases had a similar effect, suggesting that the disruption of the mrkD1P gene causes sensitivity to general proteolytic effects and indicating a role for MrkD1P in protection against host antibiofilm effectors. Our results suggest that MrkD1P allows for competition of K. pneumoniae with P. aeruginosa in a mixed-species biofilm and provides defense against microbial and host-derived proteases.
INTRODUCTION
Despite medical advances that have decreased mortality from infectious agents, recurrent and chronic wound infections are still a growing burden on the global population (1). Microorganisms that are often associated with these chronic wound infections exist in persistent sessile communities known as biofilms that consist of adherent cells contained within an extracellular polymeric substance (EPS) composed of polysaccharides, DNA, and proteins (2, 3). These communities are phenotypically distinct from their planktonic counterparts, providing the organisms with protection from various antimicrobial agents (4, 5). Biofilm development is associated with over half of all human chronic infections and is essential to the continued survival of pathogens where the host utilizes many biofilm countermeasures, including phagocytosis and secretion of effectors such as proteases, lactoferrin, oxygen radicals, and defensins, among others (6–9). Naturally occurring, biofilm-mediated chronic wound infections are typically polymicrobial in nature and can be composed of numerous species of bacteria and fungi (10). Mutualistic, commensal, and competitive relationships have been observed between various sets of organisms in mixed communities, adding to the already complex nature of the biofilm (11–13). New virulence factors that attack both the host and competing species have also been identified from these organisms (14, 15). Still, much of the research on chronic wound-related microbes has, to this point, focused on single-species models, making it likely that important virulence factors, targets, and disease mechanisms are being missed.
Klebsiella pneumoniae and Pseudomonas aeruginosa are two biofilm-producing organisms that are often found together in chronic wound infections (10). K. pneumoniae is a Gram-negative bacterium that is found ubiquitously in both terrestrial and aquatic environments (16). The organism is responsible for respiratory illnesses and urinary tract infections (UTIs) but is an increasing cause of infection at surgical sites and of chronic wound infections (17). The emergence of extended-spectrum β-lactamase (ESBL)- and carbapenemase-producing strains is a cause of growing concern in the medical community (18). Biofilm formation in K. pneumoniae is a major virulence factor used to colonize the human host (19). This activity is mediated by a type 3 fimbria that is used for adherence to various forms of extracellular matrix (ECM) proteins, such as collagen or fibronectin (19, 20). The recognition of each type of ECM protein is mediated by various forms of the fimbrial tip protein MrkD (21). For instance the MrkD1P form, found in very few strains of K. pneumoniae (but found on a stable plasmid in strain IA565, a strain whose biofilm formation is well studied), binds to type V collagen, while MrkD1C, found on the chromosome in a vast majority of strains, including IA565, binds to collagen types IV and V (22). Although not essential for biofilm production, MrkD plays a role in the physiology of the K. pneumoniae biofilm and is an important factor in pathogenesis (19, 20).
P. aeruginosa is a ubiquitous organism that is able to form biofilms on numerous biotic and abiotic surfaces and causes a wide range of infections, from lung infections with cystic fibrosis patients to urinary tract and kidney infections, which are often attributed to contaminated medical devices (23, 24). P. aeruginosa, though often found with other organisms in wound infections, maintains an antagonistic relationship with many other wound-related organisms in vitro, including Staphylococcus aureus, Staphylococcus epidermidis, and Burkholderia cepacia (15, 25, 26). This outcome is due to the large number of secreted effectors, such as phenazines, rhamnolipids, cis-2-decenoic acid, alkaline protease, exotoxins, and elastase, which are used by P. aeruginosa to outcompete other organisms (27–31). Interestingly, it has previously been demonstrated that K. pneumoniae forms stable dual-species biofilms with P. aeruginosa, suggesting the presence of defense mechanisms capable of withstanding the onslaught of these P. aeruginosa effectors (32).
Though the structure of a dual-species K. pneumoniae/P. aeruginosa biofilm has previously been studied, the interactions between the two organisms have yet to be fully understood. The aim of this study was to identify the protective properties of K. pneumoniae that allow for coexistence with P. aeruginosa in a biofilm setting. Using the reference laboratory P. aeruginosa strain PAO1 and the K. pneumoniae strain IA565, which carries both the 1P and 1C forms of the mrkD gene, we identified a protective advantage conferred by the MrkD1P protein in the dual-species biofilm setting to the LasB secreted protease of P. aeruginosa.
MATERIALS AND METHODS
Bacterial strains, plasmid construction, and culture conditions.
The K. pneumoniae and P. aeruginosa strains and recombinant plasmids are listed in Table 1. PAO1 transposon mutants were obtained from the two-allele mutant library from the Washington Genome Center. Escherichia coli strain DH5α was used for cloning, and WM3046 (a gift of William Metcalf, University of Illinois) was used for conjugation. Plasmids pBMC12 and pBMC13 were created by amplifying the kanamycin resistance marker from pKD4, using the primers FRT Up ScaI (5′-GCGCGCAGTACTGTGTAGGCTGGAGCTGCTTC-3′) and FRT Dn ScaI (5′-GCGCGCAGTACTCATATGAATATCCTCCTTA-3′), and cloning the fragment into the ScaI site found in the chloramphenicol marker in pACYC184 and pFK52, respectively. All strains were cultured in YEPD broth (10% yeast extract, 20% peptone, 20% dextrose) or on LB plates. Antibiotics were used at the following concentrations: 20 μg ml−1 or 150 μg ml−1 chloramphenicol, 15 μg ml−1 tetracycline, 50 μg ml−1 kanamycin, and 3 mg ml−1 carbenicillin.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| P. aeruginosa | ||
| PAO1 | Wild-type reference strain | 43 |
| PA0401 | Mutant PW1741, noncatalytic dihydroorotase-like protein | 37 |
| PA0697 | Mutant PW2265, hypothetical protein | 37 |
| PA3135 | Mutant PW7392, probable transcriptional regulator | 37 |
| PA2478 | Mutant PW5161, probable thiol:disulfide interchange protein | 37 |
| PA3742 | Mutant PW7303, elastase LasB | 37 |
| PA4280 | Mutant PW8208, BirA bifunctional protein | 37 |
| PA0502 | Mutant PW1924, probable biotin biosynthesis protein BirH | 37 |
| PA1684 | Mutant PW3991, 1,2-dihydroxy-3-keto-5-methylthiopentene dioxygenase MtnD | 37 |
| PA3383 | Mutant PW10408, binding protein component of ABC phosphonate transporter | 37 |
| K. pneumoniae | ||
| IA565 | Wild type containing chromosome- and plasmid-borne mrk gene cluster | 44 |
| IApc35 | Fimbriate and nonhemagglutinating strain of IA565, lacking plasmid-borne mrk gene cluster | 44 |
| BC2 | IA565 with Himar1 transposon insertion in the plasmid-borne mrkD1P gene | This study |
| UIR082 | Strain possessing mrkD1C but not mrkD1P | 36 |
| UIR207 | Strain possessing mrkD1C but not mrkD1P | 36 |
| UI917 | Strain possessing mrkD1P but not mrkD1C | 36 |
| UI925 | Strain possessing mrkD1P but not mrkD1C | 36 |
| THK12113 | Strain possessing mrkD1C but not mrkD1P | 36 |
| CAS55 | C3091 K. pneumoniae UTI clinical isolate | 45 |
| E. coli NEB10α | New England Biolabs | |
| Cloning vectors | ||
| pGEM-T Easy | Promega | |
| pACYC184 | New England Biolabs | |
| Recombinant plasmids | ||
| pFK52 | Plasmid-borne mrkD gene from K. pneumoniae IA565 cloned into pACYC184 | 46 |
| pBMC12 | pACYC plasmid with kanamycin resistance marker in place of chloramphenicol resistance marker | This study |
| pBMC13 | pFK52 plasmid with kanamycin resistance marker in place of chloramphenicol resistance marker | This study |
| pBMC14 | Chromosomal-borne mrk gene cluster from K. pneumoniae IA565 cloned into pGEM-T Easy | This study |
| pMrkD | mrkD gene from C3091 cloned into low-copy-no. vector pGB17 | 45 |
| pKEK1410 | Suicide vector pFD1 carrying a chloramphenicol marker and encoding Himar1-derived transposon and Himar1 transposase | Gift of Karl E. Klose |
Transposon mutagenesis of IA565.
The chloramphenicol-resistant derivative of pFD1 (33), pKEK1410, which encodes a Himar1-derived transposon and Himar1 transposase, was introduced into IA565 by conjugation on LB medium containing 100 μM diaminopimelic acid (DAPA) and 1 mM β-d-1-thiogalactopyranoside (IPTG). Mutants were selected on LB plates containing no DAPA with chloramphenicol. Gene disruptions in mutants defective for competition with PAO1 were identified through inverse PCR. Briefly, the chromosomal DNA of the IA565 mutant was digested with BamHI, ligated, transformed into WM3046, and plated on LB containing DAPA and chloramphenicol. Plasmids were miniprepped from the transformants and the insertion site identified by sequencing using the primer pFD1 Up (5′-ATGCATTTAATACTAGCGACGCC-3′).
CI assays.
A biofilm competitive index (CI) was generated using a mixture of K. pneumoniae and P. aeruginosa. The lacZ+-containing K. pneumoniae strains IA565, IApc35, and BC2 were each mixed with the lacZ-deficient P. aeruginosa strain PAO1 at a 1:1 ratio in a 96-well plate and allowed to form a biofilm for 24 h in YEPD medium at 37°C with shaking. The resultant biofilms were washed 3 times, resuspended in phosphate-buffered saline (PBS) by vigorous vortexing, and dilutions plated on LB containing X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) and IPTG. Blue (K. pneumoniae) and white (P. aeruginosa) colonies were counted, and the competitive index was calculated as the K. pneumoniae/P. aeruginosa output ratio divided by the input inoculum ratio. Significant differences in CI were calculated using one-way analysis of variance (ANOVA).
Bacterial supernatant preparation.
IA565 and PAO1 were grown in YEPD medium for 16 h at 37°C, cells harvested by centrifugation at 4,000 rpm for 30 min, and the resultant supernatant sterilized by filtration. The supernatants were immediately lyophilized, and a 10× stock was created by reconstitution in YEPD medium. For experiments using reconstituted supernatant, a similarly lyophilized and reconstituted YEPD was used. PAO1 transposon mutants were streaked onto LB agar and a single colony inoculated into YEPD. The mutants were grown at 37°C overnight with shaking and harvested by centrifugation, and the resultant supernatant was sterilized by filtration and used immediately. Supernatants were made fresh for each experiment.
Biofilm dispersion assays.
Static biofilm formation assays were performed in 96-well microtiter plates coated with human type I collagen (354269; BD Biosciences). K. pneumoniae strains, or E. coli DH5α carrying mrkD1P and mrkD1C vectors, were inoculated into 200 μl of YEPD medium at an initial optical density at 600 nm (OD600) of 0.05 and grown at 37°C for 16 h with shaking. Supernatants were pulled from the resultant biofilms and replaced with 200 μl reconstituted supernatants (1× concentration), filter sterilized PAO1 transposon mutant supernatant, or the following compounds dissolved in YEPD: 300 μg ml−1 purified elastase B (PE961; Elastin Products Company), 50 μg ml−1 porcine pancreatic elastase (E7885; Sigma), 50 μg ml−1 PMN elastase (BML-SE284; Enzo Life Sciences), 50 μg ml−1 porcine pancreatic trypsin (T0303; Sigma), 1 μg ml−1 bovine chymotrypsin (CHY5S; Sigma); 3 μg ml−1 human antichymotrypsin (A9285; Sigma), 100 μg ml−1 proteinase K (19131; Qiagen), 0.1% sodium dodecyl sulfate (SDS) (L6026; Sigma); 100 μg ml−1 polymyxin B (92283; Sigma), or 200 Kunitz units ml−1 DNase (79254; Qiagen). Treated biofilms were incubated for another 16 h with shaking at 37°C. Biofilms were washed 3 times with PBS solution, stained with a 0.4% solution of crystal violet, washed 3 times with PBS again, and dissolved in 33% acetic acid, and a spectrophotometer reading was taken at OD570.
Bacterial growth experiments.
Cell growth experiments on K. pneumoniae strains were performed in 96-well microtiter plates. Strains were inoculated into 200 μl of reconstituted YEPD or PAO1 supernatant at an OD600 reading of 0.05, and readings were taken every 2 h for 16 h. To evaluate the ability of the bacteria to form a biofilm in each medium, the growth medium was removed, the wells stained with crystal violet, the biofilms dissolved with 33% acetic acid, and an OD570 reading taken.
Visualization of cell morphology.
To determine the cell morphology of K. pneumoniae strains, biofilm-coated MBEC pegs (Innovotech) were fixed in 2.5% glutaraldehyde in 0.1 M PBS (pH 7.2), washed 3 times in PBS, and dehydrated through an ethanol series and hexamethyldisilazane. Samples were mounted with double-sided tape to specimen stubs, followed by gold-platinum (50:50) ion coating (108 Auto Sputter Coater; Ted Pella, Inc., Redding, CA). Samples were visualized using a Carl Zeiss EVO-40 scanning electron microscope (Oberkochen, Germany) operated at the scanning voltage of 10 kV. Cell length was measured using ImageJ 1.46r software (National Institutes of Health).
RESULTS
K. pneumoniae strains lacking the plasmid-borne type 3 fimbrial adhesin MrkD1P are at a competitive disadvantage in a dual-species biofilm with P. aeruginosa.
Though a dual-species K. pneumoniae/P. aeruginosa biofilm has previously been studied (32), the interactions between these two species have yet to be fully analyzed. To do this, we chose to look at the interaction of P. aeruginosa strain PAO1 with K. pneumoniae strain IA565, a strain whose biofilm development has been well studied (21, 32, 34, 35). To form a biofilm, IA565 utilizes type 3 fimbriae, coded for by two mrk gene clusters, one found on the chromosome and the other found on a stable plasmid (36). Each gene cluster has a markedly different mrkD gene; mrkD1C is found on the chromosome, while mrkD1P is found on the plasmid, with a 60% peptide sequence homology between the two (36). A mutant of IA565, IApc35, that has lost the plasmid-borne mrk gene cluster and carries a termination mutation in the mrkD1C gene, is capable of forming biofilm that surpasses that of the IA565 wild type in terms of biomass (20). Further, a disruption of the remaining mrkB gene, to give strain IAΔT3, results in the inability to form a functional type 3 fimbria, resulting in an inability to form a biofilm (20).
We chose to look at the competitive indexes of these mutant strains compared to that of the wild type to see what effect these mutations would have on the ability of IA565 to form a stable biofilm with PAO1. A competitive index (CI) of greater than 1.0 would indicate that the K. pneumoniae strain is found in greater numbers in the dual-species biofilm than PAO1, while a CI of less than 1.0 would indicate greater numbers of PAO1 cells. The CI of a biofilm composed of PAO1 and IA565 is around 0.25 (Fig. 1), indicating that PAO1 makes up a vast majority of the biofilm biomass compared to IA565. As expected, a CI for the biofilm-deficient mutant IAΔT3 could not be generated, as no colonies were obtained (data not shown). Interestingly, the CI of mutant IApc35, which makes a stronger monospecies biofilm than IA565, generated a CI of 0.014, suggesting a more-than-10-fold decrease in competition compared to IA565. As IApc35 is a strain composed of numerous mutations, we sought to clarify the origin of sensitivity of IApc35 to PAO1. We created a transposon library of strain IA565 and identified mutants that were also defective for biofilm formation with PAO1. The mutant identified, BC2, carried an interruption in the mrkD1P gene and exhibited a CI of 0.02 (Fig. 1), suggesting a role for the plasmid-borne fimbrial adhesin in forming a stable biofilm with P. aeruginosa.
Fig 1.

Mutants of K. pneumoniae IA565 lacking the plasmid-borne mrkD gene are unable to compete with P. aeruginosa PAO1, compared to the wild type, in an in vitro dual-species biofilm. Strains IA565, IApc35, and BC2 were each coinoculated with PAO1 in YEPD and allowed to form a biofilm for 24 h. Biofilms were washed, resuspended, and plated and the CFU of each strain determined. The competitive index (CI) was determined as output K. pneumoniae/P. aeruginosa divided by input. Data represent the mean ± standard deviation of the CI from two independent experiments. Significance was determined by one-way ANOVA (*, P < 0.01).
Supernatant from P. aeruginosa PAO1 impairs biofilm development and disperses preexisting biofilm of K. pneumoniae strains lacking mrkD1P.
To determine if the antagonistic effect of PAO1 on IApc35 and BC2 populations in dual-species biofilms was the result of a secreted effector or a consequence of cell-cell contact, we tested the ability of the K. pneumoniae strains to form and maintain a biofilm in the presence of PAO1 supernatant. IA565, IApc35, and BC2 were allowed to form biofilms in a 96-well microtiter plate and were then treated with lyophilized and reconstituted versions of YEPD, IA565 supernatant, or PAO1 supernatant for 16 h with shaking at 37°C. While YEPD and IA565 supernatant had no effect on the stability of any of the preformed biofilms, PAO1 supernatant dramatically decreased the biofilms of IApc35 and BC2, causing a >100-fold decrease in biomass (Fig. 2A). PAO1 supernatant had no effect on the stability of preformed IA565 biofilm (Fig. 2A). This activity was abolished by boiling the PAO1 supernatant for 15 min (Fig. 2A), suggesting that a heat-sensitive secreted effector from PAO1 played a significant role in the ability of P. aeruginosa to outcompete the mrkD1P-deficient mutants of IA565. The activity of the PAO1 supernatant on the IApc35 and BC2 biofilms was fast acting, reaching maximum biofilm detachment by 8 h posttreatment (Fig. 2B). The ability of IA565, IApc35, and BC2 to form biofilm in PAO1 spent medium was tested by growing the strains in the same reconstituted YEPD or PAO1 supernatant. While IA565 was able to form biofilm in both types of media, IApc35 and BC2 were not (Fig. 2D), demonstrating that the supernatant disrupted preestablished biofilms and inhibited the formation as well. In order to test if the secreted effector was bactericidal in nature, we tested the growth kinetics of IA565, IApc35, and BC2 in reconstituted YEPD or PAO1 supernatant. The supernatant of PAO1 had no significant effect on the planktonic cell growth of any of the K. pneumoniae strains (Fig. 2C), suggesting that the PAO1 secreted effector was abolishing the biofilms through detachment and not killing.
Fig 2.

A heat-sensitive effector from P. aeruginosa PAO1 supernatant disrupts established biofilms from K. pneumoniae strains IApc35 and BC2. (A) IA565, IApc35, and BC2 biofilms, preformed in YEPD in 96-well microtiter plates at 37°C with shaking, were treated by replacing the medium with fresh reconstituted YEPD or IA565 or PAO1 supernatant or with reconstituted PAO1 supernatant boiled for 15 min. Biofilms were stained with crystal violet and resuspended, and the OD570 was measured. (B) Dispersal of preexisting IApc35 and BC2 biofilms by P. aeruginosa PAO1 supernatant is completed in 8 h. Sixteen-hour biofilms of K. pneumoniae IA565, IApc35, and BC2 were treated with either YEPD or P. aeruginosa PAO1 supernatant in 96-well microtiter plates at 37°C with shaking. Biofilms were washed, stained with crystal violet, and resuspended, and OD570 readings were taken every 2 h. (C) Growth of planktonic K. pneumoniae was measured in the presence of either YEPD or PAO1 supernatant in 96-well microtiter plates at 37°C with shaking. OD600 readings were taken every 2 h. (D) After growth of the K. pneumoniae strains was assayed, the resultant biofilms were assayed using crystal violet staining.
The mrkD1P gene was also used to complement strains IApc35 and BC2, and biofilms composed of these complemented strains were tested for biofilm disruption by PAO1 supernatant. IApc35 and BC2 carrying an empty vector (pBMC12) displayed the same biofilm phenotype that is sensitive to supernatant from PAO1 (Fig. 3). Those same strains carrying a plasmid with mrkD1P (pBMC13) shared the same phenotype of resistance to biofilm disruption as the wild-type IA565 (Fig. 3), suggesting that MrkD1P, and not MrkD1C, provides IA565 with a phenotype of resistance to biofilm disruption mediated by PAO1.
Fig 3.
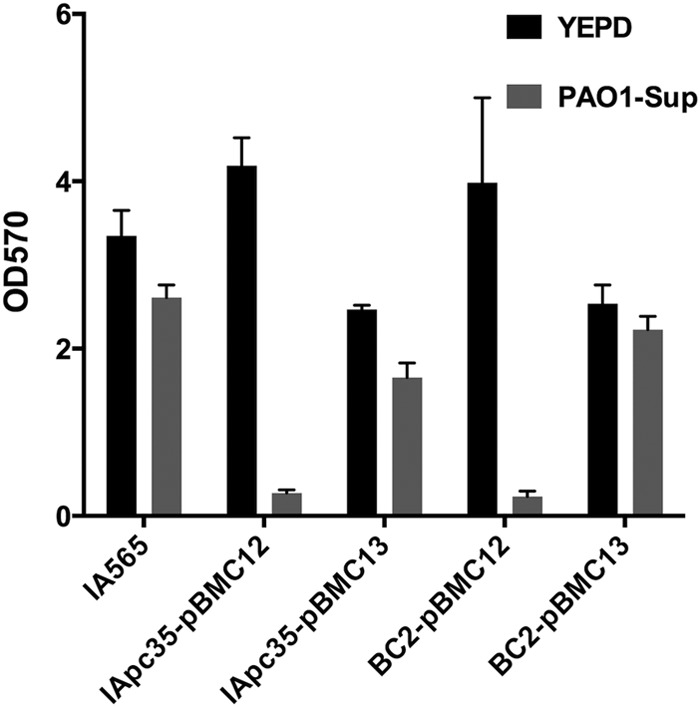
Complementation of IApc35 and BC2 with the plasmid-borne mrkD1P restores biofilm resistance to PAO1 supernatant. Preformed biofilms of wild-type IA565 or mutant forms carrying either an empty vector (pBMC12) or mrkD1P complement (pBMC13) were treated with YEPD or PAO1 supernatant for 16 h, and remaining biofilm was assayed by crystal violet stain and OD570 reading.
The secreted elastase B (LasB) from PAO1 disrupts preformed biofilm of K. pneumoniae IA565 lacking mrkD1P.
To identify the effector(s) responsible for the biofilm-disrupting ability of PAO1, the PAO1 two-allele library from the Washington Genome Center (37) was screened for mutants whose supernatants were unable to disrupt IApc35 preestablished biofilms. Strains from the library were grown in a fashion similar to that for wild-type PAO1, and the collected supernatant was used directly to assay biofilm dispersion. We identified nine mutants that were defective for the ability to disrupt IApc35 and BC2 preestablished biofilms (Fig. 4). While mutants from this library may harbor polar mutations, great care has been taken to reduce this possibility by the addition of a neomycin phosphotransferase promoter to the downstream end of the transposon (37). The disrupted genes of the identified mutants represent proteins that either have unknown function (PA0697) or have functions with unknown relationships to virulence in PAO1 (PA0502/BioH, PA1684/MtnD, and PA3383). Others identified are likely related to the transcriptional regulation of the secreted effector(s) involved in biofilm disruption (PA3135, PA0401, and PA4280). PA2478, a thiol:disulfide interchange protein, is a class of protein that is necessary for the proper folding and functionality of secreted proteins. The only secreted protein identified in the library screen was elastase B (PA3724/LasB), a zinc metalloprotease involved in host tissue damage, invasion, and immunomodulation, which has also recently been shown to play a role in the dispersal of Staphylococcus aureus biofilms. To test the effect of LasB protease on K. pneumoniae biofilms, purified LasB was mixed into YEPD medium and added to preformed IA565, IApc35, and BC2 biofilms. LasB had an effect on the mrkD1P-deficient strains of K. pneumoniae identical to that of PAO1 spent medium and also had no effect on wild-type IA565 (Fig. 5A). The results indicate that the protease LasB plays a role as an antibiofilm effector and that MrkD1P likely plays a protective role against that activity.
Fig 4.

Biofilm dispersal activity of PAO1 transposon mutants. IApc35 and BC2 biofilms, preformed in YEPD in 96-well microtiter plates at 37°C with shaking, were treated after 16 h by replacing the medium with fresh YEPD or supernatant from PAO1 or transposon mutants. Biofilm formation was measured by staining with crystal violet and OD570 measurement.
Fig 5.
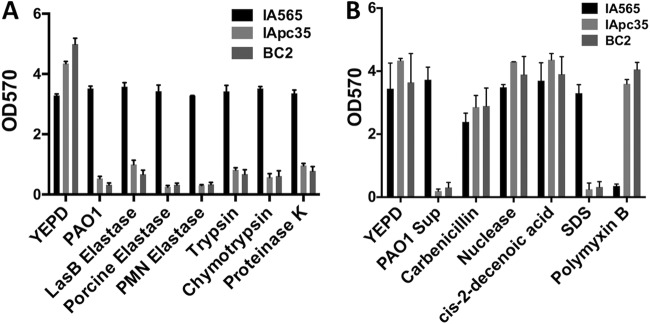
Biofilm dispersal activity of various proteases and antibiofilm agents. IApc35 and BC2 biofilms, preformed in YEPD in 96-well microtiter plates at 37°C with shaking, were treated after 16 h by replacing the medium with fresh YEPD or YEPD supplemented with the compounds listed. Biofilm formation was measured after 16 h by staining with crystal violet and OD570 measurement.
Effects of proteases and other antibiofilm agents on K. pneumoniae biofilms lacking mrkD1P.
Proteases, including LasB, have previously been shown to have biofilm-dispersant activity with various microorganism (15). To gain a better understanding of the role of LasB in the disruption of K. pneumoniae, we also tested various other proteases, including elastases of different sources, trypsin, chymotrypsin, and proteinase K. Each of these proteases had activity nearly identical to that of LasB, eradicating preformed IApc35 and BC2 biofilms while leaving the wild-type IA565 biofilm intact (Fig. 5A). Boiling of these agents destroyed the antibiofilm activity (data not shown). A mixture of antichymotrypsin and chymotrypsin in YEPD failed to disperse IApc35 and BC2 biofilms, indicating that protease activity is necessary for biofilm disruption (see Fig. S1A in the supplemental material). When IApc35 and BC2 were complemented with a mrkD1P-carrying plasmid, the resultant biofilms were protected from chymotrypsin-mediated attack (see Fig. S1B in the supplemental material). Most interesting is that mrkD1P-deficient strains of IA565 are sensitive to elastases (e.g., elastases from polymorphonuclear leukocytes) and proteases produced by the colonized host (Fig. 5A), suggesting that these mutants may be unable to properly colonize a host or cause a chronic wound infection. Further study will clarify this phenotype.
In order to fully analyze the sensitivity of mutant K. pneumoniae biofilms, we also assayed the ability of these mrkD1P mutants to maintain a biofilm in the presence of various other antibiofilm agents and antibiotics compared to that of the wild-type strain. The enzyme DNase and the fatty acid cis-2-decenoic acid, both of which have been shown to disrupt or detach biofilms (30, 38), had no effect on any of the preformed biofilms (Fig. 5B). β-Lactamase resistance in K. pneumoniae is a growing problem in the medical community (18). IA565 is highly resistant to β-lactamases such as carbenicillin, and this resistance is amplified when the organism is in a biofilm community (data not shown). Carbenicillin levels as high as 3 mg ml−1 had no effect on any of the preformed biofilms (Fig. 5B), suggesting that biofilm-mediated antibiotic resistance is unaffected by the loss of MrkD1P. However, sensitivity to SDS, a detergent, showed a pattern similar to that of LasB and other proteases (Fig. 5B). Interestingly, IApc35 and BC2 biofilms showed an increased resistance to the detergent-like polymyxin B, an antimicrobial that targets and disrupts the bacterial cell wall (Fig. 5B). The sensitivities and resistances demonstrated by the mutant strains of K. pneumoniae compared to wild-type IA565 suggest a change in the morphology and structure of the individual cell, the biofilm community, or both. To investigate these possibilities, we looked at cell morphologies of IA565, IApc35, and BC2 cells, grown under biofilm-inducing conditions, using scanning electron microscopy. The IA565 cell morphology was found to be highly elongated, creating cells of around 12 μm (Fig. 6). This morphology, however, was dramatically decreased in the mutant BC2 strain, creating cell lengths of less than 2 μm on average. Cell length was restored by complementation with the mrkD1P-carrying plasmid pBMC13 (Fig. 6), suggesting that mrkD1P may play a role in the development of an elongated cell morphology (representative micrographs are shown in Fig. S2 in the supplemental material). These changes in cell morphology likely represent a pleiotropic effect from the deletion of mrkD1P that leads to the sensitivity/resistance pattern seen in mrkD1P-deficient mutants, and elucidation of these mechanisms is still required.
Fig 6.
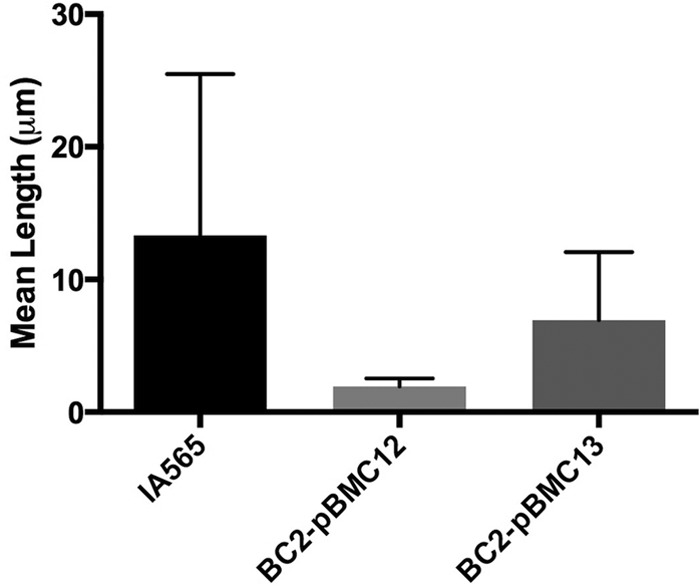
Mean cell length of K. pneumoniae wild-type strain IA565 and mrkD1P-deficient strain BC2 with and without mrkD1P complementation.
Role of mrkD in protection against protease-mediated biofilm disruption.
Biofilm morphology, cell morphology, antibiotic resistance, and various other physiological features of K. pneumoniae can exhibit large degrees of variation between two strains (39). In order to examine the roles of MrkD1C and MrkD1P in providing biofilm-mediated resistance to protease-mediated dispersion, we looked at a number of other K. pneumoniae strains, carrying either MrkD1C or MrkD1P or both (Table 1), in the presence of porcine pancreas-derived elastase. As expected, UI917 and UI925, both carrying mrkD1P but not mrkD1C, were resistant to protease-mediated biofilm disruption (see Fig. S3 in the supplemental material). However, only two strains of K. pneumoniae carrying a mrkD1C gene but no mrkD1P gene showed any sensitivity to PAO1 supernatant (Fig. 7; see Fig. S3 in the supplemental material). The sensitive strains, THK12113 and CAS55 (C3091), showed a restored resistance to protease treatment with mrkD1P-complementing plasmid pFK52 (Fig. 7). All strains were tested for planktonic growth and found to have growth kinetics similar to that of IA565 (see Fig. S4 in the supplemental material). These data suggest that in other strains, various factors besides mrkD may be involved in the protection of K. pneumoniae biofilms from protease-mediated attack.
Fig 7.
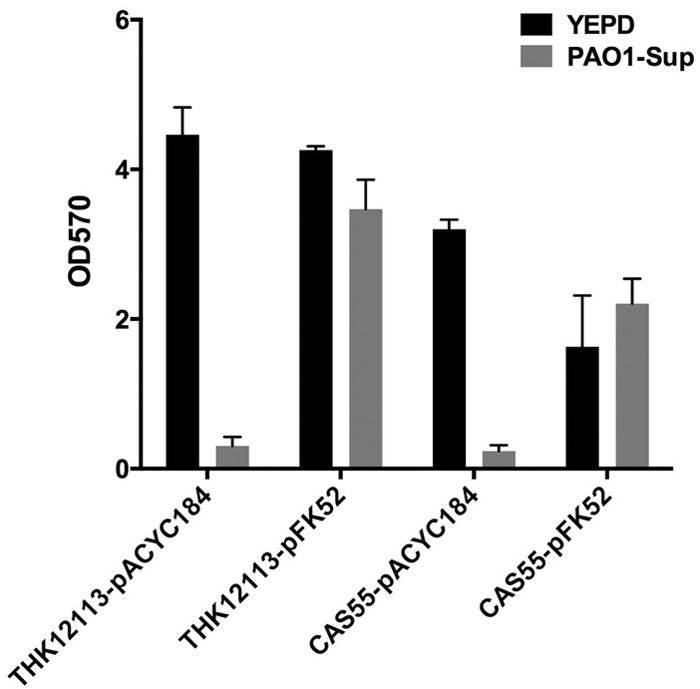
Complementation of CAS55 and THK12113 with the plasmid-borne mrkD1P restores biofilm resistance to PAO1 supernatant. Preformed biofilms of CAS55 and THK12113, carrying either an empty vector (pACYC184) or mrkD1P complement (pFK52), were treated with YEPD or PAO1 supernatant for 16 h, and remaining biofilm was assayed by crystal violet stain and OD570 reading.
DISCUSSION
Naturally occurring biofilms in the environment and the host are frequently found as multispecies communities that differ vastly in composition, structure, and antimicrobial resistance from monospecies biofilm systems (10). It is essential to look at these heterogeneous populations in order to gain a better understanding of the interactions that different microbial species have with one another in this complicated environment. Understanding these interactions will lead to identification of novel targets and therapeutics that likely would be missed otherwise. K. pneumoniae and P. aeruginosa are two organisms that are often found together in biofilm-mediated chronic wound infections (10), which are a growing burden on the medical community and cost millions of dollars every year in treatment (40). Understanding the interactions of these two species is essential in eliminating each as a common hospital-acquired infectious agent.
P. aeruginosa is an antagonistic organism that uses a number of effectors to gain a competitive advantage over other organisms, in both sessile and planktonic cultures. These effectors include LasA, LasB, N-homoserine lactone, pyocyanin, cis-2-decenoic acid, and rhamnolipids, which can kill or hinder the growth of competing bacteria or disrupt and disperse bacteria from heterogeneous biofilm communities (15). These effects have been demonstrated with various types of microorganisms, including Candida albicans, which is sensitive to phenazine (41), and Staphylococcus aureus, which is sensitive to LasB (15). However, it has been shown that K. pneumoniae is able to form a stable biofilm with P. aeruginosa (32), suggesting potential mechanisms used by K. pneumoniae to combat these effectors, which may also provide potential targets that could lead to the disruption of biofilms composed of this bacterium. The use of these potential therapeutics could lead to a drastic change in the population dynamics of the biofilm structure, which could allow for greater access of more common antimicrobial agents.
Many K. pneumoniae strains form biofilms using a type 3 fimbria that is produced and used by various members of the Enterobacteriaceae (42). These fimbriae are composed of subunits of the protein MrkA and accentuated at the tip by the fimbrial adhesion MrkD (19). Each of these, and other components, is chaperoned into place by the protein MrkB, and a knockout of the mrkB gene results in a strain lacking fimbriae and incapable of forming biofilm (20). The strain IA565 carries two copies of the mrkD gene (forms 1C and 1P) that are used for adherence to various forms of extracellular matrix proteins, such as collagen. A strain of IA565, IApc35, that lacks the plasmid-borne mrkD1P gene and has a translation termination codon in the coding sequence of the chromosomal mrkD1C actually forms a more robust biofilm than the wild type (20). However, this strain has an ∼10-fold decrease in its ability to form a biofilm with the P. aeruginosa strain PAO1, and a monospecies biofilm composed of IApc35 is almost completely dispersed by the application of spent medium from PAO1. Using a transposon screen, we identified a mrkD1P gene disruption, BC2, that produced an identical phenotype in relation to IApc35 in its reaction to the presence of PAO1 or PAO1 spent medium. Complementation of the mrkD1P gene partially restored the resistance of IApc35 and BC2 biofilms to that of the wild-type IA565, suggesting that mrkD1P, but not mrkD1C, is required for resistance to the effects of PAO1. This also suggests a potent target in K. pneumoniae that could be exploited in the future to help combat both single- and mixed-species infections. In fact, antibodies directed at MrkD were shown to protect against K. pneumoniae lung infection, providing a possible course of treatment using MrkD-specific antibodies in conjunction with other antimicrobials to help treat infection.
The ability of the PAO1 spent medium to disrupt preformed biofilms and inhibit their formation suggests that a secreted effector(s) is being used by PAO1 to attack K. pneumoniae biofilm. A screen of the PAO1 two-allele library from the Washington Genome Center identified elastase B (LasB) as the likely effector. Elastase B is a metalloprotease used by PAO1 for host invasion and biofilm dispersal in other microorganisms. The effect was not relegated only to LasB, as many other forms of proteases were able to emulate the disruptive effect. Most interestingly, leukocyte-I elastase was shown to have an effect on K. pneumoniae biofilms that was identical to that of LasB, suggesting that the human host may already be primed to fight K. pneumoniae infection. Targeting of MrkD1P in K. pneumoniae biofilms could provide a key component in the host fight against infection.
The disruption of mrkD1P also allowed for greater susceptibility to SDS, although it had no effect on the susceptibility to other biofilm-disrupting agents such as cis-2-decenoic acid or nucleases or to β-lactam antibiotics such as carbenicillin. Most interestingly, the biofilms of the mutant forms of IA565 showed increased resistance to polymyxin B, suggesting that a mechanism other than biofilm permeability may be at play in the altered resistance/susceptibility of IApc35 and BC2. Scanning electron microscopy revealed an altered cell morphology of the mrkD1P mutants of IA565, showing an inhibition of cell elongation that occurs quite frequently in the wild type.
It is possible that the absence of MrkD1P may cause a pleiotropic change in the cell surface content of IA565, either by changing cell surface dynamics or by altering the expression of cell surface-associated proteins or compounds. This may allow for decreased or increased access of antimicrobial agents, proteases, etc. These potential changes in cell surface content, extracellular matrix, and cell morphology caused by the deletion of mrkD1P may inhibit the creation of a robustly interconnected community of bacterial cells that could allow for easier access of proteases that might otherwise be excluded. In these cases, it is likely that the MrkD protein is not directly mediating protection from proteolytic cleavage but rather is altering the biofilm dynamic in such a way as to create a protective environment for the bacteria. Further study is necessary to find the root cause of this phenotype.
While MrkD1P appears to be required for competition with PAO1 and protection against protease-mediated biofilm disruption, this does not appear to be the case with various other forms of K. pneumoniae. While protected from the same protease treatments, the different forms of MrkD had no effect on the resistance phenotype. The cell morphologies and biofilm physiologies of these various strains can differ markedly, and it is likely that other factors are being used to provide the same type of resistance in various K. pneumoniae strains. Identification of these targets may also provide a similar route to the antibiofilm sensitivity found in the IA565 mutants IApc35 and BC2. It is important to understand these interactions of K. pneumoniae and P. aeruginosa further to identify better ways of preventing and treating chronic wound infections.
Supplementary Material
ACKNOWLEDGMENTS
This work was partly supported by the U.S. Army Medical Research and Materiel Command, Military Infectious Diseases Defense Health Program.
The opinions or assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the Department of the Army or the Department of Defense.
Footnotes
Published ahead of print 26 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00521-13.
REFERENCES
- 1.Aronson NE, Sanders JW, Moran KA. 2006. In harm's way: infections in deployed American military forces. Clin. Infect. Dis. 43:1045–1051 [DOI] [PubMed] [Google Scholar]
- 2.Tsuneda S, Aikawa H, Hayashi H, Yuasa A, Hirata A. 2003. Extracellular polymeric substances responsible for bacterial adhesion onto solid surface. FEMS Microbiol. Lett. 223:287–292 [DOI] [PubMed] [Google Scholar]
- 3.Flemming HC, Wingender J. 2010. The biofilm matrix. Nat. Rev. Microbiol. 8:623–633 [DOI] [PubMed] [Google Scholar]
- 4.Mah TF, Pitts B, Pellock B, Walker GC, Stewart PS, O'Toole GA. 2003. A genetic basis for Pseudomonas aeruginosa biofilm antibiotic resistance. Nature 426:306–310 [DOI] [PubMed] [Google Scholar]
- 5.Mah TF, O'Toole GA. 2001. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 9:34–39 [DOI] [PubMed] [Google Scholar]
- 6.Singh PK, Parsek MR, Greenberg EP, Welsh MJ. 2002. A component of innate immunity prevents bacterial biofilm development. Nature 417:552–555 [DOI] [PubMed] [Google Scholar]
- 7.Meyle E, Stroh P, Gunther F, Hoppy-Tichy T, Wagner C, Hansch GM. 2010. Destruction of bacterial biofilms by polymorphonuclear neutrophils: relative contribution of phagocytosis, DNA release, and degranulation. Int. J. Artif. Organs 33:608–620 [DOI] [PubMed] [Google Scholar]
- 8.Yao Y, Vuong C, Kocianova S, Villaruz AE, Lai Y, Sturdevant DE, Otto M. 2006. Characterization of the Staphylococcus epidermidis accessory-gene regulator response: quorum-sensing regulation of resistance to human innate host defense. J. Infect. Dis. 193:841–848 [DOI] [PubMed] [Google Scholar]
- 9.Ganz T. 2003. Defensins: antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 3:710–720 [DOI] [PubMed] [Google Scholar]
- 10.Dowd SE, Sun Y, Secor PR, Rhoads DD, Wolcott BM, James GA, Wolcott RD. 2008. Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol. 8:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cowan SE, Gilbert E, Liepmann D, Keasling JD. 2000. Commensal interactions in a dual-species biofilm exposed to mixed organic compounds. Appl. Environ. Microbiol. 66:4481–4485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palmer RJ, Jr, Kazmerzak K, Hansen MC, Kolenbrander PE. 2001. Mutualism versus independence: strategies of mixed-species oral biofilms in vitro using saliva as the sole nutrient source. Infect. Immun. 69:5794–5804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rao D, Webb JS, Kjelleberg S. 2005. Competitive interactions in mixed-species biofilms containing the marine bacterium Pseudoalteromonas tunicata. Appl. Environ. Microbiol. 71:1729–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cowell BA, Twining SS, Hobden JA, Kwong MS, Fleiszig SM. 2003. Mutation of lasA and lasB reduces Pseudomonas aeruginosa invasion of epithelial cells. Microbiology 149:2291–2299 [DOI] [PubMed] [Google Scholar]
- 15.Park JH, Lee JH, Cho MH, Herzberg M, Lee J. 2012. Acceleration of protease effect on Staphylococcus aureus biofilm dispersal. FEMS Microbiol. Lett. 335:31–38 [DOI] [PubMed] [Google Scholar]
- 16.Bagley ST. 1985. Habitat association of Klebsiella species. Infect. Control. 6:52–58 [DOI] [PubMed] [Google Scholar]
- 17.Rezaei E, Safari H, Naderinasab M, Aliakbarian H. 2011. Common pathogens in burn wound and changes in their drug sensitivity. Burns 37:805–807 [DOI] [PubMed] [Google Scholar]
- 18.Angel Diaz M, Ramon Hernandez J, Martinez-Martinez L, Rodriguez-Bano J, Pascual A, Grupo de Estudio de Infeccion Hospitalaria. 2009. Extended-spectrum beta-lactamase-producing Escherichia coli and Klebsiella pneumoniae in Spanish hospitals: 2nd multicenter study (GEIH-BLEE project, 2006). Enferm. Infecc. Microbiol. Clin. 27:503–510 [DOI] [PubMed] [Google Scholar]
- 19.Murphy CN, Clegg S. 2012. Klebsiella pneumoniae and type 3 fimbriae: nosocomial infection, regulation and biofilm formation. Future Microbiol. 7:991–1002 [DOI] [PubMed] [Google Scholar]
- 20.Langstraat J, Bohse M, Clegg S. 2001. Type 3 fimbrial shaft (MrkA) of Klebsiella pneumoniae, but not the fimbrial adhesin (MrkD), facilitates biofilm formation. Infect. Immun. 69:5805–5812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jagnow J, Clegg S. 2003. Klebsiella pneumoniae MrkD-mediated biofilm formation on extracellular matrix- and collagen-coated surfaces. Microbiology 149:2397–2405 [DOI] [PubMed] [Google Scholar]
- 22.Rego AT, Johnson JG, Geibel S, Enguita FJ, Clegg S, Waksman G. 2012. Crystal structure of the MrkD(1P) receptor binding domain of Klebsiella pneumoniae and identification of the human collagen V binding interface. Mol. Microbiol. 86:882–893 [DOI] [PubMed] [Google Scholar]
- 23.Mikkelsen H, McMullan R, Filloux A. 2011. The Pseudomonas aeruginosa reference strain PA14 displays increased virulence due to a mutation in ladS. PLoS One 6:e29113. 10.1371/journal.pone.0029113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harmsen M, Yang L, Pamp SJ, Tolker-Nielsen T. 2010. An update on Pseudomonas aeruginosa biofilm formation, tolerance, and dispersal. FEMS Immunol. Med. Microbiol. 59:253–268 [DOI] [PubMed] [Google Scholar]
- 25.Pihl M, Davies JR, Chavez de Paz LE, Svensater G. 2010. Differential effects of Pseudomonas aeruginosa on biofilm formation by different strains of Staphylococcus epidermidis. FEMS Immunol. Med. Microbiol. 59:439–446 [DOI] [PubMed] [Google Scholar]
- 26.Tomlin KL, Coll OP, Ceri H. 2001. Interspecies biofilms of Pseudomonas aeruginosa and Burkholderia cepacia. Can. J. Microbiol. 47:949–954 [PubMed] [Google Scholar]
- 27.Baum EZ, Crespo-Carbone SM, Morrow BJ, Davies TA, Foleno BD, He W, Queenan AM, Bush K. 2009. Effect of MexXY overexpression on ceftobiprole susceptibility in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 53:2785–2790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davey ME, Caiazza NC, O'Toole GA. 2003. Rhamnolipid surfactant production affects biofilm architecture in Pseudomonas aeruginosa PAO1. J. Bacteriol. 185:1027–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morales DK, Grahl N, Okegbe C, Dietrich LE, Jacobs NJ, Hogan DA. 2013. Control of Candida albicans metabolism and biofilm formation by Pseudomonas aeruginosa phenazines. mBio 4(1):e00526–00512. 10.1128/mBio.00526-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davies DG, Marques CN. 2009. A fatty acid messenger is responsible for inducing dispersion in microbial biofilms. J. Bacteriol. 191:1393–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Purschke FG, Hiller E, Trick I, Rupp S. 2012. Flexible survival strategies of Pseudomonas aeruginosa in biofilms result in increased fitness compared with Candida albicans. Mol. Cell Proteomics 11:1652–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stewart PS, Camper AK, Handran SD, Huang C, Warnecke M. 1997. Spatial distribution and coexistence of Klebsiella pneumoniae and Pseudomonas aeruginosa in biofilms. Microb. Ecol. 33:2–10 [DOI] [PubMed] [Google Scholar]
- 33.Rubin EJ, Akerley BJ, Novik VN, Lampe DJ, Husson RN, Mekalanos JJ. 1999. In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proc. Natl. Acad. Sci. U. S. A. 96:1645–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boddicker JD, Anderson RA, Jagnow J, Clegg S. 2006. Signature-tagged mutagenesis of Klebsiella pneumoniae to identify genes that influence biofilm formation on extracellular matrix material. Infect. Immun. 74:4590–4597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson JG, Clegg S. 2010. Role of MrkJ, a phosphodiesterase, in type 3 fimbrial expression and biofilm formation in Klebsiella pneumoniae. J. Bacteriol. 192:3944–3950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sebghati TA, Korhonen TK, Hornick DB, Clegg S. 1998. Characterization of the type 3 fimbrial adhesins of Klebsiella strains. Infect. Immun. 66:2887–2894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Held K, Ramage E, Jacobs M, Gallagher L, Manoil C. 2012. Sequence-verified two-allele transposon mutant library for Pseudomonas aeruginosa PAO1. J. Bacteriol. 194:6387–6389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang J, Kang M, Chen H, Shi X, Zhou R, Chen J, Du Y. 2011. The staphylococcal nuclease prevents biofilm formation in Staphylococcus aureus and other biofilm-forming bacteria. Sci. China Life Sci. 54:863–869 [DOI] [PubMed] [Google Scholar]
- 39.Verma V, Harjai K, Chhibber S. 2009. Restricting ciprofloxacin-induced resistant variant formation in biofilm of Klebsiella pneumoniae B5055 by complementary bacteriophage treatment. J. Antimicrob. Chemother. 64:1212–1218 [DOI] [PubMed] [Google Scholar]
- 40.Tomic-Canic M, Ayello EA, Stojadinovic O, Golinko MS, Brem H. 2008. Using gene transcription patterns (bar coding scans) to guide wound debridement and healing. Adv. Skin Wound Care 21:487–492 ((Quiz, 21:493–494) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gibson J, Sood A, Hogan DA. 2009. Pseudomonas aeruginosa-Candida albicans interactions: localization and fungal toxicity of a phenazine derivative. Appl. Environ. Microbiol. 75:504–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ong CL, Beatson SA, Totsika M, Forestier C, McEwan AG, Schembri MA. 2010. Molecular analysis of type 3 fimbrial genes from Escherichia coli, Klebsiella and Citrobacter species. BMC Microbiol. 10:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964 [DOI] [PubMed] [Google Scholar]
- 44.Hornick DB, Thommandru J, Smits W, Clegg S. 1995. Adherence properties of an mrkD-negative mutant of Klebsiella pneumoniae. Infect. Immun. 63:2026–2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stahlhut SG, Struve C, Krogfelt KA. 2012. Klebsiella pneumoniae type 3 fimbriae agglutinate yeast in a mannose-resistant manner. J. Med. Microbiol. 61:317–322 [DOI] [PubMed] [Google Scholar]
- 46.Hornick DB, Allen BL, Horn MA, Clegg S. 1992. Adherence to respiratory epithelia by recombinant Escherichia coli expressing Klebsiella pneumoniae type 3 fimbrial gene products. Infect. Immun. 60:1577–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.