

Abstract
The complexity of metazoan organisms requires precise spatiotemporal regulation of gene expression during development. We find that in the nematode Caenorhabditis elegans approximately 2,000 transcripts undergo expression oscillations synchronized with larval transitions while thousands of genes are expressed in temporal gradients, similar to known timing regulators. By counting transcripts in individual animals, we show that the pulsatile expression of the microRNA (miRNA) lin-4 maintains the temporal gradient of its target lin-14 by dampening its expression oscillations. Our results demonstrate that this insulation is optimal when pulsatile expression of the miRNA and its target is synchronous. We propose that such a miRNA-mediated incoherent feed-forward loop is a potent filter that prevents propagation of potentially deleterious gene expression fluctuations during the development of an organism.
Proper development of a multi-cellular organism relies on the faithful expression of developmental genes. However, gene expression dynamics of any individual cell is inevitably noisy due to stochastic molecular events1 and global intracellular fluctuations in cellular components due to environmental perturbation2. Such fluctuations, when miscontrolled, could be detrimental as they could potentially interfere with proper expression of important developmental genes and could create uncertainty in developmental outcomes. Surprisingly, developmental phenotypes of multi-cellular organisms under normal conditions are robustly reproduced, suggesting that these animals successfully dampen undesired expression fluctuations of genes that require precise control. However, these control mechanisms are largely unexplored.
Larval development of the nematode C. elegans is an excellent model system for investigating mechanisms that control developmental gene expression, because its physiological state changes almost discretely during four successive larval stages (L1-L4). These changes are mediated by both rhythmically and temporally graded expression of key developmental genes in the same cell or tissue that are known to be required for normal progression of developmental events3–5. By the end of each larval stage the worm passes through a quiescent sleep-like behavioral state, or lethargus6, followed by a molt7. In the hypoderm, collagen-encoding molting genes and nuclear hormone receptors exhibit periodic expression peaking once every larval stage, potentially acting as timers of molting cycle4,5. Although most somatic tissues are already differentiated upon hatching, the body plan of the animal is further finalized by expansion and migration of hypodermal, intestinal and neuronal tissues8. The germ line maturates and the worm becomes reproductively competent9. In contrast to the molting cycle, the timing of many of these developmental events is often precisely controlled by cell-fate regulators expressed in a temporal gradient in which the level of these regulators monotonically decreases as development progresses10–12. Mutations of these so-called heterochronic genes lead to precocious or retarded development (omission or reiteration of developmental events)10,11,13,14.
The transcription factor lin-14 was among the first heterochronic genes described in C. elegans3,13 and is a target of the miRNA lin-415. miRNAs are ~22 nucleotide long non-coding RNAs that guide a multi-protein complex to its complementary elements in the 3’UTR of protein coding genes and repress protein production by translational inhibition or transcript degradation16. Gene products of lin-14 trigger dosage-dependent larval stage-specific programs10. Down-regulation of the LIN-14 protein by lin-4 was found to be critical for the transition from L1 to L2-specific programs and further downregulation at the end of L2 is required for the transition to L3-specific programs. When lin-4 mediated repression is lost, LIN-14 protein level remains high and animals display a “retarded” phenotype where cells variably reiterate early developmental programs17. The importance of the gradually decreasing lin-14 expression was first inferred from genetic analyses10, which suggested that cells in wild-type animals can distinguish distinct levels of these gene's activities at specific times during the first two larval stages. Despite the fact that lin-4 and lin-14 are historically the first miRNA and target gene to be discovered and have been extensively studied, mechanisms that control the integrity of the temporal gradient have not been elucidated.
In our study, we used next generation mRNA sequencing to explore the dynamics of the C. elegans larval transcriptome with unprecedented temporal resolution to systematically determine the extent to which genes fluctuate during development. Based on these data, we classify genes into groups that either exhibit periodic expression synchronized with the molting cycle or display a temporal gradient, similar to heterochronic genes. Using computational analyses, we assess the role of miRNAs in regulating these different expression patterns. We then performed a detailed characterization of the expression dynamics of microRNA lin-4 and its target lin-14, and propose a novel regulatory role of a microRNA as a damper of target gene expression oscillations. Our findings suggest that, during C. elegans post-embryonic development, expression of lin-14 is driven by an upstream regulator that oscillates in phase with the molting cycle. The transcript levels of lin-14, however, do not oscillate in wild-type animals due to a synchronous oscillatory expression of lin-4. This gene network motif is identical to an incoherent feedforward motif18. Our mathematical model analysis suggests that this microRNA-mediated incoherent feedforward circuit is crucial for efficient dampening of expression oscillations of the microRNA target, ensuring a stable temporal gradient of lin-14.
RESULTS
Thousands of genes display periodic expression in synchrony with the molting cycle
To obtain precise dynamics of the C. elegans transcriptome during larval development, we conducted RNA-Seq experiments using poly(A)+ RNA libraries prepared from synchronized larval populations representing 20 different developmental time points ranging from 0 hr to 38 hr after hatching with 2 hr resolution at 20°C (Fig. 1a). This range spans the first three larval stages (L1, L2, and L3). In an independent experiment we prepared and sequenced libraries for the L4 stage, again with 2 hr resolution, ranging from 38 hr to 48 hr. We used the Illumina HiSeq 2000 platform to generate single-end 40-bp reads. Reads were mapped to recently published improved gene models19 and expression aggregated across all isoforms derived from a given gene locus. In our analysis, we discarded genes that had an average RPKM (reads per kilobase of exon model per million mapped reads) value20 of less than one and detected 16,776 reliably expressed annotated gene loci. We first examined how the transcriptome-wide gene expression levels varied by computing the pair-wise correlations between transcriptomes at different developmental time points (Supplementary Fig. 1a). Interestingly, we found that the pair-wise correlations did not monotonically decrease when comparing more distant time points. For example, the correlation coefficient between time-points 10 hr and 16 hr was smaller than between 10 hr and 22 hr (Supplementary Fig. 1a). To test whether this was a robust phenomenon, we cultured synchronized larval populations at 25°C. At this elevated temperature, animals develop normally but at an accelerated rate. We therefore harvested poly(A)+ RNA libraries every 1.5 hr and found strikingly similar pair-wise correlations to those obtained at 20°C (Supplementary Fig. 1a). This suggested that the C. elegans transcriptome is modulated periodically during larval development.
Figure 1. A number of genes display cycling expression dynamics in synchrony with the molting cycle during larval development.

(a) Experimental set-up. After synchronizing hatched worms by starvation, samples were collected every 2 hours (1.5 hours) during larval development at 20°C (25°C). For each sample, transcript expression was measured by poly(A)+ RNA-seq. (b-c) Normalized expression profiles of all 231 genes (grey) that co-cluster with the collagen encoding molting gene dpy-10 (red) known to exhibit periodic expression7. Cycling expression was observed at 20°C (b) and 25°C (c). The average profile is indicated by the broken black line. All curves are spline fits of the discrete expression profiles (red dots for dpy-10). Dashed lines separate larval stages.
To detect periodic signals from expression profiles of individual genes we first decomposed the temporal expression profile of each gene into a trend and a cycling component using Hodrick-Prescott filtering. Next, we assessed the significance of autocorrelation of the cycling component. Genes with a significant autocorrelation (Durbin-Watson P < 0.05) were classified as cycling genes, while all other genes were termed graded, since their temporal expression changes were dominated by the slowly changing trend component. We resolved distinct modes of expression within the groups of cycling and graded genes, and reduced the entire 16,776 profiles to reproducible clusters of gene expression profiles (see online Methods, Supplementary Fig. 1b and Supplementary Table 1). While the majority of genes exhibited graded expression patterns (14,703 genes, 88%), we were surprised to find a large number of genes with cycling expression (2,073 genes, 12%). In total, 12,695 graded genes (86%) and 1,592 cycling genes (77%) were assigned to robust clusters. As an example, we considered the cuticle collagen dpy-10, which displays periodic expression7. This gene cycles with a period length of about 12 hours and peaks once every larval stage (Fig. 1b). A 30-fold change is observed between expression minima and maxima and these pronounced expression changes occur within six hours. In Figure 1b, all 231 genes that co-cluster with dpy-10 are shown. We observed a similar behavior for this group of genes at 25°C although the period length was reduced due to an accelerated development (Fig. 1c). We found that after accounting for a homogenous ~30% speed-up of development at 25°C, genome-wide temporal gene expression profiles are indeed highly similar to independently grown worm populations at 20°C (Supplementary Fig. 2).
Cycling genes have a distinctive functional signature and are highly enriched in putative miRNA targets
From the robust gene expression clusters identified, we found six distinct clusters of co-expressed cycling genes (Fig. 2a). Remarkably, average expression profiles for all of these clusters displayed a period length of approximately 12 hours and appeared to be synchronized with the larval transitions. Four clusters (1,3,5,6) cycle with similar amplitudes but small relative shifts of the expression peaks. The two remaining clusters (2,4) display less pronounced expression peaks and the average profiles of these clusters are phase shifted by half a period length. Graded gene expression profiles were distributed among six robust clusters, with decreasing (7,10,11), increasing (8,9) or constant temporal expression (12). Notably, robust clusters of co-regulated genes with pronounced pulsatile, but noncycling expression, were not found.
Figure 2. Clusters of genes with cycling or graded expression show differential signature of post-transcriptional regulation.

(a) Average normalized expression profiles of distinct expression clusters (Supplementary Table 1) for cycling genes (left) and graded genes (right) at 20°C (blue) and 25°C (red). The transparent shading indicates the standard deviation. For developmental times larger than 38 hours at 20°C, data from an independently grown worm population were used. Developmental time at 25°C was rescaled linearly by a factor of 0.77 to account for faster development and make expression at different temperatures comparable (Supplementary Fig. 2). Broken grey lines indicate transitions between larval stages at 20°C. (b) Upper panel: Enrichment of human orthologs (blue) as a proxy for gene age, of lethal RNAi phenotypes (brown) to measure the fraction of genes essential for survival, and of predicted conserved microRNA targets to estimate the extent of post-transcriptional regulation for each cluster. Error bars are based on random counting statistics. P-values were computed by Fisher's exact test. Data are shown for all clusters and for the ensemble of all clustered genes. Lower panel: Percentage of conserved nucleotides within aligned contigs of total exonic sequence (blue), of 3’UTR exons (orange) and for 1 kilobase of upstream sequence (brown). Conservation was measured using multiple sequence alignments of C. elegans, C. briggsae and C. remanei, downloaded from the UCSC genome browser27. Unaligned sequence of C. elegans was counted as mismatches to obtain a conservative estimate. Shown are the interquartile range (box) and the median (black). Data are shown for all clusters and for the ensemble of all clustered genes. The colored lines indicate the median across all genes. P-values were computed by a two-sided t-test. (*P<0.05; **P<0.001).
In order to investigate and characterize co-expressed genes, we first computed functional enrichments in Gene Ontology (GO) terms for each of the clusters (online Methods, Supplementary Table 2). A summarized annotation is given in Table 1. Not surprisingly, cycling clusters with pronounced amplitudes (1,3,6) were associated with molting, a process that occurs at the end of each larval stage, during which the worm replaces the entire exoskeleton by proteolysis of old and generation of new cuticle7. Other cycling clusters, however, were also preferentially associated with metabolic functions (cluster 4) and germ line development (cluster 2). Hence, cycling genes are not only associated with molting but appear to exert more diverse functions.
Table 1.
Summarized functional annotation of the expression clusters based on GO term enrichment analysis. For a full list of enriched GO terms, see Supplementary Table 2.
| Cluster | Nb. of genes | Summarized enriched biological processes (P < 10−5) |
|---|---|---|
| 1 | 231 | molting cycle |
| 2 | 384 | gonad morphogenesis, protein localization |
| 3 | 116 | proteolysis |
| 4 | 544 | metabolism |
| 5 | 92 | - |
| 6 | 225 | cuticle development, proteolysis, body morphogenesis |
| 7 | 3175 | signaling, metabolism, transcription, nervous system development |
| 8 | 610 | (de-)phosphorylation, gamete generation |
| 9 | 841 | (de-)phosphorylation, gamete generation |
| 10 | 2092 | signaling, neurological system process, sensory perception, transcription |
| 11 | 932 | sensory perception, transcription |
| 12 | 5045 | embryo development, larval development, reproduction, cell cycle |
Within the graded clusters, genes with temporally decreasing expression (clusters 7,10,11) were enriched in annotations related to sensory perception and nervous system development, and were, more generally, associated with transcription and signaling. These observations probably reflect the ongoing neuronal differentiation during the first two larval stages. An enhanced number of genes with maximal expression towards the end of larval development (clusters 8,9) appear to be involved in (de-) phosphorylation and gamete generation. These genes most likely become expressed in the germ-line upon onset of spermatogenesis during the fourth larval stage9. Interestingly, a pronounced over-representation of developmental gene annotations was observed for the largest cluster (12) with constant temporal expression. Presumably, many of these genes are switched on upon specification of somatic tissues during embryonic development and maintain tissue specific expression during larval development and adulthood.
To analyze essentiality of genes with distinct temporal expression patterns for development and survival of the organism we measured enrichment in lethal RNAi phenotypes21 (Fig. 2b). Consistent with the pronounced over-representation of developmental genes, the strongest enrichment was observed for cluster 12. Interestingly, the only other clusters (2,4) with an over-representation of lethal phenotypes belonged to the class of cycling genes. We observed a strikingly similar enrichment pattern for the fraction of human orthologs (see online Methods), confirming the assumption that essential genes are preferentially evolutionarily old.
One of our major goals was to investigate the role of post-transcriptional regulation in modulating temporal gene expression patterns. We therefore screened the 3’UTRs of all genes in a given expression cluster for the presence of conserved miRNA target sites. We used target predictions of TargetScanS22 after discarding lowly conserved sites (PCT < 0.9). While three of the cycling clusters (1,2,4) were significantly enriched in conserved miRNA targets, only one of the graded clusters (7) was enriched and three graded clusters (8,9,11) were strongly depleted in miRNA targets sites. This observation suggests that post-transcriptional gene regulation could be required for mediating highly dynamic expression changes, which would be consistent with a more sensitive expression response upon increased transcript degradation triggered by miRNAs23–26. Consistently, all but one of the cycling clusters exhibit on average significantly increased 3’UTR sequence conservation (Fig. 2b), measured from multiple alignments to the related nematode species C. briggsae and C. remanei27. We also detected an overall enhanced sequence conservation of exonic sequence (i. e. coding exons and UTRs) and promoter sequence (annotated as 1 kilobase upstream of transcription start site) for the cycling clusters (Fig. 2b). In contrast, conservation in the majority of the graded clusters (8,9,10,11) was significantly reduced (Fig. 2b). Although cycling genes were overall more highly conserved than graded genes, the difference was particularly pronounced for promoter and 3’UTR sequence, where average conservation was low. Considering the under-representation of essential genes in most of the cycling clusters, it was surprising to observe enhanced conservation of regulatory sequence. These at first glance contradictory findings could potentially be reconciled by the requirement of cycling genes to host a higher density of regulatory elements within promoters and 3’UTRs in order to confer highly dynamic regulation.
Temporal expression of lin-4 targets suggests specific role of lin-4 in maintaining temporal gradients
We identified miRNAs with preferences for predicted target genes with either cycling or graded expression (Fig. 3a,b). For instance, targets of miR-230 were strongly over-represented in two cycling clusters (1,6), and targets of miR-71 were overall enriched in cycling genes (Fig. 3a). Interestingly, miR-71 is necessary for larval survival after stress conditions28 and promotes longevity29,30. Other miRNAs, like miR-1/796, have significantly increased numbers of targets in cycling and graded clusters (Fig. 3a).
Figure 3. Predicted targets of many microRNAs are enriched in temporally co-expressed genes.

(a) Fold enrichment of predicted microRNA targets in cycling expression clusters. Targets of miR-230 are strongly over-represented in cycling clusters with pronounced amplitudes, while miR-1/796 targets are enriched in cycling clusters with low amplitudes. In contrast, predicted miR-71 targets are enriched in all cycling clusters. (b) Fold enrichment of predicted targets of developmental microRNAs. Targets are either dispersed across different expression clusters (miR-51 and let-7 family) or concentrated in a single expression cluster (lin-4 family). Error bars in a and b are based on random counting statistics. P-values in a and b were computed by Fisher's exact test. (*P<0.05; **P<0.001).
In contrast, many predicted targets of the heterochronic miRNAs let-7 and lin-4 as well as many targets of the miR-51 family, one of the few miRNA families that are indispensible during embryonic development31, were found among genes with a temporal gradient (Fig. 3b). The specificity of temporal gene expression profiles was particularly pronounced for lin-4 targets, which were highly concentrated in cluster 7. This observation suggests a major role of lin-4 in maintaining a robust temporal gradient of its targets. Among these targets is the important developmental timing regulator lin-14, a classic heterochronic gene that displays a negatively graded temporal expression3,10,13.
The miRNA lin-4 shows periodic expression in synchrony with the molting cycle
To study whether lin-4 plays any role in insulating lin-14 from periodic fluctuations, we first measured mature lin-4 miRNA levels in total RNA extracts harvested from staged larvae using a quantitative real-time PCR assay for small RNA detection. To our surprise, mature lin-4 expression was also pulsatile, peaking approximately once per larval stage (Fig. 4a,b). We also investigated the activity of the lin-4 promoter by measuring the transcript levels of a lin-4 promoter-GFP fusion construct32 (Fig. 4c). The lin-4 promoter was ubiquitously active in somatic cells, but not in the germ line, in agreement with previous reports32. Strikingly, we found the promoter activity of lin-4 to alternate between high and low during post-embryonic development with a period of approximately one larval stage (Fig. 4c), suggesting that the pulsatile mature lin-4 miRNA levels (Fig. 4a,b) are transcriptionally regulated. Although it is widely accepted that the expression level profile of lin-4 miRNA exhibits a gradual “switch-like” transition25,33 at the end of L1, our sub-larval stage high-resolution data suggest the existence of intricate dynamics that has been previously unseen.
Figure 4. lin-4 microRNA expression is pulsatile.
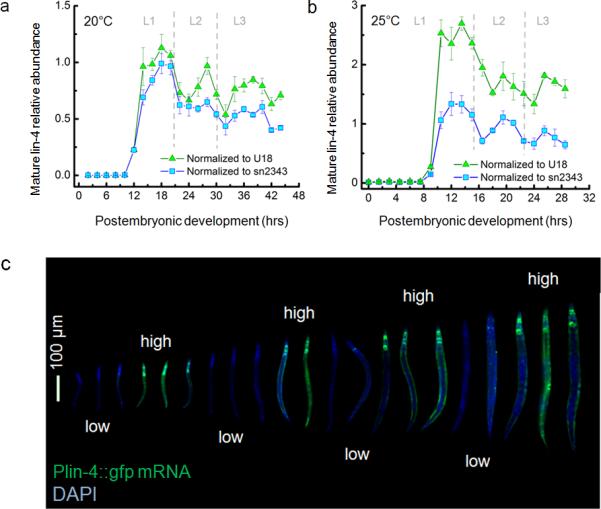
(a-b) Mean mature lin-4 level of staged larvae determined by Taqman qPCR and 20°C (a) and 25°C (b). Error bars represent standard deviation (n>3). Dashed lines separate larval stages. (c) Plin-4::gfp smFISH (green) and DAPI (blue) images of staged larvae are merged and ordered by their body lengths. Scale bar indicates 100 μm.
Transcript counting by single molecule in situ hybridization confirms temporal gradient of lin-14
Intrigued by this dynamic expression of lin-4 miRNA, we decided to obtain detailed expression dynamics of the canonical lin-4 target, and counted individual lin-14 transcripts in intact animals using a single molecule fluorescence in situ hybridization method (smFISH)34 (Fig. 5a). We obtained high temporal resolution by estimating the post-embryonic age of individual animals from body length, defined as the distance between the anterior-most nucleus hyp4 and the posterior-most nucleus hyp10 along the antero-posterior (AP) axis (Fig. 5b, Supplementary Fig. 3-5). lin-14 transcripts were detected ubiquitously throughout the body in all previously reported expressing cells3,11 of wild-type animals during all larval stages, from L1 to L4 (Fig. 5a). The exceptions were germ line and somatic gonad cells where no lin-14 transcripts were observed. We determined mRNA concentrations at each developmental time point by normalizing the total transcript count to the volume of the worm35 (Supplementary Fig. 6). We found that the lin-14 mRNA concentration forms a smooth temporal gradient during postembryonic development (Fig. 5c). To test whether these expression dynamics were observed consistently along the AP axis, we generated AP mRNA density maps (Supplementary Fig. 7). We found that, with the exception of the head, tail and germ line regions where cell densities are higher, lin-14 transcripts were equally distributed along the mid-body region and were uniformly down-regulated during larval development, recapitulating the dynamics observed in the whole animal (Fig. 5c).
Figure 5. lin-14 mRNA is homogeneously expressed in somatic tissue of worm larvae and exhibits a temporal gradient.
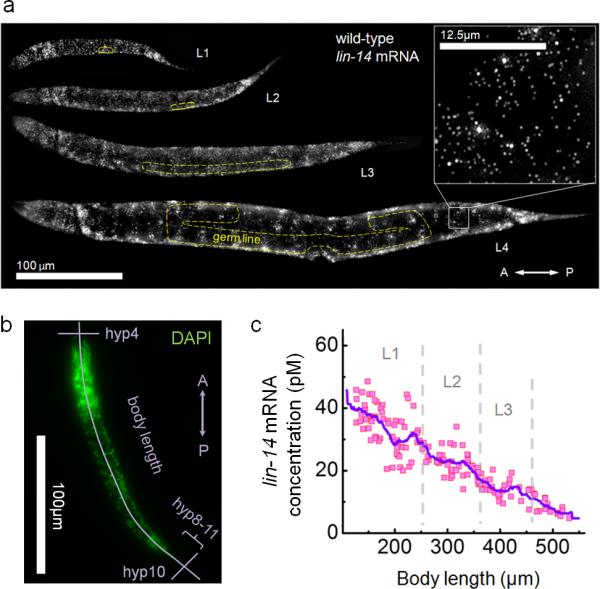
(a) Maximum z-projection of lin-14 smFISH stack images of staged larvae. Transcripts are visualized as countable fluorescent spots (inset at 8X zoom). Yellow dashed lines outline the germ line and gonad. Scale bars indicate 100 μm and 12.5 μm (inset) respectively. (b) Body length determined from DAPI staining. (c) Overall lin-14 mRNA concentration of individual animal as a function of body length. The curve indicates a moving average (bin; 36 μm). Dashed lines separate larval stages.
Expression of the lin-4 target lin-14 becomes pulsatile upon loss of lin-4 expression
The pulsatile dynamics of the lin-4 miRNA is difficult to reconcile with the smooth temporal gradient observed for lin-14, which is a direct target of lin-411,15. To explore why the pulsatile dynamics of lin-4 is not propagated to its target, we measured lin-14 mRNA concentration in lin-4(e912) knock-out mutant animals (Fig. 6a). During early L1, lin-14 transcript levels in these mutant animals were identical to that of wild-type animals. However, as larvae entered late-L1, the lin-14 concentration started to exhibit large peaks in the absence of lin-4. The peaks occurred approximately once per larval stage. Our in situ results in these mutants faithfully recapitulated our RNA-Seq results, suggesting that smFISH reliably detects RNA molecules even in C. elegans larvae with abnormal cuticles (Supplementary Fig. 8). We questioned whether this pulsatile dynamics was also present at the level of individual cells and measured lin-14 mRNA in a group of hypodermal cells, hyp8-11 (five nuclei), at the tail tip, which increase in volume but do not divide36 (Fig. 6b, Supplementary Fig. 9). The lin-14 mRNA dynamics in these cells were qualitatively similar to those observed in the entire animal. Next we generated a lin-14 transcript fold-change map and found that the de-repression in the absence of lin-4 activity was present globally along the AP axis (Fig. 6c). Each colored bin indicates the fold-change of lin-14 transcript numbers in lin-4(912) animals compared to wild-type animals at a given location along the AP body axis and at a given post-embryonic age. Recurring vertical bands in this map reflect the fact that the timings of lin-14 transcript level pulses are synchronized in every segment of the animal body along the AP axis. We examined the mid-body region of the animals which is uniformly comprised of hypodermal, intestinal and muscle cells, and indeed found a transient ~3-fold repression of lin-14 transcript level in the wild-type compared to lin-4(e912) animals during the L1 and L2 stage (Fig. 6d, Supplementary Fig. 8). The strength of the transient repression was further reduced during the L3 stage, reaching a steady ~1.5-fold repression. Because lin-4(e912) animals exhibit reiterating L1 fates (hatch→L1→L1→L1→)10,13, it is possible that the pulsatile lin-14 transcript levels are merely a repetition of L1 fate specific pulses. However, we observed similar lin-14 transcript level dynamics in lin-4(e912); lin-14(n179ts) animals of “precocious” phenotype which skips the L1 specific larval program (hatch→L2→L3→L4→)10, suggesting that the lin-14 transcript level pulses are not L1 specific (Supplementary Fig. 10).
Figure 6. lin-14 mRNA level exhibits pulsatile dynamics in the absence of lin-4 negative regulation.

(a-b) lin-14 mRNA concentration dynamics of individual lin-4(e912) mutants as a function of body length. Shown are the overall concentration (a) and the local concentration in hyp8-11 (b). Curves in a and b indicate moving averages (bin size = 36 μm) and dashed lines separate larval stages. (c) AP map of the lin-14 mRNA level fold repression in wild-types compared to lin-4(e912) mutants. AP (%) indicates relative position along AP axis (0%-head, 100%-tail). The height of each bin is 1% and a moving average (bin size = 36 μm) is applied in x-direction. (d) lin-14 mRNA fold repression in the mid-body. Each curve represents trajectory along x-axis of the heat map in c where its corresponding colorbar is located.
Dampening of lin-14 transcript fluctuations is directly mediated by lin-4 target sites in its 3’UTR
To examine whether the pulsatile lin-14 transcript levels observed in lin-4(e912) animals were caused by the absence of the direct interaction between lin-4 and the lin-14 3’UTR, we counted lin-14 transcripts in lin-14(n355gf) animals (Supplementary Fig. 11), which lack all of the seven putative lin-4 complementary elements (LCE) due to genomic rearrangements37,38. We found that lin-14 mRNA levels also exhibited pulsatile dynamics in lin-14(n355gf) animals similar to those observed in lin-4(e912) animals (Fig. 6a,b). In addition, we measured transcripts in lin-14(n536gf) animals which carry only two LCEs due to a deletion in the 3’UTR 37,38 and observed a similarly spaced train of peaks with reduced amplitudes (Supplementary Fig. 12). These data suggest that the pulsatile lin-14 expression observed in mutant animals are attributed to the loss of a direct interaction between lin-4 and the lin-14 3’-UTR.
An incoherent feed forward loop mediated by lin-4 can efficiently insulate lin-14 from pulsatile expression dynamics
To understand how the miRNA lin-4 can eliminate the pulses of its targets’ transcript levels, we compared the timing of the lin-4 and lin-14 peak expression. First, we tested for a difference in developmental timing between wild-type and lin-4(e912) animals by comparing temporal expression profiles in the two strains across all genes and measured a small delay for lin-4(e912) animals. After conversion of body length to time (Supplementary Fig. 3a) and rescaling of developmental time in lin-4(e912) animals by a factor of 0.85 to account for the developmental delay (Supplementary Fig. 13a), we could directly compare expression of lin-4 in wild-type and lin-14 in lin-4(e912) animals as a function of developmental time. We found that the peaks of lin-4 mature miRNA levels (as quantified from Fig. 4a) coincide with the peaks of lin-14 transcript levels in lin-4(e912) animals (Fig. 7a). Given that the spatial expression patterns of lin-4 and lin-14 are similar and that lin-4 acts cell autonomously39, these results suggest that lin-4 and its target lin-14 are synchronously expressed by a pulsatile cue in individual cells.
Figure 7. Synchronous expression of microRNA and its target dampens target gene expression fluctuations.

(a) Mean mature levels of lin-4 (from Fig. 4A, normalized to U18) in animals with wild-type background (red) and lin-14 mRNA concentration in lin-4(e912) mutants (black) oscillate in phase. Expression was normalized by the mean in order to plot data on the same scale. Body length was converted to developmental time using the regression in Supplementary Figure 3a for lin-4(e912) animals. Developmental time was rescaled for the lin-14 profile to account for slower development of lin-4(e912) animals (Supplementary Fig. 13a). Data for lin-14 were binned (bin size = 10 μm) before conversion and error bars represent variability across five independent replicates. (b) A biochemical model of miR-IFFL to describe target and microRNA level dynamics. The target mRNA and microRNA are coherently produced, and decay at constant rates ɣR and ɣm, respectively. microRNA-mediated mRNA degradation occurs with rate kon,, which is a function of the number of LCE (N), and microRNA is recycled. (c) kon fit values for lin-14 alleles with different N. Error bars correspond to 95% confidence interval. (d) miR-IFFL performance landscape. Damping is efficient when microRNA is expressed synchronously to its target (lin-4 microRNA relative fluctuation > 0) with an optimal value of kon (dark blue valley). Asterisks indicate lin-14 alleles with different N.
Taken together, we suggest that the pulsatile transcript levels of lin-14 are effectively dampened by a synchronized pulsatile expression of their negative regulator lin-4 in wild-type animals. The gene network motif composed of lin-4 miRNA and its target lin-14 can be classified as an “incoherent feed-forward loop (IFFL)”18 (Fig. 7b). To quantitatively understand how a miRNA-mediated IFFL (miR-IFFL) motif can efficiently dampen target mRNA oscillations, we built a mathematical model to characterize the dynamics of target gene level in the presence of miRNA activity (Fig. 7b, Supplementary Note). We assumed a synchronous periodic production of the target mRNA and miRNA, with decays described by first-order constant rates ɣR and ɣm, respectively. We also assumed irreversible second-order miRNA-mediated target transcript degradation kinetics with a rate constant kon. The rate kon depends on the strength of the miRNA-target interaction, which is proportional to the number of miRNA complementary elements N of the target transcript (thus kon(N=0) = 0 in both lin-4(e912) and lin-14(n355gf) strains). We assumed steady state dynamics and fitted this model to the experimental data (Supplementary Fig. 13b-h). This simple model describes the experimental data well and, reassuringly, we found a linear correlation between kon and the number N of lin-4 complementary elements (LCEs) in the lin-14 3’UTR (Fig. 7c).
To test the role of synchronous expression of the miRNA and its target, we explored the parameter space of the miR-IFFL as an insulator of pulsatile input signals using the model described above. We found that the relative fluctuations of lin-14 mRNA levels are minimized only when the relative fluctuations of miRNA levels are non-zero and optimally balanced with the strength of miRNA-target interaction kon (Fig. 7d, dark blue valley and Supplementary Fig. 14).
DISCUSSION
While previously only a handful of genes were shown to peak during each larval stage5,40,41 we found that approximately 2,000 genes exhibit temporal expression oscillations synchronized with the molting cycle. Most other genes are either expressed at a constant level, become up-regulated upon gametogenesis in the germ line, or display a temporal gradient, akin to the well-studied heterochronic cell-fate regulators. Two modes of timekeeping are apparently at work during nematode larval development: temporal gradients of cell fate regulators that control the transition between subsequent larval stage specific programs and periodically expressed genes that control progression of the molting cycle. Although the heterochronic pathway and the molting cycle are likely to be linked at the molecular level in order to coordinate larval transitions with interlarval molts, the temporal gradients of heterochronic cell fate regulators need to be efficiently insulated from temporal fluctuations. Our cluster analysis provides a clue for a more general involvement of miRNAs in the maintenance of oscillatory and graded expression patterns. We followed up by quantitatively examining expression dynamics of the canonical miRNA-target pair, lin-4 and lin-14, and show that miR-IFFLs actively insulate temporal-gradient genes from these global oscillations. It has been previously demonstrated that an IFFL can generate a gene expression pulse when the response time of its negative component is slow18. Our findings suggest that an IFFL can instead eliminate a gene expression pulse when it is mediated by negative components with fast response times42, such as miRNAs that are short and do not require translation to be functional. We suggest that a miR-IFFL could be a simple but powerful design motif that dynamic gene regulatory networks of multi-cellular organisms can adopt to insulate genes from highly dynamic but undesired inputs. Given its prevalence throughout different species43, this design motif might serve similar function in other species.
Online Methods
Strains
C. elegans strains used in this study were N2 (wild-type), MT873 (lin-4(e912)II), MT355 (lin-14(n355gf)X), MT536 (lin-14(n536gf)X), VT1072 (unc-119(ed3)III; maIs134[unc-119(+) + Plin-4::GFP]), MT723 (lin-4(e912)II; lin-14(n179ts)X) and PD7190 (lin-4(e912)/mC6 II; pha-1(e2123ts) III; rde-1(ne300) V; pHZ081 [pcol-10::lin-4::let-858_3’UTR]; pC1 [pha-1(+)]).
Synchronization and RNA extraction
Wild-type (N2) and mutant nematodes were synchronized by hypochlorite treatment. L1-arrested animals in starvation medium were spotted onto NGM plates with food (OP50 E. coli strain) and cultured at 20°C unless otherwise stated. Total RNA was isolated from synchronized larval populations using Trizol (Invitrogen). Each sample underwent 3 rounds of freeze-thaw cycles for enhanced efficiency.
Poly(A)+ RNA-sequencing experiment
Poly(A)+ RNA libraries were prepared from total RNA. Reads were aligned with bwa44 using default parameters to modENCODE integrated transcript models based on WormBase Release ws19019. Reads were aggregated across isoforms and expression per gene locus was calculated in reads per one million mapped reads (RPM). Whenever expression was measured in reads per one million mapped reads per kilobase of exon model (RPKM), the length of merged isoforms was used for normalization20.
Inference of expression clusters
The expression profile of each gene was normalized by the sum of expression values across all time points and log-transformed. Normalized expression profiles were topologically ordered into a two-dimensional discrete map with 20 x 20 grid points using a self-organizing map (SOM)45, implemented in R (SOM package). The SOM was then subjected to robust k-means clustering with 100 bootstraps, implemented in R (fpc package), using k=8. The value of k was optimized to obtain the best coverage of the SOM. Clusters with a Jaccard-similarity < 0.6 across bootstrap runs were not considered robust and discarded.
Identification of human orthologs
One-to-one human orthologs of C. elegans genes were identified by running blastp46 on translated protein sequences. Only significant (E < 10−10) reciprocal best hits were retained.
GO-term analysis
GO term analysis was performed in R using the GOstats package47. Over-represented GO-terms for each expression cluster were computed against the background of all genes falling into one of the robust clusters.
Single mRNA FISH
Probe design and hybridization protocol for single molecule mRNA FISH in C. elegans larvae was performed as previously described34. We used M9 to wash larvae off from plates. Animals were additionally washed twice to empty the gut filled with bacteria and were fixed in 4% formaldehyde 1X PBS for 45 minutes. Fixed animals were permeabilized in 70% ethanol overnight. All probes were coupled to either Cy5 (GE Amersham) or Alexa Fluor 594 (Invitrogen) which gave us high signal to background ratio in all larval stages. Hybridized animals were imaged using a Nikon Ti-E inverted fluorescence microscope equipped with a 100X oil-immersion objective and a Photometrics Pixis 1024 CCD camera using MetaMorph software (Molecular Devices, Downington, PA) and appropriate optical filters for Cy5, Alexa Fluor 594 and DAPI. Data analyses and model simulations were carried out semi-automatically with the aid of custom software written in MATLAB (Mathworks).
microRNA TaqMan PCR assay
TaqMan PCR assay for lin-4 microRNA quantification was performed following the instructions from the manufacturer (Applied Biosystems) using Light Cycler 480 II Real-time PCR machine (Roche). We used sn2343 and U18 for normalization controls.
Supplementary Material
ACKNOWLEDGEMENTS
We thank V. Ambros, H. R. Horvitz, G. Ruvkun and J. Gore for helpful discussions and advice. We thank S. Itzkovitz, J. P. Junker, G. Neuert, J. van Zon, A. Sahay and C. Engert for a critical reading of the manuscript. We thank the MIT BioMicro Center for Illumina sample preparation and sequencing experiments. We also thank the Biopolymers & Proteomics Core Facility of the MIT Koch Institute for Integrative Cancer Research for probe purification. This work was supported by the NIH/NCI Physical Sciences Oncology Center at MIT (U54CA143874), an NIH Pioneer award (8 DP1 CA174420-05), an ERC Advanced grant (ERC-AdG 294325-GeneNoiseControl), and a Green Cross Corp. (Korea) Mogam Science Scholarship. Several nematode strains used in this work were provided by H. R. Horvitz, A. Fire, and the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources (NCRR).
Footnotes
AUTHOR CONTRIBUTIONS
D.K. and A.v.O. conceived the project and designed the experiments. D.K. performed the experiments. D.K., D.G. and A.v.O. analyzed the data and wrote the paper.
Accession codes.
Transcript sequencing data have been deposited under GenBank Gene Expression Omnibus (GEO) accession GSE49043.
COMETING FINANCIAL INTERESTS
The author have no competing financial interests
REFERENCES
- 1.Raj A, van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008;135:216–226. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rougvie AE. Intrinsic and extrinsic regulators of developmental timing: from miRNAs to nutritional cues. Development. 2005;132:3787–3798. doi: 10.1242/dev.01972. [DOI] [PubMed] [Google Scholar]
- 3.Ruvkun G, Giusto J. The Caenorhabditis elegans heterochronic gene lin-14 encodes a nuclear protein that forms a temporal developmental switch. Nature. 1989;338:313–319. doi: 10.1038/338313a0. [DOI] [PubMed] [Google Scholar]
- 4.Johnstone L, Barry JD. Temporal reiteration of a precise gene expression pattern during nematode development. The EMBO J. 1996;15:3633–3639. [PMC free article] [PubMed] [Google Scholar]
- 5.Gissendanner CR, Crossgrove K, Kraus KA, Maina CV, Sluder AE. Expression and function of conserved nuclear receptor genes in Caenorhabditis elegans. Dev. Biol. 2004;266:399–416. doi: 10.1016/j.ydbio.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 6.Raizen DM, et al. Lethargus is a Caenorhabditis elegans sleep-like state. Nature. 2008;451:569–572. doi: 10.1038/nature06535. [DOI] [PubMed] [Google Scholar]
- 7.Page AP, Johnstone IL. The cuticle. WormBook ed. The C. elegans Research Community. 2007 doi: 10.1895/wormbook.1.138.1. http://www.wormbook.org. doi:10.1895/wormbook.1.138.1. [DOI] [PMC free article] [PubMed]
- 8.Altun ZF, Hall DH. Introduction. WormAtlas. 2009 http://www.wormatlas.org. doi:10.3908/wormatlas.1.1.
- 9.Hubbard EJA, Grenstein D. Introduction to the germ line. WormBook ed. The C. elegans Research Community. 2005 doi: 10.1895/wormbook.1.18.1. http://www.wormbook.org. doi:10.1895/wormbook.1.18.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ambros V, Horvitz HR. The lin-14 locus of Caenorhabditis elegans controls the time of expression of specific postembryonic developmental events. Genes & Development. 1987;1:398–414. doi: 10.1101/gad.1.4.398. [DOI] [PubMed] [Google Scholar]
- 11.Moss EG, Lee RC, Ambros V. The cold shock domain protein LIN-28 controls developmental timing in C. elegans and is regulated by the lin-4 RNA. Cell. 1997;88:637–646. doi: 10.1016/s0092-8674(00)81906-6. [DOI] [PubMed] [Google Scholar]
- 12.Slack FJ, et al. The lin-41 RBCC gene acts in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Mol. Cell. 2000;5:659–669. doi: 10.1016/s1097-2765(00)80245-2. [DOI] [PubMed] [Google Scholar]
- 13.Ambros V, Horvitz H. Heterochronic mutants of the nematode Caenorhabditis elegans. Science. 1984;226:409–416. doi: 10.1126/science.6494891. [DOI] [PubMed] [Google Scholar]
- 14.Ambros V. A hierarchy of regulatory genes controls a larva-to-adult developmental switch in C. elegans. Cell. 1989;57:49–57. doi: 10.1016/0092-8674(89)90171-2. [DOI] [PubMed] [Google Scholar]
- 15.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 16.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reinhart BJ, Ruvkun G. Isoform-specific mutations in the Caenorhabditis elegans heterochronic gene lin-14 affect stage-specific patterning. Genetics. 2001;157:199–209. doi: 10.1093/genetics/157.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alon U. Network motifs: theory and experimental approaches. Nat. Rev. Genet. 2007;8:450–461. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- 19.Gerstein MB, et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;330:1775–1787. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:1–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 21.Sönnichsen B, et al. Full-genome RNAi profiling of early embryogenesis in Caenorhabditis elegans. Nature. 2005;434:462–469. doi: 10.1038/nature03353. [DOI] [PubMed] [Google Scholar]
- 22.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 23.Selbach M, et al. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 24.Baek D, et al. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bagga S, et al. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell. 2005;122:553–563. doi: 10.1016/j.cell.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 26.Lim LP, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 27.Dreszer TR, et al. The UCSC Genome Browser database: extensions and updates 2011. Nucleic acids research. 2012;40:D918–23. doi: 10.1093/nar/gkr1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang X, Zabinsky R, Teng Y, Cui M, Han M. microRNAs play critical roles in the survival and recovery of Caenorhabditis elegans from starvation-induced L1 diapause. Proc. Natl. Acad. Sci. USA. 2011;108:17997–18002. doi: 10.1073/pnas.1105982108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lencastre A, et al. MicroRNAs both promote and antagonize longevity in C. elegans. Current Biology. 2010;20:2159–2168. doi: 10.1016/j.cub.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boulias K, Horvitz HR. The C. elegans microRNA mir-71 acts in neurons to promote germline-mediated longevity through regulation of DAF-16/FOXO. Cell Metabolism. 2012;15:439–450. doi: 10.1016/j.cmet.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alvarez-Saavedra E, Horvitz HR. Many families of C. elegans microRNAs are not essential for development or viability. Current Biology. 2010;20:367–373. doi: 10.1016/j.cub.2009.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez NJ, et al. Genome-scale spatiotemporal analysis of Caenorhabditis elegans microRNA promoter activity. Genome research. 2008;18:2005–2015. doi: 10.1101/gr.083055.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feinbaum R, Ambros V. The timing of lin-4 RNA accumulation controls the timing of postembryonic developmental events in Caenorhabditis elegans. Dev. Biol. 1999;210:87–95. doi: 10.1006/dbio.1999.9272. [DOI] [PubMed] [Google Scholar]
- 34.Raj A, Bogaard PVD, Rifkin SA, Oudenaarden AV, Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods. 2008;5:877–879. doi: 10.1038/nmeth.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knight CG, Patel MN, Azevedo RBR, Leroi AM. A novel mode of ecdysozoan growth in Caenorhabditis elegans. Evolution & development. 2002;4:16–27. doi: 10.1046/j.1525-142x.2002.01058.x. [DOI] [PubMed] [Google Scholar]
- 36.Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- 37.Ruvkun G, et al. Molecular genetics of the Caenorhabditis elegans heterochronic gene lin-14. Genetics. 1989;121:501–516. doi: 10.1093/genetics/121.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- 39.Zhang H, Fire AZ. Cell autonomous specification of temporal identity by Caenorhabditis elegans microRNA lin-4. Dev. Biol. 2010;344:603–610. doi: 10.1016/j.ydbio.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeon M. Similarity of the C. elegans Developmental timing protein LIN-42 to circadian rhythm proteins. Science. 1999;286:1141–1146. doi: 10.1126/science.286.5442.1141. [DOI] [PubMed] [Google Scholar]
- 41.Frand AR, Russel S, Ruvkun G. Functional genomic analysis of C. elegans molting. PLoS Biol. 2005;3:e312. doi: 10.1371/journal.pbio.0030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Basu S, Mehreja R, Thiberge S, Chen M-T, Weiss R. Spatiotemporal control of gene expression with pulse-generating networks. Proc. Natl. Acad. Sci. USA. 2004;101:6355–6360. doi: 10.1073/pnas.0307571101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsang J, Zhu J, van Oudenaarden A. MicroRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol. Cell. 2007;26:753–767. doi: 10.1016/j.molcel.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kohonen T. Self-organized formation of topologically correct feature maps. Biological Cybernetics. 1982;43:59–69. [Google Scholar]
- 46.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. of Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 47.Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23:257–258. doi: 10.1093/bioinformatics/btl567. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.