

Abstract
One of the key mechanisms linking cell signaling and control of gene expression is reversible phosphorylation of transcription factors. FOXC2 is a forkhead transcription factor that is mutated in the human vascular disease lymphedema-distichiasis and plays an essential role in lymphatic vascular development. However, the mechanisms regulating FOXC2 transcriptional activity are not well understood. We report here that FOXC2 is phosphorylated on eight evolutionarily conserved proline-directed serine/threonine residues. Loss of phosphorylation at these sites triggers substantial changes in the FOXC2 transcriptional program. Through genome-wide location analysis in lymphatic endothelial cells, we demonstrate that the changes are due to selective inhibition of FOXC2 recruitment to chromatin. The extent of the inhibition varied between individual binding sites, suggesting a novel rheostat-like mechanism by which expression of specific genes can be differentially regulated by FOXC2 phosphorylation. Furthermore, unlike the wild-type protein, the phosphorylation-deficient mutant of FOXC2 failed to induce vascular remodeling in vivo. Collectively, our results point to the pivotal role of phosphorylation in the regulation of FOXC2-mediated transcription in lymphatic endothelial cells and underscore the importance of FOXC2 phosphorylation in vascular development.
INTRODUCTION
Forkhead box (Fox) proteins are a family of transcription factors (TFs) that play an important role in development, cell cycle regulation, and other key biological processes (1). In mammals, more than 40 forkhead family members have been identified, all sharing the evolutionarily conserved forkhead DNA binding domain. Despite the similarity in their DNA-binding domains, different members of the forkhead family have evolved distinct functional roles. The forkhead transcription factor FOXC2 was first demonstrated to play a role in the morphogenesis of the cardiovascular system during embryonic development (2, 3). Subsequent studies revealed that FOXC2 is also implicated in lymphatic vascular development and disease. Mutations in FOXC2 cause lymphedema-distichiasis (LD; OMIM 153400) characterized by lymphedema and double rows of eyelashes (4). In both humans and mice, FOXC2 is highly expressed in the developing lymphatic vessels, as well as in the adult lymphatic valves (5, 6). The critical role of FOXC2 in lymphatic vascular development has been underscored by the demonstration of abnormal lymphatic patterning and failure to form lymphatic valves in Foxc2-deficient mice (5, 7, 8). LD patients develop similar defects characterized by lymph and venous reflux, indicating failure or absence of lymphatic and venous valves (9, 10). On a mechanistic level, FOXC2 genomic binding sites are enriched in NFATC1 consensus sequences, and the two transcription factors cooperate in vivo during lymphatic vascular morphogenesis (8).
Phosphorylation of transcription factors represents a rapid and reversible mechanism for dynamic regulation of transcriptional networks (11). Most of the Fox transcription factors are known to be modified by phosphorylation, but only for a few of them is there a detailed understanding of how phosphorylation modulates their function. One example is FOXO proteins whose transcriptional activity is tightly regulated by phosphorylation at multiple sites by at least nine protein kinases (12). In contrast, little is known about phosphorylation of FOXC2. The protein has been reported to be phosphorylated when overexpressed in immortalized cells (13), but the molecular details and functional significance of the phosphorylation have not been studied. Here, we show that FOXC2 is phosphorylated in primary lymphatic endothelial cells (LECs), a well-established in vitro model for the study of the lymphatic vasculature (14, 15). We identify eight evolutionarily conserved phosphorylation sites in FOXC2, seven of which are clustered within a 70-amino-acid domain of previously unknown function. Loss of phosphorylation at these sites extensively modifies the FOXC2 transcriptional program in LECs, affecting the expression of several hundred genes. While previous TF phosphorylation studies focused on the regulation of individual enhancer sites, we use genome-wide location analysis to study how phosphorylation affects FOXC2 binding to physiological targets across the whole genome. Our results show that phosphorylation differentially regulates FOXC2 recruitment to individual binding sites, suggesting a novel mechanism for selective transcriptional regulation. In agreement with our in vitro data, we further demonstrate that phosphorylation is important for the FOXC2 function in vivo.
MATERIALS AND METHODS
Cell culture, adenoviral transduction, and plasmid transfection.
Human LECs were isolated and cultured as described previously (16). Lymphatic endothelial identity was confirmed with PROX1 staining. For adenoviral experiments, LECs were transduced with 20 PFU/cell of recombinant adenoviruses expressing human FOXC2 (5), phosphorylation-deficient human FOXC2 (pmFOXC2), or LacZ (17) in serum-free medium for 1 h and harvested 48 h after transduction. HepG2, HEK 293T, and HeLa cells were transfected with plasmids constitutively expressing human FOXC2 (5) or pmFOXC2 using Lipofectamine 2000 transfection reagent (Invitrogen).
Antibodies, staining procedures, and image acquisition.
LECs were fixed with 4% paraformaldehyde (PFA), permeabilized with 0.1% Triton X-100, and blocked with 5% donkey serum, 0.5% bovine serum albumin (BSA), 0,1% Triton X-100, and 0.01% sodium azide in phosphate-buffered saline. Skins were dissected and fixed in 4% PFA, and whole-mount staining was performed as described previously (18). For immunofluorescence staining, we used mouse anti-Myc (clone 9E10; Santa Cruz Biotechnology), rabbit anti-Myc-Tag (clone 71D10; Cell Signaling Technology, used for transgene detection in vivo), rat anti-mouse PECAM-1 (BD Pharmingen), rat anti-mouse FOXC2 (19), sheep anti-human FOXC2 (R&D Systems), and goat anti-human PROX1 (R&D Systems). Alexa Fluor 488, 555, 594, and 647 fluorochrome-conjugated secondary antibodies (Molecular Probes) were used for signal detection. Samples were mounted with DAPI (4′,6′-diamidino-2-phenylindole)-containing ProLong Gold antifade reagent (Invitrogen) or Vectashield (Vector Laboratories). The samples were then analyzed at room temperature with a confocal microscope (LSM 510 Meta; Carl Zeiss) using LSM AIM software (objectives EC Plan-Apochromat 10×/0.45, 20×/0.8, and 40×/1.30; Carl Zeiss) or with a motorized Axio Imager Z1 microscope (Carl Zeiss) equipped with a charge-coupled device camera (Camera AxioCam MRC 5; Carl Zeiss) using Axiovision software (version 4.7 [Carl Zeiss]; objectives Plan-Neofluar 10×/0.3, 20×/0.50, and 40×/1.30 [Carl Zeiss]). Three-dimensional projections of whole-mount images were digitally reconstructed from confocal z-stacks using Imaris suite 6.3.1 software and processed using Adobe Photoshop CS4. Images are representative of three to five embryos per stage or per phenotype or three independent experiments in vitro. Antibodies used for immunoblotting were as follows: mouse anti-Myc (clone 9E10; Santa Cruz Biotechnology), rat anti-mouse FOXC2 (19), rabbit antiphosphoserine and rabbit antiphosphothreonine (Zymed Laboratories), mouse antiphosphotyrosine (clone 4G10; Upstate Biotechnology/Millipore), and rabbit anti-human PIN1, rabbit anti-human protein phosphatase 2A (PP2A) B subunit, rabbit anti-rat extracellular signal-regulated kinase 1/2 (ERK1/2), and rabbit anti-human phospho-ERK1/2 (Cell Signaling Technology).
Animal experiments.
Animal experiments were approved by the Veterinary Office of the Canton de Vaud and the Animal Ethics Committee. Embryonic age was determined according to the day of the vaginal plug (embryonic day 0.5 [E0.5]). Tie1-tTA (tet-Off) driver mice were generously provided by Dan Dumont, University of Toronto, Toronto, Ontario, Canada (20). For the production of responder lines TetOs-FOXC2 and TetOs-pmFOXC2, FOXC2 and pmFOXC2 cDNAs were cloned into pTetOs vector (20), and transgenic mice on FVB/N background were generated by microinjection. Mice were genotyped by PCR by using the primers 5′-TTCTTCTCGGGCGCGTTCTGG-3′ and 5′-GTCCAAACCGGGCCCCTCTGC-3′. The driver and responder transgenic mice were mated to obtain double-transgenic offspring. The transgene was repressed by administration of 1 mg of tetracycline hydrochloride (AppliChem)/ml with 5% sucrose in the drinking water of pregnant females starting at E0.5. Transgene expression was induced by discontinuing the administration of tetracycline. Single-transgenic or wild-type littermates were used as controls for double-transgenic mice.
For transgene expression analysis, total RNA from E15.5 lungs was isolated using the Qiagen RNeasy Plus minikit (Qiagen). Reverse transcriptase reaction was performed using the Transcriptor first-strand cDNA synthesis kit (Roche Diagnostics) with random hexamer primers. The resulting cDNA was amplified by PCR using a FastStart Taq DNA polymerase and dNTPack kit (Roche Diagnostics). The primer sequences are available upon request. A reverse transcription (RT) reaction containing all components, except reverse transcriptase, was used as a negative control.
Vascular quantification analysis.
For quantification of the vascular branching and density, confocal images obtained with a ×20 objective, a ×0.7 scanning digital zoom, 512 × 512 pixels were analyzed using AngioTool 0.5a (21). For quantification of branching points, vessels were outlined in Adobe Photoshop CS4 software prior to analysis. For quantification of vascular sprouting, endothelial sprouts were quantified at the angiogenic front in a minimum of nine fields and three head skin samples per group. The total number of sprouts was counted with ImageJ 1.43u software and normalized to the length of the sprouting front, which was measured and defined according to the published protocol (22).
MS.
For liquid chromatography-mass spectrometry (LC-MS) analysis, tryptic or Glu-C peptides derived from immunoprecipitated Myc-FOXC2 were separated by reversed-phase high-performance liquid chromatography on a CapLC instrument (Waters) with a C18 column (0.075 by 150 mm; Atlantis dC18, 100 Å, 3 μm; Waters), which was eluted with a linear gradient of acetonitrile (5 to 50% in 30 min) in 0.1% formic acid. The flow rate was 0.3 μl/min, and the eluent was directly injected into a quadrupole/time-of-flight hybrid mass spectrometer (Q-TOF Micro; Waters) equipped with an electrospray ionization (ESI) source. The mass spectrometer was calibrated using 2 pmol of glufibrinogenic peptide B fragments/μl as a standard. Mass spectra collected during the LC-MS separation of digested peptides were exported into ASCII text files using the DataBridge of the MassLynx software (Waters). The text files were imported into the DeCyder MS software (GE Healthcare), where different elution profiles were visualized as two-dimensional and three-dimensional graphs and different m/z values were deconvoluted into molecular masses of 500 to 6,000 Da. The ion counts of all different charge states of the same peptide were taken into account to calculate total intensity of the deconvoluted masses. The search for candidate phosphopeptides was performed both manually and using PROWL software (http://prowl.rockefeller.edu/). Tandem MS (MS/MS) fragmentation spectra of the candidate phosphopeptides were acquired by colliding the doubly or triply charged precursor ions with argon collision gas at accelerating voltages of 30 to 45 V. The de novo sequencing of fragmented phosphopeptides was performed using BioLynx peptide sequencing software (Waters).
Metabolic labeling with [32P]orthophosphate.
HepG2 cells were transfected with plasmids expressing Myc-FOXC2 or its individual phosphorylation site mutants using DharmaFECT transfection reagent (Thermo Scientific) according to the manufacturer's instructions. At 18 h after transfection, the cells were transferred to phosphate-free Dulbecco modified Eagle medium (Gibco) and metabolically labeled for 12 h with [32P]orthophosphate (8,500 to 9,120 Ci/mmol; Perkin-Elmer). Myc-FOXC2 or its mutants were immunoprecipitated using protein G-beads (Sigma-Aldrich) and anti-Myc antibody (clone 9E10; Santa Cruz Biotechnology). The immunoprecipitated proteins were separated by SDS-PAGE, transferred to nitrocellulose membranes, and either immunoblotted with sheep anti-human FOXC2 antibody (R&D Systems) or subjected to autoradiography. Band quantification was performed using ImageJ software (23).
Microarray analysis.
Each microarray experiment was carried out in triplicate. Total RNA from cultured LECs was isolated and purified with RNeasy kit (Qiagen) according to the manufacturer's instructions. RNA quantity was assessed with a NanoDropND-1000 spectrophotometer, and the RNA quality was assessed with an Agilent 2100 bioanalyzer using RNA 6000 Nano-Chips. For each sample, 100 ng of total RNA was amplified using a WT Sense-Strand target labeling kit (Affymetrix); 5.5 μg of the resulting sense cDNA was fragmented by UDG (uracil DNA glycosylase) and APE 1 (apurinic/apyrimidic endonuclease 1) and biotin-labeled with TdT (terminal deoxynucleotidyltransferase) using a GeneChip WT terminal labeling kit (Affymetrix). Affymetrix Human Gene 1.0 ST arrays were hybridized with 3 μg of biotinylated target at 45°C for 17 h, washed, and stained according to the protocol described in the Affymetrix GeneChip expression analysis manual. The arrays were scanned using a GeneChip Scanner 3000 7G (Affymetrix), and raw data was extracted from the scanned images and analyzed with the Affymetrix Power Tools software package (Affymetrix). All statistical analyses were performed using the high-level interpreted statistical language R and various Bioconductor packages (http://www.bioconductor.org). Hybridization quality was assessed using the Expression Console software (Affymetrix). Normalized expression signals were calculated from Affymetrix CEL files using RMA normalization method. Differential hybridized features were identified using Bioconductor package “limma” that implements linear models for microarray data (24). The P values were adjusted for multiple testing with Benjamini and Hochberg's method.
Validation of microarray and ChIP-chip results by real-time PCR.
RT-PCR analyses were performed on StepOnePlus or 7500-Fast Real-Time PCR systems (Applied Biosystems) using a SYBR green PCR Master Mix (Applied Biosystems). Analysis of gene expression was carried out using the comparative threshold cycle (CT; i.e., ΔΔCT) method as described by the manufacturer. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as an endogenous control. ΔCT values were calculated by subtracting the CT value for the reference gene from that of the gene of interest. The fold change in gene expression for a particular gene was calculated by using the formula for 2−ΔΔCT as described by the manufacturer. The primer sequences are available upon request.
ChIP-qPCR data analysis was performed using the differential occupancy fold change method described in the ChampionChIP PCR Primers user manual (SABiosciences/Qiagen [http://www.sabiosciences.com/support_manual.php]). Each real-time PCR analysis was carried out in triplicate. The primer sequences are available upon request.
EMSA.
The 25-μl electrophoretic mobility shift assay (EMSA) reaction mixtures contained 10 mM HEPES (pH 8.0), 3 mM Tris-HCl (pH 8.0), 50 mM KCl, 2 mM MgCl2, 10% glycerol, 1 mM dithiothreitol, 0.04 mg of poly-deoxy-inosinic-deoxy-cytidylic acid/ml, 0.4 mg of BSA/ml, γ-33P-labeled DNA probe, and cell lysates. For supershift assays, 1 μg of mouse anti-Myc antibody (clone 9E10; Santa Cruz Biotechnology) was added to the reaction mixtures. After incubation for 30 min at room temperature, probes were loaded on 5% criterion Tris-borate-EDTA polyacrylamide gel (Bio-Rad Laboratories). Electrophoresis was performed for 1 to 2 h at 10 to 20 mA/100 to 150 V. The gels were dried and visualized by autoradiography or using a phosphorimager. Mutation of the forkhead consensus sequence prevented binding of Myc-FOXC2 to DNA (see Fig. S8 in the supplemental material).
ChIP-chip.
ChIP-chip assays and data analysis were performed as described previously (8), except that mouse anti-Myc antibody (clone 9E10; Santa Cruz Biotechnology) was used for immunoprecipitations. All ChIP experiments were performed in biological triplicate. CEAS (25) annotation results are available as supplemental data set A2 (http://bcf.isb-sib.ch/data/ivanov_supp_dataset_A2/). ChIP-chip results were validated by ChIP-qPCR as described in the experimental procedures in the supplemental material. Genomic interval intersection analysis was carried out using the web-based genome analysis tool Galaxy (http://main.g2.bx.psu.edu/) (26) with default settings. ChIP-chip data were deposited in the public database of functional genomics experiments ArrayExpress (accession number E-MTAB-596).
Other assays.
Immunoprecipitation and immunoblotting were carried out as described previously (8). Covalent cross-linking of antibodies to protein G-Sepharose, immunoprecipitation coupled with enzymatic dephosphorylation, and protein digestion were performed as described previously (27). Site-directed mutagenesis was performed using standard PCR-based methods; mutagenic primer sequences are available upon request. The data are shown as mean ± the standard deviation. P values were calculated using a two-tailed Student t test, except for the sprouting quantification in Fig. 5G, where it is a one-way analysis of variance of the P value. The data were considered to be significantly different if P was <0.05.
Fig 5.

Phosphorylation regulates FOXC2 function in vivo. (A) Endothelial cell-specific gain-of-function models for the analysis of FOXC2 phosphorylation. (B) Both models express comparable levels of the transgene, as evidenced by RT-PCR analysis of the indicated mRNAs from E15.5 lungs. Transgene expression was initiated at E13.5. (C) Macroscopic appearances of FOXC2ecGOF, pmFOXC2ecGOF, and control E15.5 embryos. (D and E) FOXC2 overexpression does not affect capillary sprouting. (F) Overexpression of FOXC2 but not pmFOXC2 promotes vascular remodeling in maturing capillaries. Note the increased capillary branching and density in FOXC2ecGOF embryos. Whole-mount staining of E15.5 head skin for pan-endothelial marker CD31 (green) and the transgene (red). The transgene expression was detected using anti-Myc antibody. Scale bars: 100 μm (D), 38 μm (E), 35 μm (F). (G) Quantification of vascular branching, density, and sprouting at the vascular front in the control, pmFOXC2ecGOF, and FOXC2ecGOF embryos. n = 3 per genotype. *, P < 0.05. n.s., nonsignificant.
RESULTS
FOXC2 contains eight proline-directed Ser/Thr phosphorylation sites.
Phosphatase treatment has been previously reported to increase the electrophoretic mobility of recombinant FOXC2 overexpressed in immortalized COS-7 cells, indicating that the overexpressed FOXC2 is a phosphoprotein (13). Therefore, we first sought to determine whether endogenous FOXC2 is similarly phosphorylated in primary human LECs. In a reference experiment, we transfected HEK 293T or HepG2 immortalized cell lines with plasmids expressing Myc-tagged FOXC2 (Myc-FOXC2) or transduced them with a Myc-FOXC2-expressing recombinant adenovirus (Ad-Myc-FOXC2). We treated cell lysates with lambda protein phosphatase (λ-PPase) and analyzed the lysates by Western blotting with anti-Myc antibody. In both cell lines tested, we observed an apparent increase in Myc-FOXC2 electrophoretic mobility, indicative of dephosphorylation (Fig. 1A). More importantly, we detected a similar electrophoretic mobility shift upon λ-PPase treatment of LEC lysates expressing endogenous FOXC2 (Fig. 1A, lower panel). Thus, we concluded that endogenous FOXC2 and overexpressed FOXC2 are similarly modified by phosphorylation in both primary and immortalized cell types.
Fig 1.

Analysis of FOXC2 phosphorylation. (A) Endogenous and recombinant human FOXC2 are similarly phosphorylated in primary LECs and immortalized cell lines. Cell lysates were treated (+) or not treated (−) with lambda protein phosphatase (λ-PPase) and analyzed by Western blotting with anti-FOXC2 or anti-Myc antibodies. (B) Schematic representation of FOXC2 phosphorylation sites. FHD, forkhead domain; TA, transactivation domains (5, 34); PD, phosphorylated domain. Phosphorylation sites identified by LC-MS/MS are shaded in red; phosphorylation sites identified by mutagenesis are shaded in yellow. Peptides detected by MS in tryptic and Glu-C digests are underlined in cyan and green, respectively. Amino acid numbering is the same as in the endogenous protein (NP_005242). (C) Substitution of eight phosphorylation sites in Myc-FOXC2 with alanine abolishes the phosphorylation-dependent electrophoretic mobility shift. Lysates of cells transfected with a plasmid expressing the phosphorylation-deficient mutant Myc-pmFOXC2 were treated (+) or not treated (−) with λ-PPase and analyzed by Western blotting with anti-Myc antibody. (D) FOXC2 phosphorylation-deficient mutant (pm) has increased electrophoretic mobility compared to the wild-type (wt) protein. Shown is Western blot analysis of lysates from HepG2 cells transduced with adenoviruses expressing Myc-FOXC2 or Myc-pmFOXC2.
To determine whether FOXC2 is phosphorylated on serine/threonine or tyrosine residues, we immunoprecipitated the protein from HepG2 cells transduced with Ad-Myc-FOXC2 and analyzed the immunoprecipitate by Western blotting with phosphoamino acid-specific antibodies. Only the antibodies against phosphoserine (pS) and phosphothreonine (pT) could recognize the immunoprecipitated Myc-FOXC2, while the antiphosphotyrosine (pY) antibody did not produce a reactive band (see Fig. S1 in the supplemental material). The pS- and pT-positive bands disappeared upon phosphatase treatment, confirming the antibody specificity. Based on these findings, we concluded that FOXC2 is phosphorylated on serine and threonine but not on tyrosine residues.
To identify the phosphorylation sites in FOXC2, we performed analysis of tryptic peptides derived from immunoprecipitated Myc-FOXC2 by liquid chromatography coupled with electrospray ionization mass spectrometry (LC-ESI-MS). The initial analysis yielded 70.8% sequence coverage of trypsin-digested Myc-FOXC2 (Fig. 1B). We identified several candidate phosphopeptides among the Myc-FOXC2-derived tryptic peptides by comparing experimentally measured peptide masses to those predicted from a simulated tryptic digest. The masses of four tryptic peptides differed from predicted masses by ∼80 Da, suggesting the presence of single phosphorylation sites. For two peptides, mass differences also suggested double phosphorylations (see Table S1 in the supplemental material). To validate these results, we examined ion signal intensities of the same peptides derived from enzymatically dephosphorylated Myc-FOXC2. As shown in Table S1 in the supplemental material, we observed either a decrease in intensity or the complete disappearance of the peptide ion signals after treatment with λ-PPase. However, sequence coverage was incomplete due to the presence of a large 12-kDa peptide in the tryptic digests. To increase the sequence coverage, we digested Myc-FOXC2 with another protease, Glu-C, which cleaves proteins at the C terminus of glutamic acid. Indeed, the combination of Glu-C and trypsin digestion data brought total sequence coverage to nearly 80% (Fig. 1B). However, more importantly, digestion with Glu-C allowed us to identify a novel candidate phosphopeptide (amino acids 357 to 379) within the sequence of the large tryptic peptide, which escaped detection in our initial analysis (see Table S2 in the supplemental material).
To determine the exact positions of phosphorylated amino acid residues, we analyzed the selected tryptic and Glu-C peptides by MS/MS. The fragmentation spectra of each phosphopeptide showed the presence of dehydroalanine (dA) with a characteristic mass of 69 Da at positions corresponding to serine residues, indicative of serine phosphorylation (see Fig. S2 in the supplemental material). In total, this approach allowed us to unequivocally identify six serine phosphorylation sites in Myc-FOXC2, corresponding to S219, S232, S240, S281, S288, and S367 in endogenous FOXC2. All of the identified phosphorylation sites were immediately followed by proline, indicating that they were phosphorylated by proline-directed protein kinase(s).
To determine whether we had identified all major FOXC2 phosphorylation sites, we generated a mutant protein in which S219, S232, S240, S281, S288, and S367 were replaced by alanine and treated the mutant protein with λ-PPase. Phosphatase treatment still caused an increase in electrophoretic mobility of the mutant FOXC2 protein, suggesting the presence of other yet unidentified phosphorylation sites (see Fig. S3 in the supplemental material). To identify the remaining phosphorylation sites, we performed systematic deletion mutagenesis of Myc-FOXC2. We generated six deletion mutants spanning the phosphorylated region of FOXC2 (amino acids 219 to 366), with each mutant having the previously identified phosphorylation sites replaced by alanine. By comparing the effect of λ-PPase treatment on the electrophoretic mobility of each deletion mutant, we narrowed our search to a region between amino acids 241 and 280 (see Fig. S3A in the supplemental material), which contained only two potential proline-directed phosphorylation sites: T247 and S251. When we separately mutated each of these residues to alanine, bringing the total number of alanine substitutions to seven, the resulting proteins still underwent a mobility shift after phosphatase treatment (see Fig. S3C in the supplemental material). However, when we simultaneously mutated both residues, the protein's electrophoretic mobility remained unchanged upon phosphatase treatment (Fig. 1C; see Fig. S3C in the supplemental material), indicating that both T247 and S251 were phosphorylated in addition to S219, S232, S240, S281, S288, and S367. Thus, using the combination of MS and mutagenesis, we identified eight proline-directed phosphorylation sites in FOXC2 clustered in a relatively short region of the protein encompassing amino acids 219 to 367 (Fig. 1B).
Next, we sought to determine the contribution of each of these sites to the overall phosphorylation stoichiometry of FOXC2 in living cells. For this purpose, we generated eight individual phosphorylation site mutants of FOXC2, each having only one phosphorylated residue mutated to alanine, and compared their ability to metabolically incorporate radioactive orthophosphate. None of the individual mutations alone caused a strong decrease in radioactive phosphate incorporation, indicating that FOXC2 does not contain distinct primary phosphorylation sites (see Fig. S4 in the supplemental material).
FOXC2 interacts with PIN1, PP2A, and ERK1/2.
The observation that all phosphorylated serine and threonine residues in FOXC2 are followed by proline prompted us to investigate whether FOXC2 is recognized by peptidyl-prolyl cis/trans isomerase PIN1. PIN1 isomerizes the phosphorylated Ser/Thr-Pro bonds exclusively, thereby changing their conformation and modulating protein function (28). Coimmunoprecipitation assays with anti-Myc antibody demonstrated the physical association between Myc-FOXC2 and endogenous PIN1, whereas no interaction was observed in the control cells (Fig. 2A). This finding suggested that FOXC2 activity is regulated through conformational changes induced by the PIN1-catalyzed cis-trans isomerization of the phosphorylated Ser/Thr-Pro bonds.
Fig 2.
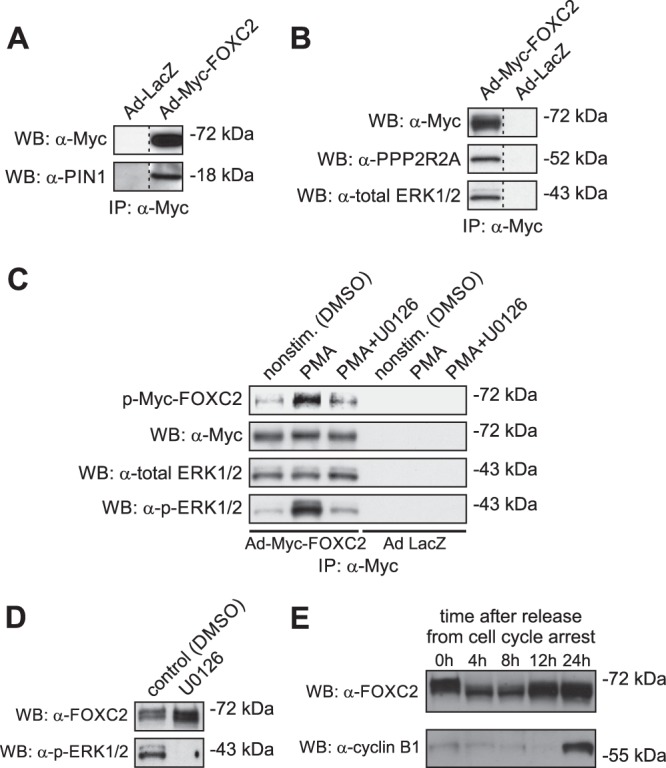
FOXC2 interacts with peptidyl-prolyl cis/trans isomerase PIN1, alpha isoform of the regulatory subunit B of the protein phosphatase PP2A (PPP2R2A), and ERK1/2 protein kinases. (A) Coimmunoprecipitation assays with anti-Myc antibody demonstrate the association of Myc-FOXC2 with endogenous PIN1 in HeLa cells transduced with recombinant Ad-Myc-FOXC2. Shown is Western blot (WB) of anti-Myc immunoprecipitates consecutively probed with anti-Myc and anti-PIN1 antibodies. Control immunoprecipitation was performed from extracts of HeLa cells transduced with recombinant adenovirus expressing bacterial β-galactosidase (Ad-LacZ). (B) Myc-FOXC2 binds to endogenous PPP2R2A and ERK1/2 in HepG2 cells transduced with recombinant Ad-Myc-FOXC2. Coimmunoprecipitation assays were performed and analyzed as in A, except that anti-PPP2R2A and anti-total ERK1/2 antibodies were used for immunoblotting. (C) Immunocomplex kinase assays demonstrate that Myc-FOXC2 is phosphorylated in vitro by the coprecipitating endogenous ERK1/2 kinases. Shown is Western blot (WB) of anti-Myc immunoprecipitates incubated in the presence of [γ-32P]ATP and phosphorimage of the corresponding membrane. The blot was consecutively probed with anti-Myc, anti-total ERK1/2 and anti-active ERK1/2 (p-ERK1/2) antibodies. The identity of ERK1/2 was confirmed by immunocomplex kinase assays with anti-Myc antibody using lysates of HepG2 cells stimulated with PMA in the presence or absence of 10 mM U0126, a selective inhibitor of upstream MEK. (D) Inhibition of ERK1/2 does not modify the electrophoretic mobility of endogenous FOXC2 in LECs. Shown is a Western blot of total LEC lysates consecutively probed with anti-FOXC2 and anti-active ERK1/2 antibodies. (E) The electrophoretic mobility of endogenous FOXC2 changes after release from serum starvation-induced cell cycle arrest in LECs, suggesting CDK involvement in FOXC2 phosphorylation.
To identify binding partners of Myc-FOXC2, we used coimmunoprecipitation coupled with MS. Using this approach, we identified the 55-kDa regulatory subunit B (alpha isoform) of the proline-directed phosphatase PP2A (PPP2R2A) as a novel interacting partner of FOXC2. The interaction was confirmed by immunoblotting with anti-PP2A B subunit antibody (Fig. 2B), suggesting PP2A as a candidate phosphatase responsible for FOXC2 dephosphorylation.
We next focused on identifying the protein kinase(s) involved in FOXC2 phosphorylation. Because ERK1/2 proline-directed kinases have been suggested to phosphorylate a related forkhead transcription factor FOXC1 (29), we investigated whether the same kinases are also responsible for FOXC2 phosphorylation. Immunoblotting with anti-ERK1/2 antibody revealed that ERK1/2 coprecipitated with Myc-FOXC2, whereas no coprecipitation was observed with extracts from the control lacZ-expressing cells (Fig. 2B). Furthermore, immunocomplex kinase assays showed that the coprecipitating kinase activity could phosphorylate Myc-FOXC2 in vitro (Fig. 2C). This kinase activity was stimulated upon activation of the mitogen-activated protein kinase (MAPK) pathway by phorbol-12-myristate-13-acetate (PMA) and attenuated by the upstream MEK inhibitor UO126, confirming its identity as ERK1/2 (Fig. 2C). However, pharmacological inhibition of the MAPK/ERK pathway with UO126 in living LECs failed to induce an electrophoretic mobility shift characteristic of the loss of FOXC2 phosphorylation (Fig. 2D), suggesting that other, yet-unidentified proline-directed protein kinases are also involved in FOXC2 phosphorylation. This finding prompted us to investigate the possible involvement of proline-directed cyclin-dependent protein kinases (CDKs) in FOXC2 phosphorylation. To this end, we analyzed the phosphorylation status of endogenous FOXC2 in LECs synchronized by serum starvation. As shown in Fig. 2E, the electrophoretic mobility of FOXC2 changed after release from cell cycle arrest, suggesting CDK involvement in FOXC2 phosphorylation. Taken together, these results suggest that FOXC2 activity is reversibly regulated through the interplay between ERK1/2, CDK(s), and PP2A and that the mechanism underlying this regulation involves conformational changes induced by PIN1-catalyzed isomerization of the phosphorylated Ser/Thr-Pro peptide bonds.
Phosphorylation regulates the FOXC2 transcriptional program in primary lymphatic endothelial cells.
Based on the above results, we next sought to determine whether phosphorylation regulates FOXC2 transcriptional activity. To address this question, we compared gene expression profiles between primary LECs overexpressing wild-type and phosphorylation-deficient FOXC2. Since LECs cannot be transfected with plasmid DNA, we generated an adenovirus expressing a phosphorylation-deficient mutant (pm) of Myc-FOXC2 in which all eight phosphorylated residues were replaced by alanine. The recombinant adenovirus, designated Ad-Myc-pmFOXC2, readily transduced both primary and immortalized cells, as evidenced by the overexpression of the phosphorylation-deficient protein with increased electrophoretic mobility (Fig. 1D). To determine whether the loss of phosphorylation could affect FOXC2 expression and/or subcellular localization, we subjected LECs transduced with Ad-Myc-FOXC2 and Ad-Myc-pmFOXC2 to immunofluorescent staining with an anti-Myc tag antibody. The staining and Western blot analysis revealed that pmFOXC2 was expressed at about the same level as wild-type FOXC2, ∼100-fold higher than the endogenous protein (Fig. 3A; see Fig. S5 in the supplemental material). Furthermore, the loss of phosphorylation had no effect on the nuclear localization of FOXC2 (Fig. 3A). These findings allowed us to directly compare gene expression profiles of LECs overexpressing wild-type and mutant FOXC2. To account for gene expression changes caused by adenoviral transduction per se, we transduced LECs with a recombinant adenovirus expressing an irrelevant gene, bacterial β-galactosidase (Ad-LacZ).
Fig 3.

Phosphorylation regulates FOXC2-mediated transcription in primary LECs. (A) Immunofluorescent staining for Myc (green), lymphatic marker PROX1 (red), and DNA (blue) of LECs transduced with adenoviruses expressing wild-type Myc-FOXC2, phosphorylation-deficient mutant Myc-pmFOXC2, or control bacterial β-galactosidase (LacZ). Note that wild-type and mutant FOXC2 have similar expression levels and subcellular localization. Bars, 20 μm. (B) Phosphorylation regulates FOXC2 transcriptional activity. Gene expression profiling was performed on the adenovirus-transduced LECs shown in panel A. Genes whose expression changed >2-fold in response to the loss of FOXC2 phosphorylation (FDR < 0.01) are shown in orange (upregulated) and purple (downregulated) in the Volcano plot of significance against the fold change in gene expression. Vertical dotted lines mark the 2-fold change limits. (C) RT-PCR validation of the gene expression profiling results. Genes upregulated or downregulated >2-fold in response to the loss of FOXC2 phosphorylation are shown in orange and purple, respectively; genes affected <2-fold are shown in gray. No change in FOXC2 expression reflects equally efficient cell transduction with Ad-Myc-FOXC2 and Ad-Myc-pmFOXC2. The data are presented as log2-transformed fold change in gene expression normalized to a housekeeping gene (GAPDH). Horizontal dotted lines mark the 2-fold change limits. Shown are the means and standard deviations of triplicate determinations in a single experiment representative of two independent experiments. (D) Heat map representation of the differences in gene expression in response to the loss of FOXC2 phosphorylation. The left heat map shows expression levels of 57 of 59 genes downregulated >2-fold (FDR < 0.01) in Ad-Myc-pmFOXC2-transduced LECs compared to Ad-Myc-FOXC2-transduced LECs. The right heat map shows expression levels of 57 out of 88 genes upregulated >2-fold (FDR < 0.01) in the same cells. Three biological replicates are shown for each condition. The color key at the lower left corresponds to the mean-centered, arctan-transformed log2-fold change in gene expression falling within the range from −π/2 to π/2. Blue denotes genes with relative decreased expression; red denotes genes with relative increased expression.
Hierarchical cluster analysis clearly distinguished between LECs overexpressing wild-type and mutant FOXC2 (see Fig. S6 in the supplemental material), revealing the presence of a subset of FOXC2-regulated genes coordinately responding to the loss of FOXC2 phosphorylation. To identify these genes, we first filtered out genes that did not vary at least 2-fold between cells overexpressing Myc-FOXC2 and LacZ or Myc-pmFOXC2 and LacZ, obtaining a list of 2,419 genes. Next, we selected genes with a >1.5-fold difference in expression between Myc-FOXC2- and Myc-pmFOXC2-overexpressing cells. The significance threshold was set to a false discovery rate (FDR) < 0.01. The identified signature consisted of 467 genes, of which 191 genes were upregulated and 276 were downregulated in response to the loss of FOXC2 phosphorylation (see Table S3 in the supplemental material). Among the 467 genes, 147 were affected more than 2-fold, with 88 genes upregulated and 59 genes downregulated (see Table S3, highlighted genes). The magnitude of change in expression of these genes and the levels of significance are graphically represented in a volcano plot (Fig. 3B). The expression profiles of 114 genes from the expression signature are also presented in the form of heat maps in Fig. 3D. To validate the expression profiling results, we selected several genes affected >2-fold by the loss of FOXC2 phosphorylation and genes with little or no change in expression and examined their mRNA levels by sq-RT-PCR. As shown in Fig. 3C, the results obtained by sq-RT-PCR were in good agreement with the microarray data. Furthermore, sq-RT-PCR showed that the levels of FOXC2 mRNA were similar in LECs transduced with Ad-Myc-FOXC2 and Ad-Myc-pmFOXC2, confirming that the expression signature was indeed associated with the loss of FOXC2 phosphorylation and not caused by differences in expression of Myc-FOXC2 and Myc-pmFOXC2. Altogether, these results demonstrate that the loss of phosphorylation modifies the FOXC2 transcriptional program and provide insight into the identity of the FOXC2 target genes regulated by phosphorylation.
To determine the functional significance of the phosphorylation-induced changes in the FOXC2 transcriptional program, we performed gene ontology enrichment (GO) analysis of the genes differentially expressed in response to the loss of FOXC2 phosphorylation. The analysis revealed significant enrichment (FDR < 0.01) for the following GO terms: cell cycle, cell division, M phase of mitotic cell cycle and mitosis (fold enrichments of 2.75, 3.48, 3.65, and 4.06, respectively). All of these terms were grouped into a single annotation cluster associated with cell cycle and cell division. Among the well-characterized cell cycle genes affected by the loss of FOXC2 phosphorylation were those encoding cyclins A1, A2, B2, and E2, cyclin-dependent kinase 1, Wee1-like protein kinase, kinesin family member 11, anaphase promoting complex subunit 1, centriolin, kinetochore protein Nuf2, and centromere protein F. These results suggest a role for FOXC2 phosphorylation in cell cycle regulation through transcriptional control of cell cycle-specific genes.
Phosphorylation differentially regulates FOXC2 recruitment to genomic binding sites in the context of native chromatin but not in vitro.
The phosphorylation-dependent regulation of the FOXC2 transcriptional program prompted us to investigate whether phosphorylation regulates FOXC2 chromatin occupancy. For this purpose, we performed comparative genome-wide location analysis of wild-type and phosphorylation-deficient FOXC2 in primary LECs after transduction with Ad-Myc-FOXC2 or Ad-Myc-pmFOXC2. We used the ChIP-chip assay, a technique that couples chromatin immunoprecipitation (ChIP) with microarray technology (chip), to systematically compare genome-wide occupancy of the two proteins. Using this technique, we have previously mapped 2,350 high-confidence binding sites for endogenous FOXC2 in primary LECs (8). To eliminate concerns over binding artifacts due to Myc-FOXC2 overexpression, we performed genomic interval intersection analysis and selected only the binding sites that overlapped those identified previously for the endogenous FOXC2. This way we would ensure that our study focused only on the physiologically relevant FOXC2 binding sites.
Analysis of the ChIP-chip data using a cutoff of FDR < 0.01 revealed a >6-fold difference in the number of high-confidence binding sites between the wild-type and phosphorylation-deficient FOXC2, although the loss of phosphorylation did not affect the recognition of the forkhead consensus motif, as determined by de novo motif discovery analysis (data not shown). Of 707 physiologically relevant binding sites detected for the overexpressed wild-type Myc-FOXC2, only 108 were identified as enriched for the phosphorylation-deficient mutant. Such a difference in binding site occupancy between the wild-type and mutant proteins indicated that the loss of phosphorylation inhibited FOXC2 recruitment to chromatin. Therefore, we next analyzed the enrichment (P < 10−5) of Myc-FOXC2 over Myc-pmFOXC2 among the physiological FOXC2 binding sites to define the repertoire of binding sites affected by the loss of phosphorylation. The list of 600 “phosphorylation-regulated” sites is presented in Data set SA1 in the supplemental material and in data set A2 at http://bcf.isb-sib.ch/data/ivanov_supp_dataset_A2/, together with their genomic coordinates (hg18 assembly) and neighboring RefSeq and miRNA genes, as well as data on evolutionary conservation, transcription factor motif enrichment, and distribution in the human genome. Among these sites, the decrease in occupancy in response to the loss of FOXC2 phosphorylation ranged from 2.2- to 11.4-fold. The variable changes in occupancy were relatively evenly distributed across all 600 sites (see Fig. S7 in the supplemental material), indicating that throughout the genome, individual binding sites had variable responses to the loss of FOXC2 phosphorylation, ranging from no response to substantial inhibition of binding (Fig. 4). These results were experimentally validated by ChIP-qPCR (Fig. 4), giving good agreement with the ChIP-chip data. Taken together, these results indicate that the loss of phosphorylation inhibits FOXC2 recruitment to chromatin, but the extent of the inhibition varies between different binding sites.
Fig 4.

Phosphorylation differentially regulates FOXC2 binding to genomic target sites in the context of native chromatin but not in vitro. We used genome-wide ChIP-chip to compare the binding of adenovirus-expressed wild-type Myc-FOXC2 and the phosphorylation-deficient mutant Myc-pmFOXC2 to physiological binding sites occupied by endogenous FOXC2 in primary LECs. Endogenous FOXC2 enrichment profiles are shown at the top of each panel. Purple peaks indicate FOXC2-enriched regions; their relative occupancies by Myc-FOXC2 and Myc-pmFOXC2 are shown in callout boxes in green and orange, respectively. Vertical axes represent MAT score. Binding sites are numbered as in Norrmén et al. (8); genomic coordinates refer to the hg18 human genome assembly. An unbound control region is shown in the lower right panel. The ChIP-chip results were validated by ChIP-qPCR with primers flanking ∼100-bp sequences within the FOXC2-enriched regions. The results are presented as the fold enrichment over the unbound control region. Green and orange bars correspond to wild-type Myc-FOXC2 and Myc-pmFOXC2, respectively. Shown are the means and standard deviations of triplicate determinations. An EMSA was used to compare the in vitro binding of adenovirus-expressed wild-type Myc-FOXC2, Myc-pmFOXC2, and deletion mutant Myc-FOXC2 D219-366 to naked dsDNA from the ChIP-enriched regions or the unbound control region. Binding specificity was controlled with adenovirus-expressed bacterial β-galactosidase (LacZ) and anti-Myc antibody. Asterisks indicate the positions of the antibody-supershifted complexes.
To determine whether native chromatin structure was important for the phosphorylation-mediated regulation of FOXC2 recruitment to DNA, we performed an EMSA with cell extracts from LECs overexpressing Myc-FOXC2, Myc-pmFOXC2, or deletion mutant Myc-FOXC2 Δ219-366. EMSA revealed that loss of phosphorylation had no effect on FOXC2 binding to naked DNA from ChIP-enriched regions (Fig. 4). Moreover, the Myc-FOXC2 Δ219-366 mutant lacking the entire phosphorylated domain, but still retaining the DNA binding domain, formed equally stable in vitro complexes with the same DNA fragments (Fig. 4). In agreement with these results, enzymatic dephosphorylation with λ-PPase had no effect on the in vitro binding of Myc-FOXC2 to a double-stranded oligonucleotide bearing two forkhead consensus sequences (see Fig. S8 in the supplemental material). Collectively, these results suggest that FOXC2 binding to DNA is regulated by phosphorylation only in the context of native chromatin but not in reconstituted in vitro systems.
We next sought to determine whether the decrease in FOXC2 recruitment to chromatin could be responsible for the changes in the FOXC2 transcriptional program induced by the loss of FOXC2 phosphorylation. Assuming that FOXC2 is a transcriptional activator, we hypothesized that among the genes whose expression was downregulated in response to the loss of FOXC2 phosphorylation, there would be genes with decreased FOXC2 recruitment to nearby enhancer sites. Indeed, examples of such genes are indicated in Table S3 in the supplemental material. The correlation between decreased gene transcription in the absence of FOXC2 phosphorylation and decreased FOXC2 recruitment to nearby sites is also graphically illustrated in Fig. S9 in the supplemental material. Taken together, these results suggest that the decrease in recruitment of FOXC2 to a subset of binding sites in the absence of phosphorylation may downregulate transcription of the neighboring direct target genes, which may in turn affect the expression of a multitude of downstream genes. Thus, we propose that phosphorylation-mediated regulation of FOXC2 recruitment to transcriptional enhancer sites is one of the mechanisms controlling the FOXC2 transcriptional program.
Phosphorylation is important for FOXC2-induced vascular remodeling in vivo.
To confirm our in vitro findings, we developed inducible endothelial cell-specific gain-of-function (ecGOF) mouse models FOXC2ecGOF and pmFOXC2ecGOF (Fig. 5A). In these models, transgene expression is induced in blood endothelial cells upon withdrawal of tetracycline. Although, ideally, overexpression in lymphatic endothelial cells should have been investigated, this approach was not possible because of the lack of lymphatic endothelial expression in Tie1-tTA driver model (data not shown). Therefore, we induced transgene expression in blood endothelial cells at E13.5 and analyzed the vascular architecture at E15.5. RT-PCR demonstrated similar levels of FOXC2 and pmFOXC2 mRNA in endothelial cells, and this observation was further confirmed by immunofluorescent staining (Fig. 5B and D). Macroscopically, all FOXC2ecGOF embryos appeared hemorrhagic (n = 5) and differed from the pmFOXC2ecGOF embryos that were morphologically similar to the control embryos (n = 9, Fig. 5C). We next investigated the development of the capillary network in the skin of the head, where both the sprouting capillary front and the more mature vascular network can be studied simultaneously. Expression of the FOXC2 transgene in capillary tip cells had no noticeable effect on the number of sprouting cells, although more subtle effects, such as changes in the number of filopodia per cell, still remain to be investigated. On the other hand, FOXC2 overexpression in maturing capillaries behind the sprouting front resulted in the formation of large syncytium-like vascular plexus, leading to the significant increase in capillary density and branching (Fig. 5D to G; see Fig. S10 in the supplemental material). In contrast, the equivalent regions of capillaries in pmFOXC2ecGOF embryos appeared similar to those in the controls, despite the high levels of transgene expression (Fig. 5E). Interestingly, in some regions of FOXC2ecGOF embryos, such as ventral and dorsal skin, we also observed blood-filled dilated lymphatic vessels (see Fig. S11 in the supplemental material). In the absence of appreciable levels of transgene expression in lymphatic vessels, this phenotype is likely secondary to the blood vessel phenotype, and it suggests that the defects of blood vascular development may lead to miscommunication between blood and lymphatic vessels. Taken together, these findings demonstrate that phosphorylation is important for the ability of FOXC2 to induce vascular remodeling in vivo, thus confirming our in vitro results on the role of FOXC2 phosphorylation in transcriptional regulation.
DISCUSSION
During development, tissue-specific transcriptional programs are established by networks of transcription factors to drive cell differentiation from a pluripotent state to one of many committed cell lineages. Until recently, we had little understanding of how such tissue-specific transcriptional programs are regulated on a genome-wide scale. The advent of ChIP coupled with microarray hybridization (ChIP-chip) or high-throughput sequencing (ChIP-seq) has brought about a new approach to dissection of transcriptional networks, known as genome-wide location analysis (GWLA). GWLA has been successfully used to study transcriptional regulation by histone posttranslational modifications and to identify cis-acting regulatory elements bound by specific TFs within a genome (30). Most of the TF studies using GWLA have thus far focused on the identification of TF binding sites in different cell types and under various environmental conditions. Although these studies represent significant progress in our understanding of the complex transcriptional regulatory networks, they also raise new questions. One such question is how the transcriptional programs are regulated by TF posttranslational modifications. These modifications may include, but are not limited to, phosphorylation, acetylation, sumoylation, and ubiquitination and represent an important means for regulating TF activity and availability. Among them, phosphorylation is particularly important due to the fact that it is rapid and reversible. Activation of signaling pathways often triggers bursts of transcription through changes in the phosphorylation state of specific TFs (11). In this way, TF phosphorylation links signaling from cell surface receptors to activation of specific transcriptional programs in the nucleus. Furthermore, TF phosphorylation also plays a critical role during the cell cycle, allowing, due to its reversible nature, the repeated resetting of oscillating transcriptional programs.
Although most of the earlier studies on TF phosphorylation have been performed in reconstituted systems lacking chromatin, the present study examines the phosphorylation-mediated transcriptional regulation in the native chromatin environment. Our data emphasize the importance of the native chromatin structure for the dynamic regulation of FOXC2 recruitment to genomic DNA. Indeed, we show that the loss of phosphorylation is able to inhibit FOXC2 binding to consensus site DNA only in the context of native chromatin but not in reconstituted in vitro systems. Unlike most previous studies, which focused on the effects of TF phosphorylation on the transcription of individual genes, this study investigates the phosphorylation-mediated transcriptional regulation on a genome-wide scale. We used global gene expression profiling to identify the gene expression signature associated with the loss of FOXC2 phosphorylation and to demonstrate that phosphorylation can induce genome-wide changes in the FOXC2 transcriptional program. We further used ChIP-chip to examine how phosphorylation affects FOXC2 recruitment to target sites across the human genome. The ChIP-chip analysis revealed decreased recruitment of FOXC2 to target binding sites in response to the loss of phosphorylation, suggesting a mechanism underlying the observed changes in gene transcription. While comparing genomic occupancies of the overexpressed Myc-FOXC2 and its phosphorylation-deficient mutant, we took special care to exclude binding artifacts caused by TF overexpression. For this purpose, we discarded all nonphysiological binding sites through genomic interval intersection analysis and focused only on sites occupied by the endogenous FOXC2 in primary LECs. Consequently, we were able to define a global repertoire of physiologically relevant FOXC2 binding sites whose occupancy is regulated by phosphorylation (see Data set SA1 in the supplemental material.
Previously, it had been thought that TF phosphorylation acts as a simple on-off switch, either activating or inhibiting transcription. More recent studies have revealed that TFs are regulated in a graded mode by progressive phosphorylation at multiple sites (11). Although in this study all FOXC2 phosphorylation sites were simultaneously mutated to alanine, the phosphorylation-deficient mutations still had variable effects on the occupancies of individual binding sites across the genome (see Fig. S7 in the supplemental material). This finding suggests that the responses of individual binding sites to the loss of FOXC2 phosphorylation are DNA context dependent, adding an additional layer of complexity to the regulation of TF recruitment to chromatin that goes beyond the previously reported graded mode of regulation by progressive phosphorylation. According to the canonical enhanceosome theory, specific complexes between enhancer DNA elements and multiple TFs and coactivators distinguish the functional TF binding sites from their nonphysiological counterparts. Consequently, TFs occupy only a small and highly specific subset of potential DNA recognition motifs in the genome, thus allowing the establishment of dedicated transcriptional programs. Our results suggest an extension of this theory, where a carefully orchestrated network of context-dependent protein-DNA and protein-protein interactions also determines whether and how the occupancy of a binding site is affected by TF phosphorylation. This previously unsuspected mode of regulation provides a potential mechanism for selective modification of transcriptional programs during the cell cycle or in response to extracellular and intracellular signals. Such a mechanism would act more like a rheostat than a binary switch to dynamically and differentially regulate TF recruitment to the functionally relevant binding sites across the genome.
In agreement with the lack of effect of phosphorylation-deficient mutations on DNA binding in vitro, all FOXC2 phosphorylation sites identified in the present study are located outside the forkhead DNA binding domain (Fig. 1B). Seven of these sites are clustered within a 70-amino-acid domain of previously unknown function in the middle of the protein, which we designate the phosphorylated domain (PD) (Fig. 1B). In line with their important role in transcriptional regulation, all seven phosphorylation sites within this domain, as well as the eighth phosphorylation site 79 amino acids downstream, are highly conserved among vertebrates. Furthermore, these residues are also conserved in the related transcription factor FOXC1, suggesting that both TFs may be similarly regulated by phosphorylation. In accordance with this possibility, half (4 of 8) of the conserved phosphorylation sites have already been experimentally confirmed in FOXC1 (29, 31, 32). These findings once again underscore the importance of phosphorylation in the regulation of FOXC2- and FOXC1-mediated transcription and further imply that the regulation involves complex molecular interactions, rather than simple addition of negative charge to the DNA binding domain. Interestingly, the phosphorylated domains in FOXC2 and FOXC1 also harbor SUMOylation sites, which negatively regulate transcriptional activity of both TFs (33). Thus, the interplay between phosphorylation and SUMOylation may add yet another layer of complexity to the functional regulation for FOXC2 and FOXC1.
Consistent with our finding that the loss of phosphorylation selectively downregulates FOXC2 recruitment to chromatin, only wild-type FOXC2, but not its phosphorylation-deficient mutant, was able to induce vascular remodeling in vivo. Notably, despite comparable expression levels, strongest effects were observed in maturing capillaries but not in sprouting capillary endothelial cells or arteries (Y. Agalarov, unpublished observations). Although other explanations are possible, we suggest that FOXC2 may be differentially phosphorylated in these vascular compartments.
The present study is not without limitations. First, we chose to map the phosphorylation sites in the overexpressed protein, because mapping of endogenous FOXC2 phosphorylation sites in LECs was not technically possible. Overexpression of FOXC2 did not significantly affect its phosphorylation status, based on the fact that rising levels of FOXC2 expression in LECs did not change the protein's electrophoretic mobility (see Fig. S5C in the supplemental material). However, we cannot rule out that there may be less pronounced differences in the phosphorylation stoichiometry between endogenous and overexpressed FOXC2, which do not have an appreciable effect on the protein's electrophoretic mobility. Further studies using more sensitive methods are needed to examine this possibility. Second, we analyzed the in vivo effects of FOXC2 overexpression only in blood endothelial cells. In the future, it will be important to extend this analysis to lymphatic vasculature.
Interestingly, the cell cycle was the most significantly overrepresented functional annotation category among the genes differentially expressed in response to the loss of FOXC2 phosphorylation. In an intriguing regulatory twist, FOXC2 phosphorylation may itself be regulated during the cell cycle. Support for this scenario comes from a high-throughput phosphoproteomics study, which identified FOXC2 as one of the proteins with increased phosphorylation during mitosis (32). In agreement with these published data, our results from synchronized cells also suggest that FOXC2 is phosphorylated by cyclin-dependent protein kinases (CDKs), which oscillate between states of low and high activity during the cell cycle. Taken together, the results of the present study underscore the functional importance of FOXC2 phosphorylation and suggest mechanisms by which FOXC2 phosphorylation may regulate gene expression during normal development and in disease.
Supplementary Material
ACKNOWLEDGMENTS
We thank D. Dumont for providing Tie1-tTA transgenic mice and K. Alitalo for FOXC2 expression vectors and adenovirus constructs. We gratefully acknowledge the technical assistance of C. Cornu, A. Parsons, T. Tainola, and S. Kajander. ChIP-chip microarray hybridization was performed at the Biomedicum Helsinki Biochip Center, and we thank O. Monni and H. Pesonen for their help with microarray processing. Expression microarray hybridization was carried out at the Lausanne Genomics Technologies Facility, and we are grateful to K. Harshman, O. Hagenbüchle, M. Dupasquier, and A. Paillusson for their help with microarray experiments. We thank F. Morgenthaler from the Cellular Imaging Facility at the University of Lausanne for assistance with image acquisition. Transgenic mice were generated at the Transgenic Animal Facility of the University of Lausanne.
This study was supported by the Swiss National Science Foundation (PPP0033-114898 and CRSII3_141811), Telethon Action Suisse, Lymphatic Research Foundation (to H.M.-E.H.), Leenaards Foundation, Academy of Finland, and European Diabetes Foundation.
Footnotes
Published ahead of print 22 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01387-12.
REFERENCES
- 1.Wijchers PJ, Burbach JP, Smidt MP. 2006. In control of biology: of mice, men, and Foxes. Biochem. J. 397:233–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iida K, Koseki H, Kakinuma H, Kato N, Mizutani-Koseki Y, Ohuchi H, Yoshioka H, Noji S, Kawamura K, Kataoka Y, Ueno F, Taniguchi M, Yoshida N, Sugiyama T, Miura N. 1997. Essential roles of the winged helix transcription factor MFH-1 in aortic arch patterning and skeletogenesis. Development 124:4627–4638 [DOI] [PubMed] [Google Scholar]
- 3.Winnier GE, Kume T, Deng K, Rogers R, Bundy J, Raines C, Walter MA, Hogan BL, Conway SJ. 1999. Roles for the winged helix transcription factors MF1 and MFH1 in cardiovascular development revealed by nonallelic noncomplementation of null alleles. Dev. Biol. 213:418–431 [DOI] [PubMed] [Google Scholar]
- 4.Fang J, Dagenais SL, Erickson RP, Arlt MF, Glynn MW, Gorski JL, Seaver LH, Glover TW. 2000. Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am. J. Hum. Genet. 67:1382–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petrova TV, Karpanen T, Norrmén C, Mellor R, Tamakoshi T, Finegold D, Ferrell R, Kerjaschki D, Mortimer P, Yla-Herttuala S, Miura N, Alitalo K. 2004. Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat. Med. 10:974–981 [DOI] [PubMed] [Google Scholar]
- 6.Dagenais SL, Hartsough RL, Erickson RP, Witte MH, Butler MG, Glover TW. 2004. Foxc2 is expressed in developing lymphatic vessels and other tissues associated with lymphedema-distichiasis syndrome. Gene Expr. Patterns 4:611–619 [DOI] [PubMed] [Google Scholar]
- 7.Kriederman BM, Myloyde TL, Witte MH, Dagenais SL, Witte CL, Rennels M, Bernas MJ, Lynch MT, Erickson RP, Caulder MS, Miura N, Jackson D, Brooks BP, Glover TW. 2003. FOXC2 haploinsufficient mice are a model for human autosomal dominant lymphedema-distichiasis syndrome. Hum. Mol. Genet. 12:1179–1185 [DOI] [PubMed] [Google Scholar]
- 8.Norrmén C, Ivanov KI, Cheng J, Zangger N, Delorenzi M, Jaquet M, Miura N, Puolakkainen P, Horsley V, Hu J, Augustin HG, Yla-Herttuala S, Alitalo K, Petrova TV. 2009. FOXC2 controls formation and maturation of lymphatic collecting vessels through cooperation with NFATc1. J. Cell Biol. 185:439–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brice G, Mansour S, Bell R, Collin JR, Child AH, Brady AF, Sarfarazi M, Burnand KG, Jeffery S, Mortimer P, Murday VA. 2002. Analysis of the phenotypic abnormalities in lymphoedema-distichiasis syndrome in 74 patients with FOXC2 mutations or linkage to 16q24. J. Med. Genet. 39:478–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mellor RH, Brice G, Stanton AW, French J, Smith A, Jeffery S, Levick JR, Burnand KG, Mortimer PS. 2007. Mutations in FOXC2 are strongly associated with primary valve failure in veins of the lower limb. Circulation 115:1912–1920 [DOI] [PubMed] [Google Scholar]
- 11.Gardner KH, Montminy M. 2005. Can you hear me now? Regulating transcriptional activators by phosphorylation. Sci. STKE 2005:pe44. [DOI] [PubMed] [Google Scholar]
- 12.Yang JY, Hung MC. 2009. A new fork for clinical application: targeting forkhead transcription factors in cancer. Clin. Cancer Res. 15:752–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berry FB, Tamimi Y, Carle MV, Lehmann OJ, Walter MA. 2005. The establishment of a predictive mutational model of the forkhead domain through the analyses of FOXC2 missense mutations identified in patients with hereditary lymphedema with distichiasis. Hum. Mol. Genet. 14:2619–2627 [DOI] [PubMed] [Google Scholar]
- 14.Mäkinen T, Veikkola T, Mustjoki S, Karpanen T, Catimel B, Nice EC, Wise L, Mercer A, Kowalski H, Kerjaschki D, Stacker SA, Achen MG, Alitalo K. 2001. Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF-C/D receptor VEGFR-3. EMBO J. 20:4762–4773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kriehuber E, Breiteneder-Geleff S, Groeger M, Soleiman A, Schoppmann SF, Stingl G, Kerjaschki D, Maurer D. 2001. Isolation and characterization of dermal lymphatic and blood endothelial cells reveal stable and functionally specialized cell lineages. J. Exp. Med. 194:797–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Norrmén C, Vandevelde W, Ny A, Saharinen P, Gentile M, Haraldsen G, Puolakkainen P, Lukanidin E, Dewerchin M, Alitalo K, Petrova TV. 2010. Liprin (beta)1 is highly expressed in lymphatic vasculature and is important for lymphatic vessel integrity. Blood 115:906–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laitinen M, Makinen K, Manninen H, Matsi P, Kossila M, Agrawal RS, Pakkanen T, Luoma JS, Viita H, Hartikainen J, Alhava E, Laakso M, Yla-Herttuala S. 1998. Adenovirus-mediated gene transfer to lower limb artery of patients with chronic critical leg ischemia. Hum. Gene Ther. 9:1481–1486 [DOI] [PubMed] [Google Scholar]
- 18.Tammela T, Saaristo A, Lohela M, Morisada T, Tornberg J, Norrmen C, Oike Y, Pajusola K, Thurston G, Suda T, Yla-Herttuala S, Alitalo K. 2005. Angiopoietin-1 promotes lymphatic sprouting and hyperplasia. Blood 105:4642–4648 [DOI] [PubMed] [Google Scholar]
- 19.Furumoto TA, Miura N, Akasaka T, Mizutani-Koseki Y, Sudo H, Fukuda K, Maekawa M, Yuasa S, Fu Y, Moriya H, Taniguchi M, Imai K, Dahl E, Balling R, Pavlova M, Gossler A, Koseki H. 1999. Notochord-dependent expression of MFH1 and PAX1 cooperates to maintain the proliferation of sclerotome cells during the vertebral column development. Dev. Biol. 210:15–29 [DOI] [PubMed] [Google Scholar]
- 20.Sarao R, Dumont DJ. 1998. Conditional transgene expression in endothelial cells. Transgenic Res. 7:421–427 [DOI] [PubMed] [Google Scholar]
- 21.Zudaire E, Gambardella L, Kurcz C, Vermeren S. 2011. A computational tool for quantitative analysis of vascular networks. PLoS One 6:e27385. 10.1371/journal.pone.0027385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pitulescu ME, Schmidt I, Benedito R, Adams RH. 2010. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat. Protoc. 5:1518–1534 [DOI] [PubMed] [Google Scholar]
- 23.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9:671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smyth GK. 2004. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3:Article3 [DOI] [PubMed] [Google Scholar]
- 25.Ji X, Li W, Song J, Wei L, Liu XS. 2006. CEAS: cis-regulatory element annotation system. Nucleic Acids Res. 34:W551–W554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goecks J, Nekrutenko A, Taylor J. 2010. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11:R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mäkinen KM, Ivanov KI. 2008. Phosphorylation analysis of plant viral proteins. Methods Mol. Biol. 451:339–359 [DOI] [PubMed] [Google Scholar]
- 28.Lu KP, Zhou XZ. 2007. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signaling and disease. Nat. Rev. Mol. Cell. Biol. 8:904–916 [DOI] [PubMed] [Google Scholar]
- 29.Berry FB, Mirzayans F, Walter MA. 2006. Regulation of FOXC1 stability and transcriptional activity by an epidermal growth factor-activated mitogen-activated protein kinase signaling cascade. J. Biol. Chem. 281:10098–10104 [DOI] [PubMed] [Google Scholar]
- 30.Hawkins RD, Ren B. 2006. Genome-wide location analysis: insights on transcriptional regulation. Hum. Mol. Genet. 15 Spec. No. 1:R1–R7 [DOI] [PubMed] [Google Scholar]
- 31.Daub H, Olsen JV, Bairlein M, Gnad F, Oppermann FS, Korner R, Greff Z, Keri G, Stemmann O, Mann M. 2008. Kinase-selective enrichment enables quantitative phosphoproteomics of the kinome across the cell cycle. Mol. Cell 31:438–448 [DOI] [PubMed] [Google Scholar]
- 32.Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. 2008. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U. S. A. 105:10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Danciu TE, Chupreta S, Cruz O, Fox JE, Whitman M, Iniguez-Lluhi JA. 2012. Small ubiquitin-like modifier (SUMO) modification mediates function of the inhibitory domains of developmental regulators FOXC1 and FOXC2. J. Biol. Chem. 287:18318–18329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujita H, Kang M, Eren M, Gleaves LA, Vaughan DE, Kume T. 2006. Foxc2 is a common mediator of insulin and transforming growth factor beta signaling to regulate plasminogen activator inhibitor type I gene expression. Circ. Res. 98:626–634 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.