

Abstract
In budding yeast, commitment to DNA replication during the normal cell cycle requires degradation of the cyclin-dependent kinase (CDK) inhibitor Sic1. The G1 cyclin-CDK complexes Cln1-Cdk1 and Cln2-Cdk1 initiate the process of Sic1 removal by directly catalyzing Sic1 phosphorylation at multiple sites. Commitment to DNA replication during meiosis also appears to require Sic1 degradation, but the G1 cyclin-CDK complexes are not involved. It has been proposed that the meiosis-specific protein kinase Ime2 functionally replaces the G1 cyclin-CDK complexes to promote Sic1 destruction. To investigate this possibility, we compared Cln2-Cdk1 and Ime2 protein kinase activities in vitro. Both enzyme preparations were capable of catalyzing phosphorylation of a GST-Sic1 fusion protein, but the phosphoisomers generated by the two activities had significantly different electrophoretic mobilities. Furthermore, mutation of consensus CDK phosphorylation sites in Sic1 affected Cln2-Cdk1- but not Ime2-dependent phosphorylation. Phosphoamino acid analysis and phosphopeptide mapping provided additional evidence that Cln2-Cdk1 and Ime2 targeted different residues within Sic1. Examination of other substrates both in vitro and in vivo also revealed differing specificities. These results indicate that Ime2 does not simply replace G1 cyclin-CDK complexes in promoting Sic1 degradation during meiosis.
Keywords: cyclin-dependent kinase, G1 cyclin, meiosis, phosphorylation, replication protein A, Sic1
1. Introduction
Progression into the DNA synthetic (S) phase of the cell cycle is a critical transition that is often defective in cancer cells. Studies in a variety of systems have revealed that precise regulation of cyclin-dependent kinase (CDK) activation controls S phase entry. In the budding yeast Saccharomyces cerevisiae, many of the molecular details underlying this regulation have been elucidated. Central components are the cyclin-CDK complexes Clb5- and Clb6-Cdk1 [1]. These protein kinases are held inactive in G1 by Sic1, a CDK inhibitor that is specific for B-type cyclin-CDK complexes [2]. At the G1/S transition, the G1 cyclin-CDK complexes Cln1- and Cln2-Cdk1 catalyze Sic1 phosphorylation at multiple CDK consensus phosphorylation sites to promote Sic1 removal through ubiquitin-mediated proteolysis [3–7]. Consequently, the Clb5- and Clb6-Cdk1 complexes become active and DNA replication ensues. These Cdk1 complexes also contribute to prevention of DNA re-replication (see [8] for review), and loss of this control ultimately activates a Mec1-dependent checkpoint pathway that responds to genotoxic stress [9].
DNA replication is not limited to proliferating cells, but also occurs during meiosis as a precursor to the two nuclear divisions that lead to generation of haploid gametes. As is the case during the mitotic cell cycle, Clb5, Clb6, and Cdk1 are required for proper meiotic S phase entry [10–12]. It is likely that meiotic Clb5- and Clb6-Cdk1 complexes act similarly to mitotic Clb5- and Clb6-Cdk1 complexes in promoting DNA replication. Whether they are also involved in preventing DNA re-replication during meiosis is not known. However, overexpression of Clb5 during meiosis stimulates DNA re-replication [13], a phenotype that would not be expected based on mitotic studies. As in proliferating cells, regulation of Clb5- and Clb6-Cdk1 activities in early meiosis appears to involve Sic1 because it disappears as cells enter meiotic S phase [10]. However, the G1 cyclins inhibit meiotic entry [14] and Cdk1 itself is not required for Sic1 removal [12]. Therefore, a different mechanism must operate to relieve Sic1 inhibition during meiosis. A simple scenario is that a meiosis-specific kinase replaces the G1 cyclin-Cdk1 complexes, catalyzing phosphorylation of the same sites to initiate Sic1 degradation. It has been proposed that the protein kinase Ime2 fulfills this role [10].
Ime2 is induced early in meiosis and is required for normal meiotic progression into S phase [15–17]. Ime2 and Cdk1 have been placed in the same general group of related protein kinases based on sequence analysis of their catalytic domains [18], perhaps suggesting that the two enzymes recognize common motifs. In the absence of Ime2, Sic1 does not disappear during meiosis; furthermore, the meiotic S phase entry defect of Ime2-deficient cells is rescued by deletion of Sic1 [10]. In addition, over-expression of a Sic1 mutant that is altered at several Cdk1 consensus phosphorylation sites prevents meiotic S phase entry [11]. Therefore, considerable evidence supports the hypothesis that Ime2 serves as a G1 cyclin-Cdk1 replacement during meiosis. However, we have recently found that Ime2 catalyzes phosphorylation of a non-Cdk1 consensus phosphorylation site in replication protein A (RPA) [19]. This observation prompted us to determine whether Ime2 catalyzes Sic1 phosphorylation in a manner similar to Cln2-Cdk1. We have found that the two enzymes display different specificities, indicating that other possible mechanisms underlying meiotic Sic1 disappearance should be considered.
2. Materials and methods
2.1. Enzyme induction
Yeast strains capable of expressing tandem fusion tagged (TFT) versions of Cln2 and Ime2 were purchased from Open Biosystems. In these cells, a GAL1 promoter drives gene expression from a URA3-containing high copy plasmid leading to generation of protein with tandem hexa-histidine (6xHis), hemagglutinin (HA), and protein A tags at the C terminus [20]. All yeast growth was conducted at 30°C. Cells were inoculated into synthetic complete medium lacking uracil containing 2% raffinose at a low density and incubated overnight until an A600 of ~1.0 was reached. Nutrient rich media was then added to bring final concentrations of yeast extract to 1%, peptone to 2%, and either glucose or galactose to 2%. Incubation was continued to achieve an approximate doubling in population.
2.2. Extract preparation and fractionation
Native crude extracts were prepared by bead beating. Harvested cell pellets were suspended in lysis buffer (LyB) (50 mM Tris-Cl pH 7.5, 0.1% NP-40, 10% glycerol, 250 mM NaCl, 0.1 mM PMSF, 1 mg/ml leupeptin, 1 mg/ml pepstatin A), and an approximately equal volume of acid-washed glass beads (Sigma) was added. The cells were disrupted by vortexing a total of 10 times for 20 sec each, with 1 min intermittent incubations on ice. Resulting lysates were clarified by centrifugation at 18,000 x g for 10 min at 4°C. Extracts were fractionated in batch with Talon Metal Affinity Resin (BD Biosciences) using imidazole for elution. Protein concentrations of lysates and purified fractions were measured by the Bradford assay [21] (Bio-Rad) with bovine serum albumin as the standard. Western blot analyses were conducted using the fluorescence-scanning Odyssey system and its associated software (LiCor). Primary antibodies included HA-11 monoclonal (Covance), rabbit anti Rfa2 polyclonal [22], and rat anti α-tubulin polyclonal (Serotec). Signals were generated with IRDye 800-conjugated goat anti-mouse or -rabbit secondary antibodies (Rockland) or Alexa Fluor 680 goat anti-rat secondary antibody (Invitrogen).
2.3. Sic1 generation and purification
Plasmids encoding Sic1 derivatives with HA and 6xHis tags at the C–terminus were kindly provided by Raymond Deshaies. One version, referred to here simply as Sic1, is degraded properly during vegetative growth; the other, referred to here as Sic1ΔP, is resistant to degradation during vegetative growth due to mutations of multiple Cdk1-targeted phosphorylation sites [5]. Both Sic1 and Sic1ΔP contain a T2A mutation, while Sic1ΔP contains additional T5GP, S33A, and S76A mutations. The two SIC1 genes were individually cloned into pIVEX-GST (Roche) for production of glutathione-S-transferase (GST) fusion proteins. Versions encoding GST-Sic1 mutated from serine to alanine at positions corresponding to Sic1 145 (S145A) or 201 (S201A) were generated from the pIVEX-GST construct containing SIC1 using QuikChange (Stratagene) methodology. The pIVEX-GST-based plasmids were introduced into E. coli BL21(DE3) and expression of the GST-tagged Sic1 derivatives was induced in these cells by exposure to IPTG. Purification was accomplished through glutathione-agarose (Pierce) affinity chromatography using glutathione for elution followed by Mono Q (Pharmacia) ion-exchange chromatography. Protein concentrations were measured as described above.
2.4. Protein and peptide kinase assays
Phosphorylation reactions designed to compare Cln2-Cdk1 and Ime2 activities in vitro were conducted with solid phase enzyme fractions and soluble substrates. For experiments shown in Figs. 2–4, a set of Talon eluates (see above) were prepared from the crude extracts analyzed in Fig. 1A and mixed with IgG Sepharose (Amersham) beads in LyB at 3.5 mg protein per ml of beads for 2 hr at 4°C. For the experiments shown in Figs. 5 and 6, Talon eluates were prepared from different crude extracts than those used previously. Based on analysis of enzyme induction and histone H1 kinase activity, we elected to maintain the above conditions for generation of the Cln2TFT-IgG fraction while incubating the Ime2TFT Talon eluate with IgG Sepharose at either 7 mg (Fig. 5 and 6) or 14 mg (Figs. 5) protein per ml beads. In all cases, the beads were then washed twice with LyB and twice with 10 mM HEPES pH 7.4, 10 mM MgCl2, 1 mM DTT. Protein kinase assays were conducted with 5 μl washed beads in a final volume of 20 μl containing 15 mM HEPES pH 7.4, 15 mM MgCl2, 12.5 μM ATP, and ~2.5 μCi [γ-32P]ATP. Where indicated, either 1 μg of calf thymus histone H1 (Sigma), GST-Sic1, GST-Sic1 P, GST-Sic1S145A, or GST-Sic1S201A was added. The samples were incubated at 30°C for 40 min and then subjected to electrophoresis through a 10% SDS-polyacrylamide gel. Proteins were stained with GelCode Blue Stain Reagent (Pierce), and incorporation of radioactive phosphate was detected by phosphorimaging analysis of the dried gel. Similar reaction conditions were used to test casein kinase II -catalyzed phosphorylation of the GST-Sic1 fusion proteins except that soluble enzyme (New England BioLabs) was used and samples were incubated at 30°C for 10 min. For peptide kinase assays, 5 μl washed beads were combined with 40 μl buffer containing 10 mM HEPES pH 7.4, 10 mM MgCl2, 1 mM DTT, 100 μM ATP, ~0.2 μCi [γ-32P]ATP, and, where indicated, 1 mM peptide SRPGSGER or peptide SRPGAGER (EZBiolab). Samples were incubated at 30°C for 30 min and the reactions were stopped by addition of 5 μl 0.5M EDTA. Incorporation of radioactive phosphate into peptides was monitored through liquid scintillation spectrometry of peptides bound to phosphocellulose paper (see ref. [23]).
Fig. 2.
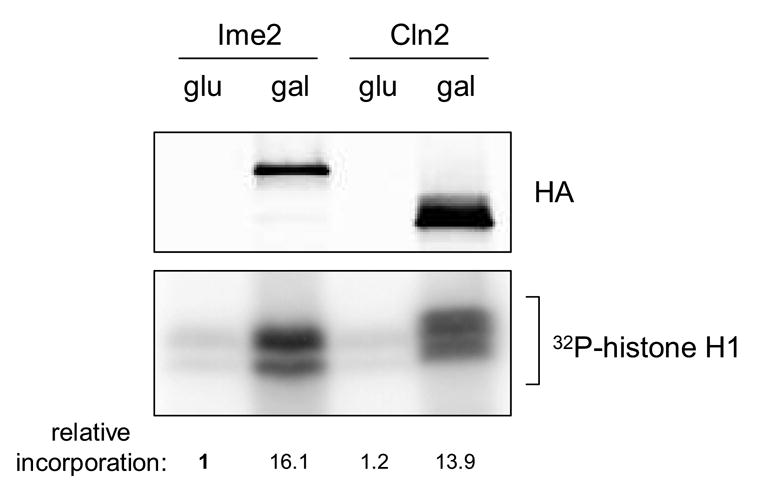
Histone H1 kinase activity. Protein kinase assays were conducted with purified histone H1 serving as substrate. Enzyme fractions were prepared from cells harboring plasmid encoding either Ime2TFT or Cln2TFT grown under non-inducing (glu) or inducing (gal) conditions. Relative levels of Ime2TFT and Cln2TFT employed in the kinase assays were analyzed through separate western blot analysis of the enzyme fractions (upper panel). Incorporation of radioactive phosphate into H1 was detected and quantified by phosphorimaging analysis (lower panel). Values shown indicate relative incorporation into histone H1 and have been normalized to the sample containing the uninduced Ime2 enzyme fraction (lane 1).
Fig. 4.
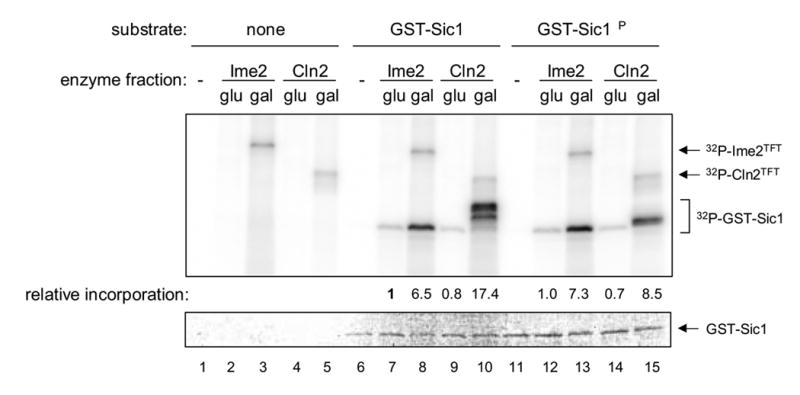
Sic1 kinase activity. Purified GST-tagged versions of Sic1 and Sic1ΔP were tested as substrates of the Ime2TFT and Cln2TFT (gal) fractions and the control (glu) fractions that had been tested for histone H1 kinase activity. Incorporation of radioactive phosphate was detected and quantified by phosphorimager analysis (upper panel). Values shown indicate relative incorporation into Sic1 derivatives and have been normalized to the sample containing the uninduced Ime2 enzyme fraction and GST-Sic1 (lane 7). Higher molecular weight radioactive species are presumed to be phosphorylated Ime2TFT and Cln2TFT, as shown. GST-Sic1 content in these samples was visualized by Coomassie staining (lower panel).
Fig. 1.
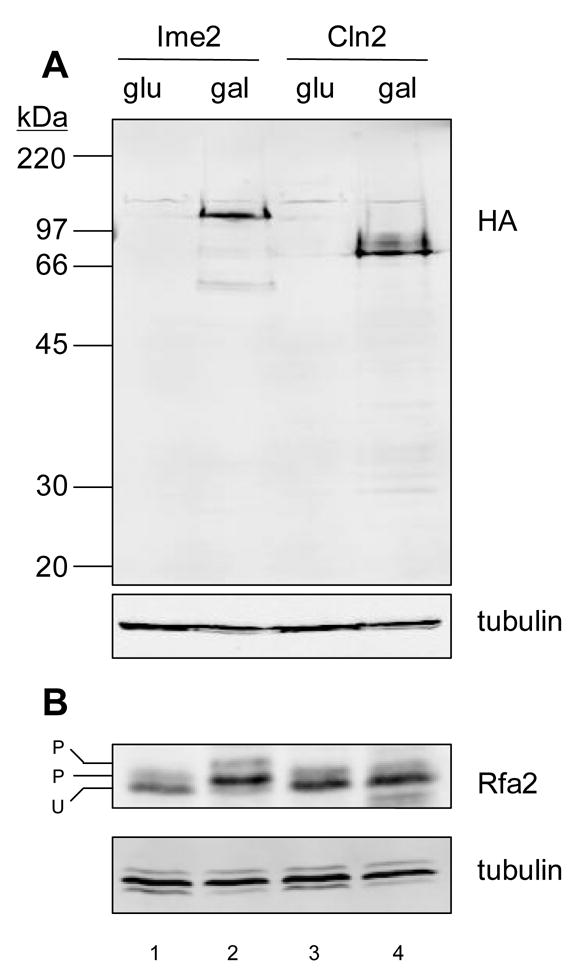
Protein expression. (A) Western blot analysis of Ime2TFT and Cln2TFT using antibody directed against HA. Crude extracts were prepared from cells grown in glucose (glu) or alternatively in galactose (gal), which induced expression of the tagged enzymes. The same membrane was probed with anti-tubulin for loading comparison. (B) Western blot analysis of Rfa2. The same extracts used in A were analyzed for Rfa2 using specialized gel conditions to resolve unphosphorylated (U) and phosphorylated (P) species [27]. As in A, the same membrane was probed with anti-tubulin for loading comparison.
Fig. 5.
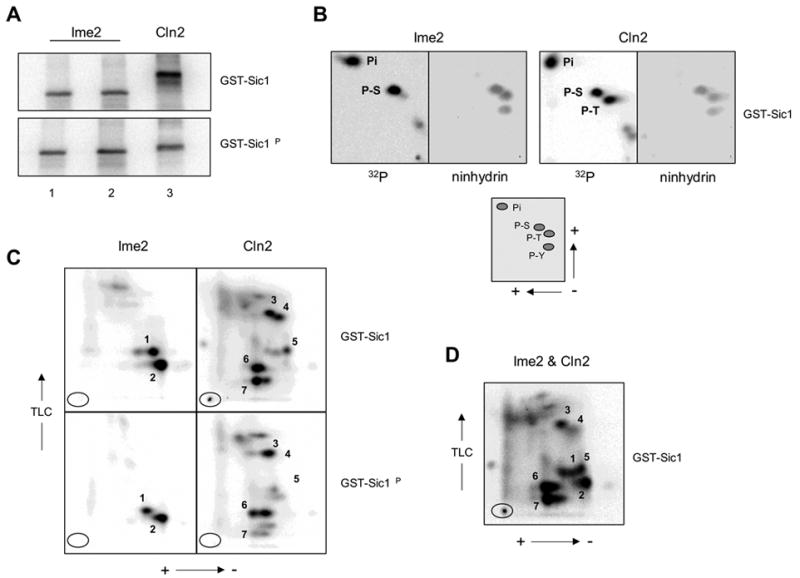
Physical analysis of phosphorylated Sic1 derivatives. (A) Phosphorylated substrates. GST-Sic1 and GST-Sic1ΔP phosphorylation reactions were conducted in vitro with either Ime2TFT or Cln2TFT fractions. Phosphoproducts were transferred to a PVDF membrane and visualized by phosphorimaging. For Ime2TFT, two concentrations of enzyme were used (lane 2 > lane 1; see Materials and methods) with slight increases in reaction efficiency. Phosphorylated substrates generated through the two Ime2TFT-mediated reactions were pooled for subsequent analysis. (B) Phosphoamino acid analyses of Ime2TFT- and Cln2TFT-Cdk1-generated phospho-GST-Sic1. The positions of the phosphoamino acid standards included with the samples during the two-dimensional separation were determined by ninhydrin staining, and the presence of 32P-labeled phosphoamino acids was determined by phosphorimaging. The lower panel indicates the directions of electrophoresis and the relative positions of inorganic phosphate (Pi), phosphoserine (P-S), phosphothreonine (P-T), and phosphotyrosine (P-Y). (C) Phosphopeptide maps of phospho-GST-Sic1 and phospho-GST-Sic1ΔP generated through Ime2TFT or Cln2TFT-Cdk1 catalysis. Tryptic fragments were separated in two dimensions and visualized by phosphorimaging. The numbers 1–7 refer to the predominant radiolabeled tryptic fragments present in the GST-Sic1 samples (see text for details). Ovals indicate origins and directions of electrophoresis and TLC are shown. (D) Phosphopeptide map of a sample containing tryptic fragments from both the Ime2TFT- and Cln2TFT-Cdk1-generated phospho-GST-Sic1 populations analyzed in C.
Fig. 6.
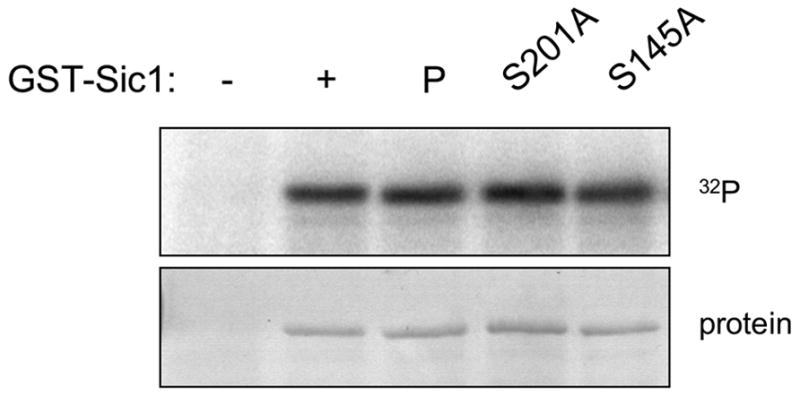
Phosphorylation of additional Sic1 mutants. GST-Sic1S145A and -Sic1S201A were tested as substrates of Ime2TFT in vitro. Radioactive incorporation was detected by phosphorimaging analysis (upper panel) of the stained gel (lower panel).
2.5. Phosphoamino acid analysis and phosphopeptide mapping
Two dimensional phosphoamino acid analysis and phosphopeptide mapping were conducted according to previously described protocols [24, 25]. Sic1 phosphoisomers generated in vitro were subjected to electrophoresis, transferred to a polyvinylidene fluoride membrane, and excised. The membranes were then incubated with 0.5% polyvinyl pyrrolidone in 100 mM acetic acid for 30 min at 37°C and washed extensively. The Sic1 phosphoisomers were then digested with 5 μg of sequencing grade modified trypsin (Promega) in 50 mM ammonium bicarbonate for 2 hr at 37°C and with an additional 5 μg of trypsin overnight. Eluted peptides were oxidized with performic acid and subsequently washed with pH 1.9 thin layer chromatography (TLC) buffer (see [24]). Aliquots of the samples were used for phosphamino acid analysis. Peptides were hydrolyzed in 6N HCl at 111°C for 1 hr and phosphoamino acids were resolved on cellulose TLC plates (Merck) by electrophoresis in two dimensions. 32P-labeled amino acids were visualised by phosphorimaging and identified by reference to ninhydrin-stained phosphoamino acid standards. Separate aliquots were used for phosphopeptide mapping. Peptides were spotted on cellulose TLC plates and separated using the Hunter thin layer electrophoresis system (CBS Scientific) in pH 1.9 buffer at 1250 V for 30 min. The plates were dried overnight and subjected to second dimension chromatographic separation in a phospho-chromatography buffer (see [24]). The plates were dried and phosphopeptide spots were visualized by phosphorimaging.
3. Results and discussion
To compare Cln2-Cdk1 and Ime2 kinase activities, we employed identically tagged versions of Cln2 and Ime2 to generate fractions enriched for these and associated proteins (see Materials and methods). Plasmids encoding these proteins (Cln2TFT and Ime2TFT) were harbored in the same strain background, and expression was controlled by the galactose-inducible GAL1 promoter. Western blot analysis revealed comparable levels of the two identically tagged proteins specifically in cells exposed to galactose (Fig. 1A). Consistent with our earlier studies [26], we observed extensive phosphorylation of the RPA middle subunit (Rfa2) upon induction of Ime2TFT. In uninduced cells, a low level of phosphorylated Rfa2 was observed (Fig. 1B, lanes 1 and 3). We have previously shown that Mec1 catalyzes Rfa2 phosphorylation during the normal cell cycle to generate this species [27]. Ime2TFT induction generated a predominant Ime2-dependent phosphorylated form of Rfa2 and a second Ime2- and Mec1-dependent form (Fig. 1B, lane 2). Such an effect was not observed upon induction of Cln2TFT (Fig. 1B, lane 4). It is noted that the version of Cln2 that we employed in these studies was fully functional based on our in vitro studies (see below). We recently reported that Ime2 directly catalyzes Rfa2 phosphorylation at a single site that is not typically recognized by CDK complexes, and that mutation of this site abolishes the alteration in Rfa2 mobility that accompanies Ime2 induction [19]. Our new results here suggest that Cln2-Cdk1 does not recognize this site in vivo, a conclusion that is supported by further data presented below.
Cln2TFT and Ime2TFT were purified from crude cell extracts through a two step procedure that took advantage of the tandem tags (see Materials and methods). The tagged proteins in the final fraction could not be detected by silver staining, but relative levels were compared by western blot analysis (Fig. 2, upper panel). For control purposes, identical fractionation was conducted with extracts from cells in which expression was not induced. The isolates containing either Cln2TFT or Ime2TFT exhibited significant histone H1 kinase activity (Fig. 2, lower panel). We repeatedly noticed a difference in the electrophoretic migration of the phosphorylated histone H1 products that were generated by Cln2TFT- and Ime2TFT-associated activities. These results suggest a disparity in the specificity of the two enzymes, given that the overall level of histone H1 phosphorylation that we observed was comparable in the two cases.
Because Rfa2 was differentially phosphorylated in vivo upon expression of Cln2TFT or Ime2TFT, we elected to examine the activities of the two enzymes using a peptide substrate relevant to Rfa2. We showed previously that the peptide SRPGSGER containing the sequence immediately surrounding Rfa2 serine 27 (S27) serves as a substrate of Ime2 while the “mutant” peptide SRPGAGER does not [19]. We tested these two peptides as substrates using the same fractions that were employed in the protein kinase assays with histone H1 and found that only the Ime2TFT-containing fraction catalyzed phosphorylation in an S27-dependent manner (Fig. 3). These results indicate that the S27 peptide does not serve as a general protein kinase substrate and provide further evidence that Cln2-Cdk1 and Ime2 recognize different sequences. It is important to add that Rfa2 S27 is recognized by Ime2 during meiosis [19]. Therefore, the Ime2TFT that we purified from mitotic cells behaved like meiotic Ime2 with respect to at least one of its natural substrates.
Fig. 3.
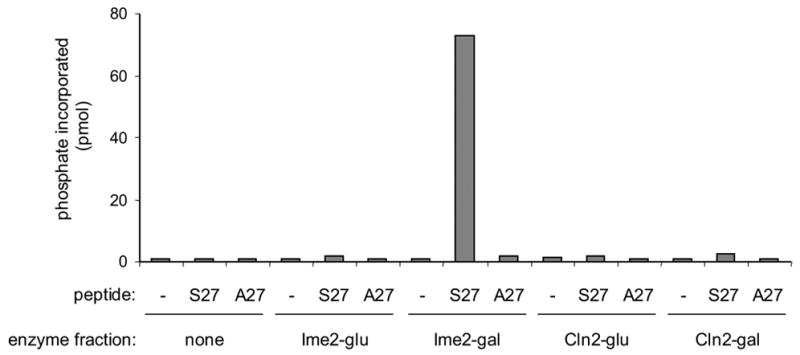
Peptide kinase activity. Enzyme fractions from the same extracts employed in the histone H1 kinase assay were tested for their ability to catalyze phosphorylation of a peptide derived from Rfa2 (S27) or its mutant derivative (A27).
To specifically determine whether Ime2 is capable of replacing Cln2-Cdk1 as a Sic1 kinase, we tested the two enzyme preparations for Sic1 phosphorylation in vitro. For this purpose, we employed two purified GST versions of Sic1: one derived from a Sic1 protein that is degraded efficiently in vegetative cells (GST-Sic1), and the other derived from a Sic1 protein that is not degraded in vegetative cells due to multiple alterations of Cdk1-targeted phosphorylation sites (GST-Sic1ΔP) (see ref. [5] and Materials and methods). Neither kinase preparation catalyzed appreciable phosphorylation of GST alone (data not shown). However, we found that Cln2TFT-associated activity catalyzed GST-Sic1 phosphorylation, leading to multiple phosphoisomers with reduced electrophoretic mobility (Fig. 4, lane 10). In contrast, GST-Sic1ΔP was phosphorylated with 50% efficiency compared to GST-Sic1 and the resulting GST-Sic1ΔP phosphoisomers had significantly higher electrophoretic mobility on average than the GST-Sic1 phoshoisomers (Fig. 4, lane 15). These differences in degree and character of Cln2TFT-mediated phosphorylation were reasonable given that the Sic1 portion of the GST-Sic1 derivative contains eight CDK consensus sites while the Sic1ΔP portion of the GST-Sic1ΔP derivative contains five. The Ime2TFT-containing fraction also catalyzed GST-Sic1 phosphorylation, but the change in electrophoretic mobility characteristic of the Cln2TFT-mediated reaction was not observed (Fig. 4, lane 8). Furthermore, the extent of the Ime2TFT-mediated reaction was less than that of the Cln2TFT-mediated reaction despite the similar histone H1 kinase activities of the two enzyme preparations (see Fig. 2). Strikingly, Ime2TFT-dependent phosphorylation of GST-Sic1ΔP was nearly identical to that of GST-Sic1 (Fig. 4, lane 13), indicating that mutation of multiple CDK consensus sites did not significantly alter recognition by Ime2TFT or an associated protein kinase. These results suggest that Ime2 directly catalyzes Sic1 phosphorylation but at a different subset of sites than those recognized by Cln2-Cdk1.
To pursue the hypothesis that the two enzymes recognize different sites within Sic1, we further analyzed phosphorylated GST-Sic1 and GST-Sic1ΔP products. Phosphoisomers were generated in vitro (Fig. 5A) and isolated as described in Materials and methods. Partial acid hydrolysis of Cln2-Cdk1-catalyzed phospho-GST-Sic1 yielded phosphoserine and phosphothreonine in nearly equal proportions, as expected (Fig. 5B, upper right). However, the same treatment of Ime2-generated phospho-GST-Sic1 yielded phosphoserine almost exclusively (Fig. 5B, upper left). Phosphopeptide mapping revealed entirely different patterns for the two phosphorylated products. Tryptic digestion of Cln2-Cdk1-catalyzed phospho-GST-Sic1 led to a complex map (Fig. 5C, upper right), as would be expected with a protein phosphorylated at numerous sites, while tryptic digestion of Ime2-catalyzed phospho-GST-Sic1 led to only two predominant phosphorylated species (Fig. 5C, upper left). Comparison of the GST-Sic1 maps with the GST-Sic1ΔP maps revealed obvious differences with Cln2TFT but not with Ime2TFT. For example, species 4, 5 and 7 generated through Cln2-Cdk1 catalysis were abolished or greatly diminished by mutation of Cdk1-targeted phosphorylation sites (Fig. 5C, right panels), whereas species 1 and 2 generated through Ime2 catalysis appeared in similar proportions regardless of Sic1 status (Fig. 5C, left panels). We generated an additional phosphopeptide map in which the GST-Sic1 phosphopeptides resulting from the two kinase reactions were mixed (Fig. 5D). Comparison of these data with the original maps (Fig. 5C, upper panels) clearly indicates that species 2 was unique to the Ime2TFT sample. It is conceivable that species 1, generated through Ime2 catalysis, and species 5, generated through Cln2-Cdk1 catalysis, comigrated. However, species 5 was abolished by mutation of Cdk1 phosphorylation sites, while species 1 was not (Fig. 5C). Therefore, we would argue that species 1 and 5 were distinct phosphopeptides. Taking these physical data together, we suggest that Cln2-Cdk1 and Ime2 catalyze phosphorylation of different sites within Sic1.
Our results with Sic1 and Rfa2 indicate that Cdk1 and Ime2 have different specificities. This result is consistent with the data from a proteomic analysis in which Cln2-Cdk1 and Ime2 were found to catalyze phosphorylation of largely non-overlapping sets of proteins [28]. However, our results are not consistent with a very recent report, published during the course of our studies, in which Ime2 and Cln2-Cdk1 are reported to recognize overlapping sets of residues within Sic1 in vitro [29]. At this point, we can only speculate as to the underlying cause of these disparate results, but it could involve differences in the nature of the reactions, including the versions of Ime2 and/or Sic1 that were employed, differences in the method of analysis, or both. As a first step in identifying the serines that were phosphorylated in our system, we purified GST-Sic1 containing Sic1 S145A or S201A mutations. These two residues were characterized as Ime2 targets in vitro in the aforementioned study, along with three threonines [29], and we expected that S145 would be recognized by Ime2 based on our experiments with Rfa2 [19]. However, neither mutation affected Ime2TFT-dependent GST-Sic1 phosphorylation in vitro (Fig. 6). Consistent with published results [30], the S201A mutation virtually abolished casein kinase II-catalyzed GST-Sic1 phosphorylation (data not shown). Therefore, S201 was accessible but not recognized by Ime2 in our system. Further experiments will be required to determine whether S145 can be characterized similarly. Regardless, our results suggest that Ime2 can catalyze phosphorylation of Sic1 serine residues that were not previously identified as targets in vitro.
The differences that we have uncovered between Cdk1 and Ime2, particularly with respect to Sic1 phosphorylation, indicate that a current model regarding meiotic destruction of Sic1 should be modified. It has been reported that meiosis-specific expression of Sic1ΔP prevents meiotic DNA replication [11], suggesting that the general mechanism of Sic1 degradation is similar during the cell cycle and during meiosis except that different kinases catalyze Sic1 phosphorylation. One possibility, as proposed previously in the literature [10], is that Ime2 is the Sic1 kinase that promotes Sic1 degradation. However, our results indicate that Ime2 and Cdk1 recognize different sites in Sic1. If Sic1 phosphorylation is in fact important for its degradation and the same sites are involved as during the cell cycle, we suggest that another protein kinase downstream of Ime2 directly catalyzes Sic1 phosphorylation during meiosis. This protein kinase would share the Cdk1 specificity and thereby serve as an effective G1 cyclin-Cdk1 replacement in early meiosis. Future experiments will be aimed at exploring this possibility and precisely defining the mechanism by which Ime2 promotes proper S phase entry in meiosis.
Acknowledgments
We thank Raymond Deshaies for providing reagents, Afreen Siddiqui and Andrea Beebe for expert technical assistance, and Grant Brown for reviewing the manuscript. This work was supported by grant GM061860 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schwob E, Nasmyth K. CLB5 and CLB6, a new pair of B cyclins involved in DNA replication in Saccharomyces cerevisiae. Genes Dev. 1993;7:1160–1175. doi: 10.1101/gad.7.7a.1160. [DOI] [PubMed] [Google Scholar]
- 2.Schwob E, Bohm T, Mendenhall MD, Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell. 1994;79:233–244. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- 3.Schneider BL, Yang QH, Futcher AB. Linkage of replication to start by the Cdk inhibitor Sic1. Science. 1996;272:560–562. doi: 10.1126/science.272.5261.560. [DOI] [PubMed] [Google Scholar]
- 4.Tyers M. The cyclin-dependent kinase inhibitor p40SIC1 imposes the requirement for Cln G1 cyclin function at Start. Proc Natl Acad Sci USA. 1996;93:7772–7776. doi: 10.1073/pnas.93.15.7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verma R, Annan RS, Huddleston MJ, Carr SA, Reynard G, Deshaies RJ. Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science. 1997;278:455–460. doi: 10.1126/science.278.5337.455. [DOI] [PubMed] [Google Scholar]
- 6.Feldman RM, Correll CC, Kaplan KB, Deshaies RJ. A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell. 1997;91:221–230. doi: 10.1016/s0092-8674(00)80404-3. [DOI] [PubMed] [Google Scholar]
- 7.Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, Sicheri F, Pawson T, Tyers M. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature. 2001;414:514–521. doi: 10.1038/35107009. [DOI] [PubMed] [Google Scholar]
- 8.Diffley JFX. Regulation of early events in chromosome replication. Current Biology. 2004;14:R778–R786. doi: 10.1016/j.cub.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 9.Archambault V, Ikui AE, Drapkin BJ, Cross FR. Disruption of mechanisms that prevent rereplication triggers a DNA damage response. Mol Cell Biol. 2005;25:6707–6721. doi: 10.1128/MCB.25.15.6707-6721.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dirick L, Goetsch L, Ammerer G, Byers B. Regulation of meiotic S phase by Ime2 and a Clb5,6-associated kinase in Saccharomyces cerevisiae. Science. 1998;281:1854–1857. doi: 10.1126/science.281.5384.1854. [DOI] [PubMed] [Google Scholar]
- 11.Stuart D, Wittenberg C. CLB5 and CLB6 are required for premeiotic DNA replication and activation of the meiotic S/M checkpoint. Genes Dev. 1998;12:2698–2710. doi: 10.1101/gad.12.17.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benjamin KR, Zhang C, Shokat KM, Herskowitz I. Control of landmark events in meiosis by the CDK Cdc28 and the meiosis-specific kinase Ime2. Genes Dev. 2003;17:1524–1539. doi: 10.1101/gad.1101503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strich R, Mallory MJ, Jarnik M, Cooper KF. Cyclin B-Cdk activity stimulates meiotic rereplication in budding yeast. Genetics. 2004;167:1621–1628. doi: 10.1534/genetics.104.029223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colomina N, Gari E, Gallego C, Herrero E, Aldea M. G1 cyclins block the Ime1 pathway to make mitosis and meiosis incompatible in budding yeast. EMBO J. 1999;18:320–329. doi: 10.1093/emboj/18.2.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith HE, Mitchell AP. A transcriptional cascade governs entry into meiosis in Saccharomyces cerevisiae. Mol Cell Biol. 1989;9:2142–2152. doi: 10.1128/mcb.9.5.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshida M, Kawaguchi H, Sakata Y, Kominami K, Hirano M, Shima H, Akada R, Yamashita I. Initiation of meiosis and sporulation in Saccharomyces cerevisiae requires a novel protein kinase homologue. Mol Gen Genet. 1990;221:176–186. doi: 10.1007/BF00261718. [DOI] [PubMed] [Google Scholar]
- 17.Foiani M, Nadjar-Boger E, Capone R, Sagee S, Hashimshoni T, Kassir Y. A meiosis-specific protein kinase, Ime2, is required for the correct timing of DNA replication and for spore formation in yeast meiosis. Mol Gen Genet. 1996;253:278–288. doi: 10.1007/s004380050323. [DOI] [PubMed] [Google Scholar]
- 18.Hunter T, Plowman GD. The protein kinases of budding yeast: six score and more. Trends in Biochemical Sciences. 1997;22:18–22. doi: 10.1016/s0968-0004(96)10068-2. [DOI] [PubMed] [Google Scholar]
- 19.Clifford DM, Stark KE, Gardner KE, Hoffmann-Benning S, Brush GS. Mechanistic insight into the Cdc28-related protein kinase Ime2 through analysis of replication protein A phosphorylation. Cell Cycle. 2005;4:1826–1833. doi: 10.4161/cc.4.12.2214. [DOI] [PubMed] [Google Scholar]
- 20.Gelperin DM, White MA, Wilkinson ML, Kon Y, Kung LA, Wise KJ, Lopez-Hoyo N, Jiang L, Piccirillo S, Yu H, Gerstein M, Dumont ME, Phizicky EM, Snyder M, Grayhack EJ. Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev. 2005;19:2816–2826. doi: 10.1101/gad.1362105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 22.Brush GS, Kelly TJ. Phosphorylation of the replication protein A large subunit in the Saccharomyces cerevisiae checkpoint response. Nucleic Acids Res. 2000;28:3725–3732. doi: 10.1093/nar/28.19.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casnellie JE. Assay of protein kinases using peptides with basic residues for phosphocellulose binding. Methods Enzymol. 1991;200:115–120. doi: 10.1016/0076-6879(91)00133-h. [DOI] [PubMed] [Google Scholar]
- 24.Boyle WJ, van der Geer P, Hunter T. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 1991;201:110–149. doi: 10.1016/0076-6879(91)01013-r. [DOI] [PubMed] [Google Scholar]
- 25.Luo K, Hurley T, Sefton B. Cyanogen bromide cleavage and proteolytic peptide mapping of proteins immoblized to membranes. Methods Enzymol. 1991;201:149–152. doi: 10.1016/0076-6879(91)01014-s. [DOI] [PubMed] [Google Scholar]
- 26.Clifford DM, Marinco SM, Brush GS. The meiosis-specific protein kinase Ime2 directs phosphorylation of replication protein A. J Biol Chem. 2004;279:6163–6170. doi: 10.1074/jbc.M306943200. [DOI] [PubMed] [Google Scholar]
- 27.Brush GS, Morrow DM, Hieter P, Kelly TJ. The ATM homologue MEC1 is required for phosphorylation of replication protein A in yeast. Proc Natl Acad Sci USA. 1996;93:15075–15080. doi: 10.1073/pnas.93.26.15075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ptacek J, Devgan G, Michaud G, Zhu H, Zhu X, Fasolo J, Guo H, Jona G, Breitkreutz A, Sopko R, McCartney RR, Schmidt MC, Rachidi N, Lee SJ, Mah AS, Meng L, Stark MJR, Stern DF, De Virgilio C, Tyers M, Andrews B, Gerstein M, Schweitzer B, Predki PF, Snyder M. Global analysis of protein phosphorylation in yeast. Nature. 2005;438:679–684. doi: 10.1038/nature04187. [DOI] [PubMed] [Google Scholar]
- 29.Sedgwick C, Rawluk M, Decesare J, Raithatha SA, Wohlschlegel J, Semchuk P, Ellison M, Yates JR, III, Stuart DT. Saccharomyces cerevisiae Ime2 phosphorylates Sic1 at multiple PXS/T sites but is insufficient to trigger Sic1 degradation. Biochem J. 2006;399:151–160. doi: 10.1042/BJ20060363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coccetti P, Rossi RL, Sternieri F, Porro D, Russo GL, di Fonzo A, Magni F, Vanoni M, Alberghina L. Mutations of the CK2 phosphorylation site of Sic1 affect cell size and S-Cdk kinase activity in Saccharomyces cerevisiae. Molecular Microbiology. 2004;51:447–460. doi: 10.1046/j.1365-2958.2003.03836.x. [DOI] [PubMed] [Google Scholar]