

Abstract
Objective:
To investigate whether cortical superficial siderosis (cSS) on MRI, especially if disseminated (involving more than 3 sulci), increases the risk of future symptomatic lobar intracerebral hemorrhage (ICH) in cerebral amyloid angiopathy (CAA).
Methods:
European multicenter cohort study of 118 patients with CAA (104 with baseline symptomatic lobar ICH) diagnosed according to the Boston criteria. We obtained baseline clinical, MRI, and follow-up data on symptomatic lobar ICH. Using Kaplan-Meier and Cox regression analyses, we investigated cSS and ICH risk, adjusting for known confounders.
Results:
During a median follow-up time of 24 months (interquartile range 9–44 months), 23 of 118 patients (19.5%, 95% confidence interval [CI]: 12.8%–27.8%) experienced symptomatic lobar ICH. Any cSS and disseminated cSS were predictors of time until first or recurrent ICH (log-rank test: p = 0.0045 and p = 0.0009, respectively). ICH risk at 4 years was 25% (95% CI: 7.6%–28.3%) for patients without siderosis; 28.9% (95% CI: 7.7%–76.7%) for patients with focal siderosis; and 74% (95% CI: 44.1%–95.7%) for patients with disseminated cSS (log-rank test: p = 0.0031). In Cox regression models, any cSS and disseminated cSS were both independently associated with increased lobar ICH risk, after adjusting for ≥2 microbleeds and age (hazard ratio: 2.53; 95% CI: 1.05–6.15; p = 0.040 and hazard ratio: 3.16; 95% CI: 1.35–7.43; p = 0.008, respectively). These results remained consistent in sensitivity analyses including only patients with symptomatic lobar ICH at baseline.
Conclusions:
Our findings indicate that cSS, particularly if disseminated, is associated with an increased risk of symptomatic lobar ICH in CAA. cSS may help stratify future bleeding risk in CAA, with implications for prognosis and treatment.
Sporadic cerebral amyloid angiopathy (CAA) is a highly prevalent, age-related small-vessel disease1 caused by amyloid-β deposition in cortical and leptomeningeal vessel walls.2 CAA is a major cause of lobar intracerebral hemorrhage (ICH), particularly in elderly patients.2–5 Spontaneous ICH is one of the most catastrophic forms of stroke, with a high risk of recurrence6–8; CAA-related lobar ICH may carry a greater risk than deep ICH presumed to be due to hypertensive arteriopathy,8,9 but this is currently difficult to predict.
Predisposing factors for lobar ICH and lobar ICH recurrence in CAA include APOE ε4 and ε2 alleles,10 hemorrhagic neuroimaging markers of CAA such as lobar cerebral microbleeds (CMBs),11 and anticoagulant or antiplatelet use.12 Little is known about cortical superficial siderosis (cSS), a recently identified neuroimaging marker of CAA,13 and the risk of subsequent ICH. cSS reflects linear blood residues in the superficial (subpial) layers of the cerebral cortex.14 One likely mechanism leading to cSS is repeated episodes of hemorrhage into the subarachnoid space from brittle superficial cortical or leptomeningeal CAA-laden vessels, potentially heralding a high risk of future lobar ICH. A recent study showed that nearly 50% of CAA patients with cSS experienced intracranial hemorrhage over a period of 35 months,15 but this study did not include patients without cSS as a control group.
We tested the hypothesis that in patients with CAA, cSS, especially involving multiple sulci (reflecting more widespread or active disease), is associated with an increased risk of future symptomatic lobar ICH in a European multicenter cohort study.
METHODS
Study population and baseline data collection.
We included consecutive patients diagnosed with CAA (according to the original Boston criteria,16 i.e., not including cSS as a criterion) at 4 stroke centers over defined time periods. The centers included University College London Hospitals NHS Foundation Trust (London) (March 2003 to September 2011), Addenbrooke's Hospital (Cambridge) (July 2002 to March 2010), Cliniques Universitaires Saint Luc (Brussels) (December 2003 to April 2010), and CHU Mont-Godinne UCL (August 2005 to March 2009). At participating centers, MRI scanning is a routine investigation for cases of suspected CAA, unless there are contraindications. Our inclusion criteria were 1) patients fulfilling the original Boston criteria for CAA,16 2) available MRI sequences including T2*-weighted gradient-recalled echo (T2*-GRE) and fluid-attenuated inversion recovery (FLAIR) MRI, and 3) available follow-up information on symptomatic ICH, confirmed by neuroimaging. We included all patients with CAA, including survivors of spontaneous lobar ICH and those who, during investigation for other symptoms, were found to have strictly lobar CMBs (or asymptomatic lobar ICH). We excluded CAA patients without adequate MRI (n = 26) or reliable follow-up data (n = 37). Excluded subjects (n = 63) did not differ significantly from those included in any baseline characteristics (all p > 0.05).
Clinical data at the time of presentation (age, sex, vascular risk factors including hypertension, use of antithrombotics, and previous symptomatic ICH) were obtained from prospective databases and by medical records review using standardized data collection forms. A clearly documented history of transient (≤24 hours) focal neurologic episodes with no known alternative explanation other than CAA (e.g., structural brain lesion, atrial fibrillation, extracranial or intracranial stenosis) was ascertained by review of medical records.
Standard protocol approvals, registrations, and patient consents.
The study received ethical approval by the National Hospital for Neurology and Neurosurgery and Institute of Neurology Joint Research Ethics Committee, the Commission d'Ethique Biomedicale Hospitalo Facultaire of the Faculte de Medicine (Université Catholique de Louvain), and the Comite d'ethique medicale of the CHU Mont-Godinne UCL.
MRI acquisition and analysis.
The MRI protocol was similar in each hospital. Imaging was at 1.5T field strength for all patients and included T1-weighted, T2-weighted, FLAIR, and axial T2*-GRE (slice thickness 5 mm, repetition time 500–1,000 milliseconds, echo time 15–70 milliseconds). Images were reviewed by a trained clinical research fellow blinded to clinical and follow-up data. The presence and distribution of CMBs were evaluated on T2*-GRE images using the Microbleed Anatomical Rating Scale.17 Asymptomatic or symptomatic prior ICH (>5 mm in diameter on T2*-GRE MRI) was also noted.18 cSS was defined as linear residues of chronic blood products in the superficial layers of the cerebral cortex showing a characteristic “gyriform” pattern of low signal on T2*-GRE images, without corresponding hyperintense signal on T1-weighted or FLAIR images (i.e., without acute subarachnoid hemorrhage). We did not include cSS contiguous with any ICH. The distribution of cSS was classified as focal (restricted to ≤3 sulci) or disseminated (>3 sulci).13 White matter changes were evaluated using the 4-step simplified Fazekas rating scale (0–3: 0 = no lesions; 1 = focal lesions; 2 = early confluent; 3 = confluent).19
Follow-up.
Follow-up data were obtained from a systematic review of multiple overlapping sources including prospective databases, medical records review (including discharge summaries, follow-up outpatient and general practitioner letters), and radiologic databases, using standardized data collection forms. We collected information on clinically symptomatic ICH, defined as a symptomatic stroke syndrome associated with neuroimaging evidence of a corresponding ICH (>5 mm in diameter),20 and death of any cause. Outcome events were assessed using all clinical, radiologic, and pathologic information available, blinded to the presence of cSS at baseline MRI. We determined whether the location of symptomatic ICH at follow-up corresponded to the anatomical distribution of cSS on baseline MRI.
Statistical analysis.
We compared clinical and imaging characteristics of CAA patients with symptomatic lobar ICH during follow-up to patients without ICH, using χ2 tests and the Fisher exact test for categorical variables, and 2-sample t tests or Mann-Whitney U tests depending on the distribution of continuous variables. The reliability of rating for presence and category rating of cSS was assessed in a sample of MRI scans from patients with probable CAA (n = 48) by calculating Cohen κ and weighted κ statistic, respectively. We also compared basic clinical and imaging characteristics of patients with vs without cSS. We determined the presence of cSS and disseminated cSS as univariate predictors of ICH risk using Kaplan-Meier plots with significance testing by the log-rank test. Survival time was calculated from date of baseline MRI scan until the date of symptomatic lobar ICH at follow-up or the last known date without the outcome event of interest. For individuals experiencing multiple lobar ICHs during follow-up, data were censored at time of first ICH. Data were also censored at the time of death from causes other than documented symptomatic ICH. Cox regression analysis was performed to calculate univariate hazard ratio (HR) as a measure of the effect size.
We estimated that our study would have a power of 88% to detect a difference in ICH risk between CAA patients with and without cSS, assuming 50% and 20% rates of ICH over 4 years, respectively11,12,15 (2-tailed test and with α values of 0.05).
According to the “rule of 10” for developing proportional hazards models, we require approximately 10 outcome events for each potential covariate in multivariable analyses.21,22 Therefore, we investigated the effect of the following prespecified potential predictors using a Cox proportional hazards model: presence of cSS or disseminated cSS, the presence of multiple CMBs (≥2), prespecified on the basis of the hypothesized effect on ICH risk from previous published series in CAA,11,12 and age, which is an important potential confounder. As sensitivity analyses, we repeated all statistical tests in CAA patients with symptomatic lobar ICH at baseline, and those who had their MRI within 6 months of the index ICH. We also undertook exploratory multivariable models incorporating previous ICH as another potential confounding factor. The proportional hazard assumption was tested using graphical checks and Schoenfeld residuals–based tests.
A p value ≤0.05 was considered to be statistically significant. All analyses were performed using STATA (version 11.2; StataCorp, College Station, TX). This report was prepared with reference to the STROBE (Strengthening the Reporting of Observational Studies in Epidemiology) guidelines.23
RESULTS
The final cohort consisted of 118 patients fulfilling the Boston diagnostic criteria16 for CAA (table 1): 8 with pathologically proven CAA, 86 with probable CAA, and 24 with possible CAA. Of these, 104 patients (88%) presented with symptomatic lobar ICH at baseline, 8 with transient focal neurologic episodes, 3 with cognitive decline, 1 with acute convexity subarachnoid hemorrhage, and 2 with ischemic stroke. The interrater agreement for the presence or absence of cSS was 89.6% (Cohen κ = 0.79) and for cSS categories was 89.6% (weighted Cohen κ = 0.75). Forty-one patients (34.7%, 95% confidence interval [CI]: 26.2%–44.6%) had cSS at baseline. The presence of cSS was strongly associated with a history of transient focal neurologic episodes (48.8% vs 8.8%; p < 0.001, respectively) and the presence of ≥5 CMBs (48.8% vs 27.3%; p = 0.019, respectively) (table e-1 on the Neurology® Web site at www.neurology.org). CAA patients with cSS were slightly older compared to patients without siderosis (mean age; 95% CI: 73.2; 70.4–76 years vs 70.3; 68.2–72.5 years, respectively; p = 0.111).
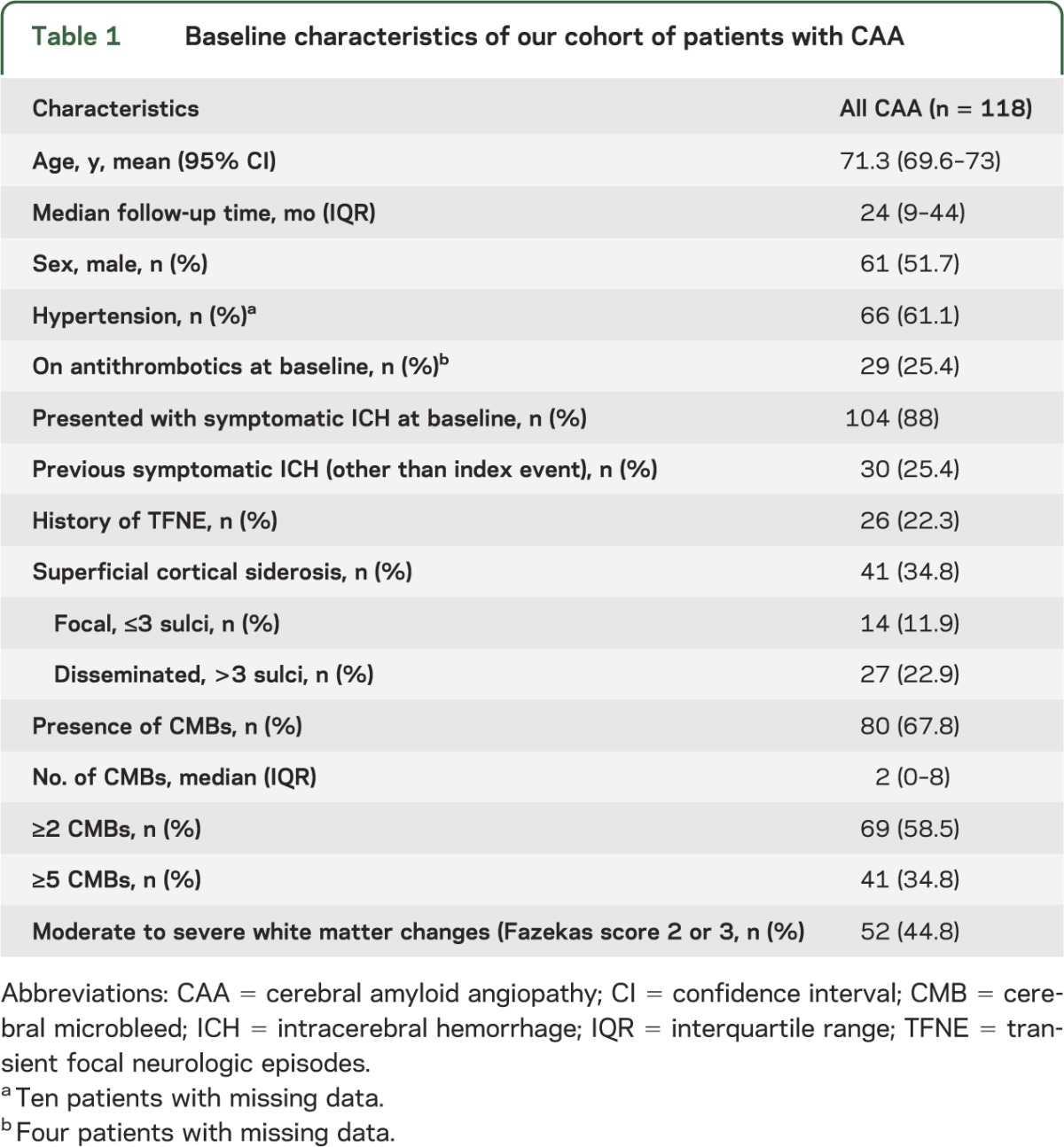
Table 1.
Baseline characteristics of our cohort of patients with CAA
During a median follow-up time of 24 months (interquartile range 9–44 months), 23 of 118 patients (19.5%, 95% CI: 12.8–27.8) experienced a symptomatic lobar ICH; 4 of these 23 patients experienced multiple (>1) sequential symptomatic lobar ICH during follow-up. The characteristics of our cohort according to subsequent lobar ICH are shown in table 2. Fourteen of 23 patients (61%) with lobar ICH during follow-up had cSS on baseline MRI: 3 had focal cSS and 11 had disseminated cSS (table 2). For 7 of these 14 patients, the subsequent ICH was anatomically correlated with the area of cSS (figure 1). Two of the 4 patients with multiple symptomatic lobar ICH at follow-up had disseminated cSS. One patient (without cSS at baseline) experienced a symptomatic deep (thalamic) ICH.
Table 2.
Characteristics and comparison of patients with CAA according to symptomatic lobar intracerebral hemorrhage at follow-up
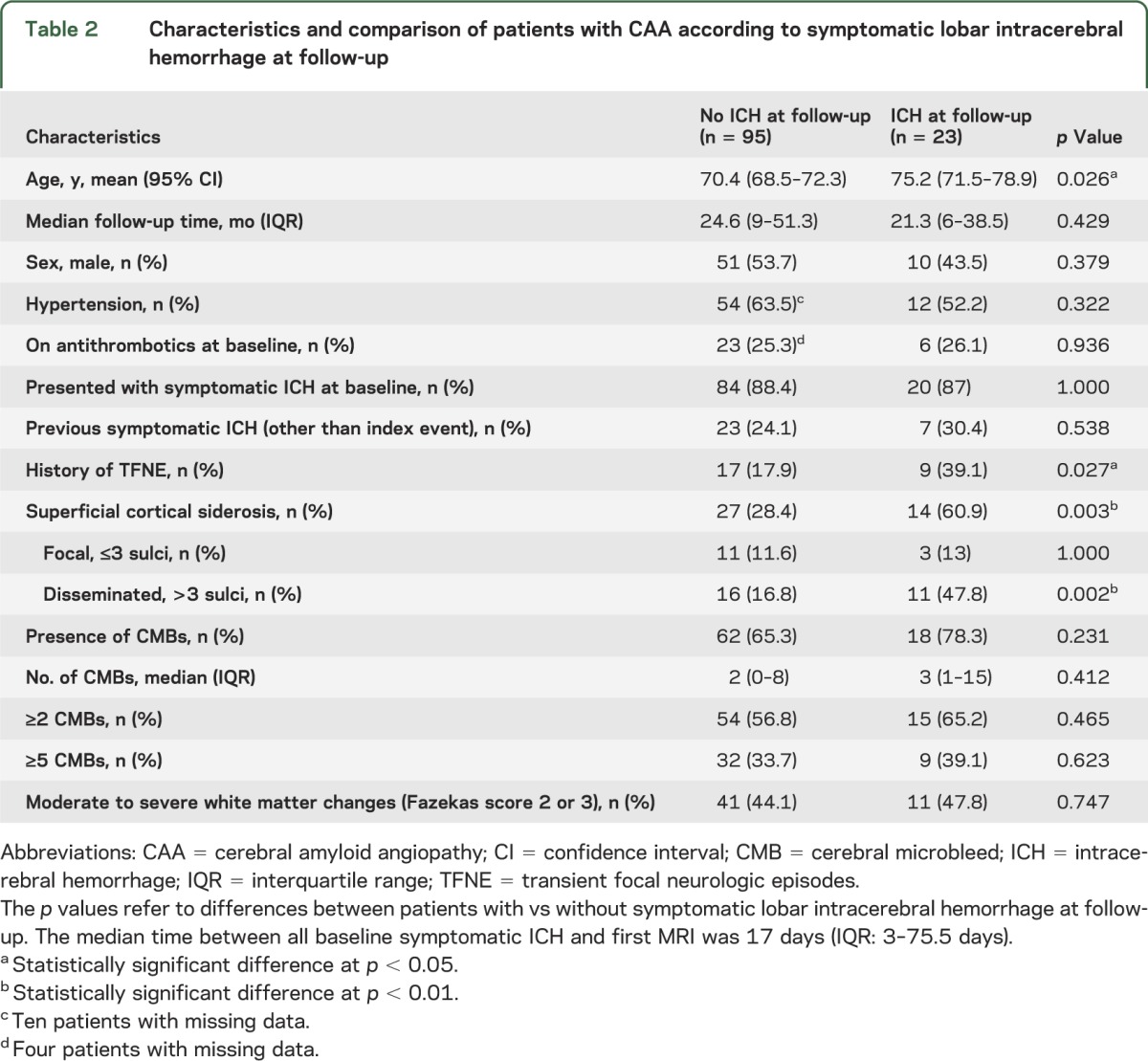
Figure 1. Cortical superficial siderosis and ICH.

Subsequent intracerebral hemorrhage (ICH) at (or close to) the site of preexisting siderosis in 2 patients with superficial siderosis. Female patient, aged 85 years; time interval between baseline imaging (A) and incident ICH (B): 19 months. Male patient, aged 80 years; time interval between baseline imaging (C) and incident ICH (D): 35 months. (A, C) Baseline MRI, T2*-weighted images. (B, D) Follow-up unenhanced CT scans. T2*-GRE = T2*-weighted gradient-recalled echo.
In the group of CAA patients without symptomatic lobar ICH at baseline (n = 14; one patient with pathologically proven CAA, 12 patients with probable CAA, and one with possible CAA), 7 had cSS, which was disseminated in 5 cases. During a median follow-up time of 15.1 months (interquartile range 7.8–39.8), 3 of these patients experienced a symptomatic lobar ICH, all of whom had disseminated cSS at baseline scans.
In Kaplan-Meier analysis, the presence of cSS and disseminated cSS at baseline scans were predictors of time until ICH (p = 0.0045 and p = 0.0009, respectively, by the log-rank test) (figure 2). The risk of symptomatic lobar ICH at 4 years of follow-up was 25% (95% CI: 7.6%–28.3%) for patients without cSS at baseline, 28.9% (95% CI: 7.7%–76.7%) for those with focal cSS, and 74% (95% CI: 44.1%–95.7%) for patients with disseminated cSS (p = 0.0031 by the long-rank test for each category increase).
Figure 2. Time to symptomatic lobar ICH during follow-up.
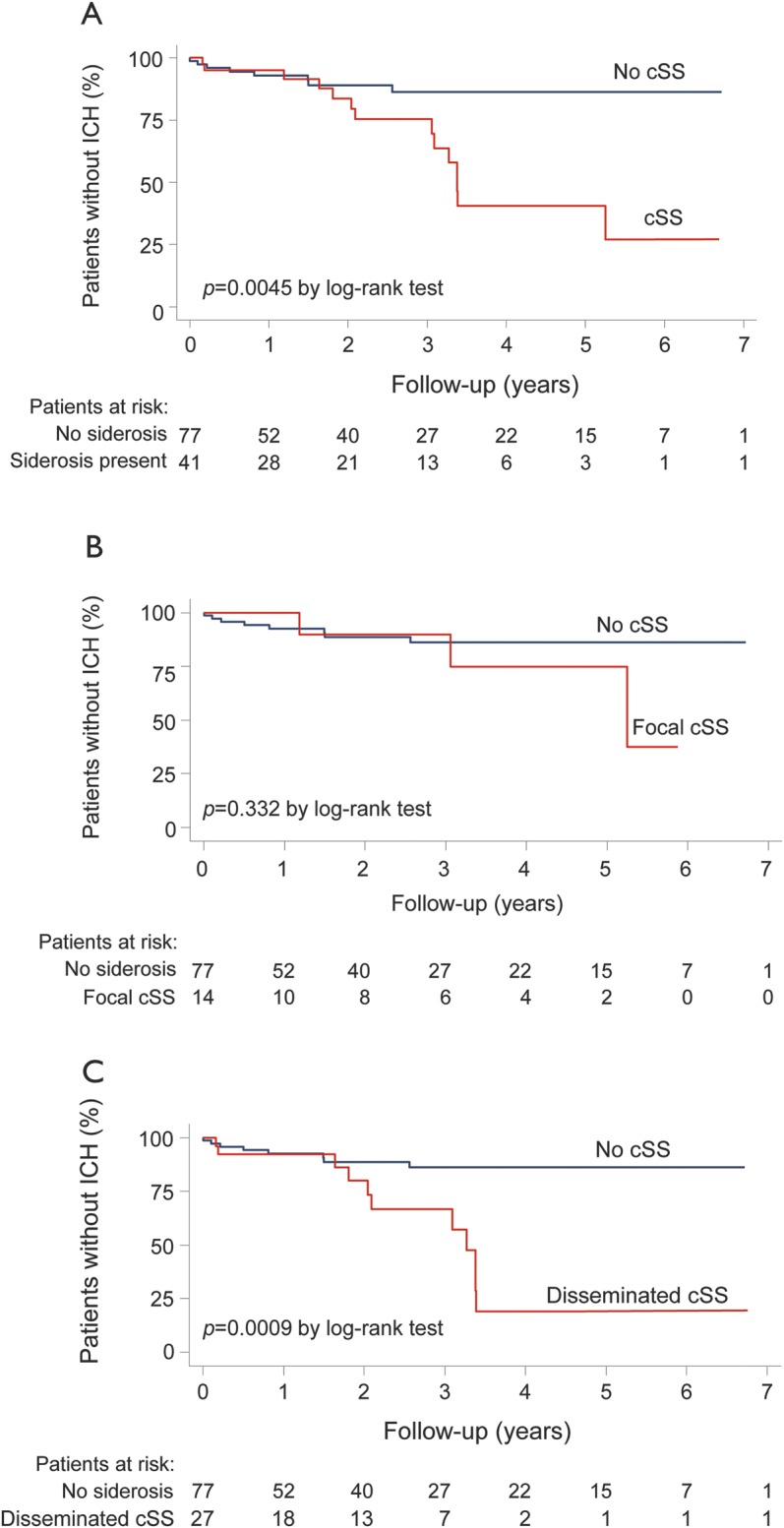
Kaplan-Meier estimates of progression to symptomatic lobar intracerebral hemorrhage (ICH) in the presence of (A) cortical superficial siderosis (cSS), (B) focal cSS, and (C) disseminated (>3 sulci) cSS in all patients with cerebral amyloid angiopathy. Testing of significance is by the log-rank test.
In univariate analysis, any cSS presence was a predictor of symptomatic lobar ICH (HR: 3.18; 95% CI: 1.37–7.39; p = 0.007), with an increased risk associated with disseminated cSS (HR: 4.07 compared with no siderosis; 95% CI: 1.66–9.96; p = 0.002). Focal cSS was associated with a nonsignificant increased hazard of subsequent ICH (HR: 1.91 compared with no siderosis; 95% CI: 0.51–7.19; p = 0.340). For each increase in category of cSS (i.e., from no siderosis, to focal, and to disseminated siderosis), the associated HR was 2.06 (95% CI: 1.31–3.24; p = 0.002). A history of symptomatic hemorrhage (before the index symptomatic ICH) (HR: 1.87; p = 0.177), presence of CMBs (HR: 2.17; p = 0.125), and presence of ≥2 or ≥5 CMBs (HR: 1.55; p = 0.318 and HR: 1.33; p = 0.507, respectively) were not associated with future symptomatic lobar ICH in univariate analysis. Only age was associated with an increased hazard of subsequent ICH (HR: 1.07; p = 0.006).
Prespecified multivariable Cox regression models demonstrated that cSS and disseminated cSS were independently associated with increased risk of symptomatic lobar ICH at follow-up, after adjusting for the presence of ≥2 CMBs and age (table 3). These effect sizes remained consistent in similar multivariable models controlling for CMBs number, CMBs presence, and ≥5 CMBs. Our main results were also consistent in sensitivity analyses including cSS, CMBs, history of symptomatic ICH, and age in multivariable models (see table e-2).
Table 3.
Prespecified multivariate analyses of predictors of symptomatic lobar intracerebral hemorrhage during follow-up in patients with cerebral amyloid angiopathy

All the results of Kaplan-Meier and prespecified Cox regression analyses remained consistent in sensitivity analyses, which included only CAA patients with symptomatic lobar ICH at baseline (figure e-1) and patients who had their MRI within 6 months of the index ICH (data not shown).
DISCUSSION
In this multicenter retrospective cohort study, we found that cSS on T2*-GRE MRI (reflecting hemosiderin deposition in the subpial superficial cortical layers) significantly increases the risk of future symptomatic lobar ICH in patients with CAA.16 The risk rate for ICH was greatest for patients with disseminated cSS, involving multiple sulci on baseline scans. These results remained consistent after adjusting for age, the presence of multiple (≥2) lobar CMBs (a hemorrhagic marker of CAA previously shown to influence the risk of ICH),12 and previous symptomatic ICH before the index inclusion event. In a subanalysis of those patients who presented with symptomatic ICH at baseline, cSS remained a predictor of ICH risk, consistent with the main analysis.
cSS is emerging as a common and characteristic feature of CAA.2,24 One study reported cSS in 60.5% (n = 38; mean age 70 ± 6.4 years) of patients with histopathologically proven CAA, compared with no control subjects with histopathologically proven non-CAA ICH (n = 22; mean age 54 ± 18 years).13 Another recent study found cSS in 40% of patients with a clinic-radiologic diagnosis of probable CAA-related ICH, but only 5% of patients with purely deep ICH, presumed to be due to hypertensive arteriopathy.25 Increasing data support the hypothesis that cSS might precede lobar ICH in patients with CAA.15,26–28 In a recent retrospective study, 51 patients with cSS and no apparent cause other than CAA were identified through a single-center systematic database search and followed up for a median of 35.3 months.15 Over this period, 24 patients (47.1%) experienced any new “intracranial hemorrhage” (ICH or acute convexity subarachnoid hemorrhage): 18 patients (35.3%) had an ICH, of which 13 were at the site of preexisting siderosis.15 This study was limited by the incomplete ascertainment of outcome intracranial bleeding events (without details of how many were symptomatic), and the lack of a comparison group without cSS at baseline.
Our larger study confirms that cSS is a risk factor for future symptomatic ICH in CAA, independent of multiple lobar CMBs. It is hypothesized that repeated episodes of hemorrhage from brittle superficial cortical or leptomeningeal CAA-affected vessels into the subarachnoid space leads to subpial hemosiderin deposition and cSS on MRI.29 The finding of cSS without previous ICH, and its tendency to occur distantly from previous ICH, favor this “primary” mechanism,13,15 rather than leakage of blood into the subarachnoid space secondary to previous lobar “macro” ICH.25 Consequently, cSS may be a marker of increased cortical and leptomeningeal small-vessel fragility, high CAA disease activity, and vulnerability to subarachnoid bleeding, which in some circumstances may extend and develop into a lobar ICH.30 Indeed, a neuropathologic series of 6 autopsy cases of subcortical hematoma caused by CAA showed that multiple leptomeningeal arteries can rupture into the subarachnoid space and the brain parenchyma.30 This hypothesis is supported by the observation that symptomatic ICH has been noted at (or close to) the site of previous siderosis,15 although we found that ICH only occurred at the site of cSS in 50% of cases. We found the highest ICH risk in patients with disseminated cSS, which may indicate widespread and numerous leptomeningeal vessels damaged by advanced CAA, providing multiple potential initiation sites for future ICH, increasing the probability of this outcome.30 Further serial MRI studies will help to unravel the sequence of events and mechanisms linking CAA, cSS, and lobar ICH, including asymptomatic bleeding.31 Assessment of the associations between APOE genotype (which was not available in our cohort) and the extent or severity of CAA-related pathology in leptomeningeal vessels may also be of interest.
Although our results suggest an increased risk of subsequent ICH in CAA patients with cSS on MRI, antithrombotic drug use probably also has a role in this risk by impairing hemostatic mechanisms.2,9,12 In our retrospective cohort, it was not possible to systematically collect data on the use of antithrombotic drugs. However, routine clinical practice in all 4 centers in our study was to avoid all antiplatelet agents (including aspirin) and anticoagulants in patients with suspected CAA, unless there was a very strong indication, so is unlikely to have contributed significantly to the outcome events in our study.
Our findings might have important implications for patients presenting with transient focal neurologic episodes (sometimes called “amyloid spells”32) resembling TIAs, migraine auras, or focal seizures,32–34 which are increasingly recognized in CAA. It was recently reported that such attacks are often related to cSS and herald a high early risk of symptomatic lobar ICH (24.5% [95% CI: 15.8%–36.9%] at 8 weeks).33 Thus, these types of attacks (which are often recurrent and stereotyped) should alert the clinician to the possibility of cSS; giving antithrombotic drugs to these patients because of misdiagnosis as TIA could significantly increase the risk of serious future ICH.31 Moreover, our current data suggest that disseminated siderosis may be a particular risk factor for future ICH in this situation.
Perhaps surprisingly, we did not find a significant association between multiple lobar CMBs and future ICH risk,11,12 which may reflect selection bias toward generally advanced disease with high prevalence of multiple lobar CMBs in our cohort. Although our study had adequate power to detect an increase in the risk of ICH in the presence of cSS, our sample size was not large enough to investigate additional potential baseline predictors of ICH (e.g., index ICH volume, APOE genotype) or to investigate ICH risk in the subset of patients without lobar ICH at baseline. Other potential limitations include the retrospective design, and the potential of bias in our sample because the requirement for MRI may exclude more severe cases of ICH. Furthermore, a proportion of otherwise eligible patients did not have reliable follow-up data. Finally, we did not have pathologic confirmation of CAA pathology, and acknowledge that the Boston criteria have imperfect specificity, especially for the “possible CAA” category.16,35
Our findings nevertheless suggest that cSS is a useful independent prognostic marker of intracerebral bleeding risk in CAA. Larger cohorts are needed to confirm our results and explore the potential implications for CAA treatment (e.g., avoiding antithrombotic agents in patients with disseminated cSS). cSS could also have implications for future disease-modifying treatments in CAA that may cause amyloid-β shifts between brain parenchyma and blood vessels (e.g., immunotherapy); CMBs have been considered a possible caution for such treatments,36 but the role of cSS in this setting remains to be determined.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Dr. Simone Gregoire for her assistance with establishing the multicenter cohort and for technical assistance with database organization.
GLOSSARY
- CAA
cerebral amyloid angiopathy
- CI
confidence interval
- CMB
cerebral microbleed
- cSS
cortical superficial siderosis
- FLAIR
fluid-attenuated inversion recovery
- GRE
gradient-recalled echo
- HR
hazard ratio
- ICH
intracerebral hemorrhage
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by A. Charidimou and Z. Fox. A. Charidimou: project design, data collection, data analysis, write-up. Z. Fox: data analysis, data interpretation. A. Peeters, Y. Vandermeeren, P. Laloux, and J.-C. Baron: data collection, critical revisions. R. Jäger: imaging analysis advice, critical revisions. D.J. Werring: project design, supervision, data interpretation, write-up, obtaining funding.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
A. Charidimou receives research support from the Greek State Scholarship Foundation, the Stroke Association, and the British Heart Foundation. A. Peeters, R. Jäger, Z. Fox, Y. Vandermeeren, P. Laloux, and J.-C. Baron report no disclosures. D. Werring receives research support from the Department of Health/Higher Education Funding Council for England (Clinical Senior Lectureship Award), the Stroke Association, and the British Heart Foundation. Part of this work was undertaken at UCLH/UCL who received a proportion of funding from the Department of Health's NIHR Biomedical Research Centres funding scheme. Go to Neurology.org for full disclosures.
REFERENCES
- 1.MRC CFAS Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Lancet 2001;357:169–175 [DOI] [PubMed] [Google Scholar]
- 2.Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatry 2012;83:124–137 [DOI] [PubMed] [Google Scholar]
- 3.Viswanathan A, Greenberg SM. Cerebral amyloid angiopathy in the elderly. Ann Neurol 2011;70:871–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vinters HV. Cerebral amyloid angiopathy: a critical review. Stroke 1987;18:311–324 [DOI] [PubMed] [Google Scholar]
- 5.Bejot Y, Cordonnier C, Durier J, Aboa-Eboule C, Rouaud O, Giroud M. Intracerebral haemorrhage profiles are changing: results from the Dijon population-based study. Brain 2013;136:658–664 [DOI] [PubMed] [Google Scholar]
- 6.Morgenstern LB, Hemphill JC, III, Anderson C, et al. Guidelines for the management of spontaneous intracerebral hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2010;41:2108–2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vermeer SE, Algra A, Franke CL, Koudstaal PJ, Rinkel GJ. Long-term prognosis after recovery from primary intracerebral hemorrhage. Neurology 2002;59:205–209 [DOI] [PubMed] [Google Scholar]
- 8.Bailey RD, Hart RG, Benavente O, Pearce LA. Recurrent brain hemorrhage is more frequent than ischemic stroke after intracranial hemorrhage. Neurology 2001;56:773–777 [DOI] [PubMed] [Google Scholar]
- 9.Viswanathan A, Rakich SM, Engel C, et al. Antiplatelet use after intracerebral hemorrhage. Neurology 2006;66:206–209 [DOI] [PubMed] [Google Scholar]
- 10.O'Donnell HC, Rosand J, Knudsen KA, et al. Apolipoprotein E genotype and the risk of recurrent lobar intracerebral hemorrhage. N Engl J Med 2000;342:240–245 [DOI] [PubMed] [Google Scholar]
- 11.Greenberg SM, Eng JA, Ning M, Smith EE, Rosand J. Hemorrhage burden predicts recurrent intracerebral hemorrhage after lobar hemorrhage. Stroke 2004;35:1415–1420 [DOI] [PubMed] [Google Scholar]
- 12.Biffi A, Halpin A, Towfighi A, et al. Aspirin and recurrent intracerebral hemorrhage in cerebral amyloid angiopathy. Neurology 2010;75:693–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Linn J, Halpin A, Demaerel P, et al. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology 2010;74:1346–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Linn J, Herms J, Dichgans M, et al. Subarachnoid hemosiderosis and superficial cortical hemosiderosis in cerebral amyloid angiopathy. AJNR Am J Neuroradiol 2008;29:184–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linn J, Wollenweber FA, Lummel N, et al. Superficial siderosis is a warning sign for future intracranial hemorrhage. J Neurol 2012;260:176–181 [DOI] [PubMed] [Google Scholar]
- 16.Knudsen KA, Rosand J, Karluk D, Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology 2001;56:537–539 [DOI] [PubMed] [Google Scholar]
- 17.Gregoire SM, Chaudhary UJ, Brown MM, et al. The Microbleed Anatomical Rating Scale (MARS): reliability of a tool to map brain microbleeds. Neurology 2009;73:1759–1766 [DOI] [PubMed] [Google Scholar]
- 18.Greenberg SM, Nandigam RN, Delgado P, et al. Microbleeds versus macrobleeds: evidence for distinct entities. Stroke 2009;40:2382–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inzitari D, Simoni M, Pracucci G, et al. Risk of rapid global functional decline in elderly patients with severe cerebral age-related white matter changes: the LADIS study. Arch Intern Med 2007;167:81–88 [DOI] [PubMed] [Google Scholar]
- 20.Kidwell CS, Greenberg SM. Red meets white: do microbleeds link hemorrhagic and ischemic cerebrovascular disease? Neurology 2009;73:1614–1615 [DOI] [PubMed] [Google Scholar]
- 21.Concato J, Peduzzi P, Holford TR, Feinstein AR. Importance of events per independent variable in proportional hazards analysis. I. Background, goals, and general strategy. J Clin Epidemiol 1995;48:1495–1501 [DOI] [PubMed] [Google Scholar]
- 22.Peduzzi P, Concato J, Feinstein AR, Holford TR. Importance of events per independent variable in proportional hazards regression analysis. II. Accuracy and precision of regression estimates. J Clin Epidemiol 1995;48:1503–1510 [DOI] [PubMed] [Google Scholar]
- 23.von Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Lancet 2007;370:1453–1457 [DOI] [PubMed] [Google Scholar]
- 24.Vernooij MW, Ikram MA, Hofman A, Krestin GP, Breteler MM, van der Lugt A. Superficial siderosis in the general population. Neurology 2009;73:202–205 [DOI] [PubMed] [Google Scholar]
- 25.Charidimou A, Jager HR, Fox Z, et al. Prevalence and mechanisms of cortical superficial siderosis in cerebral amyloid angiopathy. Neurology 2013;81:626–632 [DOI] [PubMed] [Google Scholar]
- 26.Kumar S, Goddeau RP, Selim MH, et al. Atraumatic convexal subarachnoid hemorrhage: clinical presentation, imaging patterns, and etiologies. Neurology 2010;74:893–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katoh M, Yoshino M, Asaoka K, et al. A restricted subarachnoid hemorrhage in the cortical sulcus in cerebral amyloid angiopathy: could it be a warning sign? Surg Neurol 2007;68:457–460 [DOI] [PubMed] [Google Scholar]
- 28.Kawahara I, Nakamoto M, Matsuo Y, Tokunaga Y. A case of cerebral amyloid angiopathy in which a restricted subarachnoid hemorrhage recurred in the cortical sulcus following a subcortical hemorrhage [in Japanese]. No Shinkei Geka 2010;38:551–555 [PubMed] [Google Scholar]
- 29.Koeppen AH, Dickson AC, Chu RC, Thach RE. The pathogenesis of superficial siderosis of the central nervous system. Ann Neurol 1993;34:646–653 [DOI] [PubMed] [Google Scholar]
- 30.Takeda S, Yamazaki K, Miyakawa T, et al. Subcortical hematoma caused by cerebral amyloid angiopathy: does the first evidence of hemorrhage occur in the subarachnoid space? Neuropathology 2003;23:254–261 [DOI] [PubMed] [Google Scholar]
- 31.Charidimou A, Baron JC, Werring DJ. Transient focal neurological episodes, cerebral amyloid angiopathy, and intracerebral hemorrhage risk: looking beyond TIAs. Int J Stroke 2013;8:105–108 [DOI] [PubMed] [Google Scholar]
- 32.Charidimou A, Law R, Werring DJ. Amyloid “spells” trouble. Lancet 2012;380:1620. [DOI] [PubMed] [Google Scholar]
- 33.Charidimou A, Peeters A, Fox Z, et al. Spectrum of transient focal neurological episodes in cerebral amyloid angiopathy: multicentre magnetic resonance imaging cohort study and meta-analysis. Stroke 2012;43:2324–2330 [DOI] [PubMed] [Google Scholar]
- 34.Greenberg SM, Vonsattel JP, Stakes JW, Gruber M, Finklestein SP. The clinical spectrum of cerebral amyloid angiopathy: presentations without lobar hemorrhage. Neurology 1993;43:2073–2079 [DOI] [PubMed] [Google Scholar]
- 35.van Rooden S, van der Grond J, van den Boom R, et al. Descriptive analysis of the Boston criteria applied to a Dutch-type cerebral amyloid angiopathy population. Stroke 2009;40:3022–3027 [DOI] [PubMed] [Google Scholar]
- 36.Sperling RA, Jack CR, Jr, Black SE, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimers Dement 2011;7:367–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.