

Introduction
Fluorescence correlation spectroscopy is a single-molecule fluorescence technique used to monitor molecular dynamics. When applied to living cells, FCS has been used to decipher the diffusion times of protein and lipid molecules, to determine the concentrations of a particular molecule at a specific cellular location, to determine kinetic parameters such as on/off rates constants and binding coefficients for protein/protein interactions [1].
The basic principle for FCS is that a fluorescent molecule emits photons while moving through a confocal volume illuminated by a laser (Figure 1). The number of photons that can be collected from the confocal volume depends on the diffusion time of the fluorescent molecules (which is a function of the mass of the molecule), the concentration of molecules, the quantum yield of the fluorophore attached to the molecule and finally the size of the illuminated volume, which is dependent on the instrument [2]. Typically a small diffraction limited confocal volume of 0.3μm × 1μm (∼size of an E. coli) is excited with a laser beam and then the emitted photons are collected and counted on a photodiode detector. These measurements are then used to calculate and display autocorrelation and cross correlation functions, coincidence diagrams, photon count histograms, count rate diagrams and pulse density histograms from which diffusion times, on/off rates etc can be extracted using data fitting and modeling software [3].
Figure 1.
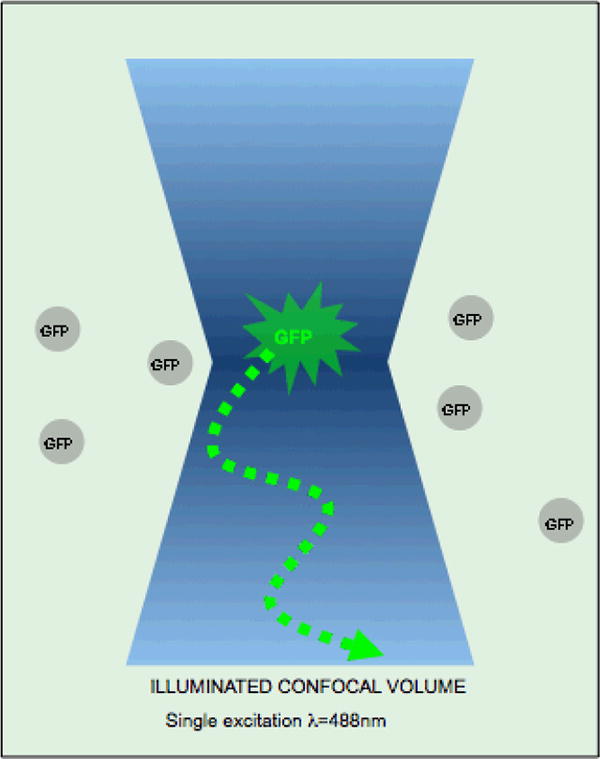
sketch of the optical setting for an FCS measurement.
In this protocol, we present the basic design of an FCS measurement in living cells, and address critical issues in experiment design (from the choice of dyes or cells to data analysis). The user must be aware that FCS requires subsequent adjustments in the experimental design as well as the data analysis to address successfully specific biological questions. Comments after the basic protocol will present classic pitfalls and troubleshooting tips to help this.
Planning an experiment
Equipment and Materials
-
LSM510-Confocor 3: a commercial system from Carl Zeiss Microimaging, which combines an LSM 510 META Laser Scanning Microscope with an FCS module to acquire high-resolution images and sensitive fluctuation analysis. The ConfoCor 3 detection allows one to quantitatively analyze the concentration, position, interaction and mobility of molecules. The new experimental capabilities provided by the ConfoCor 3 take effect in the investigation of molecule distribution in cells, diffusion processes by fluorescence correlation spectroscopy, and protein complex formations, in the detection of common movements and the quantification of receptor ligand interactions.
The software controls the detection module and allows convenient analysis of single measurements or software-controlled multi-measurements. Autocorrelation are calculated in real time, i.e. at the same time as the current measurement. And there is always the choice of using the raw data for individual analysis.
Other options include advanced software modules that incorporate the most common methods (free diffusion, anomalous diffusion and flow), provide for user-defined models, allow for global and interactive fitting with the possibility of defining start values and boundaries, and make photon counting histograms possible.
Nunc Lab-Tek® [4], Mat-tek® chambers [5] are deep-welled chambers with cover-glass bottoms onto which cells are plated. Many cells stick directly on to glass but the surfaces can be coated with poly-lysine to facilitate sticking. The cover-glass bottoms of the chambers allow high-magnification, high-resolution microscopy with water immersion objectives. Cells should be plated sufficiently in advance such that they have adhered and acclimate. For FCS experiments the cell media should be one without phenol red dye to limit background fluorescence noise. Additionally if carbon dioxide levels on the microscope stage cannot be controlled, supplement the media with HEPES buffer pH 7.2 to 25-50mM final concentration.
Objectives: High numerical aperture (N.A.) water-immersion objectives are critical components of a FCS system. Indeed, the signal/noise ratio for a FCS measurement is proportional to the square of the collected fluorescence per fluorescent object, hence the need to maximize N.A [2]. Living cells are imaged in water-based media therefore it is best to use water immersion objectives whose limited spherical aberrations insure a more accurate definition of the confocal volume from which FCS measurements are made. Typically we use a 40X/1.2 NA water immersion objective with collar adjustment for the thickness of the coverslip: it minimizes aberrations in the z-axis and provides a tight diffraction limited confocal volume of illumination inside the primarily aqueous cytoplasm of a living cell.
Fluorescent dyes for calibration of FCS instrument: fluorescent dyes (e.g. Oregon Green or FITC), fluorescently-labeled dextran or Fluospheres (Invitrogen Corp.) can all be used for calibrating the instrument prior to acquisition from cells. We typically use a single concentration of Oregon Green (typically at 100nM dilution) in PBS, placed in an 8-well LabTek chamber with #1 coverslip bottom. Calibration should be a daily routine for every experiment in order to align the detectors and maximize the number of collected photons.
Choice of labeling for molecule of study: GFP, YFP, CFP, mRFP have been used for FCS measurements. XFP-tagged proteins can be exogenously expressed in cells by transfecting a plasmid encoding the chimera at least 8 hours prior to FCS. Alternatively the fluorophores Oregon green, FITC, Rhodamine 6G etc. can be covalently linked to a protein or lipid molecule of choice and microinjected in to the cells.
Choosing Cell Types: any cell type (flat cells like fibroblasts or rounded cells like lymphocytes) can be used for FCS measurements as long as cells are adherent or can be tethered on the glass coverslip to minimize movement during fluorescence acquisition (in the latter case, proper control experiments must be performed to validate that the tethering does not perturb biological functions).
Transfection: any nucleic acid transfection agent can be used. We typically use Fugene (Roche Diagnostics Corp.) or Lipofectamine (Invitrogen Corp.). The protocols for transfection can be obtained from the manufacturers web sites.
Protein expression levels. Level of expression is an important parameter because too high levels of expression forces the experimenter to decrease laser excitation to avoid detector saturation; this decreased laser excitation in turn would substantially decrease the emission per fluorescent object and jeopardize the FCS measurements. One solution is to photobleach a large fraction of the fluorescence from the cell before making FCS measurements. In the Confocor 3, Acquisition menu, one can set the FCS acquisition to bleach for a short amount of time at a high laser intensity setting a bleach settings before switching to a lower laser power for the actual FCS acquisition.
Basic protocol 1: setting up the LSM510-FCS for living cell measurements
Below will be the steps outlined for the Zeiss LSM Confocor 3 FCS system, one of the more popular turnkey FCS systems in the market. For others, the step-by step instructions may vary.
-
1
One hour prior to FCS measurements, change the media of the cells into a medium that is optimal for keeping cells on the microscope stage. This media is typically the tissue culture media the cells are normally maintained in (containing serum), buffered with 20-50 mM HEPES pH7.3 but without phenol red. See Note 1.
-
2
Fill the Nunc Lab-Tek chambers as high as possible with the media to ensure a large volume above the cells that will not evaporate over the course of measurement time. Moreover to minimize evaporation, the inside of the chamber lid should be smeared with a coat of Vaseline (Cheeseborough Inc.) and the lid should then be fitted on the chamber. This can still allow the user to open the lid for adding various agents to the cell medium but will limit the evaporation, which can cause changes in the salt concentrations and pH of the media. Alternatively, if the cells will be left alone for long periods of time, the outside edges of the lid can be glued to the chamber.
-
3
Turn on machine according to the manufacturer's instructions. Turn on the stage temperature control and set it to the desired temperature. Wait at least 30 minutes before FCS acquisition. See Note 2.
-
3
Turn on all needed lasers. Typical Argon laser should be run at 50% of maximum output. Wait at least 30min before starting measurements to allow stabilization and to minimize fluctuations in laser output.
-
4
Go to Confocor mode, and setup the optical path: this includes choosing the laser line for excitation, choosing the laser line intensity, selecting the dichroic mirrors that will allow both the 488nm excitation and emission greater than 500nm, the emission band pass filters and the detector. For example, for Oregon green or GFP, excitation is 488nm at ∼1% intensity, a 488 dichroic mirror, and a BP filter of 500-550nm.
-
5
First place the 8-well Nunc-Lab Tek chamber containing 100nM Oregon green (or another fluorescently-labeled molecule) in PBS on the stage. Go to the Sample Carrier menu and select the correct sample carrier i.e. chamber type, (in this case a Nunc 8-well chamber). Select the exact chamber position from which the Oregon green dye acquisition will be made. Focus 200 microns above the glass coverslip into solution. One easy way to determine if you are above the glass coverslip is to run a line scan from the confocal imaging mode. Start out the line scan with the pinhole completely open and the objective far away from the bottom of the glass coverslip. As you are running the line scans move the focus wheel of the microscope such that the objective will move closer to the glass coverslip. While you are moving up the objective pay attention to the line scan window which will output a line with a hill which is the reflected excitation light not yet focused on a glass coverslip surface. As the objective hits the bottom glass coverslip surface the line will increase in amplitude, then decrease, and finally hit a maximum again when the objective reaches the top surface of the glass coverslip. At this point one can use the objective control stepper (present on most confocal systems) to go 200 microns above the top glass coverslip.
-
6
Set pinhole, from the Confocor 3 Adjust menu item, to 70μm and run pinhole alignment. The pinhole is adjusted using the Oregon green dye solution. During pinhole alignment the pinhole is moved along the x and y axis until the maximum intensity of emission is found.
-
7
Once the pinhole is aligned, take a count rate of the solution. This is the number of photons emitted by individual molecules per second. At a concentration of 100nM Oregon Green, the minimal count per molecule should be at least ∼ 5000 counts/second. If lower than this, realign pinholes either through the software for a minimal alignment or manually for more significant alignment issues (this procedure usually requires service by a professional contractor).
-
8
Switch to the chamber with cells once the system is aligned and optimized with the Oregon green sample. Perform a single XY scan of the cells (in confocal imaging mode) (Figure 2A). When appropriate cells have been identified for imaging, quick scans should be acquired while adjusting the focus, the pinhole, laser intensity, detector gain and amplifier gain, to determine the optimal image. The quality of this image will effect where you spot the beam for collecting FCS data. See Note 3
-
9
When the object is to collect correlation spectra from the plasma membrane or any other membrane, it is best to take a z-stack of 1μm and determine the z-position where the fluorescence intensity is at its peak. It is best to limit the laser excitation intensity and keep the number of z slices to a minimum to minimize bleaching the sample.
-
10
Save the image in a data base for your records and keep a copy of it on the screen.
-
11
Switch back to the FCS (or Confocor) mode.
-
12
Go to the Methods menu item. Select from either of the preexisting methods or set up a new method. To set up a new method, go to acquisition setup under measure menu, and then change the laser excitation, dichroic and band pass filter settings to ones appropriate for the fluorophore inside the cells. For a GFP expressing cells, these settings will be similar to those used to obtain count rates for Oregon Green dye. Store these settings under a new method name. Laser excitation intensity should be kept low such that the triplet fraction is <10% of the molecules. The triplet state of the molecule is when it is in an excited but dark state: where it has given off only a portion of the absorbed energy as photons. To determine the triplet fraction at particular laser intensity, one will need to pay attention to the Triplet fraction column of the data table being generated during the FCS acquisition.
-
13
Go to the Measure menu option. Switch carrier from 8-well Nunc to select LSM image. Then mark positions in the cell where you want to collect data from (Figure 2A). Pick several positions within the cell and one position outside the cell for background measurement. Do not forget to hit the Mark positions button for this will number each position.
-
14
Go to Acquisition under the Measure menu, where you can set the acquisition time from each site in the cell. Typically this time should be about 10-15 seconds or 1000 fold the diffusion time of the molecule being investigated.
-
15
Set the Repeat count. This is the number of readings taken from each site. For good data fitting you want to take at least 10 measurements from each site.
-
16
Start acquisition. During acquisition fluorescence read out will be taken from each site for a pre-defined amount of time (∼10 sec). A typical read out for GFP expressed in the cytoplasm is presented in Figure 2 including the Correlation function G(t) versus lagtime (B) and the count rate over time (C). See Note 4
Figure 2. Autocorrelation on cytosolic GFP.
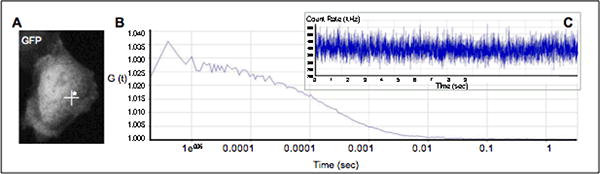
Autocorrelation function is collected from in a confocal volume chosen in the cell cytoplasm where soluble GFP is being expressed (A). Note how the baseline for the autocorrelation function is flat (between 100ms and 1s) yielding a good fit and diffusion estimate (B). The count rate is stable indicating the necessary lack of photobleaching (Inset C).
Basic protocol 2: analyzing FCS data
The most difficult task in a successful FCS measurement is the fitting and interpretation of the collected data. Although these are common tasks of any experimental measurement, there are few issues that are specific for FCS and that need to be addressed in this section.
As we pointed out in the introduction, FCS monitors any fluctuation of fluorescence within the confocal volume. Typically, any variation of fluorescence on time scales from 1μs to 1s will generate a relaxation in the autocorrelation curve that must be fitted and interpreted. To successfully process the data, the experimenter must derive an analytical formula accounting for all fluctuations. For FCS in living cells, these analytical fits may be hard to derive: that leaves the experimenter with hard-to-interpret phenomenological time scales that are still sufficient for an analytical assay in perturbation experiment (comparison of wild-type and drug-treated or gene-perturbed cells).
Simple 2D diffusion model for single specie
| (1) |
where N is the average number of fluorescent objects within the confocal volume, τD the correlation time τD=ω2/4/D where D is the diffusion coefficient of the fluorescent object and ω is the radial diameter of the confocal volume. The LSR510-Confocor software offers to use a theoretically more accurate diffusion fit including the longitudinal diffusion. But, a 3D formula does not improve the statistical accuracy of the fit, and requires careful calibration of the longitudinal dimension of the confocal volume to be valuable. A typical curve for soluble GFP diffusing with a cell cytoplasm is presented in Figure 2
Simple 2D diffusion model for multi-specie
| (2) |
where Ni is the average number of fluorescent objects labeled i within the confocal volume, τ0=w2/4/Di where Di is the diffusion coefficient of the fluorescent object labeled i and w is the radial diameter of the confocal volume. Note that the more difference there is between the diffusion of the fluorescent objects, the better the fit of the data.
Abnormal diffusion
In many living cell settings, the diffusion of fluorescence objects encompasses multi-scale, multi-timescale moves. An explicit description of such a convoluted process is formally impossible, and experimenters routinely use phenomenological “stretched diffusion” formula to fit the FCS data. An example of such a fit was used in [6]:
| (3) |
where N is the average number of fluorescent objects within the confocal volume, τD is a characteristic diffusion timescale, and α is the stretching parameter (with 0< α <1). The smaller α the more convoluted the diffusion is.
Relaxation model with 2D diffusion
When the fluorescent object under study fluctuates between quenched and fluorescent states, FCS can monitor the chemical relaxation of this process [7, 8]:
| (4) |
where N is the average number of fluorescent objects within the confocal volume, τ0 is the characteristic diffusion timescale, and τR is the characteristic chemical-relaxation timescale. Note that for a simple on-off with characteristic chemical rates kon and koff,
In practice, the dynamic range for the FCS autocorrelation amplitude β is too small to yield an accurate estimate of K: a separate measurement is then required.
More generally, the analytical fits used to analyze FCS measurements can get complicated by the rich dynamics of a system (e.g. [9]). There are reviews documenting different applications with different fitting functions for FCS measurements in living cells [2, 10].
FCS Output parameters
Here, we present a typical FCS curve for diffusing fluorescently-tagged molecules within the cytoplasm of a living cell, and the associated fit to help FCS users understand what each parameter mean (see Figure 3). Typically, the fit is:
Figure 3. typical FCS curve and relevant parameters.
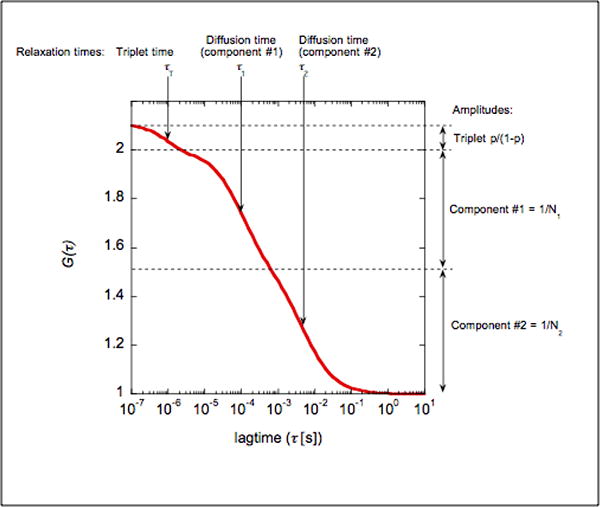
We present here the theoretical curve for the FCS measurement on a mixture of two species (component #1 and #2): Diffusion times are τ1=100μs and τ2=10ms, with N1=2 and N2=2 particles/confocal volume. The dye has a triplet time τT of 1μs and a triplet fraction at 10% (p=0.1). Note how the two diffusion times yield two relaxations that can hardly be separated.
| (5) |
Confocor fitting software generates a table with the following parameters:
Count rate [kHz] = total number of photons collected by a detector per second.
Correlation = amplitude of the correlation function G(0).
Counts per molecule [kHz] = is computed as (Count rate)*(G(0) - 1). This parameter can be very useful to estimate the state of aggregation of a fluorescent protein
Amplitude Number particles = Ni (note that when the triplet fraction p is negligible).
Triplet state Fraction [% ]: p*100.
Triplet state Relaxation time [μs] = τT.
Component i Fraction [%] =
Component i Diffusion time [μs]: τi,
Translation Structural parameter: this parameter should be fixed upon calibration of the optical setup (See Note 5). It corresponds to the aspect ratio of the confocal volume, defined as the ratio of its longitudinal direction with the transversal dimension. Typically this parameter should be set around 10.
Troubleshooting FCS measurements
1. Optimizing the signal-to-noise ratio of FCS measurements
Optimizing an FCS measurement implies optimizing the actual number of photons collected from individual fluorescent object. This parameter can be obtained from the countrate/molecule in the LSM510-Confocor software.
Typically, for GFP in living cells, with 1% of the 488nm lane of a 40mWatt Argon laser, and a collection pinhole at 70μM, one should obtain typically 15,000 photons per second (15 kHz / molecule). Once the experimental system (with a chosen fluorophore) has been configured, only three parameters [the optical alignment, the laser excitation, and the acquisition time] need to be optimized by the experimenter. LSM510-Confocor allows the experimenter to automatically align the acquisition pinhole (in x and y directions) based on maximization of collected fluorescence. The z-alignment is more difficult to optimize but factory settings are usually sufficient. The experimenter should then increase the laser intensity until fluorescence photobleaching becomes prevalent (this can be detected by seeing a non-flat baseline at long lagtimes, Figure 4). Finally, the experimenter should then increase the acquisition time to allow (the trade-off being photobleaching in the spot of interest).
Figure 4. Autocorrelation on plasma membrane associated GPI-GFP.
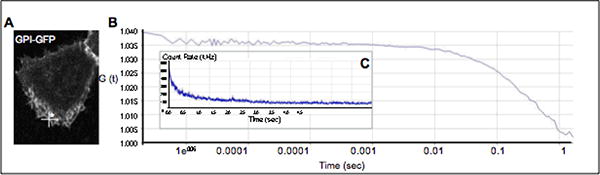
Autocorrelation function is collected from in a confocal volume chosen on the plasma membrane, where GFP is anchored via a GPI domain (A). Note how the baseline for the autocorrelation function is not flat (between 100ms and 1s) because of photobleaching of the plasma membrane-anchored GFP (B). Photobleaching is apparent as an exponential decay in the countrate (Inset C).
Note that the number of photons collected from individual fluorophores maybe limited by the saturation of the photoreceptors (theoretically 5 million counts per second, practically 1 million photons per second). Thus there are intrinsic limitations on the concentration of fluorescent objects per confocal volume that is compatible with sufficient photon collection for FCS. Typically, if one would like to collect at a count rate of 5000/s/molecule, there will be atmost 200 molecules in a confocal volume of typically 1fl, hence the maximum concentration of fluorescent objects in this experiment would be 0.4μMol. Hence is the need to select lower-expressing cells or pre-bleach the samples.
2. Photobleaching
Photobleaching of the fluorophore under consideration should not be a concern in FCS experiments when relaxation timescales below 50ms are under consideration (typical for proteins diffusing in the cytoplasm). However, for membrane proteins or for proteins bound to large biological objects (vesicles, mitochondria etc.), residency time within every confocal volume gets larger than 50ms, and one should pay special attention to the issue of photobleaching.
In typical FCS application in living cells, photobleaching appears as a large decay in the autocorrelation function with typical timescales between 100ms and 10s (Figure 4). To maintain the statistical relevance of data fitting, one must make sure that the autocorrelation baseline at long lag times is flat. The rule of thumb is then to obtain at least half a decade of flat baseline at 1.0 to be secured that photobleaching will not be an issue in the analysis of the autocorrelation function. A second rule to experimentally-control for photobleaching consists in repeating every FCS measurement with increased laser excitation: if photobleaching is irrelevant, the overall shape of the autocorrelation function should not be affected by the change in laser intensity.
3. Triplet formation and environmental variation of fluorescence
In some experiments, one would like to gain information about faster fluorescence fluctuations (with timescales below 100μs compared to typical diffusion events above 100us), in particular, to study GFP dynamics and gain information on pH environment in living cells, or to study small fluorophore diffusion. In that range of timescales, one must pay attention to potential artefacts due to the fluorophore internal dynamics or detector limitations.
Fluorophore internal dynamics in the 10μs -100μs range as measured by FCS can be informative of the biological environment (e.g. monitoring GFP quenching at low pH). On the other hand, triplet formation (a situation where the fluorophore goes from a singlet-excitable state to a triplet-quenched state) occurs with timescales between 1μs and 10μs (Figure 5). An easy solution to control for potential excitation-induced dynamics is to check the consistency of the FCS measurements for different laser intensities. Note that diffusion timescales will increase slightly as laser intensity is increased because of an increase of excitation volume.
Figure 5.
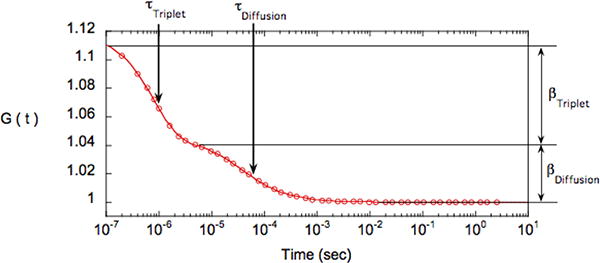
autocorrelation function (circle) and associated fit (continuous line) for a solution of FITC-labeled IgG. The fit corresponds to a single component 2D- diffusion with a triplet relaxation -equation (5). The two relaxations can easily be distinguished: the first one is associated with diffusion (τDiffusion=40μs) and the second one with triplet relaxation (τTriplet=0.8s). Their respective amplitude are βDiffusion=0.045 (hence there are 1/βDiffusion=22 fluorescent objects per confocal volume) and βTriplet=0.074 (hence pTriplet=0.62 as βTriplet= βDiffusion*(pTriplet/(1-pTriplet) or pTriplet=βTriplet/(βDiffusion+βTriplet)
Triplet formation is a problem encountered mostly with inorganic dyes (e.g. FITC) at high laser intensity. GFP and other naturally-fluorescent proteins have very low triplet conversion probability [11]. It is not recommended to use the LSM510-Confocor to decipher sub-microsecond fluctuation dynamics as detector after pulsing introduces a highly correlated signal in the autocorrelation curve in that time range [2].
4. Aligning confocal and FCS volumes
In the Zeiss Confocor 3 as well as the Leica FCS2, scanner mirrors of the confocal microscope are used for precise positioning, ensuring that the correlation spectra are recorded from the exact locations marked on the images scanned. However it is still good practice to check for any offset between the Scanners and the FCS stage positioning, especially if precision is needed <100nm in x and y. To determine the offset one can use a macro (available on the Zeiss or Leica instruments) or do it manually with an ‘ad hoc’ procedure to insure that the two confocal volumes from which fluorescence is collected are exactly aligned, such that confocal imaging and FCS measurements are in registry. Note that misregistry along the z axis is rather unlikely as this optical alignment does not vary much from the factory-settings: only x and y alignments need to be performed (before every FCS session).
Place a drop of FITC solution (1μMol in DMSO) onto a #1 coverslip.
Dry in the dark in ambient air for 30min.
Setup the LSM510-Confocor for confocal imaging and Confocor (see basic protocol 1).
Using LSM mode image the surface of the coverslip in the confocal setup. Leave the LSM image window and control screen on the computer screen.
Go back to the Confocor, Measurement menu, and mark positions on the recorded LSM image
Setup a long-time exposure (10min) for FCS measurement with maximal laser power (100% of 488nm line) to bleach the marked sites.
Re-Image the surface of the coverslip using LSM mode and measure, by drawing a line using the overlay option in the Confocor image display menu, the shift between the targeted FCS site (under the arrow) and the actual measurement site (bleach spot).
Adjust the x and y registries under the Settings in the LSM+Confocor menu item.
Repeat steps 4 to 6 to insure the accurate registry of confocal and FCS volumes
To perform offset compensation with a macro, follow directions in instrument manual.
Footnotes
The absence of phenol red pH-indicator (commonly used in tissue culture media) helps to minimize the levels of background fluorescence during FCS.
There are several options for temperature control of the microscope stage. A Nevtek Airstream incubator ASI 400 (Nevtek Corp., USA) in combination with a digital thermometer with a thermocouple (Omega Instruments, USA) whose temperature sensor is attached to the head of the condenser or to a site on the stage adjacent to the cell chamber is a convenient set-up for holding steady temp (+/- 0.5 degrees Celsius) in most applications requiring imaging up to 12 hours [12] [13]. It also allows easy access to the cell chamber for adding reagents into the cells in the course of the experiment while they are on the stage, without shaking the cell chamber or stage, both of which could result in the loss of the position of the cell being imaged. The drawback is that because of exposure to ambient CO2 and humidity and the usual degradation of the buffer, over time the media will gradually evaporate and become basic in pH. If long-term maintenance of cells is necessary then it is advisable to invest in a closed chamber set-up, one that even encloses the microscope stage and optics.
The XY scan pinhole should be adjusted to give you a high-resolution image, typically ∼1 Airy unit. To go below 1 Airy unit is usually not recommended since there is very little resolution gained to compensate for the significant loss in light collection from the sample.
Longer acquisitions from a single marked site will allow for more statistical information for long lagtimes (t>1s). This will benefit the curve fitting but it may also result in increased bleaching of the sample. Typical acquisition times are 10 seconds. In the acquisition menu one can also introduce a pre bleach time prior to FCS acquisition. Here the sample will be photobleached for a discrete amount of time before recording correlation. This is useful if the sample fluorescence is too high. Always perform measurements a second time with 5Xtimes higher power to check consistency with power amplitude.
To calibrate the size of the confocal volume, perform a FCS measurement on a solution of fluorescent dye (e.g. Oregon Green) at a known concentration c. Measure the Amplitude Number of particles (N) in the FCS measurement. Then, where Na=6.02×1023 is the Avogadro number.
In particular, this will enable you to calibrate the Translational Structure Parameter (TSM) as:
Contributor Information
Nihal Altan-Bonnet, Email: nabonnet@andromeda.rutgers.edu.
Grégoire Altan-Bonnet, Email: altanbonnet@cbio.mskcc.org.
References
- 1.Schwille P. Fluorescence correlation spectroscopy and its potential for intracellular applications. Cell Biochem Biophys. 2001;34(3):383–408. doi: 10.1385/CBB:34:3:383. [DOI] [PubMed] [Google Scholar]
- 2.Krichevsky O, Bonnet G. Fluorescence correlation spectroscopy: the technique and its applications. Report on Progress in Physics. 2002;65:251–297. [Google Scholar]
- 3.Bacia K, Schwille P. A dynamic view of cellular processes by in vivo fluorescence auto- and cross-correlation spectroscopy. Methods. 2003;29(1):74–85. doi: 10.1016/s1046-2023(02)00291-8. [DOI] [PubMed] [Google Scholar]
- 4.Nunc. Lab-tek chambers. 2008 [cited; Available from: http://www.nuncbrand.com/en/page.aspx?id=229&.
- 5.MatTek. 2008 [cited; Available from: http://www.glass-bottom-dishes.com/
- 6.Weiss M, Hashimoto H, Nilsson T. Anomalous protein diffusion in living cells as seen by fluorescence correlation spectroscopy. Biophys J. 2003;84(6):4043–52. doi: 10.1016/S0006-3495(03)75130-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonnet G, Krichevsky O, Libchaber A. Kinetics of conformational fluctuations in DNA hairpin-loops. Proc Natl Acad Sci U S A. 1998;95(15):8602–6. doi: 10.1073/pnas.95.15.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haupts U, Maiti S, Schwille P, Webb WW. Dynamics of fluorescence fluctuations in green fluorescent protein observed by fluorescence correlation spectroscopy. Proc Natl Acad Sci U S A. 1998;95(23):13573–8. doi: 10.1073/pnas.95.23.13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altan-Bonnet G, Libchaber A, Krichevsky O. Bubble dynamics in double-stranded DNA. Phys Rev Lett. 2003;90(13):138101. doi: 10.1103/PhysRevLett.90.138101. [DOI] [PubMed] [Google Scholar]
- 10.Carl Zeiss Microimaging. Confocor 3 Operating Manual Release 4.0. 2006. [Google Scholar]
- 11.Widengren J, Rigler R. Fluorescence correlation spectroscopy as a tool to investigate chemical reactions in solutions and on cell surfaces. Cell Mol Biol (Noisy-le-grand) 1998;44(5):857–79. [PubMed] [Google Scholar]
- 12.Altan-Bonnet N, Sougrat R, Liu W, Snapp EL, Ward T, Lippincott-Schwartz J. Golgi inheritance in mammalian cells is mediated through endoplasmic reticulum export activities. Mol Biol Cell. 2006;17(2):990–1005. doi: 10.1091/mbc.E05-02-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rabut G, Ellenberg J. Automatic real-time three-dimensional cell tracking by fluorescence microscopy. J Microsc. 2004;216(Pt 2):131–7. doi: 10.1111/j.0022-2720.2004.01404.x. [DOI] [PubMed] [Google Scholar]