

Abstract
Normal Pressure Hydrocephalus (NPH) is one of the causes of dementia of the elderly characterized by impaired mental function, gait difficulties and urinary incontinence. Previously, it was proposed that some of the NPH patients may develop Alzheimer’s disease (AD) like pathology. Aim of this study was to compare levels of different CSF biomarkers, including total secreted β-amyloid precursor protein (sAPP), sAPP-alpha form (sAPPα), amyloid-beta (Aβ) peptide, total-tau protein and hyperphosphorylated-tau protein in subjects from NPH and Non-NPH Control (NNC). CSF was collected from 23 NPH patients and 13 Non-NPH controls by lumber puncture. Western blot analysis was performed to measure levels of sAPP-total. ELISA was used separately to determine levels of sAPPα, Aβ peptide, total-tau and phosphor-tau proteins. We found a significant decrease in levels of total secreted APP, sAPPα and Aβ (1–42) in the CSF sample of NPH patients vs. NNC. We did not observe any change in levels of total-tau or phospho-tau in NPH vs. NNC subjects. Notably, phospho-tau level was significantly increased in the NPH patients, who were suffering from the disease for more than one year, vs. NNC. Among five biomarkers studied, decreased sAPP, sAPPα and Aβ (1–42) levels in CSF can be molecular markers to distinguish NPH cases from NNC. Disease severity can also be assessed by increased levels of CSF phospho-tau protein and the ratio of phospho-tau to Aβ (1–42), which might be a useful tool for predicting conversion of NPH individuals to other neurodegenerative disorders including Alzheimer’s disease (AD).
Keywords: NPH, Cerebrospinal fluid, Amyloid beta-peptide, Clearance, Tau, Disease progression
1. Introduction
The syndrome “Normal Pressure Hydrocephalus” (NPH), first described in 1965, is manifested by classic triad symptoms of impaired mental function, gait disturbance and urinary incontinence (Adams et al., 1965). NPH is one of the causes of treatable dementia in the elderly population (Clarfield, 1988). Even though NPH shows typical ‘triad’ of symptoms, mentioned above, diagnosis can be difficult because similar mental impairments can be found in many other disorders including Alzheimer’s disease (AD) (Chen et al., 2008). Recently, CSF biomarker studies in AD have gained importance, and various neuronal proteins, such as the amyloid-beta peptide (Aβ) consisting of either 1–40 residue Aβ (1–40) or 1–42 residue Aβ (1–42), total-tau protein and hyperphosphorylated-tau proteins (phospho-tau), have become major markers for investigation in the CSF samples of AD patients. The formation of ‘mature’ plaque primarily composed of aggregated Aβ (1–42) and (1–40) peptides, which are the cleaved products of transmembrane β-amyloid precursor protein (APP), is one of the major pathological hallmarks of AD (Selkoe, 2000). Aβ peptide is also produced in the normal brain but the exact cause of its accumulation in AD brain is yet to be determined. Deposited Aβ fibrils can be enzymatically cleaved by insulin degrading enzyme (IDE) and neprilysin (NEP) (Chander et al., 2007). Apart from that, Aβ, which is also present in the normal or aging brain, is believed to be cleared from the brain interstitial space through CSF (Chakravarty, 2004), and any alteration in this process might cause Aβ deposition. Since the reduction in craniospinal compliance and CSF outflow-absorption disturbance is the central dogma of NPH (Hamlat et al., 2006), Aβ deposition and subsequent neurodegeneration may be possible in patients suffering from NPH. The flow diagram (Fig. 1) shows abnormal CSF dynamics in AD and NPH patients and possible correlation between pathophysiologies of these two diseases.
Fig. 1.
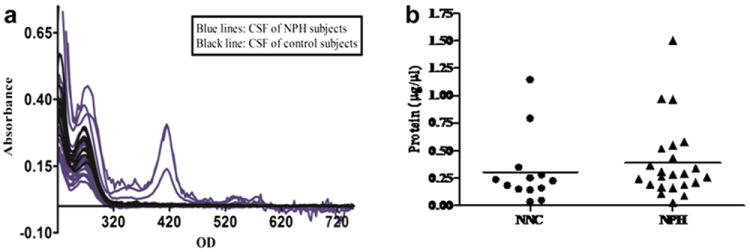
a: Effect of dilution of CSF in NPH subjects determined by UV/VIS spectra: To assess whether the CSF samples of NPH are diluted than those of NNC subjects, a wide range of UV/VIS protein spectra (from 220 nm to 750 nm) was recorded from all CSF samples (n = 36) using ‘NanoDrop V2.1.0’ machine. Spectra obtained from the CSF samples of NPH were marked in blue and those of the CSF samples of NNC subjects were marked in black, which showed no significant change in the protein concentrations of the CSF samples of both the groups. b: Effect of dilution of CSF in NPH subjects determined by Bradford protein assay: Total protein in the CSF was also measured by Bradford assay which, likewise, confirmed that total protein contents both NPH and control groups didn’t differ significantly.
Tau is a microtubule associated protein, primarily located in the axons of neuronal cells, and promotes microtubule assembly and stabilizes neurite extension (Lee and Rook, 1992; Ray et al., 2009). Postmortem analysis of AD brain tissue reveals atypical microtubule assembly in the form of neuro-fibrillary tangles (NFT) due to hyperphosphorylation of tau protein (Grundke-Iqbal et al., 1986; Iqbal et al., 1986). Tau protein can be phosphorylated at different sites. A study demonstrated that phosphorylation of tau protein at Ser262, Thr231 and Ser235 impairs microtubule binding of tau protein by 33%, 26% and 9%, respectively (Sengupta et al., 1998).
Like AD, biomarker studies in the CSF of NPH patients are also being carried out in several laboratories. It was observed that high CSF levels of total-tau, phospho-tau, and low CSF levels of Aβ (1–42) are strongly associated with AD (Hansson et al., 2006). Increase in total-tau in the CSF samples indicates severity of neuronal damage and/or loss (Blennowet al., 1995). Although an increase in CSF phospho-tau level is a distinguishing marker between AD and non-AD controls, there was no change in the CSF phospho-tau level between NPH patients vs. Non-NPH Controls (NNC) (Kapaki et al., 2007). However, some of these previous studies have certain limitations in terms of sample size and of assay conditions used to measure the protein.
The aim of our study was to perform a neuronally-selected molecular profiling of CSF samples from NPH patients and to identify a putative novel molecular marker(s) distinguishable from NNC. Our results suggest a significant decrease in levels of total secreted APP (including sAPPα isoform) in the CSF sample of NPH patients vs. NNC. There was no significant change in levels of total-tau (including phospho-tau) between these two groups. However, there was a significant increase in phospho-tau levels in the NPH patient group with illness for over a year vs. NNC. Furthermore, it was previously observed that older individual with an increased ratio of tau proteins to Aβ (1–42) levels in CSF would develop early cognitive impairment compared to those individuals with decreased ratio of tau proteins to Aβ (1–42) level in CSF(Faganet al., 2007; Smach et al., 2009). We have observed an increased ratio (~3:1) of phospho-tau to Aβ (1–42) level in the CSF of NPH patients vs. NNC indicating a possible conversion to cognitive impairment. Taken together, our results suggest that a combination of abnormal APP processing/trafficking/clearance, and tau phosphorylation characterizes the severe cases of NPH.
2. Materials and methods
2.1. Subjects and inclusion criteria
Twenty threes subjects with idiopathic NPH were included from Barrow Neurological Institute (BNI), Phoenix, AZ. Thirteen individuals who did not have NPH served as Non-NPH Controls (NNC). Fives subjects from the NNC group had multiple sclerosis, two had lymphoma and each of the remaining six subjects had rheumatoid arthritis, epilepsy, amyotrophic lateral sclerosis, myelopathy, Grave’ s disease and transient global amnesia respectively. NNC subjects were cognitively sound at the time of CSF withdrawal.
Clinical diagnosis of NPH was based on modified Ishikawa (2004) criteria for idiopathic normal hydrocephalus plus elevated CSF cineradiography (elevated CSF cerebral aqueductal flow by MRI) and gait improvement confirmed by at least 3 points using Tinetti gait testing and by caregiver after large volume spinal tap. All patients had gait difficulty, urinary incontinence, and dementia, which was established by mini mental state examination (MMSE) score of at 26 and below and formal neuropsychological testing. Each patient underwent comprehensive medical and neurological work-up at the Alzheimer’s Disease and Cognitive Disorders clinic of BNI to exclude previous history of traumatic brain injury, subarachnoid hemorrhage, CNS infection as well as systemic, urinary tract, and other neurologic conditions that may give rise to dementia, urinary incontinence and gait difficulty. Thirty five to forty ml of CSF samples obtained at large volume spinal taps were divided equally into 4 sterile polypropylene test tubes. Such tubes showed minimum binding with Aβ and tau proteins (Lewczuk et al., 2006) and hence used for the long term storage and subsequent Aβ and tau assays, which will be discussed later. The opening and closing pressures of the tubes were normal. The first three tubes were used for counting cells, chemistries and infectious studies, whereas the fourth tube was utilized for this investigation (molecular studies). CSF cells, glucose, protein and search for organisms were unremarkable.
The study was approved by St. Joseph Hospital-Barrow neurological Institute Ethics and IRB committees.
2.2. Assessment of protein content of CSF samples
A complete range of UV/VIS spectrum (220–750 nm) of all CSF samples (n = 36) was measured using ‘NanoDrop 8000. V2.1.0’ (Thermoscientific, Waltham, MA, USA). Briefly, 2 μl of each CSF sample was pipetted onto the sample pedestal and the reading was obtained through PC based software as per the manufacturer’s instruction. Simultaneously, total protein in the CSF samples was also determined by the Bradford protein assay as per the standard protocol obtained commercially (BioRad, Hercules, CA, USA).
2.3. Western immunoblotting
An equal volume of each CSF sample (15 μl) was mixed with 4 μl of Laemmli protein sample loading buffer (5×) and heated at 95 °C for 5 min. After brief centrifugation, an equal volume of each CSF sample-was analyzed in 12% Bis-Tris ‘Criterion’ gel (BioRad) at 180 V for 2 h. Proteins were electrophoretically transferred onto the PVDF membrane (BioRad) using chilled transfer buffer (25 mM Tris, 200 mM Glycine and 20% Methanol) at 100V for 1.5 h at 4 °C. The membrane was blocked in 5% nonfat milk in Tris buffered saline, (pH 7.4) containing 0.05% Tween-20 (TBST) at room temperature. After blocking, the blot was probed with monoclonal 22C11 antibody (Millipore, Billerica, MA, USA), which detects N-terminus 66–81 amino acids of APP. The antibody was diluted 1:1000 in 5% milk dissolved in TBST and probed overnight at 4 °C with gentle rocking. After incubation with the primary antibody, the membrane was washed three times with TBST, and subsequently probed with the secondary antibody (HRP-conjugated goat anti mouse IgG, Pierce, Rockford, IL, USA) for 1 h at room temperature followed by three washs with TBST. Detection of protein band signals was achieved by adding chemoluminescent buffer (GE, Buck-inghamshire, UK) to the blot, which was immediately photographed using GE chemoluminescent detection film. The same procedure was carried out with 6E10 antibody, which recognizes amino acid residues 1–16 of Aβ peptide (Covance, Princeton, NJ, USA).
2.4. ELISA of sAPPα
Levels of sAPPα were measured using the ELISA kit obtained from Immuno Biological Laboratories (IBL, Gumma, Japan). The assay was performed as per the manufacturer’s protocol. Briefly, the wells of the assay kit were pre-coated with monoclonal anti-human sAPPα antibody, (clone 2B3), an equal volume of each CSF (15 μl) was added in a separate well, incubated overnight, washed briefly, and HRP-conjugated monoclonal anti-human APP antibody (clone 10D1) was used as the detection antibody. The ELISA kit was validated to measure levels of sAPPα in human CSF samples and able to detect as low as 0.09 ng/ml of sAPPα in a typical CSF sample, with only 0.3% cross-reactivity to sAPPβ.
2.5. ELISA of Aβ (1–40) peptides
Levels of Aβ (1–40) were measured using the specific ELISA kit obtained from IBL, and the assay was performed as per the manufacturer’s protocol. Briefly, an equal volume (5 μl) of CSF was added onto the well, which was pre-coated with monoclonal anti-human Aβ (35–40) antibody (clone 1A10), and incubated overnight. HRP-conjugated monoclonal anti-human Aβ (11–28, clone 12B2) was used as the detection antibody. This assay is able to detect as low as 5 pg/ml of Aβ (1–40) peptide in a typical CSF sample with only 0.19% cross-reactivity to human Aβ (1–42).
2.6. ELISA of Aβ (1–42) peptides
Levels of Aβ (1–42) were measured using a specific ELISA kit (IBL) following manufacturer’s guidelines. In short, an equal volume (50 μl) of CSF sample was added on to the wells of the plates, which was pre-coated with the polyclonal anti-human Aβ antibody, which was raised against a C-terminal portion of human Aβ (1–42) and do not cross react with human Aβ (1–40) or Aβ (1–43). The plate was incubated overnight. HRP-conjugated monoclonal anti-human Aβ (11–28) antibody (clone 12B2) was used as the detection antibody. This ELISA kit can detect as low as 4.03 pg/ml of human Aβ (1–42) in a typical CSF sample with less than 1% cross-reactivity to human Aβ (1–40).
Prior to assaying levels of each sAPPα, Aβ (1–40) and Aβ (1–42) ELISAs in all CSF samples, initial assays were carried out separately with different volumes of CSF samples to determine the linear range for each of the sAPPα, Aβ (1–40) and Aβ (1–42) assay under our experimental conditions.
2.7. ELISA of total-tau protein
Levels of total-tau were assayed using the commercially available ELISA kit (Innogenetics, Gent, Belgium), which specifically designed to detect total-tau in human CSF. ELISA experiment was performed as per the manufacturer’s protocol. An equal volume (25 μl) of each CSF sample was added to the well of the plate, which was coated with monoclonal human anti total-tau antibody (clone AT120) as a capture antibody. Another two biotinylated human tau specific monoclonal antibodies (clone HT7 and BT2) were used as detection antibodies. Use of these three antibodies in combinations was successful in detecting different epitopes of human total-tau protein. The minimum detection limit of total-tau in a typical human CSF sample was found to be ~59.3 pg/ml.
2.8. ELISA of phospho-tau protein
The phospho-tau ELISA kit was also obtained from Innogenetics and the assay was performed as per the manufacturer’s guideline. An equal volume (75 μl) of each CSF sample was added to the well of the plate, which was coated with monoclonal HT7 antibody as a capture antibody. Biotinylated AT270bio antibody, which detects human phospho-tau (phosphorylated at threonine 181 and strongly associated with AD), was used as the detection antibody. The minimum detection limit of phospho-tau in a typical human CSF sample was found to be ~15.6 pg/ml.
Two CSF samples with high contamination from blood were excluded from tau ELISA (both total-tau and phospho-tau) as per the manufacturer’s guideline.
2.9. Determination of ELISA signals
Colorimetric signals of all ELISAs mentioned herein were read at 450 nm on a micro plate reader (BioRad; model 550). All the assays were performed in triplicate with appropriate blanks and standards.
2.10. Analysis of Western blotting data
Five randomly selected NPH and NNC samples were co-run with the rest of the samples. The blots were scanned using a Gel Documentation System (GDS; UVP Inc., Upland, CA, USA) as described previously (Alley et al., 2009). The protein band density was calculated using the NIH Scion Imaging software (Abramoff et al., 2004). Protein band signals of these common samples of each gel were quantified densitometrically and the average signal was determined. This average value was used as a ‘correction factor’ for analyzing the band signals of each lane from every gel.
3. Statistical analysis
The data were analyzed by using SPSS software (v.15). For parametric value, ‘Independent t-Test’ and for nonparametric value, ‘Mann Whitney’ tests were used. For all analyses p value of 0.05 or lower were considered significant. Figures were plotted by using GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA).
4. Results
4.1. Determination of the dilution factor in CSF samples from NPH subjects
To determine whether CSF samples obtained from NPH patients were diluted by excessive volume of discharge, protein concentrations were determined for each sample from the NPH and NNC subjects. Measurement of UV/VIS spectra of all CSF samples of both NPH and NNC subjects showed no significant change (Fig. 1a). A separate ‘Bradford’ protein assay also confirmed that there is no significant difference in the protein concentration of CSF samples of both NPH and NNC (Fig. 1b).
4.2. Levels of soluble amyloid precursor protein (sAPP-total) in the CSF samples of NPH and Control subjects
Western blotting analysis of sAPP level was carried out with equal volumes of CSF samples as described in ‘Materials and Methods’ section. A total of 36 samples was loaded in two gels. A set of 5 common samples derived from 3 NPH and 2 NNC subjects was analyzed in each gel for normalization purpose. Average signal was obtained from these five subjects. Each test sample was analyzed on the basis of the average value of these 5 common samples in each gel. This was necessary in order to correct for gel-to-gel variations. The immunoblots showed characteristic sAPP bands (95–110 kDa), which were quantified densitometrically. The results showed a significant decrease (~50%) in sAPP level in the CSF sample of NPH patients vs. NNC (p = 0.0013) (Fig. 2).
Fig. 2.
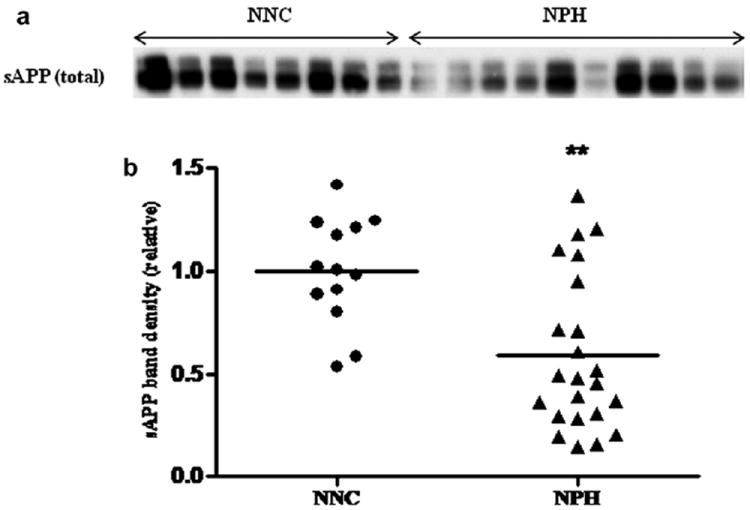
a: Levels of total sAPP: CSF levels of soluble amyloid precursor protein (APP) were determined by semi-quantitative Western blotting technique. An equal volume of CSF sample (15 μl) was loaded onto denaturing polyacrylamide gels (10%). Following PAGE and electrophoretic transfer, the PVDF membrane was probed with APP antibody (clone 22C11), as described in ‘Materials and Methods’. A representative Western blot picture with sAPP protein bands in different lanes was shown from the CSF of 11 NPH and 7 NNC subjects. b: Statistical analysis of APP band density by independent t-test shows a decrease of total sAPP level in the CSF of NPH patients (n = 23) vs. NNC (n = 13), which is statistically significant (p = 0.0013).
4.3. Levels of sAPPα form measured by ELISA
To quantify more accurately sAPPα levels, a sensitive and specific ELISA method was used. The assay showed a significant decrease in CSF-sAPPα levels in NPH patients vs. NNC population (p < 0.0001) (Fig. 3). This result is consistent with an independent experiment using the semi-quantitative Western blotting technique with 6E10 antibody (data not shown).
Fig. 3.
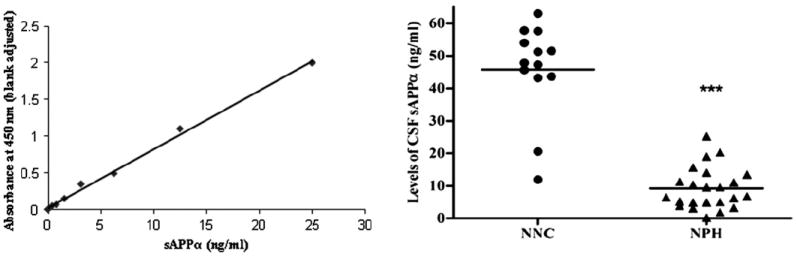
Levels of sAPPα: Measurement of sAPPα with an equal volume (15 μl) of CSF samples from each subject was performed by a specific ELISA as described in ‘Materials and Methods’. Result shows a significantly decrease in sAPPα in the CSF samples of NPH patients. The standard curve with different concentrations of human sAPPα is shown at the left.
4.4. Levels of short and long forms of the amyloid-beta (Aβ) peptides in CSF samples
4.4.1. Aβ (1–40)
Measurement of Aβ (1–40) was carried out by a specific sandwich ELISA (IBL) method. Levels of secreted Aβ (1–40) ranged from 900 pg/ml to 5900 pg/ml as determined from all the CSF samples of NPH patients (n = 23) and NNC subjects (n = 13) studied herein. Analysis showed no significant change in the levels of Aβ (1–40) in NPH patients vs. NNC (Fig. 4a).
Fig. 4.

a: Levels of Aβ (1–40): Levels of Aβ (1–40) were assayed in all CSF samples by the sensitive sandwich ELISA method. An equal volume (5 μl) of CSF samples was loaded in the antibody pre-coated 96-well ELISA plate. Samples were incubated overnight and processed as described in ‘Materials and Methods’. Independent t-test showed no change in Aβ (1–40) level in the CSF of NPH patients (p = 0.120). b: Levels of Aβ (1–42): ELISA of Aβ (1–42) with 50 μl of CSF samples was carried out as mentioned in ‘Materials and Methods’ and Fig. 4a. Statistical analysis showed a significant decreased Aβ (1–42) level in the CSF of NPH vs. NNC subjects (p = 0.017). c: Separate analysis showed a further decrease in the magnitude of CSF Aβ (1–42) in NPH patients who are suffering from more than one year, vs. NNC (p = 0.002). d: Relative proportion of Aβ (1–40) and Aβ (1–42): Ratio of levels of Aβ (1–40) and Aβ (1–42) showed no significant change in the CSF samples of NPH patients, vs. NNC (p = 0.197).
4.4.2. Aβ (1–42)
Measurement of Aβ (1–42) was carried out by the specific sandwich ELISA method, which showed a range from 42 to 607 pg/ml in the CSF samples of NPH and NNC subjects (total n = 36). Statistical analysis revealed a significant decrease (~30%) of CSF Aβ (1–42) levels in NPH patients vs. NNC subjects (p = 0.017) (Fig. 4b). Interestingly, we have seen a greater magnitude (~55%) of the decrease in CSF Aβ (1–42) level in NPH subjects have been suffering more than one year (p = 0.002) (Fig. 4c).
4.4.3. Ratio of Aβ (1–40) and Aβ (1–42)
We determined the proportion of the shorter form of Aβ peptide Aβ (1–40) to Aβ (1–42) peptides in the CSF samples from NPH and NNC subjects. We have not observed any change in the ratio of CSF Aβ (40/42) in NPH patients vs. NNC (Fig. 4d).
4.5. Levels of tau proteins in CSF samples
4.5.1. Total-tau
We detected levels of total-tau protein in the CSF sample by the sensitive human specific ELISA method as described in ‘Materials and Methods’ section. Total-tau levels in the CSF ranged from 9 to 791 pg/ml (average value of total-tau in the CSF of NPH patients was found to be 194 pg/ml ± 40.24 SEM and that for NNC was 159 pg/ml ± 32.88 SEM). We observed no significant change in the levels of total-tau in the CSF of NPH patients vs. NNC (Fig. 5a). Separate statistical analysis revealed non-significant change in levels of total-tau in CSF of NPH patients, who were suffering for more than one year, vs. NNC (p = 0.11) (Fig. 5c)
Fig. 5.
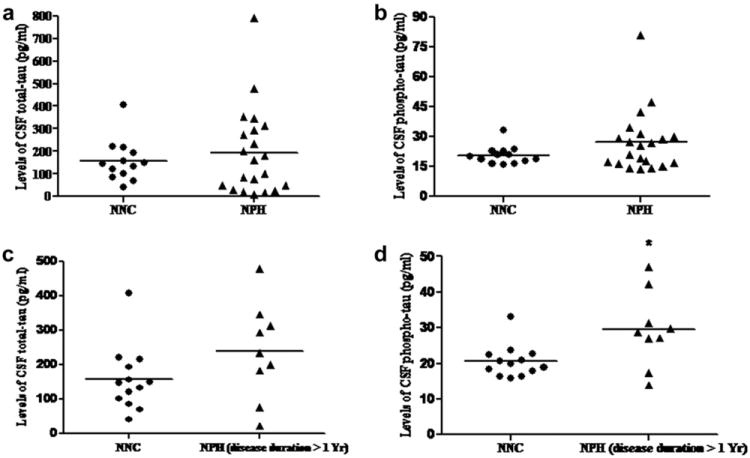
a: Levels of total-tau in the CSF of NNC vs. NPH: Human total-tau ELISA was performed with equal volume of CSF samples which showed no significant change in the CSF of NPH vs. NNC subjects (p = 0.519). b: Levels of phospho-tau in the CSF of NNC vs. NPH: Human phospho-tau ELISA was performed as described in ‘Materials and Methods’ section, which showed no significant change in phospho-tau levels in the CSF of NPH patients vs. NNC subjects (p = 0.153). c: Levels of total-tau in the CSF of NNC vs. NPH patients suffering more than one year: No significant change was observed in the levels of total-tau in the CSF samples of NNC vs. NPH subjects who were suffering from more than one year (p = 0.11). d: Levels of phospho-tau in the CSF of NNC vs. NPH patients suffering more than one year: A significant increase in phospho-tau is observed in the CSF of the patients who are suffering from the disease for more than one year (p = 0.013)(Out of twenty three NPH patients, duration of illness of seven patients was less than one year, nine patients were suffering from more than one year and data from seven patients were not obtained).
4.5.2. Phospho-tau
A similar but separate ELISA measurement of phospho-tau (with human specific antibody) in the CSF sample showed a range from 13 to 80 pg/ml (average value of phospho-tau in the CSF of NPH patients was found to be 21 pg/ml ± 3.12 SEM and that for NNC was 21 pg/ml ± 1.58 SEM). Levels of phospho-tau showed no change in the CSF of NPH patients vs. NNC (Fig. 5b). However, phospho-tau level was significantly increased in the CSF of NPH patients who were suffering from the disease for more than one year, vs. NNC (p = 0.013) (Fig. 5d).
4.5.3. Relationship of tau and Aβ peptides
Relative proportion of total-tau and Aβ (1–42) showed a non-significant increasing trend in the CSF samples of NPH patients vs. NNC (p = 0.1). Notably, the relative proportion of phospho-tau and Aβ (1–42) was significantly elevated in NPH patients vs. NNC (p = 0.008) (Fig. 6a and b).
Fig. 6.
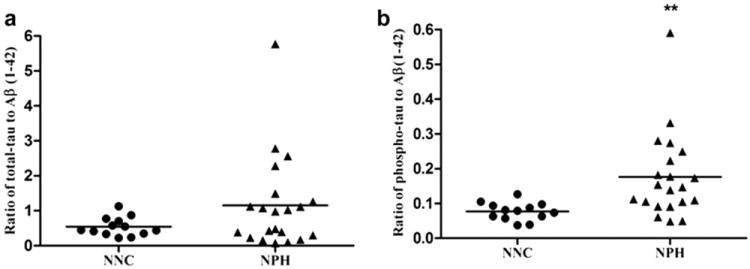
a: Relative proportion of total-tau to Aβ (1–42): Ratio of total-tau and Aβ (1–42) showed an increasing trend in the CSF samples of NPH individuals vs. NNC. However, the increase in the ratio of total-tau/Aβ (1–42) is not statistically significant (p = 0.1). b: Relative proportion of phospho-tau to Aβ (1–42): Relative proportion of phospho-tau and Aβ (1–42) showed an increase (p = 0.004) in the CSF samples of NPH vs. control, indicating more production of phospho-tau and less clearance of Aβ (1–42) in the CSF of NPH individuals vs. NNC.
5. Discussions
NPH is a disorder of CNS that primarily affects elderly people. Although the name ‘Normal Pressure Hydrocephalus’ indicates normal CSF pressure, it is considered to be a misnomer. In NPH subjects, CSF pressure may be within the normal range when measured by manometry technique at lumber puncture. However, continuous CSF pressure measurement reveals waves of increased pressure, particularly during rapid eye movement (REM) sleep (Silverberg, 2004). It has recently been hypothesized that elevated intracranial pressure can cause AD-related change in the brain (Wostyn et al., 2009). Previous studies have shown that a significant number of older NPH patients may develop AD (Golomb et al., 2000; Holm et al., 2003). In this study, we have compared the CSF protein markers of AD in the CSF sample of NPH subjects vs. NNC individuals. We also tried to evaluate relationship of these CSF protein markers with NPH disease duration, as this factor might be important in predicting disease progression. Deposition of Aβ peptide, which is a cleaved product of APP in the brain interstitial space and associated neurodegeneration, is one of the major hallmarks of AD (Ray and Lahiri, 2009). APP is a trans-membrane glycoprotein containing 695–770 amino acids residue and processed enzymatically by α-secretase and β-secretase enzymes (Ray and Lahiri, 2009). APP is cleaved by β secretase (also known as BACE-1) to produce sAPPβ and a C-terminal fragment containing 99 amino acids residue and the latter is cleaved by γ-secretase enzyme to produce Aβ peptide (Sambamurti et al., 2002). Apart from β cleavage, APP is also cleaved by α secretase to form sAPPα and C-terminal 83 amino acid containing fragment, which is considered as non-amyloidogenic (Lahiri et al., 2005). Several investigators have examined levels of CSF-total sAPP in AD patients, and they found a decreased level of CSF-total sAPP in AD patients vs. controls (Peskind et al., 1997; Van Nostrand et al., 1992). We observed a parallel finding of a significant decrease in the levels of CSF-total sAPP in NPH subjects vs. NNC. The decrease in levels of total sAPP could be due to increased proteolytic degradation (Sennvik et al., 2000) or dilution of CSF samples in NPH patients, though there is no significant difference in total protein concentration of the CSF samples between NPH and NNC subjects. The results of of CSF-sAPPα level in AD patients are rather conflicting in the literature. For example, Sennvik et al. (2000) examined CSF from 13 AD and 13 controls and reported a decreased level of CSF-sAPPα in AD patients vs. controls, suggesting a shift of APP processing towards β-secretase pathway leading to the production of Aβ. However, a recent multicenter study conducted by Lewczuk et al. (2010) examined 188 patients with early dementia or mild cognitive impairment (MCI), including 69 AD patients, and they observed a significant elevation of CSF-sAPPα levels in AD patients vs. patients with other dementias. Here we focused on NPH cases and did not test AD subjects directly. On the contrary, we observed that CSF-sAPPα levels were significantly decreased in NPH patients vs. NNC. Lack of adequate CSF production in AD patients might explain the increase of sAPPα level in the CSF of AD patients but the exact cause for decreased sAPPα levels in NPH patients remains unclear. A plausible explanation would favor a trend towards production of Aβ from APP, which leads to less production of sAPPα in NPH subjects. This finding is similar to a previous study related to AD, where Colciaghi et al. (2002) observed a significant decrease in α-secretase activity in the platelets of AD patients (Colciaghi et al., 2002).
Several isoforms of Aβ are formed during the APP cleavage process, and the major species are Aβ (1–40) and Aβ (1–42). The latter has significantly higher capacity to self-aggregate to form plaques (Citron et al., 1996). A possible correlation between AD and NPH has been suggested to be an alteration of CSF dynamics in both the diseases (Silverberg et al., 2003). There is a decreased formation of CSF in the AD patients probably due to observed morphological and functional alterations in the choroid plexus (Serot et al., 2000). In NPH, the fundamental pathology seems to be due to the decreased clearance or re-absorption of CSF, which may lead to stagnation of CSF, eventually resulting in dilatations of ventricles (Chakravarty, 2004). Thus, a combination of decreased formation and compromised re-absorption would cause a reduced clearance of Aβ peptide, resulting in increased deposition of the same in the cortex (Pirttila et al., 1996). Although we did not observe any significant change in Aβ (1–40) levels in the CSF of NPH individuals vs. controls, levels of potentially amyloidogenic Aβ (1–42) were significantly reduced in the CSF of NPH patients vs. NNC subjects (p = 0.037), which is in agreement with other studies that investigated the difference of Aβ (1–42) in the CSF of AD patients vs. controls (Formichi et al., 2006; Lins et al., 2004; Welge et al., 2009). Interestingly, the decrease in CSF Aβ (1–42) in NPH subjects vs. NNC was more intense for those NPH patients who were suffering for more than one year vs. NNC (p = 0.007). This finding might be helpful in evaluating disease progression. Further, in vivo amyloid imaging with the Pittsburgh Compound B (PIB) in non-demented subjects showed that cortical PIB positive subjects have significantly lower CSF levels of Aβ (1–42) than those who are PIB negative (Fagan et al., 2009). However, the mechanism for decrease of Aβ (1–42) in CSF of AD patients is not quite understood. It has been suggested that i) severe neuronal damage can be a cause of less production of Aβ (Andreasen et al., 1999), ii) decreased Aβ (1–42) may be the result of increased recruitment of Aβ (1–42) from the CSF to the brain and iii) decreased secretion into the CSF may be due to the fibrillar nature of Aβ (1–42) species (Samuels et al., 1999). Defective and diminished re-absorption of CSF in NPH might be a cause of decreased Aβ clearance and increased recruitment of Aβ in the cortex. An increase ratio of Aβ (1–40) to Aβ (1–42) level in the plasma of AD patients was proposed to be a sensitive biomarker in AD patients, indicating defective clearance of Aβ (1–42) from the brain (Xu et al., 2008). Furthermore, previous research in APP transgenic mice revealed that the alteration in the ratio of Aβ (1–40) to Aβ (1–42) is influenced by the presence of APOE ε4 allele in the patients (Fryer et al., 2005). We have not observed any significant increase in Aβ (1–40) to Aβ (1–42) ratio in the CSF of NPH patients vs. control (p = 0.2). Since the APOE genotype status of the majority of NPH patients was not known, separate analyses about the relationship between APOE genotype vs. CSF Aβ levels were not carried out in this study.
The formation of NFT due to phosphorylation of a microtubule binding protein tau is another important pathobiochemical event in the brain of AD patients (Grundke-Iqbal and Iqbal, 1999). Structural modifications of tau due to abnormal phosphorylation and aggregation interfere with tau function, which likely leads to neuronal dysfunction seen in AD (Lahiri et al., 2003). Elevated levels of total-tau and phospho-tau in the CSF of AD patients compared to cognitive controls have been consistent findings (Blennow and Hampel, 2003; Hampel et al., 2004; Marksteiner et al., 2008; Welge et al., 2009). In our study, we have seen a non-significant increase in levels of total-tau in the CSF samples of NPH patients, who were suffering from the disease for more than one year, vs. NNC. Interestingly, measurement of levels of phospho-tau in the CSF of NPH patients, who were suffering from more than one year, revealed a significant increase (p = 0.037) vs. NNC. As mentioned in the ‘Introduction’, increased levels of total-tau and phospho-tau are indicative of neuronal loss in the brain, our results suggest that prolong (more than one year) suffering from NPH is associated with neural destruction. Furthermore, it was suggested that increased CSF ratio of phospho-tau to Aβ (1–42) exhibits the greatest sensitivity and specificity for predicting cognitive decline (Hansson et al., 2006). In our study, we have observed a trend of increased ratio (~40%) of total-tau to Aβ (1–42) level in the CSF of NPH patients vs. NNC (p =0.076). Notably, we have seen a statistically significant increase (~55%) in the ratio of phospho-tau to Aβ (1–42) in the CSF sample of NPH patients vs. NNC (p = 0.004), indicating that NPH patients may potentially develop severe dementia in the future. From the tangle pathology perspective, it was suggested that the NFT load is more closely linked to dementia characteristic of AD than the Aβ plaque burden (Terry, 1998). However, we failed to observe such correlation of phospho-tau level with MMSE score of NPH patients suffering for more than one year. Although MMSE is a sensitive instrument for predicting moderate to severe dementia, the test’s sensitivity has been reported to be low to predict milder form of cognitive impairment (MCI) (Wind et al.,1997). Thus, we cannot rule out the possibility that these individuals might have had MCI, which may eventually progress to AD type dementia in due course of time. Leinonen et al. (2008) assessed Aβ burden in AD patients by cortical biopsy and Positron Emission Tomography (PET) with Carbon-11-labeled PIB, and some of their patients displayed AD-related pathological lesions without having severe dementia. This finding suggests that although NPH patients have similar CSF Aβ and phospho-tau profiling, it may take a longer time for them to exhibit dementia. As our results showed, a fourfold decrease of sAPPα level in the CSF of NPH patients, vs. NNC may differentiate it from patients suffering from AD and can be used as a potential biomarker.
In summary, we have used sensitive biochemical techniques and molecular approaches to study NPH cases. However, there are few caveats in the clinical aspect of our study. There is an age difference between the NNC group and NPH group of subjects. Since the prevalence of NPH in the United States is less than 0.05% (Graff-Radford, 2007), it can be assumed that the chance for any individual from the control group to develop NPH is extremely low. Less prevalence of NPH cases than AD also makes it difficult to obtain a large set of patient samples. We also have performed separate correlations analysis of all the biomarkers studied with age in both NPH and NNC population (Tables 1 and 2a) and found no correlations at all with age. Separate analyses of the protein biomarkers studied herein were performed between the NNC subjects, who are more than 60 year old, and 23 NPH patients to evaluate any ‘age’ effect, which reflected the similar results obtained in this study with all NNC and NPH patients (Table 2b). The present study meets one of the goals to find biomarkers. Thus, the present biomarker study of NPH provides an important and useful foothold and warrants further investigation by a multicenter study with larger population of NPH, AD and control subjects.
Table 1.
Demography and clinical data from NPH patients and controls. Data were presented as mean (±standard deviation) unless indicated otherwise. NPH = Normal Pressure Hydrocephalus; MMSE = Mini mental state examination. For age, independent t-test was performed which came out as significant (p < 0.05).
| Characteristics of the sample | Control (NNC)a | NPH |
|---|---|---|
| Total samples | 13 | 23 |
| Age (Mean ± SD) | 58.07 ± 9.47 | 72.78 ± 9.15 |
| Gender | Male 4; Female 9 | Male 13; Female 10 |
| Disease duration (Mean ± SD) | – | 32 months ± 63 monthsb |
| MMSE Score (Mean ± SD) | – | 23.17 ± 7.34c |
Control subjects might have other non-neurological ailments but not NPH, hence given the name Non-NPH Control (NNC). MMSE evaluation was not carried out in NNC subjects.
Data from 5 patients were not obtained; Disease duration of one patient was 276 months which caused high SD.
Data from 5 patients were not obtained.
Table 2.
a Correlation analyses of CSF biochemical markers (APP, sAPPα, Aβ (1–40), Aβ (1–42), total-tau and phospho-tau) for both NPH and Controls showed no correlation with age as indicated by R2 and significance value. This minimizes the chance of obtaining variable results because of the difference of age in control and NPH population.
| CSF protein markers | NNC (n = 13) | NPH (n = 23) |
|---|---|---|
| Aβ (1–40) | R2 = 0.151; Significance = 0.677 | R2 = 0.098; Significance = 0.657 |
| Aβ (1–42) | R2 = 0.427: Significance = 0.218 | R2 = 0.020; Significance = 0.928 |
| sAPP (total) | R2 = 0.337: Significance = 0.341 | R2 = 0.373; Significance = 0.096 |
| sAPPα | R2 = 0.234: Significance = 0.516 | R2 = 0.033; Significance = 0.882 |
| Total-tau | R2 = 0.430; Significance = 0.142 | R2 = 0.255; Significance = 0.265 |
| Phospho-tau | R2 = 0.428: Significance = 0.217 | R2 = 0.235; Significance = 0.293 |
|
b Separate analyses revealed similar result of the protein biomarkers observed in this study between all NNC and NPH patients, when those were compared between NNC who are more than 60 years old (n = 6) and NPH patients. These analyses also showed that ‘age’ had minimum influence on the protein biomarkers studied herein.
| |||
| Mean value | |||
|
| |||
| NNC (>60 yrs) n = 6 | NPH n = 23 | P value | |
| Age | 68.66 ± 7.25 | 72.78 ± 9.15 | 0.318 |
| Protein markers | |||
| sAPP (Total) | 0.982836787 | 0.588615191 | <0.0001 |
| sAPPα | 48312 ng/ml | 9.605 ng/ml | <0.0001 |
| Aβ (1–40) | 3247.5 pg/ml | 2278.8 pg/ml | 0.137 |
| Aβ (1–42) | 388.666 pg/ml | 190.956 pg/ml | 0.004 |
| Total-tau | 192.596 pg/ml | 188.244 pg/ml | 0.957 |
| Phospho-tau | 19.154 pg/ml | 26.713 pg/ml | 0.25 |
| Phospho-tau (NPH >1 yr) | 19.154 pg/ml | 29.441 pg/ml | 0.035 |
| Ratio of phospho-tau to Aβ (l–42) | 0.062049696 | 0.175746157 | 0.03 |
Acknowledgments
We thank Bryan Maloney, Jason Bailey, Cindy Morgan and Justin Long.
Role of funding sources
Debomoy K. Lahiri: Zenith Award from Alzheimer’s Associations; RO1 grants from the National Institutes of Health (AG18379 and AG18884); Patricio Reyes: Codman and Shurtleff Inc.;
Footnotes
Financial disclosure
Balmiki Ray: None.
Conflict of interest
None.
References
- Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- Adams RD, Fisher CM, Hakim S, Ojemann RG, Sweet WH. Symptomatic occult hydrocephalus with “Normal” cerebrospinal-fluid pressure. A treatable syndrome. New England Journal of Medicine. 1965;273:117–26. doi: 10.1056/NEJM196507152730301. [DOI] [PubMed] [Google Scholar]
- Alley GM, Bailey JA, Chen D, Ray B, Puli LK, Tanila H, et al. Memantine lowers amyloid-beta peptide levels in neuronal cultures and in APP/PS1 transgenic mice. Journal of Neuroscience Research. 2009;88:143–54. doi: 10.1002/jnr.22172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen N, Hesse C, Davidsson P, Minthon L, Wallin A, Winblad B, et al. Cere-brospinal fluid beta-amyloid(1–42) in Alzheimer disease: differences between early- and late-onset Alzheimer disease and stability during the course of disease. Archives of Neurology. 1999;56:673–80. doi: 10.1001/archneur.56.6.673. [DOI] [PubMed] [Google Scholar]
- Blennow K, Hampel H. CSF markers for incipient Alzheimer’s disease. Lancet Neurology. 2003;2:605–13. doi: 10.1016/s1474-4422(03)00530-1. [DOI] [PubMed] [Google Scholar]
- Blennow K, Wallin A, Agren H, Spenger C, Siegfried J, Vanmechelen E. Tau protein in cerebrospinal fluid: a biochemical marker for axonal degeneration in Alzheimer disease? Molecular and Chemical Neuropathology. 1995;26:231–45. doi: 10.1007/BF02815140. [DOI] [PubMed] [Google Scholar]
- Chakravarty A. Unifying concept for Alzheimer’s disease, vascular dementia and normal pressure hydrocephalus – a hypothesis. Medical Hypotheses. 2004;63:827–33. doi: 10.1016/j.mehy.2004.03.029. [DOI] [PubMed] [Google Scholar]
- Chander H, Chauhan A, Chauhan V. Binding of proteases to fibrillar amyloid-beta protein and its inhibition by Congo red. Journal of Alzheimer’s Disease. 2007;12:261–9. doi: 10.3233/jad-2007-12308. [DOI] [PubMed] [Google Scholar]
- Chen YF, Wang YH, Hsiao JK, Lai DM, Liao CC, Tu YK, et al. Normal pressure hydrocephalus: cerebral hemodynamic, metabolism measurement, discharge score, and long-term outcome. Surgical Neurology. 2008;70(Suppl. 1):S69–77. doi: 10.1016/j.surneu.2008.08.079. discussion S1:77. [DOI] [PubMed] [Google Scholar]
- Citron M, Diehl TS, Gordon G, Biere AL, Seubert P, Selkoe DJ. Evidence that the 42- and 40-amino acid forms of amyloid beta protein are generated from the beta-amyloid precursor protein by different protease activities. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:13170–5. doi: 10.1073/pnas.93.23.13170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarfield AM. The reversible dementias: do they reverse? Annals of Internal Medicine. 1988;109:476–86. doi: 10.7326/0003-4819-109-6-476. [DOI] [PubMed] [Google Scholar]
- Colciaghi F, Borroni B, Pastorino L, Marcello E, Zimmermann M, Cattabeni F, et al. [alpha]-Secretase ADAM10 as well as [alpha]APPs is reduced in platelets and CSF of Alzheimer disease patients. Molecular Medicine. 2002;8:67–74. [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Head D, Shah AR, Marcus D, Mintun M, Morris JC, et al. Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Annals of Neurology. 2009;65:176–83. doi: 10.1002/ana.21559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in non-demented older adults. Archives of Neurology. 2007;64:343–9. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- Formichi P, Battisti C, Radi E, Federico A. Cerebrospinal fluid tau, A beta, and phosphorylated tau protein for the diagnosis of Alzheimer’s disease. Journal of Cellular Physiology. 2006;208:39–46. doi: 10.1002/jcp.20602. [DOI] [PubMed] [Google Scholar]
- Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM, et al. Human apolipoprotein E4 alters the amyloid-beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. The Journal of Neuroscience. 2005;25:2803–10. doi: 10.1523/JNEUROSCI.5170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golomb J, Wisoff J, Miller DC, Boksay I, Kluger A, Weiner H, et al. Alzheimer’s disease comorbidity in normal pressure hydrocephalus: prevalence and shunt response. Journal of Neurology, Neurosurgery, and Psychiatry. 2000;68:778–81. doi: 10.1136/jnnp.68.6.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff-Radford NR. Normal pressure hydrocephalus. Neurologic Clinics. 2007;25:809–32. vii–viii. doi: 10.1016/j.ncl.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K. Tau pathology generated by overexpression of tau. The American Journal of Pathology. 1999;155:1781–5. doi: 10.1016/S0002-9440(10)65494-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:4913–7. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlat A, Adn M, Sid-ahmed S, Askar B, Pasqualini E. Theoretical considerations on the pathophysiology of normal pressure hydrocephalus (NPH) and NPH-related dementia. Medical Hypotheses. 2006;67:115–23. doi: 10.1016/j.mehy.2006.01.029. [DOI] [PubMed] [Google Scholar]
- Hampel H, Teipel SJ, Fuchsberger T, Andreasen N, Wiltfang J, Otto M, et al. Value of CSF beta-amyloid1-42 and tau as predictors of Alzheimer’s disease in patients with mild cognitive impairment. Molecular Psychiatry. 2004;9:705–10. doi: 10.1038/sj.mp.4001473. [DOI] [PubMed] [Google Scholar]
- Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurology. 2006;5:228–34. doi: 10.1016/S1474-4422(06)70355-6. [DOI] [PubMed] [Google Scholar]
- Holm A, Savolainen S, Alafuzoff I. Brain biopsy prior to treatment of Alzheimer’s disease. Minimally Invasive Neurosurgery. 2003;46:161–4. doi: 10.1055/s-2003-40733. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Grundke-Iqbal I, Zaidi T, Merz PA, Wen GY, Shaikh SS, et al. Defective brain microtubule assembly in Alzheimer’s disease. Lancet. 1986;2:421–6. doi: 10.1016/s0140-6736(86)92134-3. [DOI] [PubMed] [Google Scholar]
- Ishikawa M. Clinical guidelines for idiopathic normal pressure hydrocephalus. Neurologia Medico-chirurgica. 2004;44:222–3. doi: 10.2176/nmc.44.222. [DOI] [PubMed] [Google Scholar]
- Kapaki EN, Paraskevas GP, Tzerakis NG, Sfagos C, Seretis A, Kararizou E, et al. Cerebrospinal fluid tau, phospho-tau181 and beta-amyloid1-42 in idiopathic normal pressure hydrocephalus: a discrimination from Alzheimer’s disease. European Journal of Neurology. 2007;14:168–73. doi: 10.1111/j.1468-1331.2006.01593.x. [DOI] [PubMed] [Google Scholar]
- Lahiri DK, Chen DM, Lahiri P, Bondy S, Greig NH. Amyloid, cholinesterase, melatonin, and metals and their roles in aging and neurodegenerative diseases. Annals of the New York Academy of Sciences. 2005;1056:430–49. doi: 10.1196/annals.1352.008. [DOI] [PubMed] [Google Scholar]
- Lahiri DK, Farlow MR, Sambamurti K, Greig NH, Giacobini E, Schneider LS. A critical analysis of new molecular targets and strategies for drug developments in Alzheimer’s disease. Current Drug Targets. 2003;4:97–112. doi: 10.2174/1389450033346957. [DOI] [PubMed] [Google Scholar]
- Lee G, Rook SL. Expression of tau protein in non-neuronal cells: microtubule binding and stabilization. Journal of Cell Science. 1992;102(Pt 2):227–37. doi: 10.1242/jcs.102.2.227. [DOI] [PubMed] [Google Scholar]
- Leinonen V, Alafuzoff I, Aalto S, Suotunen T, Savolainen S, Nagren K, et al. Assessment of beta-amyloid in a frontal cortical brain biopsy specimen and by positron emission tomography with carbon 11-labeled Pittsburgh Compound B. Archives of Neurology. 2008;65:1304–9. doi: 10.1001/archneur.65.10.noc80013. [DOI] [PubMed] [Google Scholar]
- Lewczuk P, Beck G, Esselmann H, Bruckmoser R, Zimmermann R, Fiszer M, et al. Effect of sample collection tubes on cerebrospinal fluid concentrations of tau proteins and amyloid beta peptides. Clinical Chemistry. 2006;52:332–4. doi: 10.1373/clinchem.2005.058776. [DOI] [PubMed] [Google Scholar]
- Lewczuk P, Kamrowski-Kruck H, Peters O, Heuser I, Jessen F, Popp J, et al. Soluble amyloid precursor proteins in the cerebrospinal fluid as novel potential biomarkers of Alzheimer’s disease: a multicenter study. Mol Psychiatry. 2010;15:38–45. doi: 10.1038/mp.2008.84. [DOI] [PubMed] [Google Scholar]
- Lins H, Wichart I, Bancher C, Wallesch CW, Jellinger KA, Rosler N. Immunoreactivities of amyloid beta peptide((1–42)) and total tau protein in lumbar cerebrospinal fluid of patients with normal pressure hydrocephalus. Journal of Neural Transmission. 2004;111:273–80. doi: 10.1007/s00702-003-0075-x. [DOI] [PubMed] [Google Scholar]
- Marksteiner J, Pirchl M, Ullrich C, Oberbauer H, Blasko I, Lederer W, et al. Analysis of cerebrospinal fluid of Alzheimer patients. Biomarkers and toxic properties Pharmacology. 2008;82:214–20. doi: 10.1159/000156487. [DOI] [PubMed] [Google Scholar]
- Peskind ER, Leverenz J, Farlow MR, Ito RK, Provow SA, Siegel RS, et al. Clinico-pathologic correlations of soluble amyloid beta-protein precursor in cerebrospinal fluid in patients with Alzheimer disease and controls. Alzheimer Disease and Associated Disorders. 1997;11:201–6. [PubMed] [Google Scholar]
- Pirttila T, Mehta PD, Soininen H, Kim KS, Heinonen O, Paljarvi L, et al. Cerebrospinal fluid concentrations of soluble amyloid beta-protein and apolipoprotein E in patients with Alzheimer’s disease: correlations with amyloid load in the brain. Archives of Neurology. 1996;53:189–93. doi: 10.1001/archneur.1996.00550020105022. [DOI] [PubMed] [Google Scholar]
- Ray B, Bailey JA, Sarkar S, Lahiri DK. Molecular and immunocytochemical characterization of primary neuronal cultures from adult rat brain: differential expression of neuronal and glial protein markers. Journal of Neuroscience Methods. 2009;184:294–302. doi: 10.1016/j.jneumeth.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray B, Lahiri DK. Neuroinflammation in Alzheimer’s disease: different molecular targets and potential therapeutic agents including curcumin. Current Opinion in Pharmacology. 2009;9:434–44. doi: 10.1016/j.coph.2009.06.012. [DOI] [PubMed] [Google Scholar]
- Sambamurti K, Greig NH, Lahiri DK. Advances in the cellular and molecular biology of the beta-amyloid protein in Alzheimer’s disease. Neuromolecular Medicine. 2002;1:1–31. doi: 10.1385/NMM:1:1:1. [DOI] [PubMed] [Google Scholar]
- Samuels SC, Silverman JM, Marin DB, Peskind ER, Younki SG, Greenberg DA, et al. CSF beta-amyloid, cognition, and APOE genotype in Alzheimer’s disease. Neurology. 1999;52:547–51. doi: 10.1212/wnl.52.3.547. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Toward a comprehensive theory for Alzheimer’s disease. Hypothesis: Alzheimer’s disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Annals of the New York Academy of Sciences. 2000;924:17–25. doi: 10.1111/j.1749-6632.2000.tb05554.x. [DOI] [PubMed] [Google Scholar]
- Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Archives of Biochemistry and Biophysics. 1998;357:299–309. doi: 10.1006/abbi.1998.0813. [DOI] [PubMed] [Google Scholar]
- Sennvik K, Fastbom J, Blomberg M, Wahlund LO, Winblad B, Benedikz E. Levels of alpha- and beta-secretase cleaved amyloid precursor protein in the cerebrospinal fluid of Alzheimer’s disease patients. Neuroscience Letters. 2000;278:169–72. doi: 10.1016/s0304-3940(99)00929-5. [DOI] [PubMed] [Google Scholar]
- Serot JM, Bene MC, Foliguet B, Faure GC. Morphological alterations of the choroid plexus in late-onset Alzheimer’s disease. Acta Neuropathologica. 2000;99:105–8. doi: 10.1007/pl00007412. [DOI] [PubMed] [Google Scholar]
- Silverberg GD. Normal pressure hydrocephalus (NPH): ischaemia, CSF stagnation or both. Brain. 2004;127:947–8. doi: 10.1093/brain/awh178. [DOI] [PubMed] [Google Scholar]
- Silverberg GD, Mayo M, Saul T, Rubenstein E, McGuire D. Alzheimer’s disease, normal-pressure hydrocephalus, and senescent changes in CSF circulatory physiology: a hypothesis. Lancet Neurology. 2003;2:506–11. doi: 10.1016/s1474-4422(03)00487-3. [DOI] [PubMed] [Google Scholar]
- Smach MA, Charfeddine B, Ben Othman L, Lammouchi T, Dridi H, Nafati S, et al. Evaluation of cerebrospinal fluid tau/beta-amyloid(42) ratio as diagnostic markers for Alzheimer disease. European Neurology. 2009;62:349–55. doi: 10.1159/000241881. [DOI] [PubMed] [Google Scholar]
- Terry RD. The cytoskeleton in Alzheimer disease. Journal of Neural Transmission: Supplementum. 1998;53:141–5. doi: 10.1007/978-3-7091-6467-9_12. [DOI] [PubMed] [Google Scholar]
- Van Nostrand WE, Wagner SL, Shankle WR, Farrow JS, Dick M, Rozemuller JM, et al. Decreased levels of soluble amyloid beta-protein precursor in cerebrospinal fluid of live Alzheimer disease patients. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:2551–5. doi: 10.1073/pnas.89.7.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welge V, Fiege O, Lewczuk P, Mollenhauer B, Esselmann H, Klafki HW, et al. Combined CSF tau, p-tau181 and amyloid-beta 38/40/42 for diagnosing Alzheimer’s disease. Journal of Neural Transmission. 2009;116:203–12. doi: 10.1007/s00702-008-0177-6. [DOI] [PubMed] [Google Scholar]
- Wind AW, Schellevis FG, Van Staveren G, Scholten RP, Jonker C, Van Eijk JT. Limitations of the mini-mental state examination in diagnosing dementia in general practice. International Journal of Geriatric Psychiatry. 1997;12:101–8. doi: 10.1002/(sici)1099-1166(199701)12:1<101::aid-gps469>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Wostyn P, Audenaert K, DeDeyn PP. The Valsalva maneuver and Alzheimer’s disease: is there a link? Current Alzheimer Research. 2009;6:59–68. doi: 10.2174/156720509787313943. [DOI] [PubMed] [Google Scholar]
- Xu W, Kawarabayashi T, Matsubara E, Deguchi K, Murakami T, Harigaya Y, et al. Plasma antibodies to Abeta40 and Abeta42 in patients with Alzheimer’s disease and normal controls. Brain Research. 2008;1219:169–79. doi: 10.1016/j.brainres.2008.02.060. [DOI] [PubMed] [Google Scholar]