

Abstract
Bacterial pathogens rely on their DNA repair pathways to resist genomic damage inflicted by the host. DNA double-strand breaks (DSBs) are especially threatening to bacterial viability. DSB repair by homologous recombination (HR) requires nucleases that resect DSB ends and a strand exchange protein that facilitates homology search. RecBCD and RecA perform these functions in E. coli and constitute the major pathway of error free DSB repair. Mycobacteria, including the human pathogen M. tuberculosis, elaborate an additional error-prone pathway of DSB repair via nonhomologous end-joining (NHEJ) catalyzed by Ku and DNA ligase D (LigD). Little is known about the relative contributions of HR and NHEJ to mycobacterial chromosome repair, the factors that dictate pathway choice, or the existence of additional DSB repair pathways. Here we demonstrate that Mycobacterium smegmatis has three DSB repair pathway options: HR, NHEJ, and a novel mechanism of single-strand annealing (SSA). Inactivation of NHEJ or SSA is compensated by elevated HR. We find that mycobacterial RecBCD does not participate in HR or confer resistance to ionizing radiation (IR), but is required for the RecA-independent SSA pathway. In contrast, the mycobacterial helicase-nuclease AdnAB participates in the RecA-dependent HR pathway, and is a major determinant of resistance to IR and oxidative DNA damage. These findings reveal distinctive features of mycobacterial DSB repair, most notably the dedication of the RecBCD and AdnAB helicase-nuclease machines to distinct repair pathways.
Introduction
The maintenance of genome integrity is pivotal for cell survival, yet double-strand breaks (DSBs) are routinely induced by endogenous sources such as reactive by-products of cellular metabolism, replication across nicks, and exogenous agents like ionizing radiation and genotoxic chemicals (Lieber, 2010). Most bacteria overcome such perils by employing homologous recombination (HR) as the major DSB repair pathway, which results in faithful repair of the damaged duplex. The first step of HR is catalyzed by a helicase-nuclease that resects the DSB end to generate a 3’ single-stranded tail onto which the RecA protein is loaded to form a nucleoprotein filament. RecA mediates a homology search and strand invasion into a homologous duplex, typically of a sister chromatid. In many bacteria, including the model organism Escherichia coli, the heterotrimeric RecBCD complex serves as the major resection nuclease involved in DSB repair (Dillingham & Kowalczykowski, 2008). An alternate solution to the end resection problem is evident in pathogenic bacteria like Helicobacter pylori, Bacillus species, and other Gram-positive bacteria, which lack RecBCD but possess a heterodimeric helicase-nuclease complex, AddAB, that resects DSBs before RecA loading (Amundsen et al., 2008, Chedin & Kowalczykowski, 2002).
Mycobacteria have evolved more complex systems of DSB repair that embellish the standard HR-centric scenario with additional repair options and distinctive repair proteins. Mycobacterium tuberculosis and Mycobacterium smegmatis express a nonhomologous end-joining pathway (NHEJ) that uses Ku and DNA ligase D (LigD) to seal linear plasmids and chromosomal DSBs (Gong et al., 2005, Gong et al., 2004, Shuman & Glickman, 2007). Many of the mechanistic insights into mycobacterial NHEJ gained thus far used DSB substrates for which alternative pathways of repair were not possible because a donor sequence for homology-directed repair was not provided (Aniukwu et al., 2008, Stephanou et al., 2007). Consequently, the relative contributions of NHEJ and HR to DSB repair in a bacterium that possesses an NHEJ pathway are unknown and the determinants of DSB repair pathway choice have not been examined. In addition, the mycobacterial HR pathway is not well defined, e.g., in comparison to E. coli and Bacillus subtilis. Mycobacterium smegmatis and Mycobacterium bovis BCG lacking recA are sensitized to diverse clastogens, confirming that mycobacterial RecA (like E. coli RecA) is important for recombinational repair (Korycka-Machala et al., 2006, Pitcher et al., 2007, Sander et al., 2001, Stephanou et al., 2007). However, the helicase-nuclease responsible for DSB resection and RecA loading in mycobacteria is unknown. Mycobacterial proteomes contain RecBCD, but M. smegmatis ΔrecBCD is not sensitized to killing by UV (Stephanou et al., 2007), suggesting that alternate pathways exist in mycobacteria to process breaks and recruit RecA onto single-stranded DNA. We recently identified a distinctive DSB-resecting heterodimeric helicase-nuclease, AdnAB, that coexists in mycobacterial proteomes with RecBCD (Sinha et al., 2009). We speculated that the coexistence of RecBCD and AdnAB in mycobacteria might be analogous to the situation in yeast, in which functionally redundant nucleases participate in DSB end-resection (Zhu et al., 2008, Mimitou & Symington, 2008).
In this study we develop and use a DSB repair assay system in which tandem I-SceI-induced breaks in the chromosome, that generate noncomplementary 3’ overhangs, can be repaired by any one of three different repair options. By studying repair outcomes in wild type and mutant M. smegmatis strains, we determined the relative contributions of HR, NHEJ, and a novel single-strand annealing (SSA) pathway to repair at the reporter locus and find asymmetric interrelationships among mutagenic and faithful repair pathways. We also examined the helicase-nucleases involved in mycobacterial DSB repair and demonstrate that mycobacterial AdnAB and RecBCD are not redundant, but in fact define distinct DSB repair pathways. Whereas AdnAB functions as a presynaptic nuclease in the RecA-dependent HR pathway, RecBCD is dedicated to a RecA-independent single-strand annealing (SSA) pathway of recombination between direct repeats, not previously recognized in bacteria.
RESULTS
Development of an efficient chromosomal DSB repair assay system in mycobacteria
To study the interrelationship of NHEJ and HR in mycobacteria and determine the molecular requirements for each pathway in chromosomal DSB repair, we developed a reporter system in which repair outcomes at an I-SceI-induced DSB could be scored genetically and mapped physically. We placed two I-SceI cleavage sites, in inverted orientation and separated by 37 base pairs, within an internally deleted lacZ gene, such that cleavage at both sites by I-SceI yields a chromosomal DSB with non-complementary 3’ overhangs (lacZ(I-SceI), Figure 1A). The lacZ(I-SceI) allele does not encode a functional β-galactosidase enzyme and therefore bacteria with this construct integrated at the chromosomal attB site are white on plates containing X-gal. Kanamycin resistance is conferred by the aph gene cointegrated downstream of lacZ(I-SceI) (Figure 1A). Repair of the DSB pictured in Figure 1A can occur only by NHEJ, because a homologous donor sequence is not available elsewhere in the chromosome to serve as template for HR (presuming a duplicated sister chromosome that would be present after replication is also cut by I-SceI and therefore cannot serve as homology donor). Several elements of this system improve upon our initial use of I-SceI in mycobacteria (Stephanou et al., 2007). First, the incompatible 3’-overhang ends produced by I-SceI minimize repeated cycles of cleavage and faithful repair, because such ends are unlikely to be ligated without prior processing. Second, these constructs were integrated at the chromosomal attB site using a two-plasmid system, wherein the L5 integrase was provided in trans and then eliminated (Pena et al., 1997, Pashley & Parish, 2003) (see Methods). This maneuver eliminates cassette loss as a mechanism of survival after I-SceI cleavage because the integrase enzyme, which can catalyze excision of the integrated reporter, is no longer present in the cell. This innovation allowed us to forego kanamycin selection during the induction of DSBs, removing any bias against repair outcomes that might delete the kanamycin marker. Third, I-SceI expression under control of a constitutive promoter was initiated by plasmid transformation rather than by tetracycline induction from a regulated promoter on a pre-existing plasmid episome, thereby circumventing problems associated with basal expression of the I-SceI enzyme, which in our prior experiments caused significant cleavage and premature mutagenic repair at the I-SceI recognition site (Stephanou et al., 2007).
Figure 1. NHEJ mediated repair of a chromosomal I-SceI induced DSB with incompatible ends.
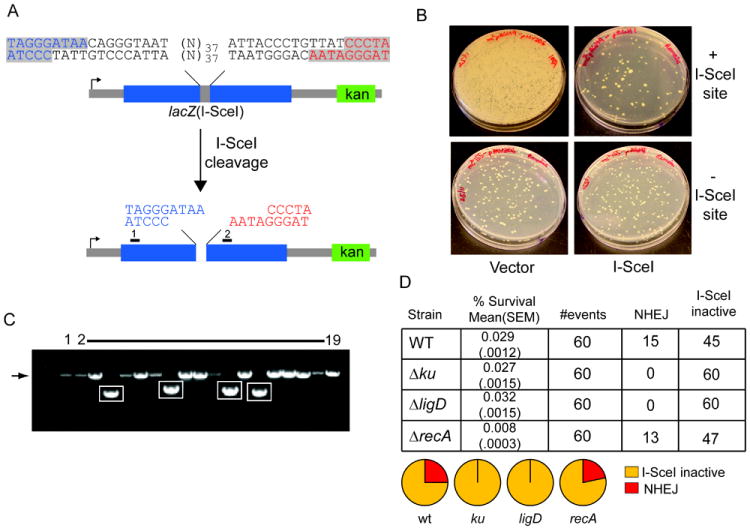
A. Schematic representation of double site I-SceI DSB substrate. Two I-SceI sites in opposite orientation produce a DSB with incompatible 3’overhangs, with the two halves of the DSB colored blue and red. The positions of primers 1 and 2, used to amplify and sequence repaired DSBs after I-SceI cleavage, are indicated by black rectangles. A kanamycin marker (green box) is 3’ of the break.
B. The I-SceI substrate pictured in (A), or a control substrate lacking the I-SceI sites, was integrated into the chromosome of M. smegmatis (see materials and methods). These strains were transformed with a plasmid conferring hygromycin resistance and directing the expression of I-SceI endonuclease (I-SceI) or vector. Shown are M. smegmatis colonies on agar plates containing hygromycin. Substantial cell death is evident in the strain with I-SceI sites transformed with I-SceI plasmid (upper right) but not in cells transformed with control plasmid (upper left), or in a strain lacking a chromosomal I-SceI cutting site (lower panels). All the cells from the entire transformation mixture (1.4 ml) were plated for the strain with chromosomal I-SceI sites (upper panels), whereas 100 μl of the transformation mixtures were plated in case of the strain lacking I-SceI sites (bottom panels).
C. Clonal repair events from 18 surviving colonies after I-SceI expression were analyzed by PCR amplification using primers 1 and 2. PCR products were separated on a non-denaturing agarose gel and visualized by ethidium bromide staining in comparison to the PCR product generated from amplification of the parent construct that had not undergone I-SceI cleavage (lane 1). The expected size of the PCR product from primers 1 and 2 (1350 bp) is indicated by an arrow to the left of the gel image, and amplicons showing deletion formation after DSB repair are boxed.
D. Repair of incompatible 3’ overhangs depends on NHEJ. The I-SceI substrate pictured in (A) was integrated into the chromosome of wild type (as in B), Δku, ΔligD and ΔrecA strains. The table shows % survival obtained after transformation with I-SceI plasmid ((mean value obtained from three experiments is shown with standard error of the mean, SEM, indicated in parentheses) and NHEJ outcomes were scored from 60 transformants, from one experiment. The pie graphs represent the fraction of each repair outcome among total survivors. I-SceI inactive refers to survivors that had inactivated the I-SceI cDNA by mutation and had unmodified I-SceI cleavage sites in the chromosome.
NHEJ dependent repair of incompatible 3’ overhang DSB
We first tested whether and how wild type M. smegmatis could repair a chromosomal DSB with incompatible 3’ overhangs induced by transformation of an I-SceI encoding plasmid that confers hygromycin resistance. When the I-SceI plasmid was introduced into an otherwise wild-type M. smegmatis strain containing a chromosomal lacZ(I-SceI), the yield of hygR survivors was 0.029% compared to when the same strain was transformed with a vector control (Figure 1B). By contrast, the I-SceI plasmid and vector control displayed equivalently high transformation efficiencies in wild type M. smegmatis cells that lacked a chromosomal I-SceI target site (Fig. 1B). These results indicate that an I-SceI-induced DSB with incompatible 3’ overhangs is toxic to mycobacteria.
To determine the mechanism of DSB repair in cells that had survived I-SceI expression, we PCR-amplified the genomic region surrounding the I-SceI sites in lacZ(I-SceI) and determined the DNA sequences of these repaired chromosomal segments from 60 independent surviving transformants. We observed two types of outcomes, whereby PCR amplification yielded either: (i) a 1350 bp fragment seemingly identical to that amplified from the uncleaved lacZ(I-SceI) locus (Fig. 1C, arrow at left); or (ii) smaller DNA fragments, reflecting deletions of varying lengths (Fig. 1C, highlighted in boxes). Sequencing of the individual PCR products confirmed two mechanisms of survival following I-SceI transformation: (1) 25% of the events entailed chromosomal deletions (of varying lengths, see below) with loss of both I-SceI cleavage sites and the intervening 37 bp segment; (2) 75% retained intact chromosomal I-SceI cleavage sites, hinting that survival was attendant on an inactivating mutation of the I-SceI endonuclease gene (Figure 1D), a suspicion that was confirmed by DNA sequencing of the I-SceI encoding plasmid and re-transformation back into the original M. smegmatis strain bearing lacZ(I-SceI) (Figure S1). These results demonstrate two mechanisms of survival following I-SceI induced DSB: stochastic inactivation of the endonuclease, or repair by site modification and ligation. We did not observe any instances in which a single I-SceI cleavage site had been mutated while the other site was intact, or in which both sites were mutated but the intervening DNA sequence (37 bp) was still present, suggesting that both sites are broken in vivo with high frequency.
In our prior experiments involving linear plasmid transformation as an assay of NHEJ, we found that ~25% of the faithful repair events at complementary 3’ overhangs were independent of Ku and LigD (Aniukwu et al., 2008). To interrogate the genetic requirements for repair of incompatible 3’ overhangs at the chromosomal lacZ(I-SceI) locus, we assayed survival and NHEJ outcomes in strains lacking ku, ligD, and recA. Whereas overall survival was unaffected in Δku and ΔligD strains, DSB repair by NHEJ was ablated so that 100% (60/60) of the hygR survivors retained intact I-SceI sites (Figure 1D). As anticipated (because no homologous donor sequence is available to direct HR), the ΔrecA strain behaved like the WT strain, showing a similar fraction of NHEJ (13/60) events among survivors, albeit with a modest decrement (3.5 fold) in survival (Figure 1D). These results prove that the “classical” mycobacterial NHEJ pathway driven by Ku and LigD is essential for repair of a chromosomal DSB with non-complementary 3’ ends.
NHEJ of incompatible 3’ overhangs involves local resection and cross-break priming
The DSB produced in the lacZ(I-SceI) reporter locus is unlikely to be repaired without end modification because the two 3’ overhangs cannot anneal directly. To define the mechanism of NHEJ-mediated repair of lacZ(I-SceI), the sequences of the repair junctions were analyzed. Two major types of repair outcomes were observed. In 7/15 events, one or both sides of the DSB were remodeled by 1-4 nt single-strand resections, followed by annealing and priming of templated fill-in DNA synthesis on one or both sides of the break (Figure 2). For instance, in the case of the Δ1/Δ1 deletion, a single 3’ nucleotide was apparently removed from both overhangs to reveal a terminal 2 nt microhomology, followed by cross-break priming to fill the single-nucleotide gap(s) prior to ligation (Figure 2). This mechanism of joining of noncomplementary overhangs has been noted in eukaryotic NHEJ (Davis et al., 2008, Nick McElhinny et al., 2005). The other class of repair events (8/15) entailed deletions into the duplex flanking the I-SceI breaks, which were of variable length and bidirectional in the majority of the cases (Figure 2). The molecular outcomes of NHEJ were similar in the ΔrecA strain. 4 out of 13 repair events analyzed exhibited local cross-break priming, and a single event involved unidirectional deletion with cross-break fill-in synthesis. The remainder of ΔrecA repair events (8/13) entailed larger deletions into the duplex flanks, mostly bidirectional and microhomology-independent, essentially as seen in the wild-type strain (Figure 2). The molecular features of NHEJ demonstrated here imply the participation of a polymerase that can catalyze cross-break DNA synthesis and one or more nucleases that can catalyze both local single strand resection and longer double strand deletions.
Figure 2. Molecular outcomes of NHEJ.
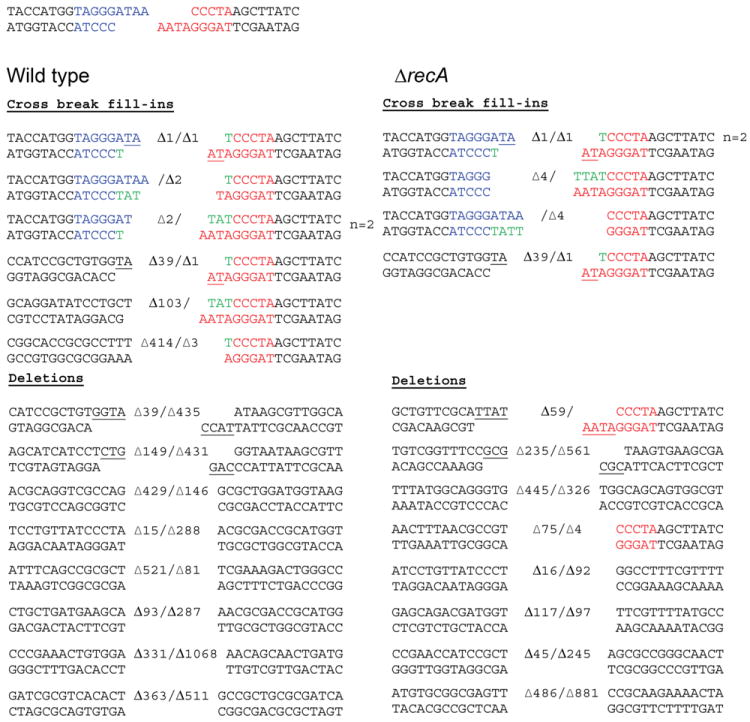
The cut I-SceI site is shown at the top of the figure with left and right ends colored blue and red respectively. Molecular outcomes from WT (n=15, left panel) and ΔrecA strain (n=13, right panel) carrying the NHEJ substrate pictured in Figure 1A are shown with the number of deleted nucleotides indicated. Nucleotides added by fill-in synthesis of the 5’ recessed ends are colored green and microhomology use is underlined.
Mycobacteria have three DSB repair pathways
A major unresolved issue in bacteria that express an NHEJ pathway concerns the relative contributions of NHEJ and HR to DSB repair. To examine this question, we constructed a chromosomal reporter substrate that not only contains lacZ(I-SceI) (described in the preceding section), but also has a second 5’-truncated lacZ copy (lacZ-ΔN) downstream of the kanamycin-resistance cassette (Figure 3A). lacZ-ΔN is missing the first 662 bp of the lacZ coding sequence, but contains the middle coding sequence absent in lacZ(I-SceI), and offers homology of 567 bp upstream and 1119 bp downstream of the DSB. Accordingly, after DSB induction, the break can either be repaired by NHEJ, or lacZ-ΔN can provide a template for homology-directed repair, resulting in the formation of a functional lacZ gene by gene conversion (GC) of lacZ(I-SceI), with retention of the intervening kanamycin-resistance gene (Figure 3A).
Figure 3. Division of labor among three DSB repair pathways in M. smegmatis.
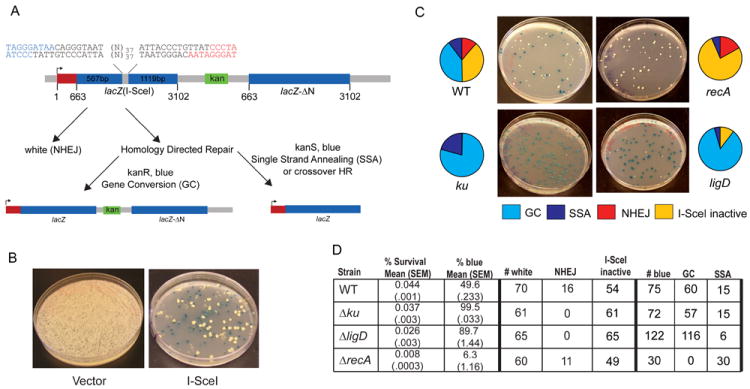
A. Recombination reporter substrates. Dual I-SceI sites are placed within a lacZ allele containing an internal deletion (lacZ(I-SceI)). Expression is directed by a promoter upstream of lacZ(I-SceI) (arrow). A segment of lacZ from nucleotides 662-3102 (lacZ-ΔN, which spans the internal deletion of lacZ(I-SceI) but lacks the N terminal 663nt (red box) is positioned 3’ of lacZ(I-SceI) with a kanamycin resistance gene (green box) between the two lacZ segments. The three potential repair outcomes after I-SceI cleavage are shown. NHEJ produces a white colony. Gene conversion of lacZ(I-SceI)using lacZ-ΔN restores a functional lacZ gene, producing a blue colony on agar plates containing X-Gal, which is kanamycin resistant. Repair by SSA or HR with crossing over (CO) restores a functional lacZ allele, but deletes the kanamycin marker, producing a blue colony on X-gal plates which is kanamycin sensitive.
B. Transformation of wild type M. smegmatis carrying the substrate pictured in A with either vector or I-SceI plasmid. Shown are hygromycin resistant transformants on media containing X-gal. Vector transformation does not induce recombination, as indicated by exclusively white colonies. I-SceI expression produces a mixture of blue and white colonies, indicating a mixture of repair outcomes.
C. Relative repair fractions for each outcome from wild type, ΔrecA, Δku, and ΔligD strains are represented by pie charts with representative hygromycin/X-gal plates shown. Each repair outcome is coded using the color key shown below the plate images. Note that the relative fraction of I-SceI inactive in the Δku strain is 0.5% and therefore not visible on the pie graph.
D. The table shows the mean % survival and % blue of three independent experiments (with standard error of the mean, SEM, indicated in parentheses). The number of repair outcomes among white and blue colonies for each strain used to calculate the percentages graphed in (C) are shown and are derived from a single representative experiment.
In mammalian and yeast cells, repair of a DSB flanked by repeats can be achieved through a third pathway, single-strand annealing (SSA) (Ivanov et al., 1996, Elliott et al., 2005). SSA involves bidirectional single-strand resection to reveal complementary repeats that anneal, followed by flap resection and ligation, deleting the DNA between the repeats. In the DSB repair reporter pictured in Figure 3A, bidirectional single strand resection could expose complementary sequences in the 567 bp segment 5’ of the I-SceI sites, which is shared in lacZ-ΔN. Repair by SSA would reconstitute the full-length lacZ gene, with deletion of the intervening kanamycin marker. This outcome could also be achieved by crossover HR (CO). Crossover HR and SSA are mechanistically distinguishable by their dependence on RecA, insofar as SSA does not require strand invasion and in yeast is independent of the yeast RecA homolog Rad51 (15).
In principle, all three potential DSB repair mechanisms can be simultaneously studied using the reporter system illustrated in Fig. 3A, and they can be lucidly distinguished from each other by simple scorable phenotypes. Colonies are first tested for blue or white color on medium containing X-gal and hygromycin (to select for transformation with the I-SceI encoding plasmid). Blue colonies are then scored for kanamycin sensitivity or resistance by transfer to agar medium containing kanamycin. Repair outcomes are scored as indicated in Figure 3A: white colonies, indicative of NHEJ or loss of I-SceI activity; blue and kanamycin-resistant colonies, representing GC/HR; blue and kanamycin-sensitive colonies, reflecting either SSA or crossover HR. After phenotypic screening, repair outcomes and NHEJ junction sequences are analyzed by PCR amplification with diagnostic primers and I-SceI digestion of the PCR products (Figure S2).
We first evaluated the frequency of NHEJ, HR, and SSA/CO in wild-type M. smegmatis by integrating the lacZ recombination substrate into the chromosome, and expressing I-SceI endonuclease by plasmid transformation to inflict the DSB (as described above). Figure 3B shows that after transformation with control vector alone, only white colonies were obtained on medium containing X-gal, thereby verifying that both lacZ loci in the chromosome were non-functional and that spontaneous recombination was rare. However, after induction of DSB, the survivors comprised an equal mixture of blue and white colonies, indicating that an active lacZ gene was reconstituted in half of the survivors. Among the blue colonies, GC and SSA/CO outcomes were discerned by screening for kanamycin resistance, and also confirmed by direct physical analysis using PCR. As predicted, blue kanamycin–sensitive colonies (SSA or CO) showed loss of the DNA between the two lacZ copies containing the kanamycin-resistance cassette (Figure S2). To fortify our inference that the kanamycin-sensitive blue colonies resulted from SSA or HR with CO, we generated another reporter strain wherein the homology donor was situated ~1.2 Mbp away from the DSB site on the chromosome (in the same orientation), with numerous essential mycobacterial genes separating the two regions of homology (Figure S3A). Following DSB induction, both blue and white colonies were observed, but all of the blue colonies were kanamycin-resistant, indicating repair by gene conversion alone, and not by SSA/CO (Figure S3B).
In wild type cells, HR, NHEJ, and SSA/CO accounted for 40%, 12%, and 10% of the survivors, respectively, with the remainder being associated with I-SceI inactivation (Figure 3C). Thus, all three pathways of repair are evident in wild type cells. HR with gene conversion is the preferred repair pathway chosen by M. smegmatis in this assay system.
Mycobacteria have a RecA independent SSA pathway
We next examined the genetic requirements for the three DSB repair pathways and the factors influencing pathway choice. To analyze differences in repair outcomes in wild type and mutant strains, we employed two methods of analysis (see methods for a detailed description). The first method calculates “absolute repair frequency” in which the frequency of each repair outcome is corrected for the observed survival. This calculation determines the frequency of each repair outcome among the originally transformed cells and the mean of three replicates for any given strain can be compared to another strain using the t-test to determine statistical significance. The second method of analysis determines the relative fraction of each repair outcome among the surviving cells (relative repair fraction). This relative repair fraction gives the frequency of a given pathway independent of survival and differences between strains can be compared statistically by using the Fisher’s exact test.
The effects of ablating HR by the ΔrecA mutation were immediately instructive. As expected, GC was abolished, such that no lacZ+ kanR survivors were obtained (Figure 3D). In contrast, the recA mutation had no effect on the relative repair frequency of NHEJ (WT vs. ΔrecA = 11.4% (17/150) vs 17.2% (76/446) of survivors, p=0.118). Similarly, we still observed a similar proportion of kanamycin sensitive blue colonies among survivors in the ΔrecA strain (WT vs. ΔrecA = 10% vs 6.3%; p=0.146 for comparison of WT (15/150) vs ΔrecA (30/446)), indicating that these events are from an SSA pathway rather than HR. We did observe a decrement in absolute repair frequency in the recA mutant for all repair outcomes, including SSA (Table S3). This decline in absolute repair frequency is due to the 5 fold decrement in survival observed in the ΔrecA strain (Figure 3D). Importantly, a similar decrement in survival was also observed in the ΔrecA reporter strain with no lacZ-ΔN homology donor (Figure 1D), indicating that the decline in survival after DSB induction in the ΔrecA strain is not due to failed recombinational repair, but likely due to pleiotropic functions of recA in DNA damage responses. Thus, the decline in survival and absolute repair frequency in the ΔrecA strain does not differentially affect one repair pathway, and therefore does not suggest a specific involvement of recA in SSA or NHEJ. Taken together, these results strongly indicate that the events classified thus far as SSA/CO do not arise by HR with crossing over (a pathway that should require RecA due to the requirement for strand invasion), but are instead formed by a bona fide SSA pathway (Ivanov et al., 1996). We conclude that mycobacteria express a previously unappreciated SSA pathway that is distinct from HR and NHEJ.
Ku suppresses repair by Homologous Recombination
Repair of the DSB by NHEJ was abolished in the Δku and ΔligD strains, as expected (Figure 3C,D). The salient finding was that the loss of NHEJ was compensated by increased relative fraction of homology directed repair (p<0.0001 for comparison of WT (75/150) vs Δku (72/72) or ΔligD (122/136) fraction of homology directed repair among surviving colonies, Figure 3C, D). The absolute frequency of gene conversion was significantly higher in the Δku strain compared to wild type (absolute GC frequency WT vs Δku = 0.017% vs 0.0287%; p=0.0039; Figure 3C,D and Table S3). In contrast, the absolute frequency of GC was not significantly higher in the ΔligD strain, despite a higher relative fraction among survivors. In Δku cells, SSA also increased by two-fold compared to wild-type M. smegmatis, suggesting that the presence of Ku on DNA ends suppressed both pathways of homology-directed repair. In contrast SSA diminished in the ΔligD strain, accounting for just 5% of blue colonies in ΔligD compared to 10% and 21% in WT and Δku cells, respectively (Figure 3C). Taken together, these experiments provide new insights to DSB repair in mycobacteria, including: (i) the existence of a recA-independent SSA pathway; (ii) the dominance of recA-dependent HR in DSB repair; (iii) the partial dependence of the SSA pathway on LigD; and (iv) an asymmetric relationship between NHEJ and HR in which inactivation of NHEJ is compensated by elevated HR, but loss of HR is not compensated by increased NHEJ.
Division of labor among helicase-nucleases in DSB repair
In E. coli, the dominant pathway of HR is initiated by the RecBCD helicase-nuclease, which generates the invading 3’ single-strand and assists with loading RecA (Dillingham & Kowalczykowski, 2008). Mycobacterial proteomes include an obvious RecBCD homolog encoded in a single operon, but prior studies showed no clastogen sensitivity or change in NHEJ deletion outcomes in M. smegmatis lacking recBCD (Stephanou et al., 2007, Aniukwu et al., 2008), either indicating redundancy with other enzymes or that RecBCD does not mediate HR in mycobacteria. We recently identified a new helicase-nuclease of mycobacteria, AdnAB, which resects DSBs and is blocked by Ku (Sinha et al., 2009). However, the in vivo function of AdnAB is not known, nor is its potential redundancy with RecBCD.
To query the functions of RecBCD and AdnAB in DSB repair, we deleted one or both of the helicase-nuclease operons and then tested the effects of the ΔrecBCD and ΔadnAB mutations on the repair of I-SceI-induced chromosomal DSBs. We first examined whether RecBCD or AdnAB are the principal agents of deletion formation during NHEJ. For the NHEJ-specific repair substrate (Figure 1A), the overall survival and relative fraction of NHEJ among survivors were unaffected by loss of recBCD, adnAB, or both (Figure 4A). We observed that the median deletion size did not diminish significantly in ΔrecBCD, ΔadnAB, or ΔrecBCD ΔadnAB strains (Figure 5, S4), indicating that RecBCD and AdnAB are unlikely to be the principal agents of duplex deletion formation during NHEJ. These results implicate other unidentified nucleases in the long tract double-strand nucleolytic resection events observed during NHEJ.
Figure 4. RecBCD and AdnAB are repair pathway specific nucleases.
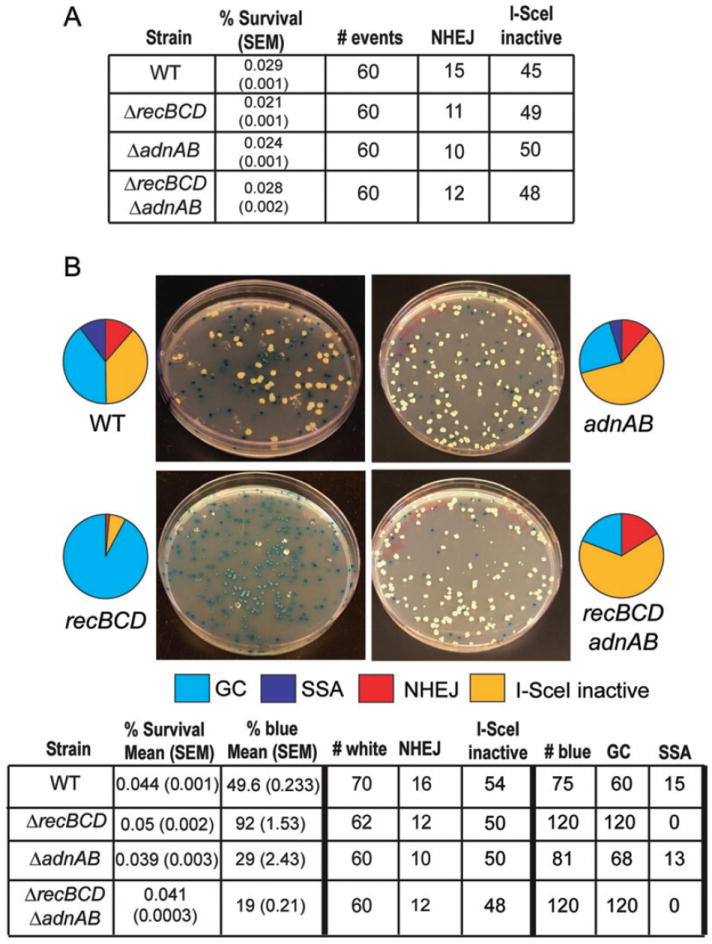
A. Repair outcomes for ΔrecBCD, ΔadnAB, and ΔadnABΔrecBCD strains carrying the no homology I-SceI repair substrate (see Figure 1A). The data for WT strain are the same as that presented in Figure 1D.
B. Repair outcomes for the indicated strains and representative hygromycin/X-gal plates using the recombination substrate pictured in Figure 3A. The table shows the mean % survival and % blue of three independent experiments (with standard error of the mean, SEM, indicated in parentheses). The number of repair outcomes among white and blue colonies for each strain used to calculate the percentages graphed in the pie graphs are shown and are derived from a single representative experiment. The data for WT strain are the same as that presented in Figure 3D.
Figure 5. Role of AdnAB and RecBCD in NHEJ deletions.
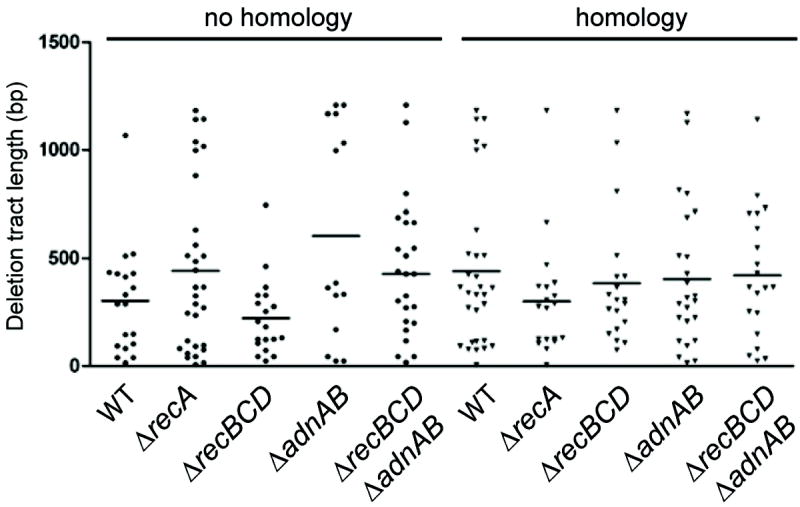
For each strain, the graph indicates the length of the duplex deletion from either end of the DSB either without or with a homology donor. The horizontal line indicates the median deletion size for each strain.
When we used the dual lacZ reporter system to test NHEJ, HR, and SSA in strains lacking recBCD, adnAB, or both, the results were surprising on several fronts. First, the ΔrecBCD mutation had no negative impact on HR (Fig. 4B). In fact, 92% of surviving colonies were blue and 100% of these had restored the lacZ coding sequence by gene conversion (Figure 4B; relative GC frequency WT= 40% (60/150) vs ΔrecBCD= 92% (120/130), p<0.0001; p<0.0001 for comparison of WT (0.017%) vs ΔrecBCD (0.0457%) absolute GC frequency, Table S3), indicating that RecBCD actually suppresses HR. By contrast, an E. coli ΔrecBCD strain is recombination-deficient for most types of recombination (Dillingham & Kowalczykowski, 2008). Second, the ΔrecBCD mutation ablated the SSA pathway of DSB repair: out of 120 blue ΔrecBCD transformants, we did not observe even one SSA repair event (Figure 4B). We conclude that mycobacterial RecBCD is specifically dedicated to the SSA pathway of DSB repair, and is dispensable for HR. These results expand the known functions of RecBCD by implicating this enzyme complex in a DSB repair pathway other than recA-dependent HR.
We next surveyed repair in the ΔadnAB strain. Both the relative fraction of GC among total survivors (WT vs ΔadnAB; 40% vs 24%, p=0.0009) and the absolute frequency of gene conversion (WT vs ΔadnAB = 0.017% vs 0.0091%; p=0.0002) were both lower by a factor of 2 in the ΔadnAB strain compared to wild type cells. These results indicate AdnAB participates in HR, but the substantial residual HR present in cells lacking adnAB points to the presence of alternative presynaptic nuclease(s). Further evidence for the participation of AdnAB in HR was gathered from analysis of DSB repair in a ΔadnAB ΔrecBCD double-deletion strain. As noted above, the relative fraction of GC in the ΔrecBCD strain is 92%, yet when adnAB is deleted this GC fraction is reduced by a factor of 4.8 (to 19%). A similar decrement is observed in the absolute GC frequency, indicating a substantial participation of AdnAB in HR (Figure 4B and Table S3; p<0.0001 for comparison of ΔrecBCD (0.0457%) vs ΔadnAB ΔrecBCD (0.0078%) absolute GC frequency). No SSA was observed among 120 lacZ+ survivors in the ΔadnAB ΔrecBCD strain, in keeping with the findings for the ΔrecBCD single mutant. Taken together, these results document a division of labor between RecBCD and AdnAB in mycobacterial DSB repair, with the former nuclease being essential for the recA-independent SSA pathway, and the latter participating in RecA-dependent HR. The data also provide support for an additional recA-dependent, adnAB-independent pathway of HR.
Role of AdnAB and RecBCD in clastogen resistance
The I-SceI induced DSB experiments described above elucidate roles for recBCD and adnAB in separate pathways of homology-directed repair of a DSB. As such, the ΔadnAB and ΔrecBCD mutants provide a tool to understand the role of each of these pathways in repair of DNA damage caused by chemical and physical agents. Accordingly, we tested these strains for sensitivity to ionizing radiation (IR), UV irradiation, hydrogen peroxide, mitomycin C, and methyl methanesulfonate (MMS). We found that inactivation of adnAB, but not recBCD, conferred UV sensitivity, albeit to a lesser degree than that observed in ΔrecA (Figure 6A). Similar results were obtained with MMS (Figure 6B) and mitomycin C (Figure 6C). In contrast, the adnAB mutant was as sensitive as ΔrecA to killing by IR, but this phenotype was not exacerbated by loss of recBCD (Figure 6D; survival of each strain at 1008 Gy compared to wild type: ΔrecA 0.001%, ΔadnAB 0.003%, ΔadnABΔrecBCD 0.002%). Similarly, either the ΔadnAB or ΔrecA mutation sensitized M. smegmatis equally to 25mM hydrogen peroxide, indicating a substantial role for AdnAB/RecA in repair of oxidative DNA damage (Figure 6E). Both the IR and hydrogen peroxide sensitivity phenotypes of the ΔadnAB and ΔadnAB ΔrecBCD mutants were rescued by reintroduction of a wild type copy of adnAB (Figure S5A,B), confirming that these phenotypes are due to loss of AdnAB function and not a spontaneous second site mutation or polar effect. We infer from these results that the RecBCD-dependent SSA pathway is not a substantial contributor to repair of DNA damage induced by a wide variety of clastogens, presumably because the number of breaks arising between appropriately spaced repeats is small. By contrast, the AdnAB/RecA pathway plays a major role in defense against hydrogen peroxide and ionizing radiation, two clastogens that induce diverse types of DNA damage, including DSBs.
Figure 6. Relative contributions of RecBCD and AdnAB to clastogen resistance.
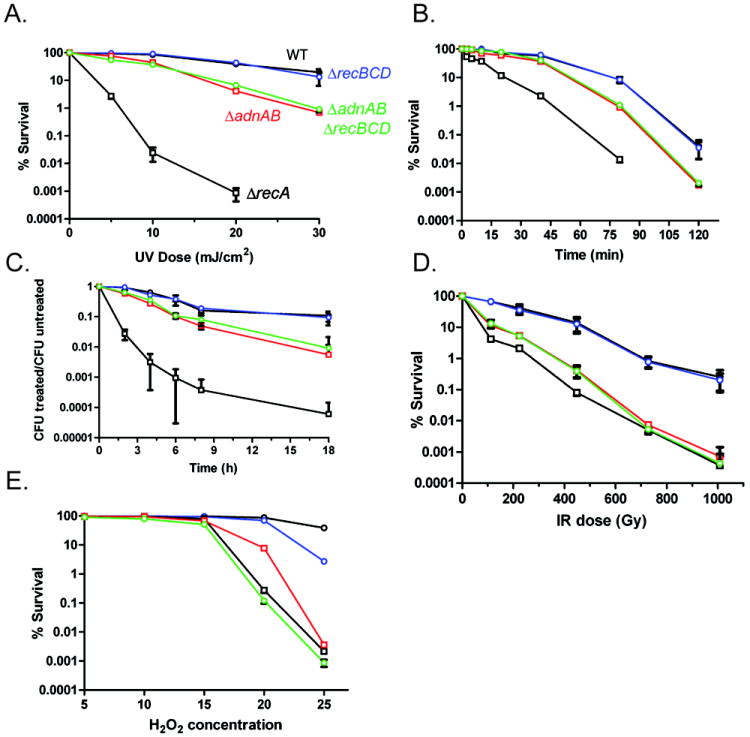
For each panel, % survival was determined by culturing serial tenfold dilutions on agar plates after exposure to the indicated dose of clastogen. Each graphed point represents the mean of biologic triplicates and % survival is graphed on a logarithmic Y axis. For all panels, strains are indicated in panel A by different colors (also see below). For all panels, error bars are SEM and when not visible are within the symbol.
A. UV radiation. Strains tested: WT (mc2155; black color with open circles), ΔrecBCD (mgm177; blue color), ΔadnAB (mgm1964; red color), ΔadnABΔrecBCD (mgm1965; green color), and ΔrecA (mgm199; black color with open squares).
B. 0.5 % MMS for the indicated time.
C. 25 ng ml-1 mitomycin C for the indicated time.
D. Ionizing radiation delivered by a cesium source.
E. Hydrogen peroxide.
Discussion
Mycobacteria have three pathways of DSB repair
Here we show that a DSB in the mycobacterial chromosome can be repaired by three genetically distinct mechanisms (Figure 7). HR is the dominant pathway in our assay system, accounting for 40% of cells that survive a DSB. NHEJ repairs the DSB in 12% of surviving cells. It was previously unappreciated that mycobacteria express an SSA pathway. In fact, mycobacterial SSA differs substantially from examples of SSA-type repair found previously in E. coli. In E. coli, an SSA-type mechanism was proposed to mediate spontaneous deletions between direct repeat sequences associated with palindromes through SbcCD mediated cleavage of a cruciform intermediate (Bzymek & Lovett, 2001a, Bzymek & Lovett, 2001b). These events differ from the mycobacterial SSA pathway in their requirement for palindromic sequences, their spontaneous occurrence without a DSB, and their independence from RecBCD. Thus, the mycobacterial recBCD-dependent SSA pathway provides a third pathway of repair (in addition to HR and NHEJ) that is available to rectify DSBs in wild type cells. The mycobacterial SSA pathway may be particularly important to repair DSBs that arise in repetitive areas of the mycobacterial chromosome.
Figure 7. Genetic requirements for three double strand break repair pathways in mycobacteria.
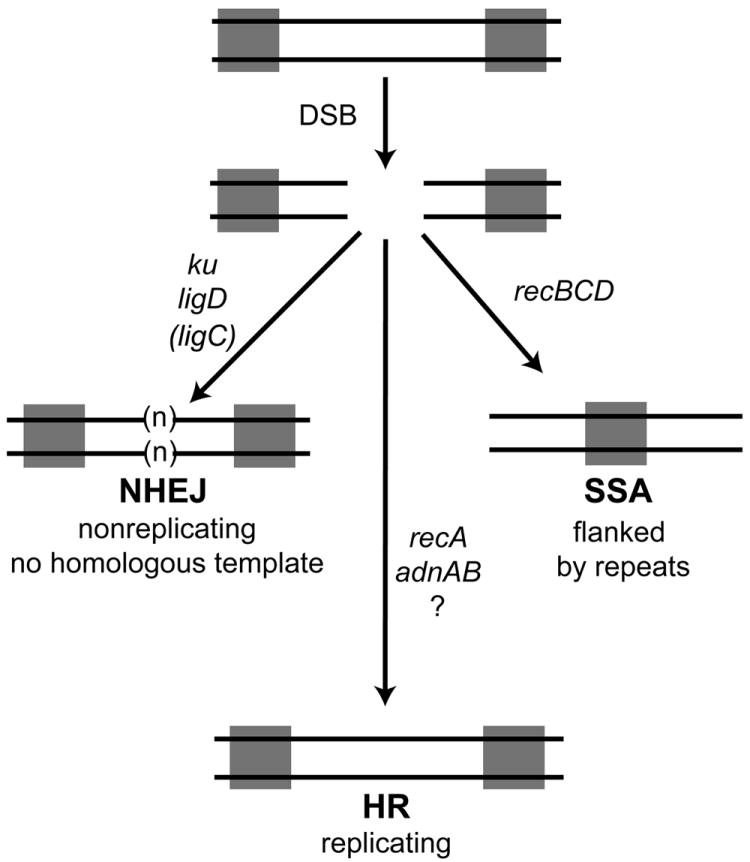
We depict a double strand break (DSB) flanked by two repeats (gray squares). The break can be repaired by NHEJ, a pathway defined by ku and ligD with a secondary role for ligC when LigD is inactivated (see Aniukwu et al., 2008). NHEJ can add additional nucleotides (n), or result in deletions into the DNA surrounding the break. NHEJ does not require a homologous template for repair and therefore can repair DSB in nonreplicating cells. Repair by HR is dependent on recA and adnAB, but is independent of recBCD. Our data indicate a additional pathway of adnAB independent, recA dependent HR (indicated by ?) evinced by the residual HR in the ΔadnAB strain. Repair by the SSA pathway is achieved by bidirectional single strand resection to reveal the complementary repeats which anneal, resulting in a deletion and retention of one copy of the repeat. Our assays indicate that the mycobacterial SSA pathway requires recBCD.
In our studies, when NHEJ is disabled in the Δku strain, but not in the ΔligD strain, HR frequency is increased, indicating that Ku suppresses HR. We envision that the end binding activity of Ku shields DSB ends from the HR initiating nucleases (AdnAB and/or others) and when Ku is absent, HR increases. In contrast, when LigD is lost, Ku is still present to block end resection, explaining the lack of HR upregulation when LigD is absent. The dynamic relationship between NHEJ and HR is unidirectional, insofar as inactivation of HR is not accompanied by increased NHEJ in the ΔrecA strain. This result suggests that mycobacteria restrain mutagenic repair by NHEJ that could be deleterious to cell viability. A similar asymmetric relationship exists between SSA and HR, whereby ablation of the SSA pathway (through inactivation of RecBCD) causes HR upregulation, but not vice versa, again suggesting that mycobacterial cells limit a mutagenic pathway while allowing upregulation of a faithful repair pathway. The role of DSB repair pathways in microbial pathogenesis is still being elucidated. Although a recA deletion mutant of Salmonella typhimurium was highly attenuated in mice (Buchmeier et al., 1993), an M. bovis BCG strain lacking recA was not attenuated in wild type or SCID mice (Sander et al., 2001). AddAB is required for H. pylori stomach colonization, indicating a role for HR in this infection (Amundsen et al., 2008). Our findings that mycobacteria express three independent DSB repair pathways raise the prospect of pathway overlap in vivo for defense against host-inflicted DNA damage. The genetic requirements for each pathway defined in this study will facilitate testing of M. tuberculosis strains deficient in HR, SSA, and NHEJ alone or in combination in animal models of infection.
The molecular outcomes of NHEJ-mediated repair involve frequent use of single-strand resection and cross-break priming to initiate fill-in of recessed 5’ ends. The LigD polymerase module is especially adept at gap filling and cross-break priming when the downstream duplex has a 5’-monophosphate terminus (Zhu & Shuman, 2010, Brissett et al., 2007), as will be the case for the I-SceI-induced breaks. We envision that this LigD Pol activity accounts for the observed NHEJ outcomes at incompatible 3’ overhangs, in which the single strand 3’ end anneals to the single strand of the opposite side of the break and primes DNA synthesis in the consequent gap. Such a mechanism of cross-break priming has been observed in studies of NHEJ in yeast (Moore & Haber, 1996) and mammalian cells (Capp et al., 2007, Capp et al., 2006). Several eukaryal polymerases can catalyze cross-break priming in vitro and in vivo, including Pol mu and lambda (Garcia-Diaz et al., 2009, Ma et al., 2004, Nick McElhinny et al., 2005). The molecular outcomes of mycobacterial NHEJ of incompatible 3’ overhangs observed here highlight a remarkable conservation of repair mechanism from bacteria to human cells (Lieber, 2010).
It is noteworthy that we observe survival of ~0.04% in our chromosomal DSB repair assays, which is comparable to the net survival seen with other homing endonuclease-induced DSB repair systems with compatible ends (Moore & Haber, 1996). We envision that multiple factors contribute to low survival in our assays. First, the DSB ends generated are incompatible 3’ overhangs and are unlikely to be sealed directly by the NHEJ machinery without prior modification. In our previous studies, the efficiency of NHEJ repair of linear plasmid substrates with cohesive or blunt ends was ~1% (Aniukwu et al., 2008). The incompatible DSB ends in the current system would decrease the efficiency of repair by NHEJ, accounting in part for the decreased survival. An additional factor likely affecting the survival in the reporter substrate with homology is the limited window of resection length that can initiate homology-directed repair. End-resection upstream to the break (at least 35 nt, to reveal the homologous sequence) is a prerequisite for repair by HR, but the deletion length cannot exceed 598 bp (in the leftward direction; Fig. 3), beyond which homology would not be available for recombination. Essential genes are located in the chromosomal DNA flanking the attB integration site of the recombination constructs (17.8 kb upstream and 1.6 kb downstream). Any resections that proceed beyond these limits will delete essential genes, in which case, the cells would not survive, even after successful DSB repair by NHEJ.
Pathway specific functions of RecBCD and AdnAB in DSB repair
A novel finding from our studies is the existence of an SSA pathway in M. smegmatis. SSA is a well-described mechanism of repair in yeast and mammalian cells when a DSB arises between direct repeats. Single-strand resection, annealing of the repeats, flap removal, gap filling, and ligation are the biochemical activities required for SSA. In yeast, SSA is Rad51-independent, but Rad52 dependent (Ivanov et al., 1996). The Rad1/Rad10 nuclease is required for yeast SSA in vivo (Ivanov & Haber, 1995) and is thought to supply the flap removal activity when heterologous DNA is present, insofar as the purified Rad1/Rad10 complex can cleave DNA at single-strand/double-strand junctions (Davies et al., 1995, Bardwell et al., 1994). Single-strand resection to expose the complementary repeats in yeast SSA is performed by the combined action of Exo1, the MRX complex, and the SgsI helicase in conjunction with Dna2 (Zhu et al., 2008, Mimitou & Symington, 2008). Rather than being dedicated to SSA, this end resection machinery is shared with the yeast HR pathway. In contrast, we find that mycobacterial RecBCD is a dedicated SSA nuclease, a surprising finding that contrasts sharply with its well-established role in HR in E. coli (Figure 7). We envision that mycobacterial RecBCD supplies the single strand resection activity required to expose the complementary repeats during SSA. Alternatively, RecBCD could also supply the nuclease activity that removes the flaps that are present after repeat annealing. Our findings expand the known functions of RecBCD in bacterial DNA repair beyond those deduced from studies in E. coli.
Mycobacteria and other Actinomycetales taxa encode the heterodimeric helicase-nuclease AdnAB. The biochemical properties of AdnAB suggested that it might be involved in resecting DSBs in vivo. Here we show that AdnAB is involved in HR, as demonstrated by the diminished gene conversion frequency in cells lacking AdnAB. Our inference that AdnAB participates in the recA dependent HR pathway is further supported by the finding that loss of adnAB causes severe sensitivity to IR and hydrogen peroxide, mimicking that of the ΔrecA strain, implying that an AdnAB/RecA pathway is the major route to rectify IR and peroxide-induced DNA damage. These results indicate the AdnAB in mycobacteria is functionally analogous to the AddAB nuclease of B. subtilis with regard to its role in HR (Chedin & Kowalczykowski, 2002, Haijema et al., 1996). However, our finding that the mycobacterial SSA pathway requires RecBCD and that AdnAB is dedicated to the HR pathway demonstrates that mycobacteria have evolved a unique solution to the end resection problem. A major question that arises out of our findings is how that choice of pathway (SSA via RecBCD or HR via AdnAB) is made. One possibility is that mycobacterial RecBCD is unable to load RecA onto single stranded DNA whereas AdnAB is able to perform this function. Although RecBCD and AdnAB could both generate the 3’ single strand ends that could form a RecA nucleoprotein filament, if mycobacterial RecBCD cannot load RecA, these resections will be directed into the SSA pathway. An explicit prediction of the data presented here is that either AdnAB or a collaborating HR protein is able to direct RecA onto single stranded DNA, thereby funneling AdnAB initiated resections into the HR pathway. An additional question raised by these studies is whether RecBCD, AdnAB, or both are regulated by Chi sites analogous to the short DNA sequences that regulate the function of E. coli RecBCD and B. subtilis AddAB (Chedin et al., 2006, Dillingham & Kowalczykowski, 2008). Currently, the putative Chi-recognition sequences for the two nucleases are unknown in mycobacteria and future investigation into these regulatory sequences in mycobacteria will provide insight into the differential regulation of these two nucleases.
The data presented here indicate that mycobacteria elaborate a unique combination of DSB repair systems compared to other bacteria previously characterized (Figure 7). In particular, the use of dedicated helicase-nucleases to distinct DSB repair pathways adds considerable new complexity to the physiology of end resection and DNA unwinding in bacteria, and expands the participation of RecBCD beyond its previously characterized role in recA dependent homologous recombination.
Experimental Procedures
Cloning of I-SceI and reporter strain construction
The cloning of I-SceI cDNA with an N-terminal hemagglutinin (HA) tag has been described previously (Stephanou et al., 2007). To clone I-SceI gene in a mycobacterial shuttle vector, a 954 bp SphI-EcoRV fragment containing HA-I-SceI fusion was mobilized into pMSG383 to create pRGM1. In pRGM1, the expression of HA-I-SceI is driven from the MOP Promoter (George et al., 1995). To generate lacZ(I-SceI), the MOP-lacZ cassette was digested with DraIII and SacI to delete 745 bp from the coding sequence of lacZ and a 219 bp fragment containing two oppositely oriented I-SceI sites separated by a 37 bp spacer was cloned at these sites. The mycobacteriophage L5 integrase gene (1244 bp) (Lee & Hatfull, 1993) was then deleted from the plasmid after a partial BamHI digestion and recircularization, resulting in pRGM9, which has the phage L5 attachment site attP for chromosomal integration, lacZ(I-SceI), and a kanamycin selectable marker. The reporter substrate with a ‘nearby’ homology donor, pRGM10, was constructed by cloning a 2.8 kb ZraI-SmaI fragment containing lacZ-ΔN (663-3102), downstream of the kanamycin gene, at a unique DraI site in pRGM9. The reporter substrates were then integrated at the bacterial attachment site, attB, of the wild-type M. smegmatis chromosome, generating the strains Mgm1601 and Mgm1602, by co-transforming pRGM9 and pRGM10 individually with a second plasmid, pDB19, that expresses the L5 integrase and bears Zeocin-resistance cassette, and selecting for kanamycin-resistance transformants. Likewise, the reporter substrates were integrated in all other strain backgrounds.
To place lacZΔN (~1.2 Mb) from the DSB site, lacZ-ΔN (663-3102) was inserted at the intergenic region of MSMEG_5848 and MSMEG_5849 in wild-type M. smegmatis by homologous recombination using the suicide plasmid pRGM5 (which contains 720 bp 3’ coding sequence of MSMEG_5848 and lacZ-ΔN), and selecting for hygromycin resistance. Colonies obtained were confirmed by sequencing of the genomic PCR of lacZ-ΔN (663-3102), and subsequently, the loxP-hyg-loxP was removed by Cre recombinase on an episomal plasmid with an unstable origin of replication, pMSG381. The strain was then cured of the kanamycin-resistant pMSG381 plasmid, and pRGM9 was integrated at the attB site of the resulting unmarked strain (as summarized above), creating the strain Mgm1603.
The M. smegmatis strain genotypes used in the study are listed in Supplementary Table 1. The adnAB operon (encoded by MSMEG_1941, MSMEG_1943; http://www.tigr.org) in M. smegmatis wild-type and the ΔrecBCD strain was disrupted by specialized transduction using a temperature-sensitive mycobacteriophage, as described (Glickman et al., 2000). The hygromycin marker (flanked by loxP sites) at the disrupted locus was subsequently excised by expressing Cre recombinase from pMSG381 as outlined above, to generate the unmarked strains Mgm1964 and Mgm1965.
I-SceI-mediated DSB repair assay
The vector or I-SceI plasmid was introduced by electroporation into 400 μl of competent M. smegmatis cells containing the reporter substrate with or without homology. The competent cells were prepared as described (Snapper et al., 1990), and electroporations were done in triplicates using 100 ng of vector and 125 ng of the I-SceI plasmid. In separate experiments, to score more survivors, the cells were transformed with higher amounts (250-500 ng) of the I-SceI plasmid. After electroporation, the cells were immediately transferred to 1 ml of LB media containing 0.1% tween-80 and recovered for 2.5 hours at 37°C, prior to plating on LB agar plates supplemented with 0.5% glycerol, 0.5% dextrose, 100 μgml-1 hygromycin and 50 μg ml-1 X-gal. In case of vector transformations, three 10-fold serial dilutions of the electroporation mixtures were plated to obtain countable cells, before plating the remaining cells (= 1.39 ml electroporation mixture) on a single plate. Colony counts and colony color (blue/white) were assessed after incubation for 3-5 days at 37°C. % survival for each reporter strain was calculated as the percentage of the number of transformants obtained with the I-SceI plasmid versus the transformants obtained with the same molar amount of the vector plasmid. % survival values from three independent experiments using different batches of competent cells for the same strain were determined and the average value is expressed in the data. To ascertain GC or SSA outcomes, representative blue colonies were checked for kanamycin-resistance by re-streaking on LB agar plates supplemented with 20 μg/ml kanamycin or 100 μg ml-1 hygromycin, and 50 μg/ml X-gal, and also confirmed by PCR amplification using diagnostic PCR primers as pictured in Figure S2.
Sequencing of repair junctions
Pairs of PCR primers flanking the two I-SceI sites in the reporter substrates were used to PCR-amplify DNA from individual hygromycin-resistant white transformants. Amplifications were performed with primers which anneal at sites located 341 bp upstream and 511 bp downstream or 836 bp upstream and 511 bp downstream from the first I-SceI site. In case these primers failed to amplify the repair junction (because NHEJ entailed deletion beyond one or both of the primer sites), a third combination of primers was used, located 836 bp upstream and 1322 bp downstream from the first I-SceI site. The PCR products were resolved by agarose gel electrophoresis, gel-purified, and sequenced using the primers used for amplification or in case of large amplicons internal primers that anneal on either side of the break were used.
Analysis of repair outcomes and statistical analysis
We utilized two different methods to analyze the repair outcomes after I-SceI induced chromosomal breaks. The first of these methods determines “relative repair fraction” by calculating the fraction of surviving cells that have undergone each of the four scored repair outcomes (NHEJ, GC, SSA, and I-SceI inactivation). To arrive at this fraction, the number of events scored for each repair outcome is divided by the total number of events scored to yield the relative fraction of that repair outcome, which is then corrected for the relative distribution of white and blue colonies. For example, for wild type outcomes in figure 3D, 49.6% of survivors are blue. Of 70 white colonies genotyped, 16 were NHEJ and 54 were I-SceI inactive. Thus, the relative fraction of NHEJ is (16/70)(50.4% white)=11.52%. The pie charts presented in Figures 1, 3, and 4 depict these fractions and the statistical test to compare the number of events in each pathway is the fisher’s exact test. The second method calculates “absolute repair frequency” for each outcome by correcting the repair distribution by the percent survival. To calculate the absolute GC frequency, the following formula was used: (%survival)×(GC events/blue events)×(fraction blue). To calculate the SSA frequency, the following formula was used: (%survival)×(SSA events/blue events)×(fraction blue). To calculate the NHEJ frequency, the following formula was used: (%survival)×(NHEJ events/white events)×(fraction white). For each genetic background, each experiment was performed in triplicate. To calculate the statistical significance of differences in absolute repair frequency between strains, we used the Student’s t-test to compare the mean repair frequencies of three replicate experiments for each strain. The calculated repair frequencies for each strain and associated p values are reported in Table S3 and are highlighted in the text.
Clastogen experiments
For all of the experiments, M. smegmatis strains were grown at 37°C in LB medium supplemented with 0.5% glycerol, 0.5% dextrose, and 0.1% Tween 80.
UV irradiation assays were performed as described (Stephanou et al., 2007). Briefly, M. smegmatis strains to be tested were grown to log-phase (A600 of 0.3 to 0.4), and 10-fold serial dilutions prepared in phosphate-buffered saline (PBS) with 0.05% Tween 80 were spotted on LB agar plates supplemented with 0.5% glycerol and 0.5% dextrose. UV exposure of the indicated doses was performed in a Stratalinker (Stratagene) fitted with 254-nm bulbs. Immediately after exposure, the plates were wrapped in foil to prevent repair by photolyase. Surviving colonies were counted after 3 days using a dissecting microscope, and % survival was calculated in comparison to the same dilution of unexposed cells from the same culture.
MMS killing assays were performed by treating M. smegmatis strains grown to log-phase (A600 of 0.3 to 0.4) with 0.5% MMS, after resuspension in PBS with 0.05% Tween 80, and doing a time-course experiment at room temperature as described previously (Sriskanda et al., 1999). After treatment with MMS, 150 μl aliquots were withdrawn at the times specified and MMS was immediately quenched by adding 150 μl of ice-cold 10% sodium thiosulphate solution. Serial 10-fold dilutions of the treated and untreated cells (from the same culture) were then spotted on LB agar plates, and after 3 days, % survival was calculated compared to untreated control cells.
IR killing assays were performed using a 137Cs source that delivers a dose rate of 11.2 Gy per minute. Log-phase (A600 of 0.3 to 0.4) cultures were collected by centrifugation and resuspended in PBS with 0.05% Tween 80. 200 μl aliquots of the resuspended cells were irradiated using a rotating platform to ensure equal exposure to each sample. Following IR exposure, serial 10-fold dilutions were plated, and surviving colonies were counted after 3 days. % survival was calculated after normalization to the colony counts obtained from the unexposed control cells.
Hydrogen peroxide killing assays were performed by treating log-phase (A600 of 0.4 to 0.5) cultures for 1 h with increasing concentrations of hydrogen peroxide (0-30 mM). Alternatively, time-course experiments were also carried out by treating the cultures with 30 mM hydrogen peroxide for different time intervals. Following treatment, serial 10-fold dilutions were plated, and % survival was calculated by determining the CFUs of the treated versus untreated cultures.
MitomycinC time-course assays
Log-phase (A600 of 0.3 to 0.4) cultures of wild-type or mutant M. smegmatis strains were treated with 25 ng ml-1 mitomycin C, and incubated at 37°C with constant shaking (150 rpm). 200 μl aliquots of the cultures were withdrawn after regular intervals for the next 18 h, and 10-fold serial dilutions were plated on LB agar plates. Survival was calculated by determining the ratio of colony forming units (CFUs) of the treated versus untreated cultures of a given strain.
Supplementary Material
Acknowledgments
This work was supported by NIH grant AI064693 to MSG. DB was partially supported by the Michael and Ethel L. Cohen Foundation. SS is an American Cancer Society Research Professor.
References
- Amundsen SK, Fero J, Hansen LM, Cromie GA, Solnick JV, Smith GR, Salama NR. Helicobacter pylori AddAB helicase-nuclease and RecA promote recombination-related DNA repair and survival during stomach colonization. Mol Microbiol. 2008;69:994–1007. doi: 10.1111/j.1365-2958.2008.06336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aniukwu J, Glickman MS, Shuman S. The pathways and outcomes of mycobacterial NHEJ depend on the structure of the broken DNA ends. Genes Dev. 2008;22:512–527. doi: 10.1101/gad.1631908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell AJ, Bardwell L, Tomkinson AE, Friedberg EC. Specific cleavage of model recombination and repair intermediates by the yeast Rad1-Rad10 DNA endonuclease. Science. 1994;265:2082–2085. doi: 10.1126/science.8091230. [DOI] [PubMed] [Google Scholar]
- Brissett NC, Pitcher RS, Juarez R, Picher AJ, Green AJ, Dafforn TR, Fox GC, Blanco L, Doherty AJ. Structure of a NHEJ polymerase-mediated DNA synaptic complex. Science. 2007;318:456–459. doi: 10.1126/science.1145112. [DOI] [PubMed] [Google Scholar]
- Buchmeier NA, Lipps CJ, So MY, Heffron F. Recombination-deficient mutants of Salmonella typhimurium are avirulent and sensitive to the oxidative burst of macrophages. Mol Microbiol. 1993;7:933–936. doi: 10.1111/j.1365-2958.1993.tb01184.x. [DOI] [PubMed] [Google Scholar]
- Bzymek M, Lovett ST. Evidence for two mechanisms of palindrome-stimulated deletion in Escherichia coli: single-strand annealing and replication slipped mispairing. Genetics. 2001a;158:527–540. doi: 10.1093/genetics/158.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bzymek M, Lovett ST. Instability of repetitive DNA sequences: the role of replication in multiple mechanisms. Proc Natl Acad Sci U S A. 2001b;98:8319–8325. doi: 10.1073/pnas.111008398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capp JP, Boudsocq F, Bertrand P, Laroche-Clary A, Pourquier P, Lopez BS, Cazaux C, Hoffmann JS, Canitrot Y. The DNA polymerase lambda is required for the repair of non-compatible DNA double strand breaks by NHEJ in mammalian cells. Nucleic Acids Res. 2006;34:2998–3007. doi: 10.1093/nar/gkl380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capp JP, Boudsocq F, Besnard AG, Lopez BS, Cazaux C, Hoffmann JS, Canitrot Y. Involvement of DNA polymerase mu in the repair of a specific subset of DNA double-strand breaks in mammalian cells. Nucleic Acids Res. 2007;35:3551–3560. doi: 10.1093/nar/gkm243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chedin F, Handa N, Dillingham MS, Kowalczykowski SC. The AddAB helicase/nuclease forms a stable complex with its cognate chi sequence during translocation. J Biol Chem. 2006;281:18610–18617. doi: 10.1074/jbc.M600882200. [DOI] [PubMed] [Google Scholar]
- Chedin F, Kowalczykowski SC. A novel family of regulated helicases/nucleases from Gram-positive bacteria: insights into the initiation of DNA recombination. Mol Microbiol. 2002;43:823–834. doi: 10.1046/j.1365-2958.2002.02785.x. [DOI] [PubMed] [Google Scholar]
- Davies AA, Friedberg EC, Tomkinson AE, Wood RD, West SC. Role of the Rad1 and Rad10 proteins in nucleotide excision repair and recombination. J Biol Chem. 1995;270:24638–24641. doi: 10.1074/jbc.270.42.24638. [DOI] [PubMed] [Google Scholar]
- Davis BJ, Havener JM, Ramsden DA. End-bridging is required for pol mu to efficiently promote repair of noncomplementary ends by nonhomologous end joining. Nucleic Acids Res. 2008;36:3085–3094. doi: 10.1093/nar/gkn164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillingham MS, Kowalczykowski SC. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol Mol Biol Rev. 2008;72:642–671. doi: 10.1128/MMBR.00020-08. Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott B, Richardson C, Jasin M. Chromosomal translocation mechanisms at intronic alu elements in mammalian cells. Mol Cell. 2005;17:885–894. doi: 10.1016/j.molcel.2005.02.028. [DOI] [PubMed] [Google Scholar]
- Garcia-Diaz M, Bebenek K, Larrea AA, Havener JM, Perera L, Krahn JM, Pedersen LC, Ramsden DA, Kunkel TA. Template strand scrunching during DNA gap repair synthesis by human polymerase lambda. Nat Struct Mol Biol. 2009;16:967–972. doi: 10.1038/nsmb.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George KM, Yuan Y, Sherman DR, Barry CE., 3rd The biosynthesis of cyclopropanated mycolic acids in Mycobacterium tuberculosis. Identification and functional analysis of CMAS-2. J Biol Chem. 1995;270:27292–27298. doi: 10.1074/jbc.270.45.27292. [DOI] [PubMed] [Google Scholar]
- Glickman MS, Cox JS, Jacobs WR., Jr A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of Mycobacterium tuberculosis. Mol Cell. 2000;5:717–727. doi: 10.1016/s1097-2765(00)80250-6. [DOI] [PubMed] [Google Scholar]
- Gong C, Bongiorno P, Martins A, Stephanou NC, Zhu H, Shuman S, Glickman MS. Mechanism of nonhomologous end-joining in mycobacteria: a low-fidelity repair system driven by Ku, ligase D and ligase C. Nat Struct Mol Biol. 2005;12:304–312. doi: 10.1038/nsmb915. [DOI] [PubMed] [Google Scholar]
- Gong C, Martins A, Bongiorno P, Glickman M, Shuman S. Biochemical and genetic analysis of the four DNA ligases of mycobacteria. J Biol Chem. 2004;279:20594–20606. doi: 10.1074/jbc.M401841200. [DOI] [PubMed] [Google Scholar]
- Haijema BJ, Venema G, Kooistra J. The C terminus of the AddA subunit of the Bacillus subtilis ATP-dependent DNase is required for the ATP-dependent exonuclease activity but not for the helicase activity. J Bacteriol. 1996;178:5086–5091. doi: 10.1128/jb.178.17.5086-5091.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov EL, Haber JE. RAD1 and RAD10, but not other excision repair genes, are required for double-strand break-induced recombination in Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:2245–2251. doi: 10.1128/mcb.15.4.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov EL, Sugawara N, Fishman-Lobell J, Haber JE. Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics. 1996;142:693–704. doi: 10.1093/genetics/142.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korycka-Machala M, Brzostek A, Rozalska S, Rumijowska-Galewicz A, Dziedzic R, Bowater R, Dziadek J. Distinct DNA repair pathways involving RecA and nonhomologous end joining in Mycobacterium smegmatis. FEMS Microbiol Lett. 2006;258:83–91. doi: 10.1111/j.1574-6968.2006.00199.x. [DOI] [PubMed] [Google Scholar]
- Lee MH, Hatfull GF. Mycobacteriophage L5 integrase-mediated site-specific integration in vitro. J Bacteriol. 1993;175:6836–6841. doi: 10.1128/jb.175.21.6836-6841.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu Rev Biochem. 2010 doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Lu H, Tippin B, Goodman MF, Shimazaki N, Koiwai O, Hsieh CL, Schwarz K, Lieber MR. A biochemically defined system for mammalian nonhomologous DNA end joining. Mol Cell. 2004;16:701–713. doi: 10.1016/j.molcel.2004.11.017. [DOI] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–774. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JK, Haber JE. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164–2173. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny SA, Havener JM, Garcia-Diaz M, Juarez R, Bebenek K, Kee BL, Blanco L, Kunkel TA, Ramsden DA. A gradient of template dependence defines distinct biological roles for family X polymerases in nonhomologous end joining. Mol Cell. 2005;19:357–366. doi: 10.1016/j.molcel.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Pashley CA, Parish T. Efficient switching of mycobacteriophage L5-based integrating plasmids in Mycobacterium tuberculosis. FEMS Microbiol Lett. 2003;229:211–215. doi: 10.1016/S0378-1097(03)00823-1. [DOI] [PubMed] [Google Scholar]
- Pena CE, Lee MH, Pedulla ML, Hatfull GF. Characterization of the mycobacteriophage L5 attachment site, attP. J Mol Biol. 1997;266:76–92. doi: 10.1006/jmbi.1996.0774. [DOI] [PubMed] [Google Scholar]
- Pitcher RS, Green AJ, Brzostek A, Korycka-Machala M, Dziadek J, Doherty AJ. NHEJ protects mycobacteria in stationary phase against the harmful effects of desiccation. DNA Repair (Amst) 2007;6:1271–1276. doi: 10.1016/j.dnarep.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Sander P, Papavinasasundaram KG, Dick T, Stavropoulos E, Ellrott K, Springer B, Colston MJ, Bottger EC. Mycobacterium bovis BCG recA deletion mutant shows increased susceptibility to DNA-damaging agents but wild-type survival in a mouse infection model. Infect Immun. 2001;69:3562–3568. doi: 10.1128/IAI.69.6.3562-3568.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuman S, Glickman MS. Bacterial DNA repair by non-homologous end joining. Nat Rev Microbiol. 2007;5:852–861. doi: 10.1038/nrmicro1768. [DOI] [PubMed] [Google Scholar]
- Sinha KM, Unciuleac MC, Glickman MS, Shuman S. AdnAB: a new DSB-resecting motor-nuclease from mycobacteria. Genes Dev. 2009;23:1423–1437. doi: 10.1101/gad.1805709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapper SB, Melton RE, Mustafa S, Kieser T, Jacobs WR., Jr Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol Microbiol. 1990;4:1911–1919. doi: 10.1111/j.1365-2958.1990.tb02040.x. [DOI] [PubMed] [Google Scholar]
- Sriskanda V, Schwer B, Ho CK, Shuman S. Mutational analysis of Escherichia coli DNA ligase identifies amino acids required for nick-ligation in vitro and for in vivo complementation of the growth of yeast cells deleted for CDC9 and LIG4. Nucleic Acids Res. 1999;27:3953–3963. doi: 10.1093/nar/27.20.3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephanou NC, Gao F, Bongiorno P, Ehrt S, Schnappinger D, Shuman S, Glickman MS. Mycobacterial nonhomologous end joining mediates mutagenic repair of chromosomal double-strand DNA breaks. J Bacteriol. 2007;189:5237–5246. doi: 10.1128/JB.00332-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Shuman S. Gap filling activities of Pseudomonas DNA ligase D (LigD) polymerase and functional interactions of LigD with the DNA end-binding Ku protein. J Biol Chem. 2010;285:4815–4825. doi: 10.1074/jbc.M109.073874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.