

It has been proposed that during the budding of COPII vesicles from transitional ER (tER) sites, Sec16 plays two distinct roles: negatively regulating COPII turnover and organizing COPII assembly. New data suggest that Sec16 does not in fact organize COPII and that regulation of COPII turnover can explain the influence of Sec16 on tER sites.
Abstract
During the budding of coat protein complex II (COPII) vesicles from transitional endoplasmic reticulum (tER) sites, Sec16 has been proposed to play two distinct roles: negatively regulating COPII turnover and organizing COPII assembly at tER sites. We tested these ideas using the yeast Pichia pastoris. Redistribution of Sec16 to the cytosol accelerates tER dynamics, supporting a negative regulatory role for Sec16. To evaluate a possible COPII organization role, we dissected the functional regions of Sec16. The central conserved domain, which had been implicated in coordinating COPII assembly, is actually dispensable for normal tER structure. An upstream conserved region (UCR) localizes Sec16 to tER sites. The UCR binds COPII components, and removal of COPII from tER sites also removes Sec16, indicating that COPII recruits Sec16 rather than the other way around. We propose that Sec16 does not in fact organize COPII. Instead, regulation of COPII turnover can account for the influence of Sec16 on tER sites.
INTRODUCTION
Transport of proteins from the endoplasmic reticulum (ER) to the Golgi is mediated by coat protein complex II (COPII)–coated vesicles (Barlowe, 1994; Barlowe and Miller, 2013). This pathway begins when the transmembrane guanine nucleotide exchange factor Sec12 recruits and activates the small GTPase Sar1, which in turn recruits the Sec23/24 complex to capture cargo and form the inner layer of the COPII coat (Barlowe and Schekman, 1993; Bi et al., 2002; Miller et al., 2003). Subsequently Sec13/31 polymerizes to form the outer layer of the coat and produce a vesicle (Fath et al., 2007; Stagg et al., 2008; Russell and Stagg, 2010). Shedding of the coat requires GTP hydrolysis, a reaction that is catalyzed by Sec23 and stimulated by Sec31 (Yoshihisa et al., 1993; Antonny et al., 2001; Bi et al., 2007). These events take place at ribosome-free transitional ER (tER) sites (also known as ER exit sites) that are surrounded by rough ER (Palade, 1975; Bannykh and Balch, 1997). The exploration of how tER sites remain distinct from the rough ER is providing insight into the phenomenon of membrane domain formation (Pfeffer, 2003). Moreover, tER sites in many eukaryotes are functionally and physically linked to Golgi stacks, so studies of tER organization are clarifying the mechanisms of Golgi biogenesis (Budnik and Stephens, 2009; Glick and Nakano, 2009).
In Saccharomyces cerevisiae, tER sites are small and numerous, but in the related budding yeast Pichia pastoris, a typical cell contains just two to five discrete tER sites, each of which is associated with a Golgi stack (Rossanese et al., 1999; Bevis et al., 2002; Shindiapina and Barlowe, 2010). P. pastoris is therefore a good experimental system for studying tER organization. A tER site in P. pastoris is ∼0.4 μm in diameter and produces multiple COPII vesicles (Mogelsvang et al., 2003). Four-dimensional (4D) video microscopy revealed that tER sites in P. pastoris are long-lived structures that form de novo, grow to a steady-state size, and fuse with one another (Bevis et al., 2002). When a fusion event creates an unusually large tER site, that site eventually shrinks back to the steady-state size. Similar behavior has been seen with mammalian tER sites (Hammond and Glick, 2000; Stephens, 2003). These observations can be explained by assuming that tER sites are maintained by a balance between growth and shrinkage (Bevis et al., 2002; Heinzer et al., 2008). Growth occurs when individual tER sites capture COPII components from the cytosol or from the rough ER and when tER sites fuse. Shrinkage occurs when budding vesicles carry COPII components out of tER sites and when GTP hydrolysis by Sar1 recycles COPII components to the cytosol.
A test of this model will require characterizing the molecules that define tER sites. Toward this end, we previously used a microscopy-based genetic screen to isolate a thermosensitive P. pastoris strain that had an altered tER pattern at 36.5°C (Connerly et al., 2005). Compared to wild-type cells, the mutant cells had smaller and more numerous tER sites, and Sec12 was delocalized from tER sites to the general ER. Those phenotypes were due to a point mutation in the SEC16 gene. The mutation caused a Pro‑1092-to-Leu (P1092L) amino acid change. Residue 1092 lies in a central conserved domain found in all known Sec16 homologues (Connerly et al., 2005; Bhattacharyya and Glick, 2007; Ivan et al., 2008). Subsequent work revealed that Sec16 homologues in mammals and Drosophila are also important for normal tER structure (Watson et al., 2006; Bhattacharyya and Glick, 2007; Iinuma et al., 2007; Ivan et al., 2008; Hughes et al., 2009).
Sec16 is essential for ER-to-Golgi transport in vivo and is concentrated at tER sites, although it is less abundant at tER sites than COPII coat proteins (Kaiser and Schekman, 1990; Espenshade et al., 1995; Connerly et al., 2005; Watson et al., 2006; Bhattacharyya and Glick, 2007; Iinuma et al., 2007; Ivan et al., 2008). This protein is relatively large—242 kDa in S. cerevisiae and 281 kDa in P. pastoris—and two-hybrid and biochemical assays identified interactions of S. cerevisiae Sec16 with the COPII coat proteins Sec23, Sec24, and Sec31 (Espenshade et al., 1995; Gimeno et al., 1996; Shaywitz et al., 1997) and with Sec12 (Montegna et al., 2012) and the Sec12 homologue, Sed4 (Gimeno et al., 1995). In addition, Sec16 interacts with Sec13 (Hughes et al., 2009; Whittle and Schwartz, 2010) and Sar1 (Nakano and Muramatsu, 1989; Supek et al., 2002; Ivan et al., 2008; Yorimitsu and Sato, 2012).
Because of its large size and multiple interaction partners, Sec16 has been proposed to act as a scaffold at tER sites (Shaywitz et al., 1997; Ivan et al., 2008; Hughes et al., 2009; Shindiapina and Barlowe, 2010). A protein scaffold binds other components and concentrates them in a specific region of the cell. In the case of Sec16, various kinds of scaffolding activity can be envisioned. A simple version of the scaffold model postulates that Sec16 links COPII coat proteins to accessory proteins, thereby tethering the accessory proteins in the vicinity of COPII vesicles. Indeed, Sec16 plays such a role in concentrating Sec12 at tER sites in P. pastoris and possibly in mammalian cells (Soderholm et al., 2004; Montegna et al., 2012). Animal cell Sec16 also binds the ER export factor TFG‑1 (Witte et al., 2011). More elaborate versions of the scaffold model postulate that Sec16 organizes COPII assembly. This idea emerged from our finding that the sec16‑P1092L mutation caused an apparent fragmentation of the tER (Connerly et al., 2005). Because P. pastoris Sec16 showed a saturable tER association, we suggested that Sec16 might bind to nascent COPII vesicles and cross-link them to form tER sites (Connerly et al., 2005). Others have gone further, proposing that Sec16 associates with the ER membrane upstream of COPII coat proteins (Ivan et al., 2008; Hughes et al., 2009; Yorimitsu and Sato, 2012). There have also been suggestions that Sec16 associates with the ER membrane upstream of Sar1, although the data in this regard are conflicting (Watson et al., 2006; Iinuma et al., 2007; Ivan et al., 2008). Finally, structural analysis of a Sec16 domain led to the idea that Sec16 is a template for assembling the COPII coat (Whittle and Schwartz, 2010). In these versions of the scaffold model, Sec16 recruits COPII to tER sites. The notion that Sec16 somehow organizes COPII has become broadly accepted (Budnik and Stephens, 2009; Barlowe and Miller, 2013; Lord et al., 2013).
Meanwhile, biochemical data suggest that Sec16 serves as a regulator. Sec16 retards GTP hydrolysis by Sar1, thereby slowing COPII turnover and stabilizing the COPII coat (Supek et al., 2002; Kung et al., 2011; Yorimitsu and Sato, 2012). These findings explain why Sec16 is dispensable in vitro for COPII vesicle budding if GTP hydrolysis by Sar1 is inhibited (Matsuoka et al., 1998; Supek et al., 2002). Thus Sec16 has been proposed to have two distinct functions: negatively regulating COPII turnover and organizing COPII assembly at tER sites.
Here we provide in vivo evidence that Sec16 is a negative regulator of COPII turnover, and we argue that this regulatory function is sufficient to explain the influence of Sec16 on tER sites. The P1092L point mutation destabilizes P. pastoris Sec16 and causes most of the Sec16 molecules to redistribute to the cytosol. Loss of Sec16 does not displace COPII coat proteins from tER sites but instead greatly accelerates tER-site formation and shrinkage in a process that requires Sar1 activity. Conversely, when COPII coat proteins are removed from tER sites using the anchor-away method (Haruki et al., 2008), Sec16 is also displaced. Our data suggest that Sec16 is recruited to tER sites, where it acts mainly to regulate rather than organize COPII.
RESULTS
P. pastoris Sec16 contains two essential regions
We began by dissecting P. pastoris Sec16 into functional domains. Our previous sequence alignments identified a central conserved domain (CCD; residues 1030–1459) that has a counterpart in every known Sec16 homologue, as well as a conserved C‑terminal region (CTR; residues 2392–2550; Connerly et al., 2005; Bhattacharyya and Glick, 2007; Ivan et al., 2008). P. pastoris Sec16 also contains 13 nearly perfect repeats of a glutamine-rich decapeptide (residues 1829–1958). These tentative domain assignments suggested boundaries for deleting portions of the protein. The experimental approach was to mutate one copy of SEC16 in a diploid while tagging the mutant allele with an ARG4 marker. This diploid was then sporulated to determine whether Arg+ haploids could be obtained.
In agreement with earlier studies of S. cerevisiae Sec16 (Espenshade et al., 1995), the CTR was essential (Figure 1A). A second sequence upstream of the CCD was also found to be essential (Figure 1A). Alignment of this upstream region of Sec16 from various yeast species revealed several conserved peptides (Supplemental Figure S1). We therefore designated the second essential sequence the upstream conserved region (UCR; residues 500–868). Although the UCR as a whole is essential, cells can tolerate separate deletions of two subregions of the UCR (residues 500–647 or 649–868), implying some redundancy of function within this region.
FIGURE 1:

Domain analysis of P. pastoris Sec16. (A) Summary of the effects of Sec16 partial deletions on cell viability. Top, the UCR (residues 500–868), CCD (residues 1030–1459), a nonconserved glutamine-rich region (Q; residues 1829–1958), and the conserved CTR (residues 2392–2550). The endogenous SEC16 gene was replaced with alleles containing the indicated deletions. Right, the ability of each mutant allele to support growth. Only the deletions marked in red caused a loss of viability. (B) Differential effects of a CCD point mutation and a CCD deletion. The indicated mutations were introduced at the SEC16 locus by gene replacement in a strain expressing Sec13-GFP. Cultures were grown at 23°C, and then half of each culture was shifted to 36.5°C for 1 h. Cells were imaged by differential interference contrast and fluorescence microscopy. Scale bar, 5 μm. (C) Yeast two-hybrid analysis of Sec16-COPII interactions. The “prey” vector encoded the indicated fragments of P. pastoris Sec16, and the “bait” vector encoded the indicated full-length P. pastoris COPII coat proteins. Growth on plates lacking histidine reflects an interaction. With this system, the UCR can self-activate when used as bait, so constructs containing the UCR were tested only as prey. The other constructs gave the same results when the Sec16 fragments were used as bait and the COPII coat proteins were used as prey (unpublished data).
Surprisingly, even though the thermosensitive P1092L mutation lies within the CCD and causes tER dispersal at 36.5°C, deletion of the CCD did not impair growth or alter tER organization at either 23 or 36.5°C (Figure 1B). As will be discussed later, this result indicates that P1092L is not a simple loss-of-function mutation for the CCD.
The conserved regions of Sec16 interact with COPII coat proteins
As the next step in the analysis, we used a yeast two-hybrid system (James, 2001) to examine interactions of the conserved regions of P. pastoris Sec16 with COPII coat proteins (Figure 1C). Interaction was observed between the CTR and Sec23. This result fits with previous studies of S. cerevisiae Sec16 (Espenshade et al., 1995; Yorimitsu and Sato, 2012) and mammalian Sec16A (Bhattacharyya and Glick, 2007). Interactions of Sec24 and Sec31 with S. cerevisiae Sec16 have been reported (Gimeno et al., 1996; Shaywitz et al., 1997; Yorimitsu and Sato, 2012) using various Sec16 fragments that we would describe as overlapping part or all of the UCR. Our tests revealed that the UCR of P. pastoris Sec16 interacts with Sec24 and Sec31. In addition, we detected a novel interaction of Sec23 with the UCR. This result was unexpected because a previous study reported that fragments from the first half of S. cerevisiae Sec16 did not interact with Sec23 (Gimeno et al., 1996). We found that in a two-hybrid analysis of S. cerevisiae proteins, the UCR showed a weak but reproducible interaction with Sec23 (Supplemental Figure S2), although this interaction remains to be confirmed by biochemical analysis. Thus the UCR interacts with at least two and possibly three different COPII coat proteins.
In the two-hybrid assay, the P. pastoris CCD interacted only with Sec13 (Figure 1C). We had predicted this interaction from a structural homology search performed with Phyre, an online protein homology/analogy recognition engine (Kelley and Sternberg, 2009). A subsequent crystallography study confirmed that the S. cerevisiae CCD interacts with Sec13 to form an ancestral coatomer element 1 structure (Whittle and Schwartz, 2010). The CCD of mammalian Sec16A also binds Sec13 (Hughes et al., 2009), so this interaction is conserved.
The UCR is the main determinant of Sec16 localization
For mammalian and Drosophila Sec16 homologues, a region upstream of the CCD is important for tER localization (Bhattacharyya and Glick, 2007; Ivan et al., 2008; Hughes et al., 2009). To determine whether the UCR of P. pastoris Sec16 has the same function, various portions of Sec16 were fused to green fluorescent protein (GFP) and expressed in cells that also contained untagged wild-type Sec16 and DsRed-tagged Sec13 (Figure 2). As a control, full-length Sec16 localized to tER sites, although some of the Sec16 puncta were significantly displaced from the COPII puncta (arrows in Figure 2). A construct lacking the UCR was primarily in the cytosol. The UCR alone conferred clear tER localization but also yielded substantial cytosolic fluorescence. A construct consisting of the UCR plus the CCD localized to tER sites as efficiently as full-length Sec16. Similar results were obtained with mammalian Sec16A and Sec16B (Hughes et al., 2009; Budnik et al., 2011; D. Bhattacharyya, personal communication) and with Drosophila Sec16 (Ivan et al., 2008). Thus a conserved property of Sec16 is that a region upstream of the CCD is necessary for full tER localization but sufficient only for partial tER localization. Full tER localization can be obtained by fusing this upstream region to the CCD.
FIGURE 2:
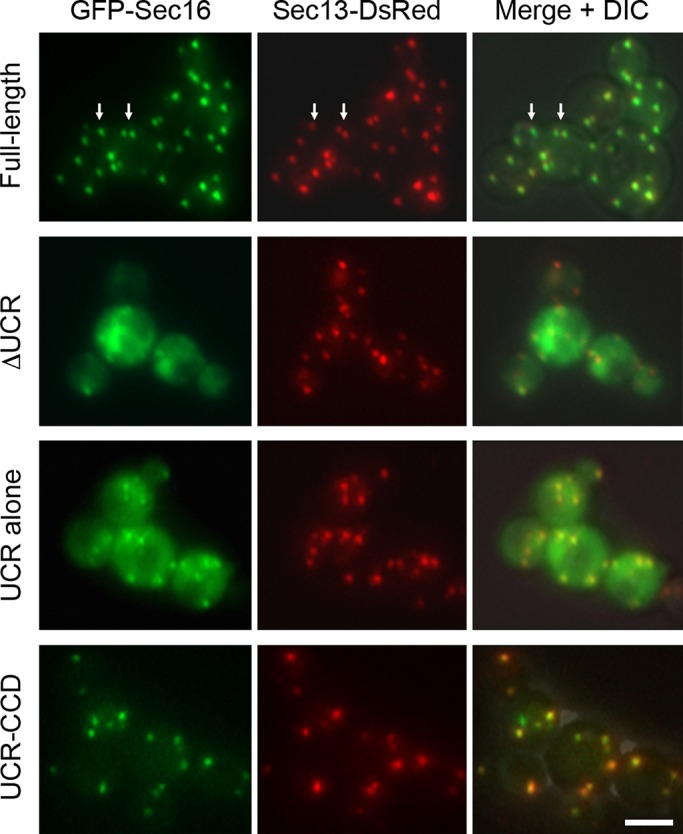
Localization of GFP-tagged fragments of P. pastoris Sec16. These fusions were expressed from the AOX1 promoter as second copies in a P. pastoris strain expressing both wild-type Sec16 and Sec13-DsRed. GFP was fused to the N‑terminus of full-length Sec16, Sec16 lacking residues 500–868 (ΔUCR), residues 500–868 only (UCR alone), or residues 500–1459 only (UCR‑CCD). Expression of the GFP fusions was induced by shifting to methanol medium for 3 h, and then the cells were imaged by fluorescence microscopy. Right, merged fluorescence and differential interference contrast images. Arrows show examples of full-length GFP-Sec16 puncta that do not entirely colocalize with Sec13-DsRed puncta. Scale bar, 5 μm.
The role of the CCD in promoting tER localization has been mysterious. We speculated that dimerization of the CCD might be relevant. The CCD forms an antiparallel dimer (Whittle and Schwartz, 2010), so fusion of the CCD to the UCR could enhance the functional affinity of the UCR for tER sites. To test this model, we replaced the CCD in a UCR-CCD construct with either wild-type FK506-binding protein (FKBP), which is monomeric, or the FKBP(F36M) mutant, which forms an antiparallel dimer (Rollins et al., 2000). The UCR-FKBP construct was mostly cytosolic, whereas the UCR-FKBP(F36M) construct showed robust tER localization (Figure 3). The combined data suggest that the UCR is the main tER localization determinant of Sec16 but that the isolated UCR has only a weak affinity for tER sites. Stronger tER localization can be obtained by dimerizing the UCR via linkage to the CCD. However, as described later, the CCD is not actually needed for tER localization in the context of full-length Sec16, implying that other interactions of Sec16 can supplement the weak tER affinity of the UCR. We conclude that specific tER localization activity resides in the UCR but not the CCD.
FIGURE 3:
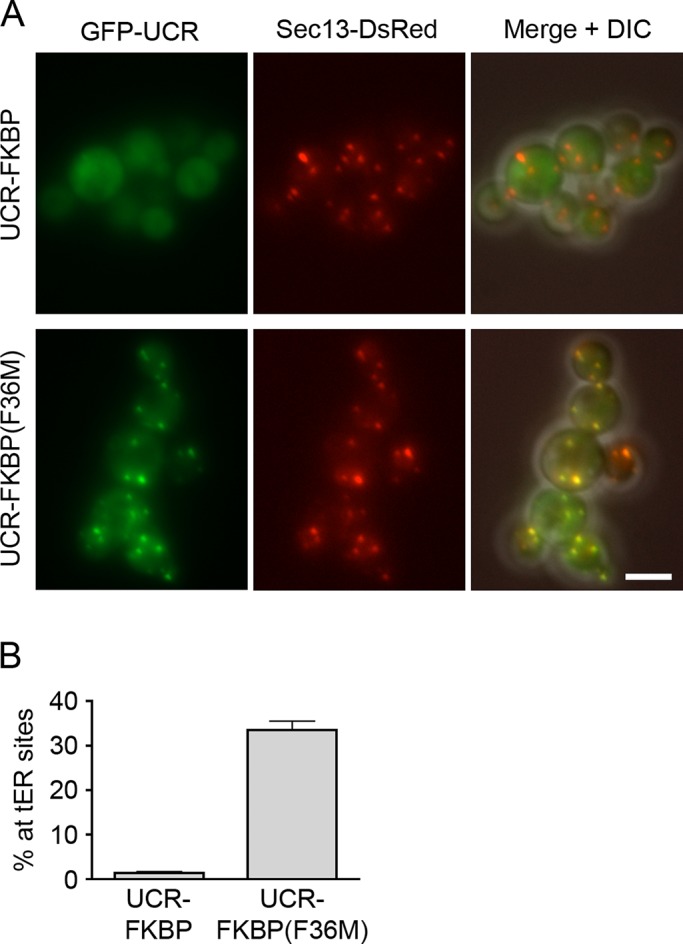
tER localization of a dimerized UCR. (A) Replacement of the CCD with monomeric or dimeric FKBP. A GFP-tagged UCR-CCD construct was modified to replace the CCD with either wild-type monomeric FKBP or the dimeric FKBP(F36M) mutant. Localization to Sec13-DsRed-labeled tER sites was then evaluated as in Figure 2. Scale bar, 5 μm. (B) Quantitation of the data from A. The percentage of the total GFP signal at punctate tER sites was measured. Plotted are mean and SEM.
Mutations that disrupt the CCD-Sec13 interaction structurally destabilize the CCD
The P1092L point mutation lies within the CCD. Based on a structural comparison with the S. cerevisiae CCD‑Sec13 complex, the P1092L mutation is unlikely to affect dimerization of the CCD, but could potentially disrupt the CCD-Sec13 interface (see Figure S3 of Whittle and Schwartz, 2010). Indeed, a yeast two-hybrid analysis suggested that binding of Sec13 to the CCD is weakened by the P1092L mutation (Figure 4A).
FIGURE 4:

Destabilization of P. pastoris Sec16 by mutations that weaken Sec13 binding. (A) Two-hybrid analysis of CCD-Sec13 interactions. The wild-type (WT) CCD of P. pastoris Sec16, or a mutant CCD carrying the P1092L or I1075D mutation was tested against P. pastoris Sec13 in a yeast two-hybrid screen (James, 2001). Where indicated, Sec13 was used as the “bait” and the CCD as the “prey” or vice versa. Interactions were detected by growth on a plate lacking histidine (–His) or, in a more stringent test, a plate lacking histidine and adenine (–His –Ade). (B) Solubility of wild-type and mutant CCD variants. In E. coli, a C-terminally FLAG-tagged wild-type or mutant CCD was either expressed alone or coexpressed with C‑terminally S peptide–tagged Sec13. The cells were lysed in detergent and centrifuged, and equivalent amounts of the pellet (P) and supernatant (S) fractions were analyzed by SDS–PAGE, followed by immunoblotting with anti-FLAG antibody. (C) Effects of CCD mutations on tER site number. For strains expressing Sec13‑GFP plus either WT or a mutant Sec16, cultures were grown at 23°C, and then a portion of each culture was shifted for 2 h to 36.5°C. The tER sites were counted in ∼100 cells from each sample. Plotted are mean and SEM.
A weakened CCD-Sec13 interaction apparently destabilizes the structure of the CCD. This effect can be seen by expressing the CCD in Escherichia coli and then fractionating the cell lysate (Figure 4B). The CCD expressed alone is found in the pellet (P), but the CCD coexpressed with Sec13 is soluble in the supernatant (S). When the CCD carries the P1092L mutation, about half of the bacterially expressed CCD molecules are in the pellet even when Sec13 is coexpressed (Figure 4B). We infer that when Sec13 is not tightly bound, the CCD becomes prone to aggregation.
This interpretation was tested by creating a separate point mutation to disrupt the CCD-Sec13 interaction. The CCD includes a β-stranded “blade,” referred to here as CCDβ, that inserts into Sec13 to complete the β-propeller fold (Whittle and Schwartz, 2010). We introduced a point mutation into CCDβ. Such a mutation should not affect CCD dimerization, which is mediated by the α‑helical portion of the CCD (Whittle and Schwartz, 2010). To identify suitable residues for mutation, we fused P. pastoris Sec13 by a linker to CCDβ from P. pastoris and determined the crystal structure of the fusion protein (Supplemental Figure S3A). Several mutations predicted to weaken the CCDβ-Sec13 interaction were introduced into SEC16 by gene replacement. The I1075D mutation was chosen for further study because it yielded viable cells. As expected, introduction of the I1075D mutation into the CCD reduced binding of Sec13 in a two-hybrid assay (Figure 4A) and also reduced solubility of CCD coexpressed with Sec13 in E. coli (Figure 4B). A P. pastoris strain carrying the sec16‑I1075D allele showed tER dispersal at 36.5°C similar to that seen with the sec16‑P1092L allele (Figure 4C and Supplemental Figure S3B). Thus mutations that weaken CCD-Sec13 interaction destabilize the CCD and alter tER structure.
How does a destabilized CCD affect Sec16 activity? The sec16‑P1092L mutation does not reduce cellular levels of the mutant Sec16 (Connerly et al., 2005). Instead, fluorescence microscopy revealed that the Sec16-P1092L protein was largely displaced from tER sites to the cytosol (Connerly et al., 2005; Figure 5). This effect was not due to disrupted CCD function because a mutant Sec16 protein lacking the CCD showed strong tER localization (Figure 5). We conclude that the P1092L mutation destabilizes the CCD and leads to mislocalization of Sec16, thereby causing thermosensitive growth even though the CCD is nonessential. Thus, in the sec16‑P1092L mutant, the altered tER structure is due to loss of Sec16 from tER sites.
FIGURE 5:
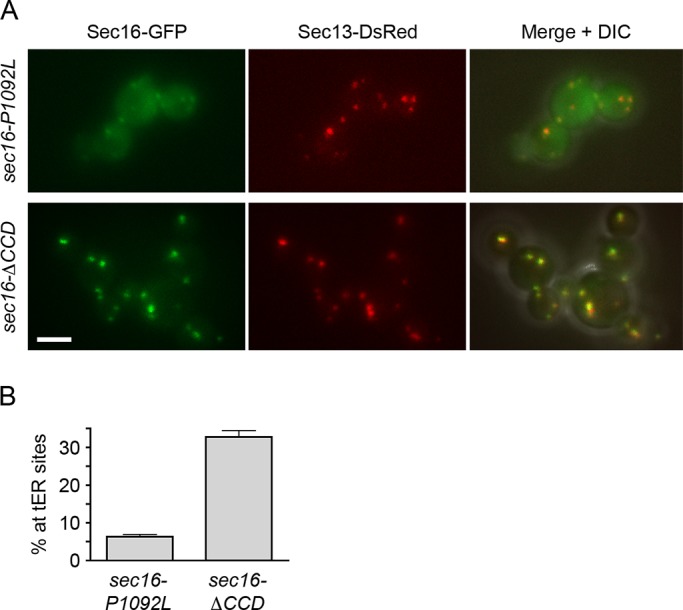
Redistribution of Sec16 to the cytosol by the P1092L mutation but not by deletion of the entire CCD. (A) Localization of Sec16 mutants. GFP-tagged Sec16‑P1092L or Sec16-ΔCCD was expressed from the SEC16 promoter in cells that also expressed wild-type Sec16 as well as Sec13-DsRed. Cells were grown at 30°C, then imaged by differential interference contrast (DIC) and fluorescence microscopy. Right, merged fluorescence and DIC images. Scale bar, 5 μm. (B) Quantitation of the data from A. The percentage of the total GFP signal at punctate tER sites was measured. Plotted are mean and SEM.
tER dynamics is accelerated in a sec16-P1092L strain
To understand how loss of Sec16 causes tER dispersal, we built on our earlier proposal that tER sites maintain a steady-state size by balancing growth with shrinkage (Bevis et al., 2002). This model suggested that the P1092L mutation might accelerate shrinkage of tER sites. As an experimental test, we took 4D movies at 36.5°C of wild-type or sec16‑P1092L cells expressing the tER marker Sec13‑GFP.
Our analysis focused on tER sites that had just undergone fusion and were therefore poised to shrink back to the steady-state size. In wild-type cells the shrinkage of newly fused tER sites was gradual, often requiring 30 min or more to reach completion (Bevis et al., 2002; Supplemental Figure S4A and Supplemental Movie S1). By contrast, in sec16‑P1092L cells the shrinkage was typically complete within 1–2 min (Figure 6A, Supplemental Figure S4A, and Supplemental Movie S2). tER sites in sec16‑P1092L cells sometimes disappeared (Supplemental Figure S4A), a phenomenon that we never observed in wild-type cells. We analyzed ∼20 fusion events for each strain. In wild-type cells the average half-time for shrinkage was 11 min, whereas in sec16‑P1092L cells it was only 20 s (Figure 6B). Faster shrinkage reduced the size of tER sites, which had fluorescence intensities 5.6-fold lower on average in sec16‑P1092L cells than in wild-type cells (Supplemental Figure S4B).
FIGURE 6:

Altered dynamics of tER sites in sec16‑P1092L mutant cells. (A) Shrinkage of tER sites after fusion events in sec16‑P1092L cells. A sec16‑P1092L strain expressing Sec13‑GFP was grown at room temperature, then warmed to 36.5°C for ∼45 min before imaging by 4D microscopy at 36.5°C. Shown are merged fluorescence and differential interference contrast (DIC) images of representative cells. The arrowhead marks a pair of tER sites that underwent fusion followed by shrinkage. Frames are taken from Supplemental Movie S2, and the time from the beginning of the movie is shown in minutes:seconds format. Scale bar, 5 μm. (B) Quantitation of tER site shrinkage in wild-type and sec16‑P1092L cells. From 4D movies of the type shown in A, ∼20 newly fused tER sites were chosen at random for WT or sec16‑P1092L cells. The half-times for shrinkage were determined from plots of the type shown in Supplemental Figure S4A. Each dot represents an individual fused tER site, and the horizontal lines represent the average half-times. (C) Quantitation of de novo tER site formation in wild-type and sec16‑P1092L cells. The cells expressed Sec13-GFP, and, where indicated, they also expressed Sar1(T34N) from the inducible AOX1 promoter. Cultures were shifted to inducing methanol medium for 3 h at room temperature, grown at 36.5°C for an additional 50 min, and then imaged by 4D microscopy at 36.5°C for either 40 min for wild-type cells or 10 min for sec16‑P1092L cells. For each culture, the number of de novo tER site formation events was recorded for ∼20 cells. Plotted are the hourly mean and SEM values. (D) Prevention of tER dispersal by expression of Sar1(T34N). Wild-type or sec16‑P1092L cells expressing Sec13-GFP were transformed with a control vector or a vector encoding Sar1(T34N) expressed from an inducible promoter. Cultures were shifted to inducing methanol medium for 2.5 h at room temperature and then grown at 36.5°C for an additional 1 h before imaging. Shown are merged fluorescence and DIC images of representative cells. Scale bar, 5 μm. (E) Quantitation of the results from D. For each of the four conditions, the tER sites were counted in ∼50 cells. Plotted are mean and SEM. (F) Cytosolic localization of Sec16-P1092L in the presence of Sar1(T34N). In a sec16-P1092L strain, the mutant sec16 gene was tagged with GFP by gene replacement. The resulting strain was transformed with a control vector or a vector encoding Sar1(T34N) expressed from an inducible promoter. Induction and imaging were performed as in D. The expression of Sar1(T34N) was confirmed by measuring growth inhibition (Connerly et al., 2005; unpublished data). Scale bar, 5 μm.
Another relevant parameter is the frequency of de novo tER-site formation. Wild-type cells had an average of 3.3 de novo formation events per hour, whereas sec16‑P1092L cells had an average of 40 de novo formation events per hour (Figure 6C). The combined results indicate that the tER dispersal seen in sec16‑P1092L cells results from greatly accelerated formation and shrinkage of tER sites.
tER dispersal caused by the sec16‑P1092L mutation requires Sar1 activity
The shrinkage of tER sites is presumably driven by Sar1 activity, which promotes both COPII vesicle formation and the recycling of COPII subunits to the cytosol. We therefore suspected that the sec16‑P1092L mutation acts by accelerating Sar1-dependent COPII dynamics. This hypothesis was tested by overexpressing the dominant inhibitory Sar1(T34N) mutant (Connerly et al., 2005). Mutations of this type in Ras-related GTPases are often assumed to create GDP-locked proteins, but the effects are variable and more accurately described as interfering with nucleotide binding (Feig, 1999; Macia et al., 2004). In P. pastoris, overexpression of Sar1(T34N) inhibits cell growth while preserving the localization of COPII at tER sites (Connerly et al., 2005), suggesting that the net effect is to slow the Sar1 GTPase cycle. Sar1(T34N) was overexpressed from an inducible promoter in a wild-type or sec16‑P1092L strain, and then the cells were shifted to 36.5°C. With the wild-type strain, two to five tER sites per cell were seen in the absence or presence of Sar1(T34N) (Figure 6D, top). With the sec16‑P1092L strain, the tER was dispersed in the absence but not the presence of Sar1(T34N) (Figure 6D, bottom). Quantitation revealed that expression of Sar1(T34N) before the temperature shift completely suppressed dispersal of tER sites that were labeled with either Sec13-GFP (Figure 6E) or Sec23-GFP (Supplemental Figure S5A). Moreover, expression of Sar1(T34N) dramatically inhibited de novo tER site formation events (Figure 6C). In an alternative protocol, sec16‑P1092L cells were incubated at 36.5°C to cause tER dispersal, and then Sar1(T34N) expression was induced. Expression of Sar1(T34N) completely reversed tER dispersal (Supplemental Figure S5B). Thus the tER dispersal seen in the sec16‑P1092L mutant apparently results from elevated Sar1 activity.
These data suggest that in sec16‑P1092L cells, Sar1(T34N) blocks tER dispersal by counteracting the loss of Sec16 from tER sites. An alternative possibility is that Sar1(T34N) somehow restores tER localization of the mutant Sec16‑P1092L protein. To rule out such an effect, we compared the localization of GFP-tagged Sec16-P1092L at 36.5°C in the absence or presence of Sar1(T34N). Expression of Sar1(T34N) did not prevent redistribution of Sec16‑P1092L to the cytosol (Figure 6F). Thus sec16-P1092L cells expressing Sar1(T34N) have large tER sites even though most of the Sec16 is in the cytosol.
Anchoring away Sec16 causes tER dispersal but not loss of tER-associated COPII
To confirm that loss of Sec16 alters tER sites by accelerating tER dynamics, we took advantage of the anchor-away method (Haruki et al., 2008). A rapamycin-resistant P. pastoris strain was engineered as described in Materials and Methods. This strain was further engineered by appending four copies of FKBP to the ribosomal protein Rpl17, which has a solvent-exposed C‑terminus (del Alamo et al., 2011). Sec16 was then tagged with the FKBP-rapamycin–binding domain (FRB). On addition of rapamycin to create an FKBP-rapamycin-FRB complex, Sec16 molecules that cycle between tER sites and the cytosol should be inactivated by becoming trapped on ribosomes.
As a control to visualize ribosomal anchoring, we tagged Sec16 by gene replacement with an FRB-GFP dual tag. Within 5–10 min after rapamycin addition, most of the Sec16-FRB-GFP molecules redistributed from tER sites to a diffuse cytosolic localization (Figure 7A). Similar results were obtained when Sec16 was tagged with two copies of FRB followed by GFP (FRBx2-GFP; unpublished data). Addition of rapamycin inhibited growth of the Sec16-FRB-GFP strain but not of the parental strain (Supplemental Figure S6A). These results confirm that FRB-tagged Sec16 can be functionally inactivated by anchoring on ribosomes.
FIGURE 7:

Removal of Sec16 from tER sites by anchoring on cytosolic ribosomes. (A) Drug-induced displacement of Sec16 from tER sites to ribosomes. Endogenous Sec16 was modified with an FRB-GFP dual tag to visualize Sec16 at tER sites in the absence of rapamycin (–Rap). Rapamycin was then added to 1 μg/ml, and cells were imaged after 10 min of incubation with the drug (+Rap). Fluorescence and differential interference contrast images (DIC) were combined, with the same exposure times for both panels. (B) tER dispersal induced by anchoring away Sec16. Gene replacement was used to tag Sec16 with two tandem copies of FRB and to tag Sec31 with GFP. tER sites were then visualized in the absence of rapamycin or after incubation for 10 min in the presence of 1 μg/ml rapamycin. Fluorescence and DIC images were combined, with the same exposure times for both panels. The tER sites per cell were counted in ∼50 cells from each sample. Plotted are mean and SEM. Scale bars, 5 μm.
The next step was to visualize COPII after anchoring away Sec16. Our prediction was that loss of Sec16 would produce tER sites that were smaller, more numerous, and more dynamic, as seen in the sec16-P1092L mutant. For this experiment, Sec31 was tagged with GFP to mark tER sites, and Sec16 was tagged with FRBx2. Within minutes after addition of rapamycin to inactivate Sec16, the Sec31-GFP pattern changed dramatically, yielding multiple small tER sites (Figure 7B). Four-dimensional video microscopy confirmed that these small tER sites showed abnormally rapid dynamics (unpublished data). No effect of rapamycin was seen in an isogenic strain lacking the FRB tag on Sec16 (unpublished data). Thus removal of Sec16 did not displace COPII from tER sites but instead altered tER behavior.
Anchoring away COPII displaces Sec16 from tER sites
The complementary experiment was to anchor away COPII and ask whether Sec16 remained at tER sites. For this purpose, we tagged the inner COPII coat subunit Sec23 with FRB. As a control to visualize ribosomal anchoring, we tagged Sec23 by gene replacement with FRB-GFP. Within 5–10 min after rapamycin addition, most of the Sec23-FRB-GFP molecules redistributed from tER sites to a diffuse cytosolic location, although a weak punctate signal was still visible in some cells (Figure 8A). P. pastoris also contains a nonessential, tER-localized Sec23 homologue called Shl23 (Esaki et al., 2006), so we tagged Shl23 with FRB as well. As expected, anchoring away Sec23-FRB and Shl23-FRB strongly inhibited cell growth (Supplemental Figure S6B). In a strain containing Sec23-FRB and Shl23-FRB, addition of rapamycin should trap the inner COPII coat subunits on ribosomes, thereby displacing both layers of the COPII coat from tER sites. When the outer COPII coat protein Sec31-GFP was visualized in a strain containing Sec23-FRB and Shl23-FRB, most of the Sec31‑GFP molecules were displaced from tER sites within 5–10 min after rapamycin addition (Figure 8B). These results indicate that rapamycin-induced anchoring removed both layers of the COPII coat from tER sites.
FIGURE 8:

Displacement of Sec16 by removal of COPII coat proteins from tER sites. (A) Drug-induced displacement of Sec23 from tER sites to ribosomes. The procedure was the same as in Figure 7A, except that Sec23 was tagged with FRB-GFP. (B) Drug-induced displacement of Sec31 from tER sites. Gene replacement was used to tag Sec23 and Shl23 with FRB and to tag Sec31 with GFP. The Sec31-GFP pattern was then visualized in the absence of rapamycin (–Rap) or after incubation for 10 min in the presence of 1 μg/ml rapamycin (+Rap). Fluorescence and differential interference contrast images were combined, with the same exposure times for both panels. (C) Drug-induced displacement of Sec16 from tER sites. The procedure was the same as in B, except that Sec16 was tagged with GFP. Scale bars, 5 μm.
When Sec16-GFP was visualized in a strain containing Sec23-FRB and Shl23-FRB, most of the Sec16-GFP molecules were displaced from tER sites within 5–10 min after rapamycin addition (Figure 8C). No effect of rapamycin was seen in an isogenic strain lacking the FRB tags on Sec23 and Shl23 (unpublished data). After Sec23 and Shl23 were anchored away, we did observe a weak punctate Sec16-GFP signal in some cells. Our favored explanation is that Sec16 is significantly less abundant than COPII (Connerly et al., 2005), so even a residual amount of tER-localized COPII may suffice to retain a fraction of the Sec16-GFP molecules at tER sites. The key result is that removal of COPII from tER sites also largely removed Sec16, indicating that COPII is important for Sec16 localization.
DISCUSSION
Sec16 influences tER structure, but the mechanism has been obscure. An important clue came from biochemical evidence that Sec16 acts as a negative regulator of Sar1 GTPase activity (Supek et al., 2002; Kung et al., 2011; Yorimitsu and Sato, 2012). The prediction was that inactivating Sec16 should accelerate Sar1-dependent COPII turnover, leading to faster dynamics of tER sites. Live-cell imaging confirms this prediction and provides strong support for a negative regulatory function of Sec16.
Our experiments focused on the P. pastoris sec16-P1092L mutant, which has dispersed tER sites at 36.5°C due to a mutation in the nonessential CCD of Sec16 (Connerly et al., 2005). The CCD binds to Sec13 (Hughes et al., 2009; Whittle and Schwartz, 2010), and we find that the P1092L mutation weakens the CCD-Sec13 interaction. As a result, the CCD becomes structurally unstable, as indicated by a tendency to aggregate. The sec16‑P1092L mutation causes most of the Sec16 molecules to redistribute from tER sites to the cytosol. Redistribution of the destabilized protein may be due to the formation of small aggregates or association with cytosolic chaperones. This analysis explains how a mutation in a nonessential domain can cause lethality (because the entire Sec16 protein is displaced from tER sites) and why the mutation is recessive (because wild-type Sec16 molecules in the same cell can fold and function normally). We used a structure-guided approach to engineer a second sec16 mutant with a weakened CCD-Sec13 interaction and, as expected, the phenotype was similar to that of the sec16‑P1092L strain. Other mutations in the CCD of S. cerevisiae Sec16 may not directly inhibit Sec13 binding but are likely to destabilize CCD structure (Whittle and Schwartz, 2010). Indeed, the S. cerevisiae sec16-2 allele, which disrupts ER export and tER structure (Espenshade et al., 1995; Shindiapina and Barlowe, 2010), was recently shown to cause loss of Sec16 from tER sites at the nonpermissive temperature (Yorimitsu and Sato, 2012). We infer that conformational perturbations of the CCD can block Sec16 function even though the CCD itself is nonessential.
The combined data indicate that in the sec16‑P1092L mutant, tER dispersal occurs because Sec16 is functionally inactivated. To verify this interpretation, we used the anchor-away method to trap Sec16 on cytosolic ribosomes. Loss of Sec16 caused tER dispersal similar to that seen in the sec16‑P1092L mutant.
Insight into how loss of Sec16 causes tER dispersal came from video microscopy. Compared to wild-type cells, sec16‑P1092L mutant cells show a 10- to 20‑fold acceleration of both tER site shrinkage and tER site formation. Qualitatively similar results were seen when Sec16 was anchored on cytosolic ribosomes. Presumably, tER dynamics is accelerated due to enhanced GTPase activity by Sar1 (Supek et al., 2002; Kung et al., 2011; Yorimitsu and Sato, 2012). A faster Sar1 GTPase cycle should increase the rate of COPII recycling to the cytosol, thereby accelerating tER site shrinkage while increasing the pool of soluble COPII components available to nucleate new tER sites (Heinzer et al., 2008). To test this idea, we inhibited Sar1 activity by overexpressing the Sar1(T34N) mutant. Overexpression of Sar1(T34N) was fully effective at blocking or reversing the tER dispersal seen in sec16‑P1092L cells. We conclude that Sec16 normally restrains the Sar1 GTPase cycle and that loss of this restraining activity alters tER structure by accelerating COPII turnover.
The next question was whether Sec16 also plays an organizing role by recruiting COPII to tER sites. We grew skeptical of this idea after observing that redistribution of Sec16 to the cytosol did not displace COPII from tER sites. To clarify how Sec16 influences tER sites, we performed a functional dissection of P. pastoris Sec16.
The CCD is the most conserved part of Sec16 (Connerly et al., 2005; Bhattacharyya and Glick, 2007), and the complex of the CCD with Sec13 forms an ancestral coatomer element 1 structure (Hughes et al., 2009; Whittle and Schwartz, 2010). An attractive speculation was that the CCD-Sec13 complex serves as a template to initiate COPII assembly (Whittle and Schwartz, 2010). However, this model is hard to reconcile with the finding that deletion of the CCD has little effect on the growth of S. cerevisiae (Whittle and Schwartz, 2010; Yorimitsu and Sato, 2012) or P. pastoris. Moreover, we find that the P. pastoris CCD is dispensable for tER localization of Sec16 and also for normal tER structure. Therefore, despite its conservation, the CCD is not crucial for the core function of Sec16.
Upstream of the CCD, yeast Sec16 proteins contain a region with limited sequence conservation that we dubbed the UCR. Although the sequence similarity does not extend to animal cells, regions corresponding to the UCR in mammalian and Drosophila Sec16 proteins have been identified as essential for tER localization (Bhattacharyya and Glick, 2007; Ivan et al., 2008; Hughes et al., 2009; Budnik et al., 2011). Similarly, tER localization of P. pastoris Sec16 requires the UCR. This tER localization activity probably explains why the UCR is essential for the viability of S. cerevisiae (Yorimitsu and Sato, 2012) and P. pastoris.
There has been disagreement about whether the UCR is sufficient for tER localization. Other groups have reported that in mammalian and Drosophila cells, the UCR must be paired with the CCD to obtain tER localization (Ivan et al., 2008; Hughes et al., 2009; Budnik et al., 2011). In our hands, the UCR of mammalian Sec16A or Sec16B is sufficient to confer tER localization (Bhattacharyya and Glick, 2007), although the localization is weaker with the UCR alone than with a UCR-CCD construct (D. Bhattacharyya, personal communication). Similarly, the UCR of P. pastoris Sec16 confers weak but detectable tER localization, whereas a UCR‑CCD construct shows strong tER localization. Thus the CCD enhances the tER localization activity of the UCR.
Our data suggest a mechanism for this enhancement. The CCD-Sec13 complex dimerizes in an antiparallel manner (Whittle and Schwartz, 2010). We conjectured that because of CCD-driven dimerization, the functional affinity for tER sites should be higher with a UCR-CCD construct than with the UCR alone. To test this idea, we modified a UCR-CCD construct by replacing the CCD either with FKBP, which is monomeric, or the FKBP(F36M) mutant, which forms an antiparallel dimer (Rollins et al., 2000). The UCR-FKBP construct showed minimal tER localization, whereas the UCR‑FKBP(F36M) construct localized to tER sites as strongly as the UCR-CCD construct. For unknown reasons, the situation is more complex with mammalian cells because UCR-CCD constructs for Sec16A and Sec16B localize to tER sites, whereas UCR-FKBP(F36M) constructs show nonspecific aggregation (D. Bhattacharyya, personal communication). The mammalian CCD may have a role in maintaining the structure of the UCR. Nevertheless, on the basis of the clear-cut results with P. pastoris, we conclude that dimerization of the CCD accounts for its ability to enhance UCR-mediated tER localization.
As noted, deletion of the CCD from full-length P. pastoris Sec16 does not significantly reduce tER localization. Thus, even though the CCD can promote tER localization in the context of a UCR-CCD fragment, the CCD is not needed for tER localization of full-length Sec16. Perhaps another region of Sec16 has oligomerization activity that substitutes for CCD-mediated dimerization. Alternatively, binding of the CTR to Sec12 (Montegna et al., 2012) may promote association of Sec16 with the ER, thereby enhancing UCR-mediated tER localization.
In addition to promoting tER localization, the UCR binds to COPII coat proteins. Previously identified interactions of yeast Sec16 with Sec24 and Sec31 (Gimeno et al., 1996; Shaywitz et al., 1997; Kung et al., 2011; Yorimitsu and Sato, 2012) can now be ascribed to the UCR. Our two-hybrid analysis suggests that the UCR also interacts with Sec23 in both P. pastoris and S. cerevisiae. Of interest, these various COPII interactions may be partially redundant. We find that although the UCR as a whole is essential for life, P. pastoris cells can survive deletion of either of two sequences that together span the entire UCR.
Because the UCR mediates tER localization and also binds COPII, we propose that UCR‑COPII interactions recruit yeast Sec16 to tER sites. This model explains why the association of P. pastoris Sec16 with tER sites is saturable (Connerly et al., 2005). A direct test was performed using the anchor-away method. Inner COPII coat subunits were anchored on cytosolic ribosomes to displace COPII coat proteins from tER sites. Under these conditions, Sec16 also redistributed to the cytosol. Our data imply that in P. pastoris, tER-localized COPII proteins bind the UCR to recruit Sec16 for its essential function.
This essential function of Sec16 is apparently mediated by the CTR, which displays sequence conservation between fungi and mammals (Bhattacharyya and Glick, 2007). In our hands, the CTR is the only part of Sec16 that could not be deleted without killing the cells. The CTR has a conserved interaction with Sec23 (Espenshade et al., 1995; Bhattacharyya and Glick, 2007). This interaction was recently shown to prevent Sec31 from stimulating the GTPase-activating activity of Sec23, so the net effect of the Sec16–Sec23 interaction is to stabilize Sar1-GTP in the COPII coat (Kung et al., 2011; Yorimitsu and Sato, 2012). Figure 9A presents an overview of the known interactions and functions of the various regions of Sec16.
FIGURE 9:

Models for Sec16 function. (A) Diagram summarizing the conserved regions of P. pastoris Sec16 and their inferred functions. See the text for details. (B) Speculative diagram of a cross section through a nascent COPII vesicle. Sar1 has largely dissociated from the interior of the coat lattice due to Sec23- and Sec31-catalyzed GTP hydrolysis. However, a ring of Sar1-GTP is maintained at the edge of the lattice because Sec16 binds to the newly assembled COPII coat subunits and slows Sar1 GTPase activity. Sec16 also recruits Sec12 to the vicinity of the budding vesicle, leading to enhanced local production of Sar1-GTP. See the text for further details.
Our analysis suggests that the main role of Sec16 at tER sites is regulatory rather than organizational. By modulating COPII dynamics, Sec16 influences the size, number, and stability of tER sites. This interpretation contravenes the generally accepted idea that Sec16 defines tER sites and recruits COPII (Ivan et al., 2008; Budnik and Stephens, 2009; Yorimitsu and Sato, 2012). The strongest argument against the COPII organization model comes from anchor-away experiments. According to the COPII organization model, anchoring away Sec16 should displace COPII from tER sites, whereas anchoring away COPII should leave Sec16 at tER sites. Yet we see the opposite: anchoring away Sec16 does not displace COPII from tER sites, whereas anchoring away COPII displaces Sec16 to the cytosol. We therefore postulate that COPII proteins by themselves can associate with the ER and assemble to form coats (Matsuoka et al., 1998) and tER sites. The membrane-bound COPII components then recruit Sec16.
How can this new interpretation be reconciled with earlier data? We suggest that some of the experimental evidence for the COPII organization model is ambiguous because the tER pattern changes upon Sec16 depletion (Bhattacharyya and Glick, 2007; Iinuma et al., 2007). For example, the dispersed COPII pattern seen in Drosophila cells depleted of Sec16 was interpreted as loss of tER sites (Ivan et al., 2008) but may actually reflect changes in COPII dynamics similar to those observed with the P. pastoris sec16‑P1092L mutation. The chronic effects seen after gradual depletion of Sec16 in animal cells may be less revealing than the acute effects seen when P. pastoris Sec16 is rapidly redistributed to the cytosol. Nevertheless, the evidence is consistent with the idea that animal Sec16 can associate with the ER and form clusters even when COPII is largely absent (Ivan et al., 2008; Hughes et al., 2009; Hughes and Stephens, 2010). To understand this possible difference between the P. pastoris and animal Sec16 proteins, it will be important to determine how animal Sec16 localizes to tER sites and whether the UCR of animal Sec16 binds COPII.
Although Sec16 acts mainly to regulate COPII dynamics, this protein seems to have additional functions. For example, Sec16 recruits Sec12 to tER sites in P. pastoris (Montegna et al., 2012), and Sec16 interacts with the ER export factor TFG‑1 in animal cells (Witte et al., 2011). Sec16 can therefore act as a scaffold in the limited sense of recruiting accessory proteins to COPII-containing regions of the ER. Future work will determine how the various functions of Sec16 are coordinated.
Still missing is a molecular picture of how Sec16 regulates COPII vesicle formation. It is noteworthy that Sec16 does not entirely colocalize with COPII coat proteins at tER sites. This phenomenon has been documented for mammalian and Drosophila Sec16 proteins (Ivan et al., 2008; Hughes et al., 2009) and now for P. pastoris Sec16 as well. Sec16 is believed to remain at the ER surface, whereas COPII-coated vesicles bud from the membrane (Hughes et al., 2009). A speculative model incorporating this idea is shown in Figure 9B. Sec16 is selectively recruited to the edge of a nascent vesicle by binding to newly assembled COPII coat subunits. As the coat polymerizes, Sar1 hydrolyzes GTP, thereby allowing depolymerization. However, we postulate that depolymerization occurs at the edge of the coat lattice, so the interior of the coat remains stable even after Sec23- and Sec31-catalyzed hydrolysis of GTP. The proposed role of Sec16 is to maintain a protective ring of Sar1-GTP at the edge of the coat. Maintenance of Sar1‑GTP could be achieved both by slowing Sar1 GTPase activity (Kung et al., 2011; Yorimitsu and Sato, 2012) and recruiting Sec12 to the vicinity of the budding vesicle (Futai et al., 2004; Sato and Nakano, 2005; Montegna et al., 2012). When coat assembly is complete, Sec16 ensures that Sar1-GTP remains at the vesicle neck to promote membrane scission (Bielli et al., 2005; Lee et al., 2005). If Sec16 is absent, the Sar1‑GTP ring is not maintained, so the coat depolymerizes prematurely and the vesicle fails to form. The requirement for Sec16 can be bypassed in vitro by blocking Sar1 GTPase activity (Supek et al., 2002). This model accommodates the biochemical and cell biological data, and it explains why Sec16 remains associated with the ER membrane whereas COPII coat proteins partition onto budding vesicles (Hughes et al., 2009).
Our analysis suggests a basic molecular mechanism for Sec16 function. Interactions of the UCR with COPII coat proteins recruit Sec16 to nascent vesicles, allowing the CTR to stabilize Sar1‑GTP. Sec16 recruitment and activity are probably sensitive to various inputs (Guo and Linstedt, 2006; Farhan et al., 2008, 2010; Zacharogianni et al., 2011), which include UCR-Sec24 interactions (Kung et al., 2011). The net effect is that Sec16 adds a layer of regulation to the core vesicle budding machinery.
This synthesis leaves a couple of prominent questions unanswered. First, what is the role of the CCD? This domain is present in all known Sec16 homologues, but our assays have not yet uncovered the reason. Second, if Sec16 does not recruit and organize COPII, then how are tER sites established? Perhaps the search for a COPII-organizing protein is misguided. Instead, tER sites may form in the context of a larger self-organizing tER–Golgi unit (Glick, 2013).
MATERIALS AND METHODS
Strains and plasmids
The P. pastoris strains used in this study were derivatives of PPY12 (his4 arg4) (Gould et al., 1992) and PPY12/SEC13-DsRed (his4) (Connerly et al., 2005). Unless otherwise indicated, yeast were grown in rich glucose medium (yeast extract/peptone/dextrose [YPD]) or minimal glucose medium (SD; Sherman, 1991). DNA manipulations were simulated and recorded using SnapGene software (GSL Biotech, Chicago, IL). Annotated sequence files for 44 of the plasmids used in this study are included in the Supplemental Material and can be opened with the free SnapGene Viewer (www.snapgene.com/products/snapgene_viewer/).
N-terminal deletion constructs for Sec16 were generated by PCR mutagenesis of pLY100, a derivative of pUC19-ARG4 (Rossanese et al., 1999) containing the 5′ control region of SEC16 followed by the first 4383 base pairs of the coding sequence, followed by a second copy of the 5′ control region. For transformation, these constructs were linearized at a unique BssHII restriction site at the end of the coding sequence. C-terminal deletion constructs for Sec16 were generated by primer-directed mutagenesis of pME005, which was constructed by inserting a SEC16 fragment spanning from position 3532 of the coding sequence to 411 base pairs downstream of the stop codon into the SmaI site of pUC19-ARG4. For transformation, these constructs were linearized within the SEC16 coding sequence at a unique site such as BstEII. Internal deletion constructs for Sec16 were generated using similar procedures in which plasmids were linearized and then integrated to achieve gene replacement at the SEC16 locus.
To define the regions of Sec16 essential for cell growth, one copy of SEC16 in the P. pastoris diploid strain PPY12D (Soderholm et al., 2004) was replaced with a mutant allele using an ARG4 construct as described. Integration at the SEC16 locus was confirmed by selection on minimal medium lacking arginine, followed by colony PCR. After sporulation of the resulting strain, the fraction of the haploids that were arginine prototrophs was determined.
The constructs for overexpressing N-terminally GFP-tagged Sec16 fragments were created as follows. SEC16 sequences were PCR amplified and inserted downstream of the AOX1 promoter in a derivative of pIB4 (Sears et al., 1998) encoding enhanced GFP (EGFP). The P1092L mutation was introduced where indicated using primer-directed mutagenesis. Constructs were transformed into a strain expressing DsRed-tagged Sec13 (Connerly et al., 2005). To induce expression from the AOX1 promoter, P. pastoris cells were grown in glycerol-containing SYG medium and then transferred to methanol-containing SYM medium (Sears et al., 1998; Soderholm et al., 2004).
To express second-copy SEC16 genes encoding C-terminally GFP-tagged wild-type and mutant Sec16, we used an in vitro ligation and transformation method (Connerly et al., 2005) because full-length P. pastoris SEC16 is toxic to E. coli cells. Briefly, two fragments were ligated in vitro to generate a linear DNA molecule that was equivalent to a linearized plasmid carrying a full-length wild-type or mutant SEC16 gene. This construct was then integrated at the HIS4 locus in a strain expressing DsRed-tagged Sec13 (Connerly et al., 2005). Correct transformants were confirmed by colony PCR.
Sec16, COPII coat proteins, and Rpl17 were modified with C-terminal tags by pop-in gene replacement as previously described (Rossanese et al., 1999; Connerly et al., 2005). Integrants were selected using marker plasmids containing a variety of markers, including the S. cerevisiae ARG4 and HIS4 genes, as well as the kanMX6 cassette encoding G418 resistance (Wach, 1996) and genes encoding resistance to hygromycin B or nourseothricin (Goldstein and McCusker, 1999). Drug-resistant clones were selected on plates containing 0.5 mg/ml G418, 0.2 mg/ml hygromycin B, or 25 μg/ml nourseothricin. GFP tagging was performed using either EGFP or monomeric EGFP (mEGFP) carrying the A206K mutation (Zacharias et al., 2002). Plasmids encoding FRB and FKBPx2 were obtained from ARIAD Pharmaceuticals (Cambridge, MA). FRB was mutagenized to revert the destabilizing T2098L mutation (Stankunas et al., 2007). These constructs were used to generate FRB-mEGFP, FRBx2-mEGFP, FRBx2, and FKBPx4 cassettes.
To express the CCDβ-Sec13 chimera in bacteria, we fused residues 1030–1076 of P. pastoris Sec16 via a 10-residue flexible linker (GSGGGSGGGS) to the N-terminus of P. pastoris Sec13 (residues 13–300). This construct was produced as an N-terminally hexahistidine-tagged fusion protein using the expression plasmid pET28a in Rosetta E. coli cells (EMD Biosciences, Billerica, MA).
Fluorescence and video microscopy
For standard fluorescence microscopy, P. pastoris strains were grown at 30 or 36.5ºC in SD or SYM media. Cells were visualized directly without fixation on a Zeiss Axioplan2 epifluorescence microscope (Carl Zeiss, Jena, Germany) using a 100×/numerical aperture 1.4 Plan Apo objective. Image Z-stacks were captured with a Hamamatsu (Hamamatsu, Japan) digital camera. Photoshop (Adobe, San Jose, CA) and ImageJ (National Institutes of Health, Bethesda, MD) were used to merge and colorize the images, adjust brightness and contrast, and adjust levels where appropriate to reduce nonspecific background.
The 4D movies were taken as previously described using Bioptechs (Butler, PA) ΔT culture dishes on a Leica (Wetzlar, Germany) SP5 scanning confocal microscope (Losev et al., 2006), except that the optical sections were 0.3 μm apart. Complete Z-stacks were collected at time intervals of 2 s for the sec16‑P1092L mutant or 4 s for the wild-type strain. The 4D data sets were processed as previously described (Losev et al., 2006). To induce thermosensitive phenotypes, cultures were grown at room temperature and then shifted to 36.5ºC for ∼1 h before imaging, and the microscope stage was heated to 36.5ºC. To image cells after rapamycin treatment, cultures were grown at room temperature, and then rapamycin was added to 1 μg/ml for 10 min before imaging. Data analysis was performed using ImageJ.
To quantify the fraction of a GFP-tagged construct that was at tER sites, we modified a previously described method (Levi et al., 2010) as follows. All manipulations were performed with ImageJ. A stack of widefield fluorescence images was average projected. The average background outside the cells was measured, and this value was subtracted from the projection. A further correction was performed by measuring and then subtracting the average intracellular green fluorescence from a control strain lacking a GFP-tagged construct. For a given cell, the total cellular GFP signal was measured from the background-corrected projection. A binary mask was then created from the background-corrected projection as follows. Using the Subtract Background command, the rolling ball radius was adjusted so that punctate tER sites remained visible while diffuse cellular fluorescence was removed. The Adjust Threshold command was then used to create a mask in which tER sites had value 0 and the remainder of the image had value 255. This binary mask was subtracted from the background-corrected projection to obtain the GFP signal at tER sites. The GFP signal at tER sites was divided by the total cellular GFP signal to obtain the fraction of the cellular GFP signal at tER sites. This process was performed for ∼30–40 randomly chosen cells, and the results were averaged.
Two-hybrid protein–protein interaction assays
Protein–protein interactions were tested in the yeast two-hybrid system (James, 2001). Interactions were tested between proteins fused to the Gal4 DNA-binding domain and Gal4 activation domain. SEC13, SEC31, SEC23, SEC24, and fragments of SEC16 were PCR amplified from P. pastoris or S. cerevisiae genomic DNA, with restriction sites added to both ends of each amplified fragment. After digestion, the fragments were inserted into pGBDU (“bait”) and pGAD (“prey”) vectors (James, 2001) digested with the same restriction enzymes. Whenever possible, interactions were tested using both combinations. Plasmids were transformed into S. cerevisiae strain PJ69‑4A using a high-efficiency lithium acetate method (Gietz and Woods, 2002). Transformants were selected on SD‑Leu-Ura plates. Interactions were tested by plating the transformants on SD-Leu, Ura, His or SD-Leu, Ura, His, Ade plates.
Functional inactivation of proteins with the anchor-away method
A PPY12 derivative suitable for anchor-away experiments was constructed as follows. First, the TOR1 gene was modified to create a dominant rapamycin-resistant allele (Helliwell et al., 1994). For this purpose, two synthetic oligonucleotides were annealed to create a 99–base pair double-stranded DNA fragment centered around the Ser‑1919 codon, which is equivalent to Ser‑1972 in S. cerevisiae TOR1. The oligonucleotides contained degenerate bases at the positions corresponding to codon 1919. This mixture of annealed degenerate oligonucleotides was introduced into PPY12 cells by electroporation, and rapamycin-resistant transformants were selected on YPD plates containing 1 μg/ml rapamycin. PCR amplification and sequencing of the mutated TOR1 alleles revealed that strong rapamycin resistance could be conferred by mutating Ser‑1919 to various amino acids, including Leu, Ile, His, Asn, Asp, and Glu. A Ser-to-Arg mutant was chosen for further work. The FPR1 gene, which encodes an FKBP homologue (Heitman et al., 1991), was then deleted by replacing the open reading frame with kanMX6. Finally, the ribosomal protein Rpl17 was C-terminally tagged with FKBPx4 by pop-in gene replacement.
For functional inactivation, a COPII protein was tagged with one or two copies of FRB in the TOR1 mutant strain carrying the fpr1Δ and RPL7-FKBPx4 alleles. A logarithmically growing culture in minimal medium was then supplemented with 1 μg/ml rapamycin, and cells were imaged within 5–10 min as described.
Protein solubility and immunoblotting analyses
Pairs of proteins were coexpressed in E. coli at 30ºC for 3.5 h using the pET-Duet system (EMD Biosciences). Bacteria were lysed using BugBuster (EMD Biosciences) in the presence of the Complete Mini EDTA-free protease inhibitor cocktail (Roche, Madison, WI) and Benzonase nuclease (EMD Biosciences). The lysate was separated into pellet and supernatant fractions by centrifugation for 5 min at 12,000 × g. Equivalent amounts of the pellet and supernatant fractions were separated by SDS–PAGE, followed by transfer to a polyvinylidene difluoride membrane. For immunodetection, an anti-FLAG antibody (catalogue number A8592; Sigma-Aldrich, St. Louis, MO) was used at 0.5 ng/ml, or an anti–S peptide antibody (catalogue number 69047; EMD Biosciences) was used at 1:10,000 dilution. Bound antibodies were detected by chemiluminescence using the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific, Waltham, MA).
Protein structure analysis
The online Phyre program (www.sbg.bio.ic.ac.uk/∼phyre/) was used to predict protein structures for the CCD from P. pastoris Sec16 and human Sec16A. Model images were generated using the PyMol Molecular Viewer (www.pymol.org/).
To produce the CCDβ-Sec13 chimera, E. coli cells carrying the CCDβ-Sec13 expression plasmid were grown at 37°C to an OD600 of 0.6. Expression was then induced with 1 mM isopropyl β-d-1-thiogalactopyranoside, and cells were grown at 23°C for an additional 12 h. The cells were centrifuged, resuspended in lysis buffer (20 mM Tris, pH 7.6, 250 mM NaCl, 5 mM imidazole, 1 mM dithiothreitol), and lysed using an Emulsiflex-C5 homogenizer (Avestin, Ottawa, Canada). The lysate was supplemented with phenylmethylsulfonyl fluoride and Protease Inhibitor Cocktail (Sigma-Aldrich) to prevent degradation. Initial purification from the clarified cell lysate was performed using Ni2+-charged IMAC Sepharose 6 Fast Flow resin (GE Healthcare, Waukesha, WI). The chimeric protein was released from the resin by digestion with thrombin at 4°C to cleave the hexahistidine tag. Further purification was performed using ion exchange (SourceQ 10/10; GE Healthcare) followed by size exclusion (Superdex200 10/300 GL; GE Healthcare) chromatography. The purified protein was flash frozen in liquid nitrogen and stored at −80°C.
For structure determination, crystals of the chimeric CCDβ-Sec13 protein were obtained at 23°C using the sitting-drop vapor-diffusion method. The protein solution (20 mM Tris, pH 7.6, 100 mM NaCl, 1 mM dithiothreitol) and well solution (100 mM Tris, pH 7.5, 200 mM CaCl2, 200 mM NaSCN, 23% [wt/vol] polyethylene glycol [PEG] 3350) were mixed 1:1. Crystals grew in space group P21 with cell dimensions a = 70.5 Å, b = 49.3 Å, c = 90.9 Å, α = 90°, β = 111.7°, and γ = 90°. Each asymmetric unit contained two polypeptide chains designated A and B. Crystals were cryoprotected by brief soaking in 25% (vol/vol) ethylene glycol, 22% (wt/vol) PEG 3350, 100 mM Tris, pH 7.5, 200 mM CaCl2, and 200 mM NaSCN. Native data were collected at National Synchrotron Light Source beamline X25 to a maximum resolution of 1.55 Å (Supplemental Table S1).
The structure was determined by molecular replacement using the program PHASER (Storoni et al., 2004) and a model of S. cerevisiae Sec13 from chain A of Protein Data Bank entry 3JRP (Fath et al., 2007). Refinement was performed using the riding hydrogen model for the protein at a maximum resolution of 1.6 Å using the program PHENIX (Adams et al., 2010). Refinement statistics are given in Supplemental Table S1. Electron density maps showed clear density for the CCDβ blade inserted into Sec13. For chain A but not chain B, three residues from the purification tag linker and the first eight residues of CCDβ formed part of the crystal lattice. Three calcium ions from the crystallization medium were also involved in crystal lattice interactions. In addition, two chloride ions and five ethylene glycol molecules were placed in the model. Chain A has unmodeled coordinates for disordered loops at residues 1038–1041 of Sec16, residues 3–8 in the linker, and residues 50–52 and 169–171 of Sec13, with modeled coordinates extending to residue 299 of Sec13. Chain B extends from residue 1043 of Sec16 to 298 of Sec13, with unmodeled coordinates for disordered loops at residues 1064–1069 of Sec16 and 142–144 and 169–171 of Sec13. The two crystallographically distinct molecules in the asymmetric unit superimpose with a root-mean-square deviation of 0.67 Å for 298 Cα atoms. Minor deviations between the two polypeptides occur in loops as a result of crystal packing interactions. This structure has been deposited in the Protein Data Bank under code 4L9O.
Supplementary Material
Acknowledgments
We thank members of the Glick lab for constructive feedback and the staff of National Synchrotron Light Source beamline X25 for assistance with data collection. This work was supported by National Institutes of Health Grants R01 GM061156 (B.S.G.) and R01 GM071574 (F.M.H.).
Abbreviations used:
- CCD
central conserved domain
- CCDβ
β-blade portion of the CCD
- COPII
coat protein complex II
- CTR
conserved C-terminal region
- DTT
dithiothreitol
- FRB
FKBP-rapamycin–binding protein
- FKBP
FK506-binding protein
- tER
transitional ER
- UCR
upstream conserved region
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-04-0185) on September 4, 2013.
*These authors contributed equally to this work.
REFERENCES
- Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonny B, Madden D, Hamamoto S, Orci L, Schekman R. Dynamics of the COPII coat with GTO and stable analogues. Nat Cell Biol. 2001;3:531–537. doi: 10.1038/35078500. [DOI] [PubMed] [Google Scholar]
- Bannykh SI, Balch WE. Membrane dynamics at the endoplasmic reticulum-Golgi interface. J Cell Biol. 1997;138:1–4. doi: 10.1083/jcb.138.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe C. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 1994;77:895–907. doi: 10.1016/0092-8674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- Barlowe C, Schekman R. SEC12 encodes a guanine-nucleotide-exchange factor essential for transport vesicle budding from the ER. Nature. 1993;365:347–349. doi: 10.1038/365347a0. [DOI] [PubMed] [Google Scholar]
- Barlowe CK, Miller EA. Secretory protein biogenesis and traffic in the early secretory pathway. Genetics. 2013;193:383–410. doi: 10.1534/genetics.112.142810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevis BJ, Hammond AT, Reinke CA, Glick BS. De novo formation of transitional ER sites and Golgi structures in Pichia pastoris. Nat Cell Biol. 2002;4:750–756. doi: 10.1038/ncb852. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya D, Glick BS. Two mammalian Sec16 homologs have nonredundant functions in ER export and transitional ER organization. Mol Biol Cell. 2007;18:839–849. doi: 10.1091/mbc.E06-08-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi X, Corpina RA, Goldberg J. Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature. 2002;419:271–277. doi: 10.1038/nature01040. [DOI] [PubMed] [Google Scholar]
- Bi X, Mancias JD, Goldberg J. Insights into COPII coat nucleation from the structure of Sec23.Sar1 complexed with the active fragment of Sec31. Dev Cell. 2007;13:635–645. doi: 10.1016/j.devcel.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielli A, Haney CJ, Gabreski G, Watkins SC, Bannykh SI, Aridor M. Regulation of Sar1 NH2 terminus by GTP binding and hydrolysis promotes membrane deformation to control COPII vesicle fission. J Cell Biol. 2005;171:919–924. doi: 10.1083/jcb.200509095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik A, Heesom KJ, Stephens DJ. Characterization of human Sec16B: indications of specialized, non-redundant functions. Sci Rep. 2011;1:77. doi: 10.1038/srep00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik A, Stephens DJ. ER exit sites—localization and control of COPII vesicle formation. FEBS Lett. 2009;583:3796–3803. doi: 10.1016/j.febslet.2009.10.038. [DOI] [PubMed] [Google Scholar]
- Connerly PL, Esaki M, Montegna EA, Strongin DE, Levi S, Soderholm J, Glick BS. Sec16 is a determinant of transitional ER organization. Curr Biol. 2005;15:1439–1447. doi: 10.1016/j.cub.2005.06.065. [DOI] [PubMed] [Google Scholar]
- del Alamo M, Hogan DJ, Pechmann S, Albanese V, Brown PO, Frydman J. Defining the specificity of cotranslationally acting chaperones by systematic analysis of mRNAs associated with ribosome-nascent chain complexes. PLoS Biol. 2011;9:e1001100. doi: 10.1371/journal.pbio.1001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esaki M, Liu Y, Glick BS. The budding yeast Pichia pastoris has a novel Sec23p homolog. FEBS Lett. 2006;580:5215–5221. doi: 10.1016/j.febslet.2006.08.058. [DOI] [PubMed] [Google Scholar]
- Espenshade P, Gimeno RE, Holzmacher E, Teung P, Kaiser CA. Yeast SEC16 gene encodes a multidomain vesicle coat protein that interacts with Sec23p. J Cell Biol. 1995;131:311–324. doi: 10.1083/jcb.131.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhan H, Weiss M, Tani K, Kaufman RJ, Hauri HP. Adaptation of endoplasmic reticulum exit sites to acute and chronic increases in cargo load. EMBO J. 2008;27:2043–2054. doi: 10.1038/emboj.2008.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhan H, Wendeler MW, Mitrovic S, Fava E, Silberberg Y, Sharan R, Zerial M, Hauri HP. MAPK signaling to the early secretory pathway revealed by kinase/phosphatase functional screening. J Cell Biol. 2010;189:997–1011. doi: 10.1083/jcb.200912082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fath S, Mancias JD, Bi X, Goldberg J. Structure and organization of coat proteins in the COPII cage. Cell. 2007;129:1325–1336. doi: 10.1016/j.cell.2007.05.036. [DOI] [PubMed] [Google Scholar]
- Feig LA. Tools of the trade: use of dominant-inhibitory mutants of Ras-family GTPases. Nat Cell Biol. 1999;1:E25–27. doi: 10.1038/10018. [DOI] [PubMed] [Google Scholar]
- Futai E, Hamamoto S, Orci L, Schekman R. GTP/GDP exchange by Sec12p enables COPII vesicle bud formation on synthetic liposomes. EMBO J. 2004;23:4146–4155. doi: 10.1038/sj.emboj.7600428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- Gimeno RE, Espenshade P, Kaiser CA. SED4 encodes a yeast endoplasmic reticulum protein that binds Sec16p and participates in vesicle formation. J Cell Biol. 1995;131:325–338. doi: 10.1083/jcb.131.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno RE, Espenshade P, Kaiser CA. COPII coat subunit interactions: Sec24p and Sec23p bind to adjacent regions of Sec16p. Mol Biol Cell. 1996;7:1815–1823. doi: 10.1091/mbc.7.11.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick BS. Integrated self-organization of transitional ER and early Golgi compartments. BioEssays. 2013 doi: 10.1002/bies.201300131. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick BS, Nakano A. Membrane traffic within the Golgi stack. Annu Rev Cell Dev Biol. 2009;25:113–132. doi: 10.1146/annurev.cellbio.24.110707.175421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein AL, McCusker JH. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast. 1999;15:1541–1553. doi: 10.1002/(SICI)1097-0061(199910)15:14<1541::AID-YEA476>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Gould SJ, McCollum D, Spong AP, Heyman JA, Subramani S. Development of the yeast Pichia pastoris as a model organism for a genetic and molecular analysis of peroxisome assembly. Yeast. 1992;8:613–628. doi: 10.1002/yea.320080805. [DOI] [PubMed] [Google Scholar]
- Guo Y, Linstedt AD. COPII-Golgi protein interactions regulate COPII coat assembly and Golgi size. J Cell Biol. 2006;174:53–63. doi: 10.1083/jcb.200604058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond AT, Glick BS. Dynamics of transitional endoplasmic reticulum sites in vertebrate cells. Mol Biol Cell. 2000;11:3013–3030. doi: 10.1091/mbc.11.9.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, Laemmli UK. The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol Cell. 2008;31:925–932. doi: 10.1016/j.molcel.2008.07.020. [DOI] [PubMed] [Google Scholar]
- Heinzer S, Wörz S, Kalla C, Rohr K, Weiss M. A model for the self-organization of exit sites in the endoplasmic reticulum. J Cell Sci. 2008;121:55–64. doi: 10.1242/jcs.013383. [DOI] [PubMed] [Google Scholar]
- Heitman J, Movva NR, Hiestand PC, Hall MN. FK 506-binding protein proline rotamase is a target for the immunosuppressive agent FK 506 in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1991;88:1948–1952. doi: 10.1073/pnas.88.5.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helliwell SB, Wagner P, Kunz J, Deuter-Reinhard M, Henriquez R, Hall MN. TOR1 and TOR2 are structurally and functionally similar but not identical phosphatidylinositol kinase homologues in yeast. Mol Biol Cell. 1994;5:105–118. doi: 10.1091/mbc.5.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes H, et al. Organisation of human ER-exit sites: requirements for the localisation of Sec16 to transitional ER. J Cell Sci. 2009;122:2924–2934. doi: 10.1242/jcs.044032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes H, Stephens DJ. Sec16A defines the site for vesicle budding from the endoplasmic reticulum on exit from mitosis. J Cell Sci. 2010;123:4032–4038. doi: 10.1242/jcs.076000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iinuma T, Shiga A, Nakamoto K, O'Brien MB, Aridor M, Arimitsu N, Tagaya M, Tani K. Mammalian Sec16/p250 plays a role in membrane traffic from the endoplasmic reticulum. J Biol Chem. 2007;282:17632–17639. doi: 10.1074/jbc.M611237200. [DOI] [PubMed] [Google Scholar]
- Ivan V, de Voer G, Xanthakis D, Spoorendonk KM, Kondylis V, Rabouille C. Drosophila Sec16 mediates the biogenesis of tER sites upstream of Sar1 through an arginine-rich motif. Mol Biol Cell. 2008;19:4352–4365. doi: 10.1091/mbc.E08-03-0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James P. Yeast two-hybrid vectors and strains. Met Mol Biol. 2001;177:41–84. doi: 10.1385/1-59259-210-4:041. [DOI] [PubMed] [Google Scholar]
- Kaiser CA, Schekman R. Distinct sets of SEC genes govern transport vesicle formation and fusion early in the secretory pathway. Cell. 1990;61:723–733. doi: 10.1016/0092-8674(90)90483-u. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Kung LF, et al. Sec24p and Sec16p cooperate to regulate the GTP cycle of the COPII coat. EMBO J. 2011;31:1014–1027. doi: 10.1038/emboj.2011.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MC, Orci L, Hamamoto S, Futai E, Ravazzola M, Schekman R. Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell. 2005;122:605–617. doi: 10.1016/j.cell.2005.07.025. [DOI] [PubMed] [Google Scholar]
- Levi SK, Bhattacharyya D, Strack RL, Austin JRI, Glick BS. The yeast GRASP Grh1 colocalizes with COPII and is dispensable for organizing the secretory pathway. Traffic. 2010;11:1168–1179. doi: 10.1111/j.1600-0854.2010.01089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C, Ferro-Novick S, Miller EA. The highly conserved COPII coat complex sorts cargo from the endoplasmic reticulum and targets it to the Golgi. Cold Spring Harb Perspect Biol. 2013;5:a013367. doi: 10.1101/cshperspect.a013367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losev E, Reinke CA, Jellen J, Strongin DE, Bevis BJ, Glick BS. Golgi maturation visualized in living yeast. Nature. 2006;22:1002–1006. doi: 10.1038/nature04717. [DOI] [PubMed] [Google Scholar]
- Macia E, Luton F, Partisani M, Cherfils J, Chardin P, Franco M. The GDP-bound form of Arf6 is located at the plasma membrane. J Cell Sci. 2004;117:2389–2398. doi: 10.1242/jcs.01090. [DOI] [PubMed] [Google Scholar]
- Matsuoka K, Orci L, Amherdt M, Bednarek S, Hamamoto S, Schekman R, Yeung T. COPII-coated vesicle formation reconstituted with purified coat proteins and chemically defined liposomes. Cell. 1998;93:263–275. doi: 10.1016/s0092-8674(00)81577-9. [DOI] [PubMed] [Google Scholar]
- Miller EA, Beilharz TH, Malkus PN, Lee MC, Hamamoto S, Orci L, Schekman R. Multiple cargo binding sites on the COPII subunit Sec24p ensure capture of diverse membrane proteins into transport vesicles. Cell. 2003;114:497–509. doi: 10.1016/s0092-8674(03)00609-3. [DOI] [PubMed] [Google Scholar]
- Mogelsvang S, Gomez-Ospina N, Soderholm J, Glick BS, Staehelin LA. Tomographic evidence for continuous turnover of Golgi cisternae in Pichia pastoris. Mol Biol Cell. 2003;14:2277–2291. doi: 10.1091/mbc.E02-10-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montegna EA, Bhave M, Liu Y, Bhattacharyya D, Glick BS. Sec12 binds to Sec16 at transitional ER sites. PLoS One. 2012;7:e31156. doi: 10.1371/journal.pone.0031156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano A, Muramatsu M. A novel GTP-binding protein, Sar1p, is involved in transport from the endoplasmic reticulum to the Golgi apparatus. J Cell Biol. 1989;109:2677–2691. doi: 10.1083/jcb.109.6.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palade G. Intracellular aspects of the process of protein synthesis. Science. 1975;189:347–358. doi: 10.1126/science.1096303. [DOI] [PubMed] [Google Scholar]
- Pfeffer S. Membrane domains in the secretory and endocytic pathways. Cell. 2003;112:507–517. doi: 10.1016/s0092-8674(03)00118-1. [DOI] [PubMed] [Google Scholar]
- Rollins CT, et al. A ligand-reversible dimerization system for controlling protein-protein interactions. Proc Natl Acad Sci USA. 2000;97:7096–7101. doi: 10.1073/pnas.100101997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossanese OW, Soderholm J, Bevis BJ, Sears IB, O'Connor J, Williamson EK, Glick BS. Golgi structure correlates with transitional endoplasmic reticulum organization in Pichia pastoris and Saccharomyces cerevisiae. J Cell Biol. 1999;145:69–81. doi: 10.1083/jcb.145.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell C, Stagg SM. New insights into the structural mechanisms of the COPII coat. Traffic. 2010;11:303–310. doi: 10.1111/j.1600-0854.2009.01026.x. [DOI] [PubMed] [Google Scholar]
- Sato K, Nakano A. Dissection of COPII subunit-cargo assembly and disassembly kinetics during Sar1p-GTP hydrolysis. Nat Struct Mol Biol. 2005;12:167–174. doi: 10.1038/nsmb893. [DOI] [PubMed] [Google Scholar]
- Sears IB, O'Connor J, Rossanese OW, Glick BS. A versatile set of vectors for constitutive and regulated gene expression in Pichia pastoris. Yeast. 1998;14:783–790. doi: 10.1002/(SICI)1097-0061(19980615)14:8<783::AID-YEA272>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Shaywitz DA, Espenshade PJ, Gimeno RE, Kaiser CA. COPII subunit interactions in the assembly of the vesicle coat. J Biol Chem. 1997;272:25413–25416. doi: 10.1074/jbc.272.41.25413. [DOI] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. Methods Enzymol. 1991;194:3–21. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- Shindiapina P, Barlowe C. Requirements for transitional endoplasmic reticulum site structure and function in Saccharomyces cerevisiae. Mol Biol Cell. 2010;21:1530–1545. doi: 10.1091/mbc.E09-07-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderholm J, Bhattacharyya D, Strongin D, Markovitz V, Connerly PL, Reinke CA, Glick BS. The transitional ER localization mechanism of Pichia pastoris Sec12. Dev Cell. 2004;6:649–659. doi: 10.1016/s1534-5807(04)00129-7. [DOI] [PubMed] [Google Scholar]
- Stagg SM, LaPointe P, Razvi A, Gürkan C, Potter CS, Carragher B, Balch WE. Structural basis for cargo regulation of COPII coat assembly. Cell. 2008;134:474–484. doi: 10.1016/j.cell.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankunas K, Bayle JH, Havranek JJ, Wandless TJ, Baker D, Crabtree GR, Gestwicki JE. Rescue of degradation-prone mutants of FK506-rapamycin binding (FRB) protein with chemical ligands. ChemBioChem. 2007;8:1162–1169. doi: 10.1002/cbic.200700087. [DOI] [PubMed] [Google Scholar]
- Stephens DJ. De novo formation, fusion and fission of mammalian COPII-coated endoplasmic reticulum exit sites. EMBO Rep. 2003;4:210–217. doi: 10.1038/sj.embor.embor736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr. 2004;66:213–221. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- Supek F, Madden DT, Hamamoto S, Orci L, Schekman R. Sec16p potentiates the action of COPII proteins to bud transport vesicles. J Cell Biol. 2002;158:1029–1038. doi: 10.1083/jcb.200207053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast. 1996;12:259–265. doi: 10.1002/(SICI)1097-0061(19960315)12:3%3C259::AID-YEA901%3E3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Watson P, Townley AK, Koka P, Palmer KJ, Stephens DJ. Sec16 defines endoplasmic reticulum exit sites and is required for secretory cargo export in mammalian cells. Traffic. 2006;7:1678–1687. doi: 10.1111/j.1600-0854.2006.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle JRR, Schwartz TU. Structure of the Sec13-Sec16 edge element, a template for assembly of the COPII vesicle coat. J Cell Biol. 2010;190:347–361. doi: 10.1083/jcb.201003092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte K, Schuh AL, Hegermann J, Sarkeshik A, Mayers JR, Schwarze K, Yates JR, 3rd, Elmer S, Audhya A. TGF-1 function in protein secretion and oncogenesis. Nat Cell Biol. 2011;13:550–558. doi: 10.1038/ncb2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorimitsu T, Sato K. Insights into structural and regulatory roles of Sec16 in COPII vesicle formation at ER exit sites. Mol Biol Cell. 2012;23:2930–2942. doi: 10.1091/mbc.E12-05-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihisa T, Barlowe C, Schekman R. Requirement for a GTPase-activating protein in vesicle budding from the endoplasmic reticulum. Science. 1993;259:1466–1468. doi: 10.1126/science.8451644. [DOI] [PubMed] [Google Scholar]
- Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- Zacharogianni M, Kondylis V, Tang Y, Farhan H, Xanthakis D, Fuchs F, Boutros M, Rabouille C. ERK7 is a negative regulator of protein secretion in response to amino-acid starvation by modulating Sec16 membrane association. EMBO J. 2011;30:3684–3700. doi: 10.1038/emboj.2011.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.