

Abstract
OBJECTIVE:
This study aimed to determine the effect of manuka honey on the oxidative status of middle-aged rats.
METHOD:
Twenty-four male Sprague-Dawley rats were divided into young (2 months) and middle-aged (9 months) groups. They were further divided into two groups each, which were either fed with plain water (control) or supplemented with 2.5 g/kg body weight of manuka honey for 30 days. The DNA damage level was determined via the comet assay, the plasma malondialdehyde level was determined using high performance liquid chromatography, and the antioxidant enzyme activities (superoxide dismutase, glutathione peroxidase, glutathione peroxidase and catalase) were determined spectrophotometrically in the erythrocytes and liver. The antioxidant activities were measured using 1,1-diphenyl-2-picrylhydrazyl and ferric reducing/antioxidant power assays, and the total phenolic content of the manuka was analyzed using UV spectrophotometry and the Folin-Ciocalteu method, respectively.
RESULTS:
Supplementation with manuka honey reduced the level of DNA damage, the malondialdehyde level and the glutathione peroxidase activity in the liver of both the young and middle-aged groups. However, the glutathione peroxidase activity was increased in the erythrocytes of middle-aged rats given manuka honey supplementation. The catalase activity was reduced in the liver and erythrocytes of both young and middle-aged rats given supplementation. Manuka honey was found to have antioxidant activity and to have a high total phenolic content. These findings showed a strong correlation between the total phenolic content and antioxidant activity.
CONCLUSIONS:
Manuka honey reduces oxidative damage in young and middle-aged rats; this effect could be mediated through the modulation of its antioxidant enzyme activities and its high total phenolic content. Manuka honey can be used as an alternative supplement at an early age to improve the oxidative status.
Keywords: Manuka Honey, Antioxidant Enzymes, DNA Damage, Malondialdehyde, Aging
INTRODUCTION
The oxidative damage of DNA (1), proteins and lipid in cells and organs is caused by free radicals that are continuously produced in the body during aerobic respiration (2). Our body defense system is equipped with antioxidants to scavenge free radicals that provide protection from oxidative damage. However, a reduction in the body's antioxidant defense together with increased free radical production with age results in oxidative stress, which has been reported to cause aging (1,3) and degenerative diseases (4) such as cancer, heart disease and diabetes (5). Oxidative damage to macromolecules causes cells to undergo apoptosis or transform into cancer cells (6).
Although the aging process is under genetic control, it is also affected by environmental and lifestyle determinants such as diet in which this process was found to be capable of manipulating the rate of free radical production (2). Many studies found that dietary supplements prevent oxidative damage via aging despite the differences in the ages of the populations studied (1,7). Antioxidant supplementation from a young age has been suggested to confer better protection against potential oxidative damage later in life (7). This protective effect is a likely result as age-related changes begin in early adulthood and most organs start to deteriorate at the age of 30 (8).
Compared with other antioxidant supplements composed primarily of vitamins and minerals, honey is rich in both enzymatic and non-enzymatic antioxidants, including glucose oxidase, catalase, ascorbic acid, flavonoids, phenolic acids, carotenoid derivatives, organic acids, Maillard reaction products, amino acids and proteins (9). The antioxidative properties in honey have been proven to be effective in aiding the healing process of wounds and burns, diabetic ulcers, cancer, cardiovascular disease and neural diseases (10).
In this study, manuka honey from the New Zealand manuka tree (Leptospermum scoparium) is used as an intervention to reduce the oxidative stress in middle-aged rats. Manuka honey has been widely researched regarding its antibacterial activity and has been recommended for wound treatment (11). Previous studies reported that manuka honey consisted of a high amount of phenolic compounds such as flavonoids, methyl syringate and a methoxylated benzoic acid, a structural isomer of syringic acid (12). The phenolic compounds are more potent antioxidants than the non-phenolic antioxidants in honey (13). Manuka honey also contains a bioactive fraction called methylglyoxal that exhibits non-peroxide antibacterial activity (14). Methyl syringate in manuka honey has been identified as a potent superoxide scavenger that could be expected to reduce free radical activity (15).
MATERIALS AND METHODS
Honey
Raw unblended manuka honey, a product of Papakura (Graham Cammell Ltd, Clevedon, Papakura, New Zealand) was purchased locally from Guardian Pharmacy, Malaysia.
Total phenolic content
The Folin-Ciocalteu method as modified by Beretta et al. (16) was used to determine the total phenolic content in the honey. Manuka honey (1 g) was diluted to 10 ml with distilled water, and 100 μl of this solution (corresponding to 10 mg of fresh manuka honey) was added to 1 ml of 10% Folin-Ciocalteu reagent. The mixture was vortexed for 2 min. After incubation for 20 min at 25°C, the absorbance was determined at 750 nm using a UV 160A spectrophotometer (Shimadzu, Japan). Gallic acid (0–200 mg/ml) was used as a standard to derive the calibration curve. The total phenolic content was expressed as mg of gallic acid per kg of manuka honey.
Determination of free radical scavenging activity
The free radical scavenging activity of the honey was estimated using the 1,1-diphenyl-2-picrylhydrazyl (DPPH) method of Brand-Williams et al. (17) with a slight modification by Aljadi and Kamaruddin (18). Briefly, a honey sample was dissolved in water at a concentration from 0 to 100 mg/ml, and 0.75 ml of each solution was mixed with 1.5 ml of 0.09 mg/ml DPPH reagent dissolved in absolute methanol. The mixtures were shaken vigorously and incubated for 10 min at room temperature in the dark, after which the absorbance of the remaining DPPH was measured at 517 nm against a blank to eliminate the influence of the color of the honey. The antioxidant activity was expressed as the percentage inhibition of the DPPH radical and was determined using the following equation (5):
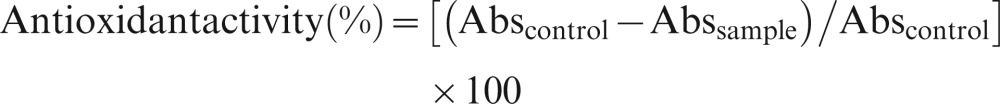 |
Determination of the total antioxidant power
The ferric reducing/antioxidant power (FRAP) assay developed by Benzie and Strain (19) was used with some modifications by Mohamed et al. (20). This method is based on the reduction of a ferric 2,4,6-tripyridyl-s-triazine complex (Fe3+-TPTZ) to its ferrous, colored form (Fe2+-TPTZ) in the presence of antioxidants. The FRAP reagent contains 2.5 ml of a 10 mM TPTZ solution in 40 mM HCl, 2.5 ml of 20 mM FeCl3 and 25 ml of 0.3 M acetate buffer, pH 3.6. Sample aliquots of 40 μl were mixed with 1.2 ml of the FRAP reagent and incubated at 37°C for 4 min before the absorbance of the reaction mixture was measured spectrophotometrically at 593 nm against the sugar analog. Aqueous standard solutions of FeSO4 6H2O (100–2,500 μM) were used for the calibration curve, and the results were expressed as the FRAP value (μM Fe [II]) of the 10% honey solution.
Animals
A total of 24 male Sprague-Dawley rats were obtained from the Animal Resources Unit, Medical Faculty, The National University of Malaysia (UKM). The rats were divided into two groups; young (2 months, weighing from 200 to 250 g) and middle-aged (9 months, weighing from 350 to 450 g). The rats were housed in a well-ventilated controlled room on a 12 h light/12 h dark schedule at room temperature. The rats were kept in the room at least 1 week prior to the treatment and were fed with standard commercial rat pellets; drinking water was made available ad libitum. The experimental protocol was approved by The National University of Malaysia Animal Ethics Committee (UKMAEC; PP/BIOK/2010/ZAKIAH/20-JANUARY/288-MARCH-2010-APRIL-2010). Each group (young, n = 12 and middle-aged, n = 12) was further divided into 2 groups consisting of 6 rats each; group 1 was fed with plain water (2.5 ml/kg body weight) and group 2 was fed with manuka honey (2.5 g/kg body weight). Both the honey and plain water were given orally using oral gavage, and the duration of the supplementation was 30 days. The 2.5 g/kg body weight of manuka honey used was equivalent to 1 teaspoon of honey, as recommended.
Sample preparation
Following 30 days of supplementation, the rats were anaesthetized with ether and approximately 6 ml of blood was collected via the orbital sinus route. A small amount of fresh whole blood was used for the comet assay, and the remainder was centrifuged at 3,000 rpm at 4°C for 10 min. The obtained plasma was divided into aliquots and stored at −80°C for malondialdehyde (MDA) determination. Erythrocytes were washed 3 times with physiological saline (0.9% NaCl), separated into aliquots and stored at −80°C for the enzyme assay. After the rats were euthanized, the livers were removed, rinsed in ice cold 1.15% NaCl, pH 7.2 (Sigma, St. Louis, USA), washed to remove any residual blood and weighed. The liver was cut into small pieces, and homogenates were prepared in 1.15% NaCl at 3 ml/g (w/v) using an Ultra Turrax T25 Homogenizer (IKA Labortechnik, Germany). The homogenates were centrifuged at 9,000 x g for 20 min at 4°C using a Sorvall RC-5B. The supernatant was placed into the centrifuge tube and centrifuged at 105,000 x g for 1 h using a Beckman L-60 ultracentrifuge (Beckman Coulter, USA). The cytosolic supernatant was frozen at −80°C for the antioxidant enzyme assays.
Detection of DNA damage via the Comet Assay
The comet assay was performed as described by Singh et al. (21) with slight modifications from Chin et al. (1). Briefly, 5 μL of whole blood was suspended in 0.6% low melting point agarose in phosphate-buffered saline, pH 7.4, in a microcentrifuge tube at 37°C, rapidly pipetted onto the first agarose layer and covered using a coverslip. The low melting point agarose was then allowed to solidify on an ice-cold flat tray. After removal of the coverslips, the slides were immersed into ice-cold lysing solution (2.5 M NaCl, 100 mM ethylene-diaminetetra-acetic acid, 10 mM Tris [pH 10], 1% Triton X-100, 1% sodium N-lauroyl sarcosinate and 10% dimethylsulfoxide) at 4°C for 1 h to lyse the cells and remove cellular protein. The slides were then removed from the lysing solution and placed side-by-side in a horizontal electrophoresis chamber filled with freshly prepared cooled alkaline electrophoresis buffer (1–10°C) to a depth of approximately 0.25 cm above the agarose layer for approximately 20 min to allow for the unwinding of the DNA. The electrophoresis was conducted for 20 min at 25 V with the current adjusted to 300 mA via a change of buffer volume. Following electrophoresis, the slides were rinsed 3 times with neutralization buffer and allowed to dry. Each slide was then stained with 30 μl of ethidium bromide and covered with a new coverslip. All slides were examined immediately at 200x magnification using a fluorescent microscope (Olympus Corp., Shibuya-Ku, Tokyo, Japan). Five hundred randomly selected non-overlapping cells on each slide were counted, and grades were assigned on an arbitrary scale of 0 to 4 based on the perceived comet tail length migration and relative proportion of DNA in the comet tail (cells without a tail and no damage, 0; cells with a tiny tail, 1; cells with a dim tail, 2; cells with a clear tail, 3; and only a tail, 4. For the final analysis, a total damage score of each slide was calculated using the following formula:
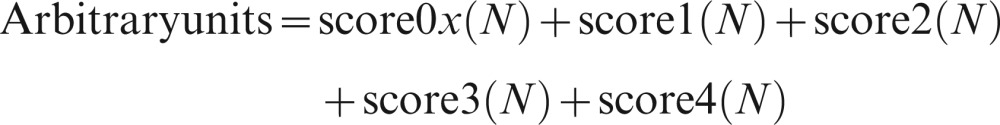 |
where N = the number of cells assigned to each damage grade.
The sum of all the grades provided a minimum possible score of 0, which corresponds to 500 cells at grade 0, and a maximum possible score of 2,000, which corresponds to 500 cells at grade 4.
Malondialdehyde level
The plasma MDA was determined using high performance liquid chromatography (HPLC) based on the derivatization of MDA with 2,4-dinitrophenylhydrazine (DNPH) (Sigma, St. Louis, USA) using a photo diode array detector (Shimadzu Corporation, Kyoto, Japan) as described by Pilz et al. (22) with some modifications. Briefly, samples (50 μl) were mixed with 200 μl of 1.3 M NaOH (Merck, Germany) and incubated at 60°C for 30 min in a water bath. After cooling the mixture to room temperature, 100 μl of 35% (v/v) HCIO4 was added to precipitate the proteins, and the mixture was centrifuged at 10,000 x g for 10 min. The supernatant of the samples (300 μl) was transferred to a 1.5-ml HPLC tube, and 5 mM DNPH solution (50 μl) was added to the mixture and incubated for 30 min at room temperature. The derivatized samples (40 μl) were then injected into the HPLC.
Analytical HPLC separation was performed using a Shimadzu Chromatographic System version 6.1 LC (Shimadzu Corporation, Kyoto, Japan) equipped with an auto injector and operated at 310 nm on a 150 mm × 3.9 mm, 5-μm alphaBond C18 column (Alltech Associated, Inc., IL, USA). The samples and standards were eluted with the mobile phase consisting of 380 ml of acetonitrile and 620 ml of distilled water, acidified with 0.2% (v/v) acetic acid and degassed at a flow rate of 0.6 ml/min. The plasma MDA level was calculated from a calibration curve prepared via acidic hydrolysis of 1,1,3,3-tetraethoxypropane (TEP) (Sigma, St. Louis, USA). The amount of MDA was expressed as the nmol of MDA per ml of plasma.
Determination of superoxide dismutase (SOD) activity
SOD was assayed using the method developed by Beyer and Fridovich (23). Briefly, 1.0-ml aliquots of a mixture containing 0.1 mM phosphate buffer pH 7.8, 57 μM nitro blue tetrazolium (Sigma, St. Louis, USA), 9.9 mM L-methionine (Sigma, St. Louis, USA) and 0.025% Triton-X (Sigma, St. Louis, USA) were added to the test tubes, followed by the addition of 20 μl of lysate or liver cytosol and 10 μl of a solution containing 4.4 mg/100 ml riboflavin (Sigma, St. Louis, USA). The tubes were illuminated for 7 min in an aluminum foil-lined box containing two 20-W Sylvania GroLux fluorescent lamps. The absorbance was then measured at a wavelength of 560 nm. A stock hemolysate was prepared by adding an equal volume of distilled water. One unit of SOD was defined as the amount of enzymes required to inhibit nitro blue tetrazolium reduction by 50% per min per ml of lysate or cytosol. The enzyme activity was expressed as the units per mg of Hb or protein (U/mg Hb or mg protein).
Determination of glutathione peroxidase (GPx) activity
GPx was determined using the method developed by Paglia and Valentine (24). The reaction mixture contained 0.05 M phosphate buffer pH 7.0, 8.4 mM NADPH (Sigma, St. Louis, USA), 1.125 M sodium azide (Hopkin & William, England), 5 mM reduced glutathione (GSH) (NADPH; Sigma, St. Louis, USA) and 3 U/ml glutathione reductase (Sigma, St. Louis, USA). The hemolysate was prepared by adding an equal volume of distilled water to the RBC pellet and allowing it stand for 1 h at 4°C. Four parts by volume of distilled water was then added. Finally, double strength Drabkin's reagent (Eagle Diagnostics, Japan) was added to yield the final hemolysate. The reaction was initiated by adding 0.1 ml of 2.2 mM H2O2 (Merck, Darmstadt, Germany). The conversion of NADPH to NADP+ was followed by measuring the change in Abs/min at 340 nm, and 1 unit of GPx was defined as the amount of enzyme required to oxidize 1 μmol of NADPH/min per ml of lysate or liver cytosol. The enzyme activity was expressed as milliunits per mg of Hb or protein (mU/mg Hb or mg protein).
Determination of catalase (CAT) activity
CAT was assayed using the method of Aebi (25). The reaction mixture consisted of 50 mM phosphate buffer pH 7.0 and 30 mM hydrogen peroxide. A stock hemolysate containing 5 g Hb/100 ml was prepared by adding four parts by volume of distilled water to the sample. A 1:500 dilution of this concentrated hemolysate was prepared by adding 50 mM phosphate buffer pH 7.0 immediately before running the enzyme assay. For liver tissue preparation, 1% (v/v) triton X-100 in the phosphate buffer was used in the preparation of the stock homogenate. For each gram of organ weight, 9 ml of 1% triton X-100 was added, and this stock homogenate was further diluted in phosphate buffer, pH 7.0 (v: v; 1:100). The reaction was then initiated by adding 1 ml of 30 mM H2O2. The absorbance was measured at a wavelength of 240 nm, and one unit of catalase enzyme was defined as the amount of enzyme that liberates half the peroxide oxygen from the H2O2 solution during 30 s at room temperature. The enzyme activity was expressed as units per mg of Hb or protein (U/mg Hb or mg protein). Hemoglobin in the hemolysate was measured using the Eagle diagnostic kit (Japan). Protein in the liver cytosol was determined using the Bradford method (26).
Statistical analyses
All data are expressed as the means ± S.D. (n = 6) because the data were normal, and differences between groups were statistically analyzed via one-way ANOVA. Correlations were obtained via Pearson's correlation coefficient (r) in bivariate linear correlations. Differences were considered to be statistically significant if p<0.05. All statistical analyses were performed using SPSS for Windows, version 17.0.
RESULTS
Total phenolic content and antioxidative activity of manuka honey
Manuka honey was found to have a high total phenolic content (mggallic acid/kg of honey). The antioxidant activities of manuka honey (determined via the DPPH and FRAP assays [Table 1]) were found to correlate significantly with the total phenolic content (Figure 1).
Table 1.
Total phenolic content, FRAP value and antiradical power (1,1-diphenyl-2-picrylhydrazyl) of manuka honey.
| Parameter | Manuka |
| Total phenolic content (mggallic acid/kg honey) | 201.00±35.92 |
| FRAP value (μM Fe[II]) | 215.71±50.00 |
| DPPH (%) | 46.64±5.95 |
Figure 1.

A) Correlation between the total phenolic content (TPC) and ferric reducing/antioxidant power (FRAP value) of manuka honey. B) Correlation between the total phenolic content and radical scavenging activity (% inhibition) using the DPPH of manuka honey.
Analysis of the baseline value of oxidative damage and its changes with honey supplementation
The baseline value showed that there were no differences in the level of DNA damage and MDA between the young and middle-aged groups (Table 2). Supplementation with manuka honey reduced the level of DNA damage and MDA in both groups (Figure 2).
Table 2.
Baseline values of DNA damage and antioxidant enzyme activities of the young (2 months) and middle-aged (9 months) groups.
| Young (2 months) | Middle-aged (9 months) | ||
| DNA damage | Arbitrary unit | 159±61.1 | 171.9±72.1 |
| MDA level | nmol/ml plasma | 7.58±0.45 | 8.29±0.59 |
| Erythrocytes | |||
| SOD | U/mg Hb | 3.96±2.16 | 2.77±1.68 |
| GPx | mU/mg Hb | 0.22±0.01 | 0.41±0.15 |
| CAT | U/mg Hb | 2.77±0.75 | 1.87±0.46 |
| Liver | |||
| SOD | U/mg protein | 1.59±0.45 | 1.86±0.24 |
| GPx | mU/mg protein | 70.73±27.26 | 78.0±17.27 |
| CAT | U/mg protein | 2.76±0.46 | 3.25±0.55 |
Data are expressed as the means ± SD for 6 animals.
Figure 2.

Effect of manuka honey supplementation on the A) DNA damage and B) MDA levels of the young (2 months) and middle-aged (9 months) groups. The data are expressed as the means ± SD for 6 animals. * indicates a significant difference compared with the control group (p<0.05).
Analysis of the baseline value of antioxidant enzyme activities and its changes with honey supplementation
The baseline values for the antioxidant enzymes activities also did not show any significant differences between the two age groups (Table 2). Supplementation with manuka honey significantly increased the GPx activity and reduced the CAT activity in the erythrocytes of the young and middle-aged groups (Figure 3). The changes in the GPx and CAT activities were more noticeable in the young group than in the middle-aged group. Manuka honey supplementation significantly reduced the GPx and CAT activities (Figure 4) in the livers of both the young and middle-aged groups but increased the SOD activity only in the middle-aged group.
Figure 3.

A) SOD activity, B) GPx activity and C) CAT activity in the erythrocytes of young (2 months) and middle-aged (9 months) rats with honey supplementation. The data are expressed as the means ± SD for 6 animals. * indicates a significant difference compared with the control group (p<0.05).
Figure 4.

A) Liver SOD activity, B) GPx activity and C) CAT activity of the young (2 months) and middle-aged (9 months) rats with manuka honey supplementation. The data are expressed as the means ± SD for 6 animals. * indicates a significant difference compared with the control group (p<0.05).
DISCUSSION
The accumulation of DNA damage and increased lipid peroxidation leads to aging and age-related diseases. Supplementation beginning at young and middle ages may provide protection against the further accumulation of oxidative damage later in life. Similar to our findings, honey supplementation has also been shown to reduce oxidative stress in the liver tissues (27) and kidneys of stress-induced rats (10).
Honey has been suggested to protect against lipid peroxidation by reducing the production of lipid hydroperoxides (28). The antioxidant property of honey may be due to its phenolic or non-phenolic antioxidant contents, such as vitamin C, vitamin E and β-carotene. Similar to previous studies, the manuka honey used in this study was found to contain a high total phenolic content and high antioxidant activity (12), which can scavenge anion superoxide radicals. In addition, the phenolic content of manuka was found to correlate significantly with the values of FRAP and DPPH, which are major components responsible for the antioxidant activity of this honey. This result is in line with a previous study by Hussein et al. (29), who found similar correlations between phenolic contents and FRAP as well as DPPH values in irradiated honeys. In addition to phenolics, manuka honey contains high amounts of quercetin, luteolin, quercetin 3-methyl ether, luteolin, chrysin, gallic acid and abscisic acid (12). Phenolic acids such as gallic acid and non-phenolic acids such as vitamin E have free radical scavenging activities that can prevent oxidative damage (30). Gallic acid is one of the strongest free radical scavengers reported in manuka honey (12). Phenolic acid and flavonoids in honey have been shown to protect endothelial cell lines against oxidative damage by peroxyl radicals and hydrogen peroxide activity (31).
In this study, honey supplementation increased GPx activity and reduced CAT activity in the erythrocytes of both young and middle-aged rats, similar to the observations of Lee et al. (7). A previous study on Portuguese unifloral honeys suggested that the selenium content of manuka honey is influenced by the botanical source of the honey (32). Because selenium functions as a cofactor for GPx, the supplementation with honey that contains selenium may explain the increased GPx activity observed in this study. As shown in this study, the increased GPx activity subsequently caused reduced CAT activity in erythrocytes because GPx acts on similar substrates such as hydrogen peroxide and organic peroxide (33). GPx hydrolyzes hydrogen peroxide to form water and oxidized glutathione (GSSH), whereas CAT reduces hydrogen peroxide to water and oxygen. A previous study reported that GPx is more active than CAT as a defense enzyme; thus, the low amount of CAT observed in this study was likely related to its peroxidatic activity (33).
The marked increase in erythrocyte GPx activity after manuka honey supplementation reflected the presence of a higher level of hydrogen peroxide. According to a previous study, honey with a high peroxide level has higher glucose oxidase and lower catalase activity (34). Generally, the predominant role of hydrogen peroxide in honey is as an antimicrobial agent; however, studies have reported that manuka honey exhibited non-peroxide antibacterial activity (14). Methylglyoxal in manuka honey was identified to be responsible for its non-peroxide antibacterial activity, and researchers have identified this as its unique activity (14). A previous study by Adams et al. (35) reported that methylglyoxal formation is a non-enzymatic conversion of dihydroxyacetone and is accelerated by protein. Therefore, further studies should be performed to determine whether the supplementation of manuka honey increases both selenium and hydrogen peroxide levels in the blood, leading to the modulation of GPx activity.
Manuka honey supplementation reduced the DNA damage and MDA levels in young and middle-aged rats. The honey conferred better protection against DNA damage in the young group than in the middle-aged group. Analyses of both the free radical scavenging activity (DPPH) and total antioxidant activity (FRAP) were found to have strong correlations with the total phenolic content. These analyses showed that the phenolic content in manuka honey may be responsible for protecting these cells from oxidative damage.
ACKNOWLEDGMENTS
This research was supported by grants from the Medical Research and Industry Secretariat, UKM Medical Centre (UKMMC), UKM (FF-087-2010). The authors would like to thank all lecturers and staff of the Department of Biochemistry, Faculty of Medicine, UKMMC UKM, who contributed to this study.
Footnotes
No potential conflict of interest was reported.
REFERENCES
- 1.Chin SF, Hamid NA, Latiff AA, Zakaria Z, Mazlan M, Yusof YA, et al. Reduction of DNA damage in older healthy adults by Tri E® tocotrienol supplementation. Nutrition. 2008;24(1):1–10. doi: 10.1016/j.nut.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Harman D. The aging process. Proc Natl Acad Sci USA. 1981;78(11):7124–8. doi: 10.1073/pnas.78.11.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hasty P. The impact of DNA damage, genetic mutation and cellular responses on cancer prevention, longevity and aging: observation in humans and mice. Mech Ageing Dev. 2005;126(1):71–7. doi: 10.1016/j.mad.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 4.Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39(1):44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Serafini M. The role of antioxidants in disease prevention. Medicine. 2006;34(12):533–5. [Google Scholar]
- 6.Jacob RA, Burri BJ. Oxidative damage and defense. Am J Clin Nutr. 1996;63(suppl):985S–90S. doi: 10.1093/ajcn/63.6.985. [DOI] [PubMed] [Google Scholar]
- 7.Lee KY, Razak SLA, Ismail N, Ng CF, Asgar MHAM, Sharif NM, et al. Malaysian gelam honey reduces oxidative damage and modulates antioxidant enzyme activities in young and middle aged rats. J Med Plant Res. 2011;5(23):5618–25. [Google Scholar]
- 8.Harman D. Aging: Overview. Ann NY Acad Sci. 2001;928:1–21. doi: 10.1111/j.1749-6632.2001.tb05631.x. [DOI] [PubMed] [Google Scholar]
- 9.Baltrušaityt V, Venskutonis PR, Ceksteryt V. Radical scavenging activity of different floral origin honey and bee bread phenolic extracts. Food Chem. 2007;101:502–14. [Google Scholar]
- 10.Erejuwa OO, Sulaiman SA, Wahab MSA, Sirajudeen KNS, Salleh MSM, Gurtu S. Antioxidant protective effect of glibenclamide and metformin in combination with honey in pancreas of streptozotocin-induced diabetic rats. Int J Mol Sci. 2010;11(5):2056–66. doi: 10.3390/ijms11052056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molan PC. The role of honey in the management of wounds. J Wound Care. 1999;8(8):415–8. doi: 10.12968/jowc.1999.8.8.25904. [DOI] [PubMed] [Google Scholar]
- 12.Stephens JM, Schlothauer RC, Morris BD, Yang D, Fearnley L, Greenwood DR, et al. Phenolic compounds and methylglyoxal in some New Zealand manuka and kanuka honeys. Food Chem. 2010;120:78–86. [Google Scholar]
- 13.Estevinho L, Pereira AP, Moreira L, Dias LG, Pereira E. Antioxidant and antimicrobial effects of phenolic compounds extracts of Northeast Portugal honey. Food Chem Toxicol. 2008;46(12):3774–9. doi: 10.1016/j.fct.2008.09.062. [DOI] [PubMed] [Google Scholar]
- 14.Adams CJ, Boult CH, Deadman BJ, Farr JM, Grainger MNC, Manley-Harris M, et al. Isolation by HPLC and characterisation of the bioactive fraction of New Zealand manuka (Leptospermum scoparium) honey. Carbohydr Res. 2008;343(4):651–9. doi: 10.1016/j.carres.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 15.Oelschlaegel S, Gruner M, Wang PN, Boettcher A, Koelling-Speer I, Speer K. Classification and characterization of manuka honeys based on phenolic compounds and methylglyoxal. J Agric Food Chem. 2012;60(29):7229–37. doi: 10.1021/jf300888q. [DOI] [PubMed] [Google Scholar]
- 16.Beretta G, Granata P, Ferrero M, Orioli M, Facino RM. Standardization of antioxidant properties of honey by a combination of spectrophotometric/fluorimetric assays and chemometrics. Anal Chim Acta. 2005;533(2):185–91. [Google Scholar]
- 17.Brand-Williams W, Culivier ME, Berset C. Use of a free radical method to evaluate antioxidant activity. LWT-Food Sci Tech. 1995;28:25–30. [Google Scholar]
- 18.Aljadi AM, Kamaruddin MY. Evaluation of the phenolic contents and antioxidant capacities of two Malaysian floral honeys. Food Chem. 2004;85:513–8. [Google Scholar]
- 19.Benzie IF, Strain JJ. The ferric reducing ability of plasma (FRAP) as a measure of “Antioxidant Power” : The FRAP assay. Anal Biochem. 1996;239(1):70–6. doi: 10.1006/abio.1996.0292. [DOI] [PubMed] [Google Scholar]
- 20.Mohamed M, Sirajudeen K, Swamy M, Yaacob NS, Sulaiman SA. Studies on the antioxidant properties of Tualang honey of Malaysia. Afr J Tradit Complement Altern Med. 2010;7(1):59–63. doi: 10.4314/ajtcam.v7i1.57256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175(1):184–91. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- 22.Pilz J, Meineke I, Gleite CH. Measurement of free and bound malondialdehyde in plasma by high-performance liquid chromatography as the 2,4-dinitrophenylhydrazine derivative. J Chrom B Biomedic Sci. 2000;742(2):315–25. doi: 10.1016/s0378-4347(00)00174-2. [DOI] [PubMed] [Google Scholar]
- 23.Beyer WF, Fridovich I. Assaying for superoxide dismutase activity: some large consequences of minor changes in condition. Anal Biochem. 1987;161(2):559–66. doi: 10.1016/0003-2697(87)90489-1. [DOI] [PubMed] [Google Scholar]
- 24.Paglia DF, Valentine WN. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med. 1967;70(1):158–69. [PubMed] [Google Scholar]
- 25.Aebi H. Catalase in vitro. In: Packer L, editor. Oxygen radicals in biological systems. London: Academic Press; 1984. pp. p.121–6. [Google Scholar]
- 26.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 27.Mahesh A, Shaheetha J, Thangadurai D, Rao DM. Protective effect of Indian honey on acetaminophen induced oxidative stress and liver toxicity in rat. Biologia. 2009;64(6):1225–31. [Google Scholar]
- 28.Alvarez-Suarez JM, Giampieri F, Damiani E, Astolfi P, Fattorini D, Regoli F, et al. Radical-scavenging activity, protective effect against lipid peroxidation and mineral contents of monofloral Cuban honeys. Plant Foods Hum Nutr. 2012;67(1):31–8. doi: 10.1007/s11130-011-0268-7. [DOI] [PubMed] [Google Scholar]
- 29.Hussein SZ, Yusoff KM, Makpol S, Yusof YA. Antioxidant capacities and total phenolic contents increase with gamma irradiation in two types of Malaysian honey. Molecules. 2011;16(8):6378–95. doi: 10.3390/molecules16086378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cai YZ, Mei Sun, Jie Xing, Luo Q, Corke H. Structure–radical scavenging activity relationships of phenolic compounds from traditional Chinese medicinal plants. Life Sci. 2006;78(25):2872–88. doi: 10.1016/j.lfs.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 31.Beretta G, Orioli M, Facino RM. Antioxidant and radical scavenging activity of honey in endothelial cell cultures (EA.hy926) Planta Med. 2007;73(11):1182–9. doi: 10.1055/s-2007-981598. [DOI] [PubMed] [Google Scholar]
- 32.Costa-Silva F, Maia M, Matos CC, Calcada E, Barros AIRNA, Nunes FM. Selenium content of Portuguese unifloral honeys. J Food Comp Anal. 2011;24(3):351–5. [Google Scholar]
- 33.Antunes F, Han D, Cadenas E. Relative contributions of heart mitochondria glutathione peroxidase and catalase to H2O2 detoxification in in vivo conditions. Free Radic Biol Med. 2002;33(9):1260–7. doi: 10.1016/s0891-5849(02)01016-x. [DOI] [PubMed] [Google Scholar]
- 34.White JW, Subers MH, Schepartz AI. The identification of inhibine, the antibacterial factor in honey, as hydrogen peroxide and its origin in a honey glucose-oxidase system. Biochim Biophys Acta. 1963;73:57–70. doi: 10.1016/0006-3002(63)90359-7. [DOI] [PubMed] [Google Scholar]
- 35.Adams CJ, Manley-Harris M. Molan PC. The origin of methylyoxal in New Zealand manuka (Leptospermum scoparium) honey. Carbohydr Res. 2009;344(8):1050–3. doi: 10.1016/j.carres.2009.03.020. [DOI] [PubMed] [Google Scholar]