

Abstract
In treating cancer with clinically approved chemotherapies, the high systemic toxicity and lack of selectivity for malignant cells often result in an overall poor response rate. One pharmacological approach to improve patient response is to design targeted therapies that exploit the cancer milieu by reductively activating prodrugs, which results in the selective release of the free drug in the tumor tissue. Previously, we characterized prodrugs of seco-CBI-indole2 (CBI-indole2) designed to be activated in hypoxic tumor microenvironments, wherein the tumor maintains higher concentrations of “reducing” nucleophiles capable of preferentially releasing the free drug by nucleophilic attack on a weak N-O bond. Of these prodrugs, BocNHO-CBI-indole2 (BocNHO) surpassed the efficacy of the free drug, CBI-indole2, when examined in vivo in the murine L1210 leukemia model and demonstrated reduced toxicity suggesting a targeted or sustained release in vivo. Herein, we further examine the biological activity of the BocNHO prodrug in murine breast cancer, as well as human prostate and lung cancer cell lines, in vitro. Notably, BocNHO manifests potent antiproliferative and cytotoxic activity in all three tumor cell lines. However, in comparison to the activity observed in the murine cancer cell line, the human cancer cell lines were less sensitive, especially at early timepoints for cytotoxicity. Based on these findings, BocNHO was tested in a more clinically relevant orthotopic lung tumor model, revealing significant efficacy and reduced toxicity compared with the free drug. The data suggests that this pharmacological approach to designing targeted therapies is amenable to human solid tumors.
Keywords: duocarmycin, prodrug, breast, prostate, lung, cancer, orthotopic tumor model
Introduction
The naturally occurring antitumor agents CC-1065 and duocarmycin SA are the parent members of a class of exquisitely potent compounds that manifests picomolar (pM) IC50 values for their ability to selectively alkylate duplex DNA and induce cell death.1-18 Mechanistically, this selective DNA alkylation is achieved through the forced adoption of a helical conformation upon binding to the minor-groove AT-rich regions of DNA, which disrupts the stabilizing vinylogous amide and activates the cyclopropane for nucleophilic attack.19,20 These natural products are not clinically viable due to either severe adverse events, including lethal hepatotoxicity and extreme myelotoxicity, or a lack of in vivo activity.1,21,22 As a result of the high systemic toxicity and reduced selectivity of this class of compounds, a new strategy has emerged to develop prodrugs that selectively release the free drug in the tumor. This strategy, known as in situ activation, seeks to design systems where an intrinsically reactive species is formed from a comparatively stable precursor at or near the site of action, thereby selectively activating a compound in the presence of its molecular target.23 The hypoxic tumor microenvironment is ideal for designing prodrugs that can be reductively activated without the requirement of enzymatic release. Previously, we utilized reductive activation, as a mechanism of in situ activation, to design and characterize analogs of the duocarmycins and CC-1065.24,25
BocNHO-CBI-indole2 (BocNHO) is a reductively activated phenol prodrug from the CC-1065 and duocarmycin class of compounds.24 BocNHO was not designed for reversible or enzymatic activation but rather for irreversible reductive activation by cleavage of a weak N-O bond by way of reducing nucleophiles. The premise was to utilize the high reducing capacity of the hypoxic tumor microenvironment (i.e., thiols) to activate the prodrug, making the cancer cells and surrounding stroma more sensitive to the prodrug treatment. The BocNHO prodrug was tested for antiproliferative activity in vitro, and it revealed equal potency to the parent compound, seco-CBI-indole2 (CBI-indole2), against a panel of cancer cell lines indicating release of the free drug.24 When examined in vivo, BocNHO revealed improved efficacy and reduced toxicity, which was attributed to a controlled or selective release of the free drug to the tumor.24 However, the model used in this in vivo study, as well as in several others examining this class of compounds, was a mouse leukemia model (L1210).
Ascites models, like the L1210 model, have identified drugs active against leukemia and lymphoma; yet, they are not predictive of activity for solid tumors.26-28 Traditional drug discovery programs frequently screen experimental therapies against a panel of human ectopic tumor models implanted subcutaneously (s.c.) in immunodeficient mice, even though s.c. xenografts are not reflective of the orthotopic tumor site.29 It is well established that the organ environment can overwhelmingly dictate how tumors behave with regard to metastasis, enzymes, neovascularization, differentiation and the expression of genes.30-33 The biological influence of the tumor microenvironment can affect the ability to evaluate experimental therapies. Indeed, significant evidence exists demonstrating the same tumor cell line has an improved response to chemotherapy when the cells are implanted subcutaneously compared with orthotopic implantation of cells.34 Thus, for proper evaluation of novel therapies, it is critical to implant cancer cells in the appropriate environment. Furthermore, in a clinical setting, patients are often being treated for advanced metastatic disease, while s.c. xenograft models are treated within 1–14 d of tumor inoculation and rarely develop metastases.35 Thus, orthotopic tumor models are more clinically relevant for assessing drug efficacy, as they more accurately represent the clinical growth characteristics of primary tumor and metastatic disease.34,36-38 To further advance BocNHO as a clinical candidate, we examined this novel prodrug for activity against breast, prostate and lung solid tumor cell lines in a series of in vitro experiments and then selected one type of cancer for an in vivo orthotopic tumor efficacy study. Herein, we reveal the potent anti-cancer properties of the BocNHO prodrug against solid tumor cell lines in vitro and demonstrate superior in vivo efficacy in an aggressive orthotopic non-small cell lung cancer model.
Results
BocNHO prodrug manifests potent antiproliferative activity in mouse and human cancer cell lines comparable to CBI-indole2
The human prostate PC3-MM2 (Fig. 1A), mouse breast EMT6 (Fig. 1B) and human lung A549 (Fig. 1C) cancer cell lines were screened using dose response over time (24, 48 and 72 h) to determine IC50 potency. Both BocNHO and CBI-indole2 have similar potency values at all time-points ranging from 1–30 nM at 24 h to 280–980 pM at 72 h. Interestingly, at 72 h, cell proliferation is nearly 100% inhibited in all three cell lines following treatment. The A549 cell line appears to be the most sensitive to these compounds followed by EMT6 and PC3-MM2. Table 1 lists the individual IC50 values for the BocNHO and CBI-indole2 compounds for each cell line at the different timepoints. The structures of BocNHO and CBI-indole2 are shown in Figure 1D. Since a major goal of these studies was to conduct an in vivo efficacy study and with an understanding that most drugs are significantly cleared from mice in 24 h or less, we focused all subsequent studies to characterize BocNHO on the 24-h timepoint.

Figure 1. Antiproliferative effects of BocNHO and CBI-indole2 in prostate, breast and lung cancer cell lines. Prostate PC3-MM2 (A), breast EMT6 (B) and lung A549 (C) cells were treated with vehicle (▲), BocNHO (●) and CBI-indole2 (■) for 24, 48 and 72 h. The BocNHO prodrug manifests equipotency compared with the parent molecule, CBI-indole2, indicating free release of the active drug. Potency and efficacy increase in a dose- and time-dependent manner across all three cell lines. The structures of BocNHO and CBI-indole2 are shown in (D).
Table 1. Anti-proliferative IC50 values.
| |
24 h |
48 h |
72 h |
|||
|---|---|---|---|---|---|---|
| BocNHO | CBI-indole2 | BocNHO | CBI-indole2 | BocNHO | CBI-indole2 | |
| PC3-MM2 |
2.571E−08 |
2.061E−08 |
8.543E−10 |
1.529E−09 |
5.855E−10 |
9.832E−10 |
| EMT6 |
1.65E−09 |
2.585E−09 |
8.507E−10 |
8.217E−10 |
4.27E10 |
6.596E−10 |
| A549 | 1.548E−09 | 1.385E−09 | 7.905E−10 | 6.132E−10 | 3.807E−10 | 2.837E−10 |
Prostate (PC3-MM2), breast (EMT6) and lung (A549) solid tumor cell lines were examined over a 72-h time-course for antiproliferative activity following treatment with the BocNHO prodrug or the parent molecule, CBI-indole2. IC50 values are reported as log [drug] (Molar) for each timepoint.
Cytotoxic effects of BocNHO and CBI-indole2
To determine if BocNHO has cytotoxic activity comparable to its potent antiproliferation properties (Fig. 1), both compounds were examined in the trypan blue assay. In agreement with the potent antiproliferation that was observed at 1–30 nM with these compounds, significant cytotoxicity is observed at similar concentrations in a dose-dependent trend (Fig. 2A–C, left panels). Moreover, when total cells are counted and compared with cell numbers at the start of dosing (time zero, t0), significant antiproliferative activity is observed at doses as low as 50–500 pM depending on cell type (Fig. 2A–C, right panels). This data further supports the accuracy of the IC50 values generated in Figure 1 across all three cancer cell lines.

Figure 2. Trypan blue cytotoxicity of BocNHO and CBI-indole2 in prostate, breast and lung cancer cell lines. PC3-MM2 (A), EMT6 (B) and A549 (C) cells were treated with vehicle, BocNHO and CBI-indole2 at the indicated doses for 24 h. Significant cytotoxic effects in EMT6 and A549 cell lines are noted at doses starting at 50 pM [(B), left] and 500 pM [(C), left), respectively, while a limited cytotoxic effect is seen in PC3-MM2 cells at doses < 1 µM (A, left). Moreover, pronounced antiproliferative effects are noted at doses as low as 500 pM in all three cell lines [(A–C), right]. Single (*) and double (**) asterisks represent significant unpaired t-test p values of < 0.05 and < 0.01, respectively, as compared with vehicle-treated cells.
BocNHO and CBI-indole2 induce apoptosis in breast, prostate and lung cancer cell lines
To determine whether these cells are dying of necrosis or programmed cell death, cancer cells were labeled with Annexin V and propidium iodide (PI). Cells positive for PI alone indicate death by necrosis, cells positive for Annexin V alone indicate early-stage apoptosis and cells positive for both indicate late-stage apoptosis. PC3-MM2 cells reveal little indication of apoptosis (Fig. 3A); while EMT6 cells demonstrate a dose-dependent increase in early- and late-stage apoptotic cells (Fig. 3B). The percent cytotoxicity, mostly apoptosis, corresponds nicely to the viability data of both cell lines in Figure 2A and B (left panels). BocNHO and CBI-indole2 both elicit early- and late-stage apoptosis in A549 cells that appear to peak at 10 nM and then diminish with increasing dosage (Fig. 3C). No significant necrosis is observed following treatment with these compounds, suggesting most of the cells are dying by programmed cell death that is likely induced by cell cycle arrest.
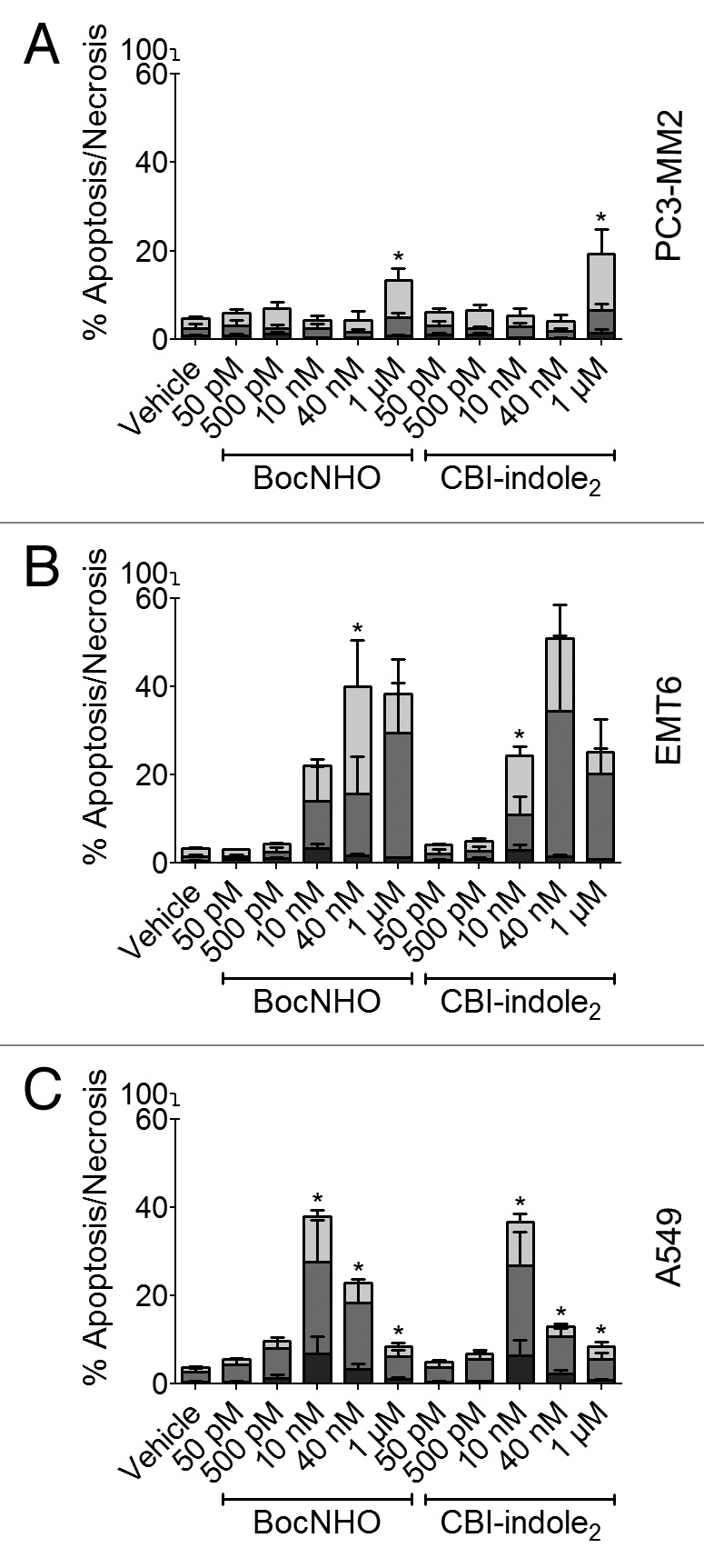
Figure 3. Induction of apoptosis in prostate, breast and lung cancer cell lines treated with BocNHO and CBI-indole2. PC3-MM2 (A), EMT6 (B) and A549 (C) cells were treated with vehicle, BocNHO and CBI-indole2 for 24 h. Bar graphs represent flow cytometry data that identifies cells undergoing necrosis (black), early-stage apoptosis (medium gray) and late-stage apoptosis (light gray) by measuring the populations of Annexin V and propidium iodide positive cells as a percent of the parent population. Significant apoptosis is observed in both the EMT6 (B) and A549 (C) cells following treatment with BocNHO and CBI-indole2 with minimal necrosis, indicating programed cell death. PC3-MM2 (A) cells are less sensitive to the induction of apoptosis with both compounds. Single (*) and double (**) asterisks represent significant unpaired t-test p values of < 0.05 and < 0.01, respectively, as compared with vehicle-treated cells.
Induction of S phase and G0/G1 cell cycle arrest following treatment with BocNHO and CBI-indole2
DNA alkylation and subsequent cell cycle arrest is the mechanism of action for this class of compounds, which results in antiproliferative activity and cytotoxicity by way of programmed cell death. Thus, the effects on cell cycle of the BocNHO prodrug were compared with those of the parent molecule, CBI-indole2, across all three solid tumor cell lines. The mouse EMT6 breast cancer cells demonstrate S phase arrest at 50 pM then subsequently shift to G0/G1 cell cycle arrest at concentrations starting as low as 500 pM (Fig. 4B). As the dose increases, approximately the same percentage of cells are arrested in G0/G1; however, there is an increase in the sub-G0 population of cells, which is a marker of apoptosis and corresponds to the data in Figure 3B. Interestingly, the human prostate PC3-MM2 and lung A549 cells display a different pattern of cell cycle arrest. At low doses (50–500 pM), both cell lines demonstrate a significant increase in S phase arrest compared with EMT6 cells (Fig. 4A and C). However, at higher doses (10 nM-1 µM), G0/G1 arrest is more prominent with an increase in the sub-G0 population.
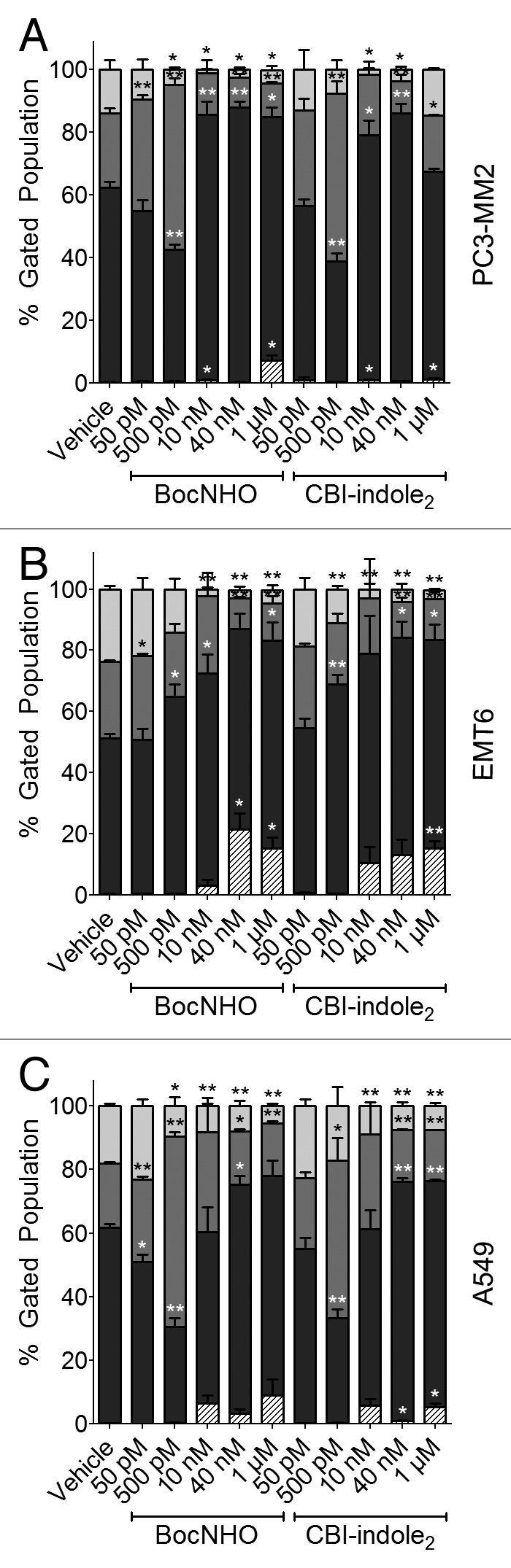
Figure 4. Cell cycle arrest in prostate, breast and lung cancer cell lines treated with BocNHO and CBI-indole2. PC3-MM2 (A), EMT6 (B) and A549 (C) cells were treated with vehicle, BocNHO and CBI-indole2 for 24 h. Bar graphs represent cell cycle arrest data that identifies cells in G2/M (light gray), S (medium gray), G0/G1 (black) and sub-G0 (striped) phases. PC3-MM2 and A549 cell lines demonstrate a dose-dependent shift from the G2/M and G0/G1 phases to S phase arrest at doses of < 500 pM; however, at concentrations > 10 nM, there is vast G0/G1 arrest in both cell lines with a subtle increase in the sub-G0 percentage of A549 and PC3-MM2 cells. In EMT6 cells, a dose-dependent increase in G2/M arrest is observed along with a significant increase in the sub-G0 population. Single (*) and double (**) asterisks represent significant unpaired t-test p values of < 0.05 and < 0.01, respectively, as compared with vehicle-treated cells.
Acute exposure (washout) of BocNHO and CBI-indole2 maintains pharmacological activity over 72 h
Prior to conducting an in vivo efficacy study, a pilot pharmacokinetic study was performed in mice to determine the plasma half-life and clearance of the BocNHO prodrug. In these studies, a majority of BocNHO was cleared from the mouse by 6 h following intraperitoneal administration (data not shown). All previous in vitro studies, described above, characterizing the BocNHO prodrug were performed at ≥ 24 h continuous exposure. Therefore, to address the issue of a six-hour exposure time in an in vivo efficacy study, in vitro washout experiments were performed following 6 h exposure of the BocNHO or CBI-indole2 compounds to mimic in vivo drug exposure. For these studies, the A549 cell line was used since this lung cancer cell line was selected for the in vivo efficacy study; it was more sensitive in the aforementioned experiments compared with the PC3-MM2 cell line, and a human tumor model was more desirable than a mouse tumor model. Treatment of A549 cells for 6 h with these two compounds, followed by drug washout, significantly decreases cell viability at 24 h that decreases further at 72 h (Fig. 5A). Remarkably, a six-hour BocNHO treatment followed by washout produces nearly identical cell viability results to those following continuous exposure at 24 h (Fig. 2C, left panel). Moreover, the BocNHO prodrug appears to have superior efficacy at 10–40 nM following 72-h washout treatment compared with CBI-indole2 (Fig. 5A, right panel). In similar experiments, antiproliferative activity was assessed by determining the total number of cells following treatment compared with the number of cells at t0. Six-hour exposure of BocNHO demonstrates a nearly 100% antiproliferative response at concentrations ≥ 10 nM (24 and 72 h), while CBI-indole2 has little response at 10 nM suggesting superior efficacy of the BocNHO prodrug (Fig. 5B). The induction of apoptosis was examined following a six-hour drug treatment with washout in A549 cells, revealing a significant increase in late-stage apoptosis and, to a lesser extent, early-stage apoptosis in these experiments (Fig. 5C). The 72-h washout results are comparable to the 24-h continuous exposure results (Fig. 3B) in terms of response but require higher concentrations (10 nM vs. 40 nM) to achieve the response. In the cell cycle arrest washout experiments, a similar tendency is observed with an S phase arrest at 10 nM and G0/G1 arrest occurring at 40 nM (Fig. 5D) in comparison to the 24-h continuous exposure (Fig. 4C). Intriguingly, while other experiments demonstrate an increase in activity over time (Fig. 5A–C), the cell cycle arrest studies are nearly identical at both 24 and 72 h following washout (Fig. 5D).

Figure 5. Drug washout studies of lung cancer cells treated with BocNHO and CBI-indole2. Following treatment of A549 cells for 6 h with vehicle, BocNHO and CBI-indole2, the cells were rinsed and incubated in drug-free media for the remainder of the 24- [(A–D), left)] or 72-h timepoint [(A–D), right]. Cytotoxicity was determined by trypan blue assay (A) and reveals a dose-dependent increase in cytotoxicity that is more pronounced with time in the absence of continuous drug treatment. The antiproliferative effects of these two drugs also remain intact over time, as total cell counts remain at t0 levels for both timepoints at concentrations > 10 nM (B). Bar graphs depict the percentage of cells undergoing necrosis (black), early-stage apoptosis (medium gray) and late-stage apoptosis (light gray) following drug washout treatment (C). Of note, increasing apoptosis and necrosis is observed over time with both drugs, demonstrating continuous treatment is not necessary for continued drug response. Cell cycle arrest bar graphs represent flow cytometry data that identifies cells in G2/M (light gray), S (medium gray), G0/G1 (black) and sub-G0 (striped) phases (D). Consistent with the 24-h continuous exposure data, S phase arrest is observed at lower doses (10 nM BocNHO and 40 nM CBI-indole2), while G0/G1 arrest is observed at higher doses (> 40 nM BocNHO and 1 µM CBI-indole2). Single (*) and double (**) asterisks represent significant unpaired t-test p values of < 0.05 and < 0.01, respectively, as compared with vehicle-treated cells.
In vivo therapeutic efficacy of BocNHO and CBI-indole2 in an orthotopic lung tumor model
To determine the efficacy of the BocNHO prodrug, mice were orthotopically inoculated with A549 cells expressing luciferase. Mice were assigned to one of nine treatment groups: 60, 100, 250 or 500 µg/kg BocNHO, 60, 100, 250 or 500 µg/kg CBI-indole2 or vehicle (DMSO). Treatment was initiated on Day 4, and mice were initially scheduled to receive a total of six doses (study days 4, 8, 12, 16, 20 and 24). However, following the third dose (Day 12), gross toxicity was observed, and the mice either were euthanized or died from toxicity in all treatment groups except 60 µg/kg BocNHO and vehicle. Subsequently, the fourth dose (Day 16) was withheld and two more doses were administered at Days 21 and 25 to the remaining animals in the study. Table 2 depicts the number of surviving animals in the 60 µg/kg BocNHO, 60 µg/kg CBI-indole2 and vehicle groups at the completion of the study on Day 32. In the BocNHO treatment group, 70% of the animals survived the study compared with 0% in the CBI-indole2 group at the 60 µg/kg dose, demonstrating reduced toxicity of the BocNHO prodrug. With regard to efficacy, bioluminescence imaging at Days 20 and 32 revealed a statistically significant reduction in the percent change in total tumor burden in mice treated with BocNHO compared with vehicle-treated mice (Fig. 6B). Representative images demonstrating the reduction in total tumor burden of mice treated with BocNHO or vehicle are shown in Figure 6A. While BocNHO appears to be less toxic than its CBI-indole2 counterpart, a statistically significant reduction in body weight was observed as early as day 5 in mice treated with BocNHO compared with vehicle (Fig. 6C).
Table 2. Number of surviving animals following treatment with BocNHO and CBI-indole2.
| Day | BocNHO | CBI-indole2 | Vehicle |
|---|---|---|---|
| 0 |
9 |
9 |
10 |
| 11 |
8 |
7 |
10 |
| 20 |
7 |
0 |
10 |
| 32 | 7 | 0 | 9 |
Following treatment of lung-tumor-burdened mice with BocNHO or CBI-indole2, increased survival was observed in the 60 µg/kg BocNHO and vehicle treatment groups compared with 60 µg/kg CBI-indole2, suggesting reduced toxicity of the BocNHO prodrug (Fig. 6A).

Figure 6. In vivo efficacy of BocNHO in an orthotopic lung tumor model. Bioluminescence imaging, as measured in total flux (photons/second, p/s), was performed on Day 3 to establish the baseline tumor burden for each mouse. Subsequent bioluminescence imaging of mice treated with 60 µg/kg BocNHO (●) or vehicle (■) on Days 11, 20 and 32 was used to quantify percent change in tumor burden relative to the baseline tumor burden over time (B). A statistically significant decrease in percent tumor burden was observed at Days 20 and 32 following treatment with BocNHO compared with vehicle-treated animals. Bioluminescence images revealing decreased tumor burden of mice treated with the BocNHO prodrug [(A), top] compared with vehicle-treated mice [(A), bottom] are shown from Day 32. As a measure of gross toxicity, body weights of mice treated with BocNHO (●) or vehicle (■) were compared (C). As early as day 5, a significant decrease in body weight is observed in mice treated with the BocNHO prodrug compared with vehicle-treated mice. Single (*) asterisks represent significant unpaired t-test p values of < 0.05.
Discussion
The CC-1065 and duocarmycin SA class of compounds is a group of natural products that has been extensively examined as very potent cytotoxic agents that selectively alkylate N3 in AT-rich regions of the DNA minor groove resulting in cell death.39 These natural products are not suitable for clinical use in cancer therapy due to their general toxicity and lack of selectivity for cancer cells. To overcome these pitfalls, novel analogs with reduced toxicity have been designed and have reached clinical trials, demonstrating the potential of this synthetic approach for the treatment of cancer.40-42 However, these newer analogs all failed in clinical trials due to severe myelotoxicity.43-46 Subsequently, a new approach of designing prodrugs that selectively release free drug in the tumor by enzymatic activation or conjugation to tumor-specific antibodies has been examined to further reduce toxicity.15,47 Previously, we revealed our approach to improving the tumor selectivity of this class of compounds by designing prodrugs that take advantage of tumor-specific reducing nucleophiles to release the free drug via reductive activation.24,25 The purpose of these studies was to examine the activity of the BocNHO prodrug, designed to undergo non-enzymatic reductive activation, against human and murine solid tumor cell lines, as well as determine its efficacy in a clinically relevant human orthotopic tumor model.
In solid tumor cell lines, the BocNHO prodrug manifested potent antiproliferative activity that was comparable to CBI-indole2, indicating free-release of the parent molecule. In trypan blue cell viability studies, cytotoxicity was observed across all three cell lines which corresponded to the induction of apoptosis, suggesting the mechanism of action was programmed cell death. Supporting this data, when total cells were examined as another indication of antiproliferation compared with total cells at t0, pronounced antiproliferation was observed across all three cell lines. The EMT6 cells were the most sensitive, with nearly 100% of the cells non-proliferative at concentrations > 40 nM (Fig. 2, right panels). However, with regard to the human cancer cell lines, a percentage of the cells were still proliferating at all concentrations, as the total number of cells was greater than at t0. In similar studies, A549 cells treated for 6 h followed by washout of BocNHO or CBI-indole2 revealed some significant differences to the studies performed under continuous drug exposure. Surprisingly, 100% antiproliferation was achieved with 10 nM BocNHO compared with 40 nM CBI-indole2 demonstrating a 4-fold increase in potency of BocNHO (Fig. 5B). A similar trend was observed in the drug washout apoptosis assays examining the mechanism of cell death, wherein 40 nM BocNHO was more potent at inducing apoptosis than an equivalent dose of CBI-indole2 at 24 h (Fig. 5C). Conversely, in continuous exposure studies, there was little difference in the response observed between the two compounds (Fig. 2C). This trend continued at the 72-h timepoint, in which BocNHO was more potent in induction of apoptosis at 10 and 40 nM compared with CBI-indole2. This data highlighted important pharmacological differences between BocNHO and CBI-indole2, in that BocNHO was more effective at a shorter duration of exposure (Fig. 5B and C).
In the cell cycle arrest studies, continuous exposure studies at 24 h demonstrated similar effects on S phase and G0/G1 arrest with both compounds. However, in washout studies (Fig. 5D), the BocNHO prodrug manifested increased potency for S phase arrest (10 nM) and G0/ G1 arrest (40 nM) compared with CBI-indole2 (40 nM and 1 µM, respectively). This data further supported the increased potency of the BocNHO prodrug observed in the other drug washout studies (Fig. 5B and C), suggesting BocNHO required a shorter duration of exposure to produce equivalent efficacy compared with the parent molecule, CBI-indole2.
In vivo efficacy of BocNHO was examined in comparison to CBI-indole2 in an aggressive orthotopic lung tumor model using the A549 non-small cell lung cancer line. Upon inoculation of tumor cells into the left lung of mice, these cells disseminated to the right lung and pleural cavity, and the mice became moribund in 30–35 d. Following administration of 60 µg/kg BocNHO prodrug, significant anti-tumor activity was observed at Day 20 (Fig. 6B). In addition to reduced tumor burden, no metastases were observed in treated animals (Fig. 6A, top panel) compared with vehicle-treated animals as evidenced by the bioluminescence signal specific to tumor burden outside the pleural cavity (Fig. 6A, bottom panel). In addition to the pronounced in vivo efficacy observed in this study, a decrease in body weight was revealed for the BocNHO-treated mice compared with vehicle control mice (Fig. 6C). It is unclear if this drop in body weight was a result of drug toxicity or if increased tumor burden in control mice had an impact on body weight. In in vitro drug washout studies (Fig. 5), significant activity was observed 72 h after a single 6 h treatment. Future studies should focus on extending this time course following a single treatment to determine the optimal dosing schedule in vitro, as well as testing lower doses of the BochNHO, in vivo.
From these studies, it is clear that prodrugs of this class of compound, given their exquisite potency, only need to reach a specific cellular concentration and duration of exposure to exert their cytotoxic mechanism of action. The studies described herein provide further evidence supporting the approach of developing prodrugs that are selectively activated to release potent chemotherapies by exploiting the tumor microenvironment. However, it is important to note that prodrugs designed to be released inside tumor-specific microenvironments should be tested for in vivo activity in models that have tumors in their relevant organ environment to improve the clinical relevance of these studies.
Materials and Methods
Cell culture
PC3-MM2 human prostate cancer cell line was obtained from MD Anderson Cancer Center and cultured in MEM Eagle media (Sigma Aldrich, M4655) with 10% FBS, MEM vitamins, non-essential amino acids and penicillin/streptomycin (100 IU/mL/100 mg/mL). A549 human lung cancer cell line was obtained from American Type Culture Collection (ATCC) and cultured in F-12K media (ATCC, 30–2004) with 10% FBS and penicillin/streptomycin (100 IU/mL/100 mg/mL). For the in vivo efficacy study, A549 cells were subsequently transfected using a lentivirus containing a FUW-mcherry-luc2 construct. Positively transfected cells were selected using 5 µg/mL puromycin. EMT6 murine breast cancer cell line was obtained from ATCC and cultured in Waymouth’s MB media (Gibco, 11220–035) with 15% FBS and penicillin/streptomycin (100 IU/mL/100 mg/mL). All three cell lines were maintained at 37°C with 5% CO2.
Antiproliferative assay
CellTiter 96 AQueous One Solution (Promega, G3581) was used per manufacturer directions. Cells were seeded into 96 well plates at densities of 3,500, 2,500 and 1,500 PC3-MM2 cells per well; 4,000, 3,000 and 2,000 A549 cells per well; and 2,000, 1,500 and 1,000 EMT6 cells per well for the 24-, 48- and 72-h timepoints, respectively. After a 24-h incubation, the cells were treated with BocNHO or CBI-indole2 for 24, 48 or 72 h. The plates were developed as instructed by the manufacturer protocol and read on the Synergy 4 plate reader (BioTek) at 490 nm. The data was normalized to the absorbance of wells containing media without cells (0%) and vehicle with cells (100%). IC50 values were calculated using GraphPad Prism with nonlinear fit and variable slope curves. Normalized data are presented as the mean ± SEM from two independent MTS assays (n = 2) performed in duplicate.
Trypan blue cytotoxicity
PC3-MM2, A549 and EMT6 cells were seeded at a density of 3 × 105 cells in T25 flasks and incubated for 24 h prior to being treated for the indicated timepoints. All cells (floating and adherent) were collected and pelleted by centrifugation at 250 × g at 4°C. The cells were washed with DPBS (Gibco, 14190–144) and re-pelleted by centrifugation. Viability and total cell counts were determined using the Vi-Cell Cell Viability Analyzer (Beckman Coulter). The results shown are the mean ± SEM from three independent experiments (n = 3). Data was analyzed in GraphPad Prism by unpaired t-test to determine statistical significance.
Annexin V apoptosis
PC3-MM2, A549 and EMT6 cells were seeded at a density of 3 × 105 cells in T25 flasks and incubated for 24 h. The cells were treated for the indicated timepoints, and then both the floating and adherent cells were collected for Annexin V and PI staining using the Annexin V FITC Apoptosis kit (Invitrogen, PHN1018) per the manufacturer instructions. Data for these samples was collected using the LSRII flow cytometer (BD Biosciences) and analyzed with FACSDiva software. The results shown are the mean ± SEM from three independent experiments (n = 3), with the exception of the 1 µM dose where one outlier point existed for each drug and was excluded (n = 2). Data was analyzed in GraphPad Prism by unpaired t-test to determine statistical significance.
Cell cycle
PC3-MM2, A549 and EMT6 cells were seeded at a density of 5 × 105 cells in T75 flasks, then the following day, the cells were serum-starved overnight using media containing 0.25% FBS. Just prior to dosing the cells, the media was replaced again with complete media. Upon treatment of the cells for the indicated timepoints, both the floating and adherent cells were collected and pelleted by centrifugation. The pellet was suspended in DPBS and fixed overnight in an equal portion of ice cold 70% ethanol. The samples were washed twice with DPBS, suspended in a PI solution containing 80 µM PI (Invitrogen, P1304MP), 6.6 mg/L RNase A (Fermentas, EN0531) and 0.5% (v/v) Tween-20 in DPBS and incubated in the dark for 15 min at room temperature. Data for these samples was collected using the LSRII flow cytometer and analyzed with FACSDiva software. The results shown are the mean ± SEM from three independent experiments (n = 3). Data was analyzed further in GraphPad Prism by unpaired t-test to determine statistical significance.
Washout experiments
The washout experiments were performed as described above; however, 6 h after the initiation of drug treatment, the media was decanted from each flask containing cells undergoing drug treatment, the cells were rinsed and fresh media was added to each flask. The cells were incubated in drug-free media for the remainder of the 24- or 72-h timepoint. The results shown are the mean ± SEM from three independent experiments (n = 3). Data was analyzed in GraphPad Prism by unpaired t-test to determine statistical significance.
In vivo orthotopic lung tumor efficacy study of BocNHO
Nu/nu mice (Charles River Laboratories International) were orthotopically inoculated in the left lung with 1 × 106 A549-luc2 (expressing luciferase) cells in PBS and 25% matrigel. Briefly, the animal was anesthetized with pentobarbital (50 mg/kg) and placed in the lateral recumbency. Without opening the thoracic cavity, a small incision was made over the left side of the thorax just behind the lung. Adipose tissue was removed to expose the lung and, using the space between ribs as the access point, the cells (1 × 106 in a volume of 75 µL) were injected into the lung. The incision was closed using surgical staples. Bioluminescence imaging using the IVIS Spectrum (PerkinElmer, Waltham, MA) was performed prior to treatment to determine baseline tumor burden in all the animals on Day 3. Mice were assigned to one of nine treatment groups (10 mice per group): 60, 100, 250 or 500 µg/kg BocNHO, 60, 100, 250 or 500 µg/kg CBI-indole2 or vehicle (DMSO). Treatment was initiated on Day 4, and mice were initially scheduled to receive a total of six doses (Days 4, 8, 12, 16, 20 and 24) by intraperitoneal administration. However, following the third dose (Day 12), gross toxicity was observed in all treatment groups except 60 µg/kg BocNHO, 60 µg/kg CBI-indole2 and vehicle. All mice in these groups either died from drug toxicity or were euthanized due to a moribund condition. Subsequently, the remaining mice received two additional doses at Days 21 and 25 for a total of five doses. The remaining mice on study were sacrificed at Day 32 due to control animals reaching a near-moribund condition. Throughout the study bioluminescence imaging was performed on Days 11, 20 and 32 to determine tumor burden as determined by total flux. Animals were injected with potassium salt of D-Luciferin (150 mg/kg, GoldBio, LUCK-1G). Following isofluorane-induced anesthesia, animals were imaged in the ventral orientation 10 min post-luciferin administration. Images were quantified using Living Image software. Region of interest (ROI) was drawn around the entire body of each animal (excluding the tail). Data from all timepoints (Days 0, 11, 20, 32) were normalized to baseline tumor burden for each individual animal and expressed as percent increase in tumor burden. Tumor burden data was analyzed in GraphPad Prism by unpaired t-test to determine statistical significance. Body weights were collected throughout the study and considered an indicator of drug toxicity along with clinical symptoms. All data are represented as mean ± SEM. Changes in body weight were analyzed by two-way ANOVA using GraphPad Prism to determine statistical significance. The in vivo efficacy study was approved by the University of Kansas Medical Center Institutional Animal Care and Use Committee (IACUC).
Acknowledgments
We would like to thank Dr Andrew L. Kung at the Dana Farber Cancer Institute for providing the FUW-Luc-mCherry-puro construct. We gratefully acknowledge the financial support of the Kansas Bioscience Authority.
Glossary
Abbreviations:
- BocNHO
BocNHO-CBI-indole2
- CBI-indole2
seco-CBI-indole2
- s.c.
subcutaneous
- nM
nanomolar
- pM
picomolar
- PI
propidium iodide
- µM
micromolar
- t0
time zero
Submited
01/02/2013
Revised
03/15/2013
Accepted
03/18/2013
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/24348
References
- 1.Baraldi PG, Bovero A, Fruttarolo F, Preti D, Tabrizi MA, Pavani MG, et al. DNA minor groove binders as potential antitumor and antimicrobial agents. Med Res Rev. 2004;24:475–528. doi: 10.1002/med.20000. [DOI] [PubMed] [Google Scholar]
- 2.Boger DL, Johnson DS. CC-1065 and the duocarmycins: unraveling the keys to a new class of naturally derived DNA alkylating agents. Proc Natl Acad Sci U S A. 1995;92:3642–9. doi: 10.1073/pnas.92.9.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boger DL, Johnson DS, Yun W, Tarby CM. Molecular basis for sequence selective DNA alkylation by (+)- and ent-(-)-CC-1065 and related agents: alkylation site models that accommodate the offset AT-rich adenine N3 alkylation selectivity. Bioorg Med Chem. 1994;2:115–35. doi: 10.1016/S0968-0896(00)82007-6. [DOI] [PubMed] [Google Scholar]
- 4.Boger DLJ, Johnson DS. CC-1065 and the Duocarmycins: Understanding their Biological Function through Mechanistic Studies. Angew Chem Int Ed Engl. 1996;35:1438–74. doi: 10.1002/anie.199614381. [DOI] [Google Scholar]
- 5.Chidester CGD, Krueger WC, Miszak SA, Duchamp DJ, Martin DG. The structure of CC-1065, a potent antitumor agent and its binding to DNA. J Am Chem Soc. 1981;103:7629. doi: 10.1021/ja00415a035. [DOI] [Google Scholar]
- 6.Ichimura M, Ogawa T, Takahashi K, Kobayashi E, Kawamoto I, Yasuzawa T, et al. Duocarmycin SA, a new antitumor antibiotic from Streptomyces sp. J Antibiot (Tokyo) 1990;43:1037–8. doi: 10.7164/antibiotics.43.1037. [DOI] [PubMed] [Google Scholar]
- 7.Igarashi Y, Futamata K, Fujita T, Sekine A, Senda H, Naoki H, et al. Yatakemycin, a novel antifungal antibiotic produced by Streptomyces sp. TP-A0356. J Antibiot (Tokyo) 2003;56:107–13. doi: 10.7164/antibiotics.56.107. [DOI] [PubMed] [Google Scholar]
- 8.MacMillan KS, Boger DL. An additional spirocyclization for duocarmycin SA. J Am Chem Soc. 2008;130:16521–3. doi: 10.1021/ja806593w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parrish JP, Kastrinsky DB, Wolkenberg SE, Igarashi Y, Boger DL. DNA alkylation properties of yatakemycin. J Am Chem Soc. 2003;125:10971–6. doi: 10.1021/ja035984h. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi I, Takahashi K, Ichimura M, Morimoto M, Asano K, Kawamoto I, et al. Duocarmycin A, a new antitumor antibiotic from Streptomyces. J Antibiot (Tokyo) 1988;41:1915–7. doi: 10.7164/antibiotics.41.1915. [DOI] [PubMed] [Google Scholar]
- 11.Tichenor MS, Boger DL. Yatakemycin: total synthesis, DNA alkylation, and biological properties. Nat Prod Rep. 2008;25:220–6. doi: 10.1039/b705665f. [DOI] [PubMed] [Google Scholar]
- 12.Tichenor MS, Kastrinsky DB, Boger DL. Total synthesis, structure revision, and absolute configuration of (+)-yatakemycin. J Am Chem Soc. 2004;126:8396–8. doi: 10.1021/ja0472735. [DOI] [PubMed] [Google Scholar]
- 13.Tichenor MS, MacMillan KS, Trzupek JD, Rayl TJ, Hwang I, Boger DL. Systematic exploration of the structural features of yatakemycin impacting DNA alkylation and biological activity. J Am Chem Soc. 2007;129:10858–69. doi: 10.1021/ja072777z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tichenor MS, Trzupek JD, Kastrinsky DB, Shiga F, Hwang I, Boger DL. Asymmetric total synthesis of (+)- and ent-(-)-yatakemycin and duocarmycin SA: evaluation of yatakemycin key partial structures and its unnatural enantiomer. J Am Chem Soc. 2006;128:15683–96. doi: 10.1021/ja064228j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tietze LF, Krewer B. Novel analogues of CC-1065 and the duocarmycins for the use in targeted tumour therapies. Anticancer Agents Med Chem. 2009;9:304–25. doi: 10.2174/1871520610909030304. [DOI] [PubMed] [Google Scholar]
- 16.Trzupek JD, Gottesfeld JM, Boger DL. Alkylation of duplex DNA in nucleosome core particles by duocarmycin SA and yatakemycin. Nat Chem Biol. 2006;2:79–82. doi: 10.1038/nchembio761. [DOI] [PubMed] [Google Scholar]
- 17.Warpehoski MA, Hurley LH. Sequence selectivity of DNA covalent modification. Chem Res Toxicol. 1988;1:315–33. doi: 10.1021/tx00006a001. [DOI] [PubMed] [Google Scholar]
- 18.Yasuzawa T, Muroi K, Ichimura M, Takahashi I, Ogawa T, Takahashi K, et al. Duocarmycins, potent antitumor antibiotics produced by Streptomyces sp. structures and chemistry. Chem Pharm Bull (Tokyo) 1995;43:378–91. doi: 10.1248/cpb.43.378. [DOI] [PubMed] [Google Scholar]
- 19.Boger DL, Garbaccio RM. Catalysis of the CC-1065 and duocarmycin DNA alkylation reaction: DNA binding induced conformational change in the agent results in activation. Bioorg Med Chem. 1997;5:263–76. doi: 10.1016/S0968-0896(96)00238-6. [DOI] [PubMed] [Google Scholar]
- 20.Boger DLG, Garbaccio RM. Shape-Dependent Catalysis: Insights into the Source of Catalysis for the CC-1065 and Duocarmycin DNA Alkylation Reaction. Acc Chem Res. 1999;32:1043–52. doi: 10.1021/ar9800946. [DOI] [Google Scholar]
- 21.McGovren JP, Clarke GL, Pratt EA, DeKoning TF. Preliminary toxicity studies with the DNA-binding antibiotic, CC-1065. J Antibiot (Tokyo) 1984;37:63–70. doi: 10.7164/antibiotics.37.63. [DOI] [PubMed] [Google Scholar]
- 22.Nagamura S, Asai A, Kobayashi E, Gomi K, Saito H. Studies on duocarmycin SA and its derivatives. Bioorg Med Chem. 1997;5:623–30. doi: 10.1016/S0968-0896(96)00276-3. [DOI] [PubMed] [Google Scholar]
- 23.Wolkenberg SE, Boger DL. Mechanisms of in situ activation for DNA-targeting antitumor agents. Chem Rev. 2002;102:2477–95. doi: 10.1021/cr010046q. [DOI] [PubMed] [Google Scholar]
- 24.Jin W, Trzupek JD, Rayl TJ, Broward MA, Vielhauer GA, Weir SJ, et al. A unique class of duocarmycin and CC-1065 analogues subject to reductive activation. J Am Chem Soc. 2007;129:15391–7. doi: 10.1021/ja075398e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lajiness JP, Robertson WM, Dunwiddie I, Broward MA, Vielhauer GA, Weir SJ, et al. Design, synthesis, and evaluation of duocarmycin O-amino phenol prodrugs subject to tunable reductive activation. J Med Chem. 2010;53:7731–8. doi: 10.1021/jm1010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schein PS, Scheffler B. Barriers to efficient development of cancer therapeutics. Clin Cancer Res. 2006;12:3243–8. doi: 10.1158/1078-0432.CCR-06-0329. [DOI] [PubMed] [Google Scholar]
- 27.Suggitt M, Bibby MC. 50 years of preclinical anticancer drug screening: empirical to target-driven approaches. Clin Cancer Res. 2005;11:971–81. [PubMed] [Google Scholar]
- 28.Schuh JC. Trials, tribulations, and trends in tumor modeling in mice. Toxicol Pathol. 2004;32(Suppl 1):53–66. doi: 10.1080/01926230490424770. [DOI] [PubMed] [Google Scholar]
- 29.Bibby MC. Orthotopic models of cancer for preclinical drug evaluation: advantages and disadvantages. Eur J Cancer. 2004;40:852–7. doi: 10.1016/j.ejca.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 30.Radinsky R, Beltran PJ, Tsan R, Zhang R, Cone RD, Fidler IJ. Transcriptional induction of the melanocyte-stimulating hormone receptor in brain metastases of murine K-1735 melanoma. Cancer Res. 1995;55:141–8. [PubMed] [Google Scholar]
- 31.Gohji K, Nakajima M, Fabra A, Bucana CD, von Eschenbach AC, Tsuruo T, et al. Regulation of gelatinase production in metastatic renal cell carcinoma by organ-specific fibroblasts. Jpn J Cancer Res. 1994;85:152–60. doi: 10.1111/j.1349-7006.1994.tb02076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fabra A, Nakajima M, Bucana CD, Fidler IJ. Modulation of the invasive phenotype of human colon carcinoma cells by organ specific fibroblasts of nude mice. Differentiation. 1992;52:101–10. doi: 10.1111/j.1432-0436.1992.tb00504.x. [DOI] [PubMed] [Google Scholar]
- 33.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 34.Killion JJ, Radinsky R, Fidler IJ. Orthotopic models are necessary to predict therapy of transplantable tumors in mice. Cancer Metastasis Rev. 1998;17:279–84. doi: 10.1023/A:1006140513233. [DOI] [PubMed] [Google Scholar]
- 35.Kerbel RS. Human tumor xenografts as predictive preclinical models for anticancer drug activity in humans: better than commonly perceived-but they can be improved. Cancer Biol Ther. 2003;2(Suppl 1):S134–9. [PubMed] [Google Scholar]
- 36.Hoffman RM. Orthotopic metastatic mouse models for anticancer drug discovery and evaluation: a bridge to the clinic. Invest New Drugs. 1999;17:343–59. doi: 10.1023/A:1006326203858. [DOI] [PubMed] [Google Scholar]
- 37.Fidler IJ. Orthotopic implantation of human colon carcinomas into nude mice provides a valuable model for the biology and therapy of metastasis. Cancer Metastasis Rev. 1991;10:229–43. doi: 10.1007/BF00050794. [DOI] [PubMed] [Google Scholar]
- 38.Fidler IJ. Rationale and methods for the use of nude mice to study the biology and therapy of human cancer metastasis. Cancer Metastasis Rev. 1986;5:29–49. doi: 10.1007/BF00049529. [DOI] [PubMed] [Google Scholar]
- 39.Boger DLaJ. D.S. CC-1065 and the duocarmycins: understanding their biological function through mechanistic studies. In: Int. AC, ed., 1996:1438-74. [Google Scholar]
- 40.Walker DL, Reid JM, Ames MM. Preclinical pharmacology of bizelesin, a potent bifunctional analog of the DNA-binding antibiotic CC-1065. Cancer Chemother Pharmacol. 1994;34:317–22. doi: 10.1007/BF00686039. [DOI] [PubMed] [Google Scholar]
- 41.Li LH, DeKoning TF, Kelly RC, Krueger WC, McGovren JP, Padbury GE, et al. Cytotoxicity and antitumor activity of carzelesin, a prodrug cyclopropylpyrroloindole analogue. Cancer Res. 1992;52:4904–13. [PubMed] [Google Scholar]
- 42.Weiland KL, Dooley TP. In vitro and in vivo DNA bonding by the CC-1065 analogue U-73975. Biochemistry. 1991;30:7559–65. doi: 10.1021/bi00244a027. [DOI] [PubMed] [Google Scholar]
- 43.Pitot HC, Reid JM, Sloan JA, Ames MM, Adjei AA, Rubin J, et al. A Phase I study of bizelesin (NSC 615291) in patients with advanced solid tumors. Clin Cancer Res. 2002;8:712–7. [PubMed] [Google Scholar]
- 44.Pavlidis N, Aamdal S, Awada A, Calvert H, Fumoleau P, Sorio R, et al. Carzelesin phase II study in advanced breast, ovarian, colorectal, gastric, head and neck cancer, non-Hodgkin’s lymphoma and malignant melanoma: a study of the EORTC early clinical studies group (ECSG) Cancer Chemother Pharmacol. 2000;46:167–71. doi: 10.1007/s002800000134. [DOI] [PubMed] [Google Scholar]
- 45.Alberts SR, Erlichman C, Reid JM, Sloan JA, Ames MM, Richardson RL, et al. Phase I study of the duocarmycin semisynthetic derivative KW-2189 given daily for five days every six weeks. Clin Cancer Res. 1998;4:2111–7. [PubMed] [Google Scholar]
- 46.Shamdas GJ, Alberts DS, Modiano M, Wiggins C, Power J, Kasunic DA, et al. Phase I study of adozelesin (U-73,975) in patients with solid tumors. Anticancer Drugs. 1994;5:10–4. doi: 10.1097/00001813-199402000-00002. [DOI] [PubMed] [Google Scholar]
- 47.Ducry L, Stump B. Antibody-drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjug Chem. 2010;21:5–13. doi: 10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]