


Abstract
Despite studies that have investigated the interactions of double-stranded RNA-binding proteins like Staufen with RNA in vitro, how they achieve target specificity in vivo remains uncertain. We performed RNA co-immunoprecipitations followed by microarray analysis to identify Staufen-associated mRNAs in early Drosophila embryos. Analysis of the localization and functions of these transcripts revealed a number of potentially novel roles for Staufen. Using computational methods, we identified two sequence features that distinguish Staufen’s target transcripts from non-targets. First, these Drosophila transcripts, as well as those human transcripts bound by human Staufen1 and 2, have 3′ untranslated regions (UTRs) that are 3–4-fold longer than unbound transcripts. Second, the 3′UTRs of Staufen-bound transcripts are highly enriched for three types of secondary structures. These structures map with high precision to previously identified Staufen-binding regions in Drosophila bicoid and human ARF1 3′UTRs. Our results provide the first systematic genome-wide analysis showing how a double-stranded RNA-binding protein achieves target specificity.
INTRODUCTION
RNA-binding proteins (RBPs) direct many co- and post-transcriptional processes. There are a number of different classes of RBPs that are defined by the presence of different RNA-binding domains (RBDs) (1). One class of RBP is double-stranded RBPs (dsRBPs), defined by the presence of one or more double-stranded RBDs (dsRBDs). dsRBDs are characterized by a conserved αβββα fold (2–4) and bind specifically to double-stranded RNA (dsRNA) (5,6). Proteins containing dsRBDs have roles in diverse processes and include Escherichia coli RNase III, Xenopus laevis Xlrbpa, a dsRBP associated with cellular RNAs and ribosomes, the dsRNA-dependent protein kinase PKR, dsRNA-dependent adenosine deaminases (ADARs), and Dicer, an important component of the RNA interference (RNAi) machinery.
One of the best-characterized dsRBPs is Staufen, an evolutionarily conserved protein first identified in Drosophila (7,8). In Drosophila, Staufen is essential for localization and translation of oskar mRNA at the posterior of the oocyte (9-11), for the anchoring of bicoid mRNA at the anterior of the early embryo (12,13) and for asymmetric localization of prospero mRNA in dividing embryonic neuroblasts (14–17).
Mammals possess two Staufen homologs, Staufen1 and Staufen2, both of which function in developing and adult neurons (18–21). Staufen2 has also been shown to segregate asymmetrically during mammalian neural stem cell divisions and to regulate that lineage (22,23). Staufen1 and 2 have been shown to direct degradation of target RNAs (24,25), and Staufen1 enhances the translation of its targets (26), regulates pre-mRNA splicing (27) and facilitates export of target mRNAs from the nucleus (28). The Caenorhabditis elegans Staufen homolog has been implicated in exogenous RNAi (29).
How dsRBPs like Staufen recognize specific mRNA targets in vivo is not well understood. Drosophila Staufen contains five dsRBDs, three of which (dsRBD1, dsRBD3 and dsRBD4) bind to dsRNA in vitro (9), and one of which (dsRBD3) binds optimally in vitro to a stem-loop containing 12 uninterrupted base pairs (bp) when compared against other stem loops (30). However, 12 uninterrupted base pairs are present in only one of the three regions of the bicoid 3′ untranslated region (UTR) to which Staufen binds in vivo (13,31). In mammals, a 19 bp stem is required for Staufen1 binding to ARF1 mRNA, its best-characterized target (32), but comparable structures have not been detected in other targets of Staufen1 (24,32,33). Intermolecular RNA–RNA interactions may also be important for target recognition by dsRBPs: loop–loop interactions between bicoid mRNAs (29) and interactions between long non-coding RNAs and Alu elements in human targets (34) or B elements in rodent targets (35) have been shown to be important for Staufen binding.
To understand how Staufen recognizes its targets in vivo, as well as identify new biological roles for Drosophila Staufen, we have performed RNA co-immunoprecipitations (RIPs) followed by microarray analysis (RIP-Chip) to identify Staufen mRNA targets in early Drosophila embryos. We used an anti-green fluorescent protein (GFP) antibody to immunoprecipitate transgenic GFP-tagged Staufen (16) as well as a synthetic anti-Staufen antibody (36) to immunoprecipitate endogenous Staufen from wild-type embryos. These experiments identified numerous novel Staufen-associated mRNAs, with a high degree of overlap between the Staufen targets identified by each approach. The functions and localization patterns of these targets support previously known Staufen functions and suggest novel roles for Staufen in early embryos. Using computational methods, we identified secondary structures that are enriched among Staufen targets and are highly specific to Staufen-bound transcripts.
MATERIALS AND METHODS
Drosophila stocks
Drosophila stocks used were as follows: w1118, GFP-Staufen transgenics (14) (line: GFP311) and staufen mutants with the genotypes w; stauD3 cn sp/CyO; GFP-Stau2.2FL/TM3 and TM6B, w; stauD3/CyO (37), and P[ry11] staury9 cn1/CyO; ry506 (8).
RNA co-immunoprecipitations
For RIPs with synthetic anti-Staufen antibody, synthetic antibodies were expressed and purified as Fabs, and immunoprecipitations were as described (36) with only minor modifications. For anti-GFP-Staufen immunoprecipitations for western blots, RIP-Chip and quantitative PCR (qPCR) validation experiments, protein G magnetic beads (Invitrogen Cat. # 10004D) were first blocked (38), and immunoprecipitations were then performed using a protocol adapted from Invitrogen’s Dynabeads® Protein G protocol and Roche’s immunoprecipitation protocol for anti-GFP (Roche Cat. # 11814460001). The RNA retrieved from these immunoprecipitations was isolated using TRIzol (Invitrogen) following the manufacturer’s protocol and concentrated using RNA clean and concentrator 5 columns (Zymo Research Cat #R1015). For the comparison of synthetic antibody to anti-GFP RIPs (Figure 2C), a slightly different protocol was used, with the anti-GFP RIP protocol modified to be as similar as possible to the synthetic antibody RIP protocol. Details can be found in the Supplementary Materials and Methods.
Figure 2.
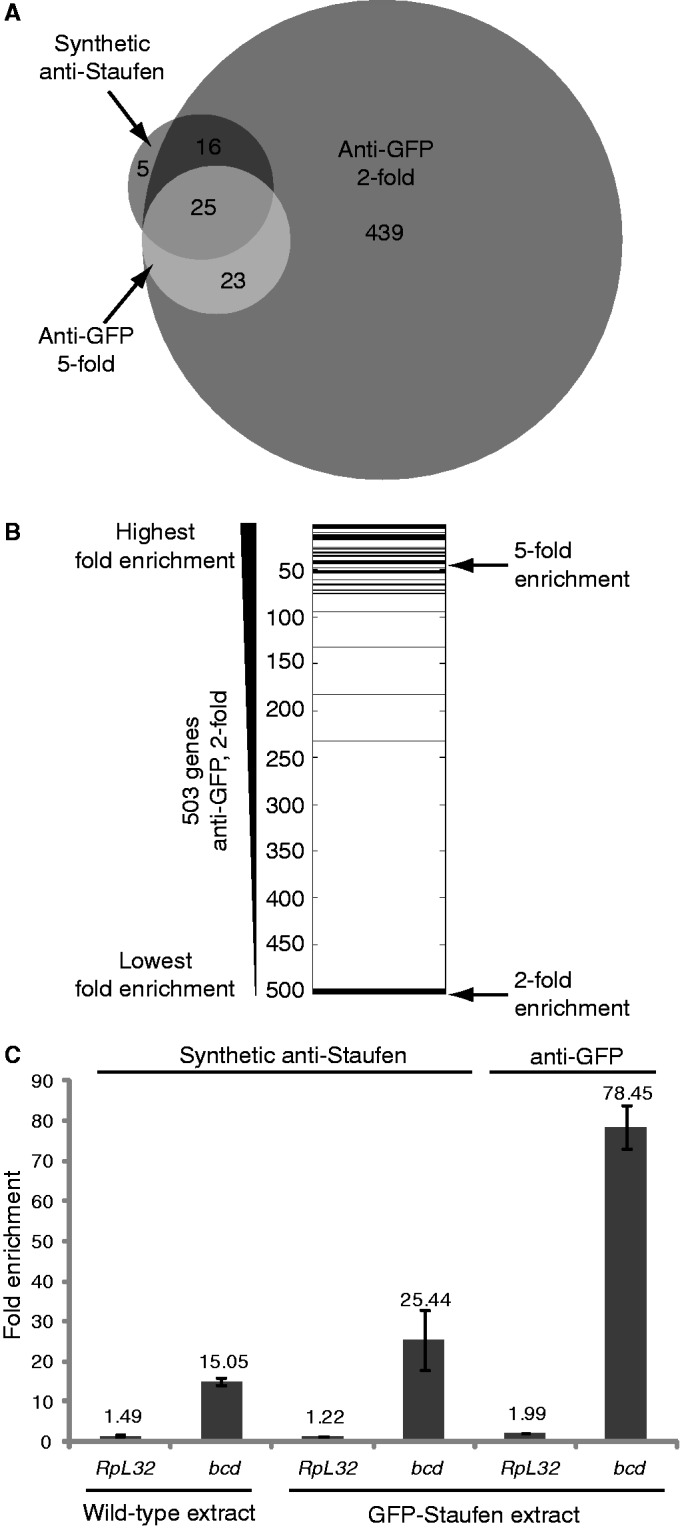
Comparison of the synthetic anti-Staufen and anti-GFP-Staufen RIPs. (A) A Venn diagram [generated using the BioVenn web application (48)] shows overlap of Staufen targets from the synthetic anti-Staufen RIP with a fold enrichment cut-off of at least two (dark grey) and the transgenic anti-GFP RIPs with fold enrichment cut-offs of at least two (medium grey) or five (light grey). (B) The 503 genes from the anti-GFP 2-fold list were ranked according to decreasing fold enrichment and the 41 overlapping genes from the synthetic anti-Staufen 2-fold list were then overlaid in black showing that they represent genes with some of the highest fold-enrichments. (C) RT-qPCR analysis of the enrichment of the target mRNA bicoid and the reference mRNA RpL32 in Staufen RIPs conducted using wild-type extract and synthetic anti-Staufen, transgenic GFP-Staufen extract and synthetic anti-Staufen, and transgenic GFP-Staufen extract and anti-GFP. Each bar represents the average fold enrichment of the respective transcript in the anti-Staufen RIPs relative to the appropriate control. Error bars represent the standard error of the mean for n = 3 biological replicates.
Microarrays
For microarray analysis, double-stranded cDNA was prepared following the protocol described in the NimbleGen Array User’s Guide (Gene Expression Arrays, version 5.0) with minor modifications. For all samples, 500 ng of double-stranded cDNA was labelled with Cy3- or Cy5-tagged random nonamers following the Roche NimbleGen protocol. Labelled cDNA was then hybridized to custom-designed Drosophila 4 × 72K NimbleGen arrays (GEO platform number: GPL10539). Microarray data were analyzed using the Significance Analysis of Microarrays (39) function available in the MultiExperiment Viewer (MeV) software application (40,41). Details can be found in the Supplementary Materials and Methods, and see Supplementary Figures S1 and S4.
Data access
The data reported in this study have been deposited in NCBI’s Gene Expression Omnibus (42) and are accessible through GEO Series accession number GSE43418 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE 43418).
Reverse transcription-quantitative PCR
For reverse transcription-quantitative PCR (RT-qPCR), RNA isolated from immunoprecipitates was reverse transcribed with random hexamer primers and Superscript II reverse transcriptase (Invitrogen). The resulting single-stranded cDNA was subjected to real-time PCR with SYBR green PCR master mix (ABI) using a CFX384 Real-Time System (Bio-Rad). Relative levels of different transcripts were determined using a standard curve.
Gene set annotation enrichment analysis
The Database for Annotation, Visualization and Integrated Discovery (DAVID) functional annotation tool web server (43,44) was used. Terms or features enriched at a false discovery rate (FDR) of ≤10% and/or a Benjamini P-value of <0.1 were considered significant. Details can be found in the Supplementary Materials and Methods.
Localization pattern enrichment analysis
The subcellular localization of Staufen-associated transcripts as annotated in the Fly-FISH database (http://fly-fish.ccbr.utoronto.ca/) was analyzed to ask whether these transcripts were enriched for particular localization patterns (Fly-FISH annotations up to date as of November 2012 were analyzed). Enrichment was determined using Fisher’s exact test, and P-values were adjusted for multiple comparisons using the Benjamini–Hochberg method to estimate FDRs. Localization terms enriched at an FDR ≤ 10% were considered significant. Details can be found in the Supplementary Materials and Methods.
Source of transcript sequences for assessment of UTR and open reading frame lengths and motif finding
The Drosophila melanogaster (BDGP5) and Homo sapiens (GRCh37.p6) transcript sequences were downloaded from Ensembl using BioMart (http://www.ensembl.org/biomart/martview/eea40c9db7c1002506d5c766e8772c08) in August 2012. We downloaded all cDNA sequences and defined 3′UTRs as the portion of the cDNA 3′ to the coding sequence, as defined by Ensembl. When there were multiple isoforms for a gene, we used the longest isoform to represent its mature mRNA sequence.
Definitions of secondary structure terms
Throughout this article, in reference to a specific secondary structure: we use the term ‘paired’ to refer to RNA bases that participate in a canonical base pair (i.e. a Watson–Crick base pair or a G-U wobble); we use the term ‘mismatch’ to indicate two bases that are found across from one another in a secondary structure but are not canonical base pairs; we use the term ‘unpaired’ to refer to bases that do not have a corresponding partner base on the other strand of a stem. Mismatches only occur in internal loops and unpaired bases occur in either bulge loops or internal loops, although the latter need not contain any unpaired bases. For example, the right-hand internal loop indicated in the schematic in Figure 5A contains two mismatches and one unpaired base, whereas the bulge in Figure 5A contains three unpaired bases and no mismatches.
Figure 5.

Schematic of the in silico assays for discovery of Staufen’s binding preferences. (A) Structural annotations used in the manuscript. (B) Examples of the 15 of 19 motif. Black represents a 15 of 19 motif, whereas light grey indicates partners of this motif. (C) Examples of the [19,15] structure. (D) Examples of the [19,15,4] structure. (E) Examples of the [19,15,4]&[12,10,2] SRS. (see ‘Materials and Methods’ and ‘Results’ sections for details).
Defining N of M motif hits
We estimated the ensemble probability that a region of M bases will contain at least N paired bases using the ensemble probabilities that individual nucleotides within these regions will be paired. First, for the entire 3′UTR, we computed the single nucleotide base-pairing probability using RNAplfold (45), with parameter settings W = 200, L = 150 and U = 1 as recommended (46). When folding the 3′ UTR, we also included the 150 nt that comprise the 5′ flanking sequence of the 3′UTR (i.e. the 3′-most 150 nt of the coding region) so that the folding window was not truncated at the 5′-end of the 3′UTR. We then estimated the probability that N bases in an M nucleotide region are paired using the M-N+1th lowest single-nucleotide probability from the region. Specifically, the pairing probability of the N of M motif was estimated using the lowest single-nucleotide probability in the M-mer, after removing the nucleotides with the lowest M-Nth single-nucleotide probability among all the nucleotides except the 5′- and 3′-closing bases. When M = N, this estimate is an upper bound on the probability that the entire region is paired; for other values of N, it is a convenient estimate. For each N of M pair, which we call a ‘motif’ because it corresponds to a contiguous sequence of bases, we deemed ‘hits’ to be those regions in the top 1% of the N of M probabilities across all 3′UTRs. When scoring other regions of the transcript [5′UTR or open reading frame (ORF)], we used the 1% cut-offs defined on 3′UTRs to select motif hits.
Discovery of N of M motifs that predict Staufen binding
To assess how well the N of M motif hits distinguish the Staufen targets from the co-expressed non-targets, we calculated Wilcoxon rank sum P-values and corresponding area under the receiver operating characteristic (ROC) curves (AUROCs) for all values of N and M where M ranged from 1 to 22 and the allowed number of mismatched or unpaired bases ranged from zero to either 4 or M divided by 4, whichever was less. The Wilcoxon rank sum P-values and the AUROCs were computed based on transcript ‘N of M motif hit scores’, which, for a given N and M, were the sum of the probabilities of all N of M motif hits in the transcript’s 3′UTR. These Wilcoxon rank sum P-values and AUROCs were compared with those derived from the 3′UTR length as a baseline. Least absolute shrinkage and selection operator (LASSO) sparse logistic regression was performed to further identify the optimal motifs for Staufen binding. The likelihood ratio test was performed to assess the significance of the improved goodness-of-fit of a regression model containing the selected motifs—over 3′UTR length alone—at classifying transcripts according to whether they were bound by Staufen. In some instances, we also used these methods to assess the goodness-of-fit of regression models based on motif hits in 5′UTRs and coding regions; in those cases, we replaced the 3′UTR-based transcript score with summed probabilities of motif hits in the appropriate region.
Defining [12,10] and [19,15] structures
For each 10 of 12 and 15 of 19 motif hit, we input the region and 150 nt of flanking sequence on either side into Sfold (47). We used Sfold to compute both the centroid structure of the input sequence as well as 1000 samples from the structural ensemble. In each structure, we then identified stems that had either a 10 of 12 or a 15 of 19 motif hit as one side of the stem and deemed them [12,10] and [19,15] structures, respectively. For a motif hit to be deemed a valid structure, it had to satisfy three criteria: (i) at least N of its M bases had to be paired, including the first and last bases; (ii) its ‘partner region’, which is the transcript sequence between the bases that pair with the first and last bases of the N of M motif hit, had to pair only with bases in that hit (i.e. contain no hairpins); (iii) the motif hit had to pair only with bases in its partner region. We found that motif hits that corresponded to valid structures in the centroid also corresponded, in nearly every case, to valid structures in the majority of the ensemble samples. We, therefore, used the centroid to represent the ensemble as this simplified subsequent analysis. We removed any motif hit from consideration that did not correspond to an appropriate valid structure in the centroid. Structures in which both sides of the stem corresponded to motif hits were only represented once in subsequent analyses. These steps are diagrammed in Figure 5B and C.
Identification of additional features of Staufen-recognized [12,10] and [19,15] structures
We examined additional features of [12,10] and [19,15] structures to determine whether any of these distinguished the target and non-target sets. They were as follows: (i) number of mismatches; (ii) number of unpaired bases; (iii) number of bulge loops; (iv) number of internal loops; (v) maximum loop size—which is the maximum loop size among all bulge and internal loops in the stem spanned by the [12,10] or [19,15] structure where the size of a loop is the length of the longest side of the loop; and (vi) the distance between the two paired regions which, depending on the relative position of the motif hit and its partner region, is either the distance between the 3′-end of the motif hit and the 5′-end of its partner region or vice versa. We compared the distribution of these feature values in the target and non-target sets at both the level of individual structures and at the transcript level using cumulative distribution function plots and Wilcoxon rank sum tests. The feature value assigned to each transcript was the minimum value for that feature for all corresponding structures in the transcript.
Using Staufen-recognized structures to predict Staufen targets and non-targets
The ‘Results’ section defines Type I, II and III Staufen-recognized structures (SRSs), and a Type II SRS is diagrammed in Figure 5C. To assess the predictive value of SRSs, we ranked transcripts based on the presence of any of the three types of SRSs in their 3′UTRs. The relative enrichment for transcripts containing the three SRSs is described in the ‘Results’ section: it decreases from Type I to Type III. We therefore assigned the highest rank to transcripts containing a Type I SRS, the second highest to those with a Type II but not a Type I SRS, the next highest to those that only contained a Type III SRS, and the lowest rank to those without any SRSs. We then plotted an ROC curve that demonstrates the ability of this ranking to distinguish target and non-target transcripts and computed the AUROC.
Scoring of the precision of motif mapping
To compute the precision of the mapping of the SRSs, we computed the proportion of nucleotides in motif hits that are in experimentally defined Staufen-binding regions in the 3′UTR-region of interest: the entire 3′UTR in the case of bicoid (13,31) and an experimentally defined 300 nt subset of the ARF1 3′UTR (32). ‘Baseline precision’ is the proportion of the 3′UTR (bicoid) or 3′UTR subset (ARF1) that is in experimentally defined Staufen-binding regions. In the case of Drosophila Staufen’s binding region in the bicoid 3′UTR, the union of the experimentally defined sites (13) was used in the calculation.
Defining bound and unbound sets for the human Staufens
To define the Staufen targets and co-expressed non-targets in human cells, we re-analyzed the published human Staufen RIP-Chip data sets (33). Details can be found in the Supplementary Materials and Methods.
RESULTS
Genome-wide identification of Staufen-associated mRNAs
To identify mRNAs associated with Staufen in early Drosophila embryos, we performed RIP-Chip using two complementary approaches. First, we carried out RIP-Chip of endogenous Staufen from wild-type 0–3 h old embryos using a synthetic antibody, anti-Staufen 2A5, that we previously showed immunoprecipitates Staufen protein along with bicoid mRNA (36). As a negative control, we performed immunoprecipitations using a control antibody (C1) derived from the same synthetic antibody library as anti-Staufen 2A5 (36). We identified 46 genes whose mRNAs were enriched at least 2-fold in Staufen immunoprecipitates compared with negative control immunoprecipitates and had an FDR of <5% (Figure 1A and B and Table 1; Supplementary Figures S1 and S2; Supplementary Table S1; see ‘Materials and Methods’ section for details). All three previously identified Staufen target mRNAs, bicoid, oskar and prospero, were among these 46, and bicoid mRNA was the most highly enriched target identified. Validation experiments using RT-qPCR are presented in Supplementary Table S2.
Figure 1.

Enrichment of expressed transcripts in Staufen RIPs using synthetic anti-Staufen and anti-GFP-Staufen antibodies. The average, across three biological replicates, of the log2 microarray signal intensities of each expressed transcript in the anti-Staufen and control RIPs were plotted against each other (A, C, E). Highlighted in red and shown at the individual replicate level in the adjacent boxplots (B, D, F) are the transcripts that were found, through Significance Analysis of Microarray two-class analysis, to be significantly enriched in the anti-Staufen versus the control RIPs. Those transcripts with an FDR < 5% and passing a fold enrichment cut-off of at least two in the synthetic anti-Staufen experiments are shown in panels (A) and (B), and those with an FDR < 5% and passing fold enrichment cut-offs of at least two and five in the transgenic anti-GFP experiments are shown in panels (C, D) and panels (E, F), respectively. The three previously identified targets of Staufen (bicoid, oskar and prospero) are labelled in each scatter plot and further denoted by crosses. In (A, C, E), solid diagonal lines represent no enrichment, and dotted diagonal lines represent 2-fold (A, C) or 5-fold (E) enrichment or depletion.
Table 1.
Staufen-associated mRNAs identified by synthetic anti-Staufen RIP-Chip (fold enrichment ≥2) and anti-GFP-Staufen RIP-Chip (fold enrichment ≥5)
| Targets identified by synthetic anti-Staufen (FDR <5%, fold enrichment ≥2) and anti-GFP-Staufen RIP-Chip (FDR < 5%, fold enrichment ≥ 5) | Additional targets identified by synthetic anti-Staufen RIP-Chip (FDR < 5%, fold enrichment ≥ 2) | Targets identified by anti-GFP-Staufen RIP-Chip only (FDR < 5%, fold enrichment ≥ 5) |
|---|---|---|
| bicoid | CR14578 | CG5830 |
| dacapo | Phosphoenolpyruvate carboxykinase | ocelliless |
| capping protein beta | Mms19 | CG32756 |
| CR18854 | 825-Oak | no on or off transient A |
| CG4068 | CG12519 | lethal (3) neo38 |
| CG32212 | sequoia | Roughened |
| CG17724 | Vacuolar H[+]-ATPase 26kD E subunit | nubbin |
| CG3523 | Histone demethylase 4B | Flotillin-2 |
| falafel | Ribosomal protein S27 | small wing |
| CR32207 | split ends | bves |
| CG32214 | CG4788 | CG32767 |
| Vacuolar H[+] ATPase accessory protein AC45 | virilizer | Sprouty-related protein with EVH-1 domain |
| prospero | kugelkern | Tob |
| Peroxiredoxin 6005 | vielfaltig/Zelda | CG43736 |
| Vacuolar H[+] ATPase subunit PPA1-1 | CG9977 | fusilli |
| partner of paired | Neurofibromin 1 | CG33932 |
| oskar | CG14915 | pasilla |
| CG14100 | Mediator complex subunit 14 | punt |
| Protein phosphatase 19C | CG32267 | CG10777 |
| CG18273 | CG17270 | CG5966 |
| staufen | CR18166 | cAMP-dependent protein kinase R1 |
| Dorsal switch protein 1 | vein | |
| squeeze | TNF-receptor-associated factor 6 | |
| Polycomb | ||
| CG31688 |
To complement the synthetic antibody RIP-Chip, we also performed RIP-Chip using flies expressing GFP-tagged Staufen (16) immunoprecipitated with a commercially available anti-GFP antibody. Western blotting showed that this antibody successfully immunoprecipitated the fusion protein from transgenic embryo extract (Supplementary Figure S3A) and that GFP-Staufen is present at 1.5–2.0-fold higher levels in GFP-Staufen extract than endogenous Staufen in wild-type extract (Supplementary Figure S3B). This anti-GFP RIP-Chip identified 503 genes (of 6151 expressed) whose mRNAs were enriched at least 2-fold in the anti-GFP immunoprecipitates compared with an anti-FLAG control and had an FDR of <5% (Figure 1C and D; Supplementary Figures S4 and S5; Supplementary Table S3). As with the synthetic antibody, all three previously known Staufen mRNA targets were identified, and bicoid was again the most highly enriched target. RT-qPCR validation experiments are presented in Supplementary Table S2.
In all, 41 of the 46 genes (89%) identified as Staufen targets using the synthetic antibody were also identified by the anti-GFP RIP-Chip (Fisher’s exact test, P < 3 × 10−16; Figure 2A). Moreover, 36 of the 46 targets identified by the synthetic antibody lay within the 100 most highly enriched targets in the anti-GFP experiment (Figure 2B). Therefore, there is a high degree of similarity between the results of the two experiments.
However, the anti-GFP RIP-Chip identified 10-fold more targets than the synthetic antibody RIP-Chip. This discrepancy is likely attributable to differences both in antibody affinities and in expression levels of endogenous versus transgenic Staufen. For example, RIPs using anti-GFP antibody from GFP-Staufen extract gave a 3-fold higher enrichment of bicoid mRNA than synthetic anti-Staufen antibody from the same extract (Figure 2C), most likely as a result of more efficient pull-down of Staufen by the anti-GFP antibody. However, RIPs using the synthetic antibody from GFP-Staufen extract yielded 1.5–2-fold higher enrichment of bicoid mRNA than RIPs using the same antibody from wild-type extract (Figure 2C). As GFP-Staufen is present at 1.5–2.0-fold higher levels in GFP-Staufen extract than endogenous Staufen in wild-type extract (Supplementary Figure S3B), overexpression of GFP-Staufen also appears to contribute to binding to a larger number of mRNAs.
This effect of the overexpression of GFP-Staufen raises the possibility that a subset of the 503 targets identified by the anti-GFP RIP-Chip may not be natural Staufen targets and may only be bound in the context of its overexpression. We therefore created a high-confidence list of 48 targets by applying a more stringent 5-fold enrichment cut-off to the anti-GFP data (Figure 1E and F; Table 1; Supplementary Table S3). These included bicoid, oskar and prospero and shared a total of 25 transcripts with the synthetic-antibody-identified targets (Figure 2A and B), a highly significant overlap (Fisher’s exact test, P < 3 × 10−16).
To avoid excluding low-affinity but real targets of Staufen identified in the anti-GFP RIP-Chip with a 2-fold enrichment cut-off, while also ensuring particular consideration of the high-confidence targets identified using the synthetic antibody and the anti-GFP RIP-Chip with a 5-fold cut-off, our subsequent analyses were conducted on all three lists of Staufen-associated transcripts.
Functional analysis of Staufen-associated transcripts
To gain insight into potentially novel functions for Staufen in early embryos, we analyzed Staufen-associated mRNAs by gene set annotation enrichment analysis using the DAVID functional annotation tool (43,44). Two stringencies were applied: the standard FDR cut-off (≤10%) or the more stringent ‘Benjamini’ P-value (≤0.1) (Table 2 and Supplementary Tables S4–S6). This revealed a number of functions for Staufen in early embryos.
Table 2.
Gene set annotation enrichment analysis results for Staufen targets
| Enriched term or featurea | Enriched with FDR ≤ 10% among targets identified byb |
Enriched with ‘Benjamini’ adjusted P ≤ 0.1 among targets identified byb |
||||
|---|---|---|---|---|---|---|
| Synthetic anti-Staufen | Anti-GFP 5-fold | Anti-GFP 2-fold | Synthetic anti-Staufen | Anti-GFP 5-fold | Anti-GFP 2-fold | |
| Cell fate determination/commitment | √ | √ | √ | – | √ | √ |
| Alternatively spliced | √ | √ | √ | – | √ | √ |
| Transcription regulator activity/Homeodomain-containing | √ | √ | – | – | √ | – |
| Reproductive process-related/Oogenesis | – | √ | √ | – | √ | – |
| Pattern specification process | – | √ | √ | – | √ | √ |
| Cognition | – | √ | √ | – | √ | √ |
| Neuroblast differentiation/Neurogenesis | – | √ | √ | – | – | – |
aOnly terms or features enriched in at least two of the three Staufen target lists (synthetic anti-Staufen, anti-GFP 2-fold, anti-GFP 5-fold) are shown. For simplicity, terms with similar meanings are clustered or represented with a single Gene Ontology term, SwissProt keyword or UniProt feature.
bEnrichment analysis was performed using the DAVID functional annotation tool.
First, our results suggest a more general role than previously suspected for Staufen in controlling the spatial and/or temporal aspects of transcription in early embryos by binding and regulating mRNAs encoding transcriptional regulators. Along with the previously known targets, bicoid and prospero, which encode homeodomain-containing transcriptional regulators, the newly identified targets included two additional homeodomain-encoding transcripts (ocelliless and nubbin), Mediator complex subunit 14 of the basal transcription machinery and several known or potential epigenetic regulators (Polycomb, Dorsal switch protein 1 and Histone demethylase 4B). Consistent with this more general role in transcriptional regulation, annotation terms related to ‘transcription regulator’ or the keyword ‘homeobox’ were significantly enriched among targets (Table 2 and Supplementary Tables S4–S6).
Second, all three of Staufen’s previously identified targets control cell fate either via transcriptional (bicoid, prospero) or post-transcriptional (oskar) regulation. Our results suggest a more general role for Staufen in regulation of cell fate and patterning at these levels as well as at the level of signal transduction and the cell cycle. Staufen’s newly identified targets include mRNAs encoding signaling molecules, cell-cycle regulators, additional transcriptional regulators and additional post-transcriptional regulators with roles in cell fate determination. Accordingly, GO terms related to cell fate were significantly enriched among all three lists of targets (Table 2 and Supplementary Tables S4–S6).
Third, Staufen is expressed and functions both during oogenesis and in the early embryo. Many of its targets are maternal mRNAs that encode proteins that are likely to function at both of these developmental stages. Thus, GO terms related to ‘gamete generation’, ‘oogenesis’ and various aspects of ‘reproductive process’ were significantly enriched among the targets (Table 2 and Supplementary Tables S4–S6). The fact that GO terms related to ‘reproductive process’ are also enriched in the targets recently identified for a Staufen homolog in C. elegans (29), suggests a conserved role during oogenesis and in early embryos.
Fourth, the SwissProt keyword ‘alternative splicing’ and/or the UniProt sequence feature ‘splice variant’, both of which denote genes with alternatively spliced isoforms, were significantly enriched among Staufen targets (Table 2 and Supplementary Tables S4–S6). This suggests a novel role for Drosophila Staufen: binding to transcripts that are alternatively spliced and/or regulation of alternative splicing. The latter function would be consistent with a recently reported role for mammalian Staufen1 in regulation of alternative splicing (27).
Finally, GO terms related to nervous system development (i.e. ‘neuroblast differentiation’, ‘neurogenesis’) and function (‘cognition’) were significantly enriched among Staufen targets (Table 2 and Supplementary Tables S4–S6). Ninety-five percent (42/44) of the genes from the anti-GFP 2-fold list in the categories ‘neuron differentiation’ and/or ‘cognition’ are expressed maternally in early embryos (according to the Fly-FISH database (49), the BDGP in situ database (50,51) and/or the modENCODE temporal expression data (52) available on FlyBase) as well as later in the developing or adult nervous system. Given that Staufen is expressed in the developing and adult nervous system, our data suggest that Staufen may regulate these targets at two different stages of development.
Sub-cellular localization of Staufen-associated transcripts
Given Staufen’s well-established and conserved role in mRNA localization, we analyzed the Staufen-associated mRNAs for enrichment of subcellular localization patterns reported by the Fly-FISH database (49) (http://fly-fish.ccbr.utoronto.ca/) (see ‘Materials and Methods’ section for details).
Analysis of the embryonic stage 1–3 as well as the stage 4–5 localization patterns of Staufen targets showed a striking enrichment for categories related to posterior localization (FDR < 1% for anti-GFP 2-fold targets; Figure 3 and Supplementary Tables S7–S12), consistent with the fact that Staufen is concentrated in the posterior of the early embryo (8). The ∼30 posterior-localized Staufen targets include nanos, oskar, arrest, lost, IGF-II mRNA-binding protein (Imp) and oo18 RNA-binding protein (orb) mRNAs, all of which encode well-known post-transcriptional regulators, and several of which are known to function in the germ plasm.
Figure 3.

Staufen targets are enriched for specific annotated embryonic localization patterns. A search of the Fly-FISH database was performed to identify localization categories that are enriched among Staufen target genes for (A) embryonic stages 1–3 and (B) embryonic stages 4–5. For both (A) and (B), categories in closed boxes were tested for enrichment. The grey shading indicates the localization categories that were enriched among anti-GFP 2-fold targets, FDR ≤10%. No categories were enriched among synthetic antibody or anti-GFP 5-fold targets with an FDR < 10%, likely due to the small sizes of these lists. See Supplementary Tables S7–S12 for detailed results.
Staufen targets were also significantly enriched for the stage 1–3 localization category ‘cell division apparatus’ and the stage 4–5 category ‘apical enrichment’ (Figure 3 and Supplementary Tables S7–S12). These categories are intriguing, as they are consistent with the previous observation that on injection into early embryos of in vitro synthesized bicoid 3′UTR RNA, Staufen localizes to mitotic spindle poles during mitosis and to the cortex of the embryo in association with the migrating syncytial nuclei (13).
Finally, analysis of the stage 4–5 patterns of Staufen-associated mRNAs showed highly significant enrichment (FDRs ranging from 0.004 to 8.3% for anti-GFP 2-fold targets) for categories describing zygotic transcription (Figure 3 and Supplementary Tables S7–S12). Enrichment for targets in the process of being transcribed is intriguing in light of the enrichment among Staufen’s targets for alternatively spliced transcripts (discussed earlier in the text) and the reported role of mammalian Staufen1 in regulation of alternative splicing (27).
Drosophila and human Staufen targets have unusually long 3′UTRs
We next assessed features of the transcript sequence that might distinguish Staufen-associated mRNAs from co-expressed non-target mRNAs (hereafter referred to simply as ‘non-targets’). First, we compared 5′UTR, ORF and 3′UTR lengths and found that the median 3′UTR length of targets was 3–4-fold greater than that of the non-targets for all three target lists (synthetic anti-Staufen, anti-GFP-Staufen 2-fold and anti-GFP-Staufen 5-fold) with a Bonferroni-corrected Wilcoxon rank sum of P < 10−4 (Figure 4A; Supplementary Table S13). The 5′UTRs of targets on both anti-GFP lists were also significantly longer than non-targets, although to a lesser extent (2–3-fold; Bonferroni-corrected Rank sum P < 10−5), and this was not the case for the synthetic anti-Staufen targets (fold = 1.78; Bonferroni-corrected Rank sum P = 0.11). The median length of target ORFs on the anti-GFP 2-fold list was marginally longer (Bonferroni-corrected Rank sum P = 0.04), but this was not true for the other two lists.
Figure 4.
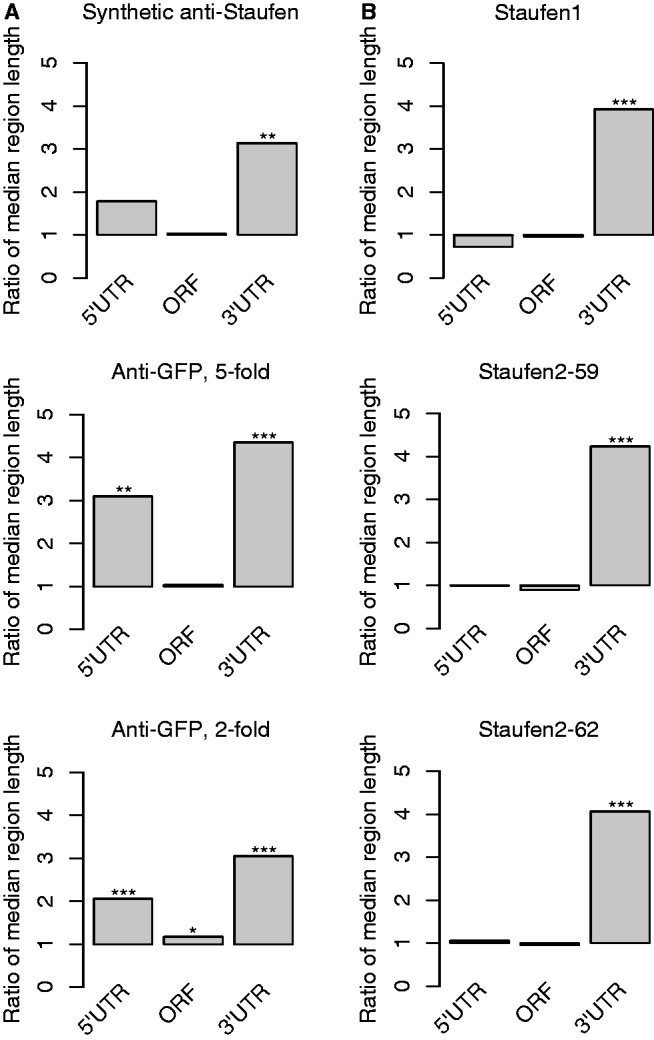
Drosophila and human Staufen targets have unusually long 3′UTRs. (A) Bar plots showing the ratios of the median length of the 5′UTR, ORF and 3′UTR of Drosophila Staufen targets to the median length of the co-expressed non-targets. (B) The ratios of median lengths of human Staufen target transcripts to the median lengths of the co-expressed non-targets. The human Staufen1 targets were identified using anti-HA RIP-Chip from HEK293T cells transfected with Stau155-HA expressor (33) (with five-fold enrichment cut-off, FDR < 5%). The human Staufen2 targets were identified using anti-HA RIP-Chip from HEK293T cells transfected with Stau259-HA expressor or Stau262-HA expressor (33) (with 5-fold enrichment cut-off, FDR < 5%). The statistical significance of the differences between the lengths of targets and non-targets was assessed using Bonferroni-corrected Wilcoxon Rank sum P-values: *P < 0.05, **P < 0.001, ***P < 10−6. Exact numbers are given in Supplementary Table S13.
We next re-analyzed three previously published RIP-Chip experiments identifying human Staufen1 and 2 targets (33) and found that they also had ∼ 4-fold longer 3′UTRs than co-expressed non-targets (Bonferroni-corrected Rank sum P < 10−7) (Figure 4B and Supplementary Table S13; see ‘Materials and Methods’ section). There was no significant difference in 5′UTR or ORF length for these targets (Figure 4B and Supplementary Table S13).
In summary, 3′UTR length is a major feature that distinguishes both the Drosophila and human Staufen targets from non-targets. To determine whether long 3′UTRs were unique to Staufen targets, we asked whether targets of several single-stranded RBPs also have long 3′UTRs. We calculated the ratios of median 3′UTR lengths of single-stranded RBP targets to the median lengths of the 3′UTRs of co-expressed non-targets identified using RIP-Chip for Drosophila PUM (53), human ELAVL1 (54), human PTB (55) and human PUM1 (56). In all cases, the mRNAs in these target sets also had long 3′UTRs; however, the fold increase was less than for Staufen targets: 1.5–2.5-fold rather 3.0–4.5-fold (Supplementary Table S13 and Supplementary Figure S6).
Thus, although long 3′UTRs are a feature of the targets of both double-stranded and single-stranded RBPs, Staufen targets exhibit particularly long 3′UTRs.
High-confidence Drosophila Staufen target mRNAs are enriched for paired regions of specific lengths in their 3′UTRs
We next asked whether we could identify specific structural motifs in the 3′UTRs of Staufen targets that would distinguish them from non-targets. Specifically, we searched for double-stranded stems of varying length, ranging from 1 to 22 bp, and with varying degrees of imperfect pairing, and asked whether any such structures were enriched among Staufen targets compared with non-targets. Local folding of mRNA is a better predictor of its secondary structure and protein interaction than global folding (46,57) but local folding tools, such as RNAplfold, only output the probability that a single base is paired. As such, we adopted a hybrid strategy to identify the characteristics of stems bound by Staufen: we first identified regions of the mRNA sequence likely to be paired; then, we assessed Staufen’s preferences for paired regions of various sizes (1–22 nt) and, finally, filtered the preferred regions for those that were actually stems. Figure 5 diagrams the steps in this process.
To identify 3′UTR regions likely to be in stems, for all M-mers (M = 1 to 22 contiguous nucleotides), we estimated the probability that a number, N, of these M contiguous bases was paired using RNAplfold. This is a necessary but not sufficient condition for the M-mer to be in a stem with N of its bases paired. For each M-mer, we set a range for the allowed number of mismatched or unpaired bases from zero to either four or M divided by four, whichever was less (e.g. where M-N = 0 to min[4,M/4]). As an example, based on this definition, an M-mer of 19 with from 15 to 19 paired bases (where the lowest number of paired bases is designated N) is given the designation 15 of 19 (i.e. N of M). We did not allow unpaired bases at the first and last position of the M-mer (see ‘Materials and Methods’ section). We then scanned each 3′UTR for each N of M combination and scored a region as containing a particular N of M ‘hit’ if the probability that the region contained N of M paired bases was found in the top 1% of the N of M probabilities across all 3′UTRs (see Supplementary Table S14 for the cut-offs). We refer to each N of M as a different ‘motif’ because each corresponds to a contiguous sequence of bases. Figure 5B diagrams several motif hits. We assigned each transcript an N of M ‘motif hit score’ equal to the sum of the probabilities of all hits in that transcript’s 3′UTR (see ‘Materials and Methods’ section for details) and asked whether these scores were significantly higher in the Staufen target transcripts versus the non-targets using a one-sided Wilcoxon rank sum test. In addition, to ensure that any significant increase in N of M motif scores of Staufen targets was not solely a result of the differences in 3′UTR length, we set the Rank sum P-value of the 3′UTR length as the baseline. We used AUROC to indicate the effective size of the enrichment, as it has a linear relationship with the Wilcoxon rank sum test statistic; in the vast majority of cases, the feature (i.e. N of M motif score or 3′UTR length) associated with a higher AUROC on a data set also has a more significant P-value. AUROC is also a measure of classification accuracy; in this context, if a feature has a higher AUROC, its scores are better predictors of Staufen binding in these data.
We performed this analysis on each of the three sets of Drosophila Staufen targets described earlier in the text as well as, for comparison, Pumilio targets (53). Analysis of the Staufen targets identified by anti-GFP RIP-Chip with a 2-fold enrichment cut-off and of the Pumilio targets did not reveal any N of M motifs with higher AUROC than 3′UTR length (Supplementary Table S15). In contrast, analyses of the targets identified using either synthetic anti-Staufen or anti-GFP-Staufen with 5-fold cut-off revealed AUROCs above baseline for several N of M motifs (Figure 6; Supplementary Table S15): for the synthetic antibody targets, these included values of M ranging from 4 to 21, with the highest peak at the 10 of 12 motif (Figure 6A, Supplementary Table S15); for the anti-GFP 5-fold targets, these included values of M ranging from 8 to 22, with the highest peak at the 16 of 19 motif (Figure 6C, Supplementary Table S15).
Figure 6.

Specific double-stranded structures are enriched in Staufen target transcript 3′UTRs. (A, C) Wilcoxon rank sum P-values were used to assess how well a particular double-stranded stem could distinguish between Staufen targets and co-expressed non-targets that were defined by (A) synthetic antibody RIP-Chip (with 2-fold enrichment cut-off, FDR ≤ 5%) or (C) anti-GFP RIP-Chip (with 5-fold enrichment cut-off, FDR ≤ 5%). The test was performed on stems of varying length ranging from 1 to 22 base pairs (indicated on the x-axis) and with varying degrees of imperfect pairing ranging from 0 to 4 mismatches (indicated by the different colours and symbols). Among these, only the stems with more significant P-value than 3′UTR length (the baseline) are shown and were tested in the analysis shown in (B) and (D). (B, D) Using the stems identified in (A) and (B) as well as the 3′UTR length as the features, LASSO regression was trained to select the features most relevant to Staufen target prediction (i.e. features with non-zero weights). Compared with the training model using 3′UTR length only, the LASSO-selected features significantly improved the prediction of Staufen binding: likelihood ratio test P < 10−7 (B) and P < 10−11 (D). Exact numbers are given in Supplementary Table S15.
Many of these motifs differed by only one in M or in N, raising the possibility that some of the motifs performed well simply because they were imperfect predictors of the presence of a related motif. Indeed, the values of the transcript scores for similar motifs were extremely highly correlated (Supplementary Figure S7). Therefore, to select a core non-redundant set of N of M motifs that collectively explained Staufen’s binding preferences, we performed LASSO logistic regression on the synthetic antibody and anti-GFP 5-fold lists. As potential features to distinguish Staufen targets from non-targets, we used 3′UTR length and all of the N of M motifs identified by us as individually having AUROCs above baseline. This sparse regression analysis assigns weights to each feature, with the most important features being assigned the greatest weights and the least important features receiving weights of zero. For both data sets, LASSO assigned non-zero weights to 10 of 12 (which had the greatest weights: 0.34 and 0.43) and to 15 of 19 (which had a weight of 0.13 for both target sets) (Figure 6B and D). Non-zero weights were also given to 9 of 10, 11 of 11, 14 of 16 and 18 of 20. The logistic regression models containing these collections of motifs had a significantly better fit to the Staufen-binding data from their corresponding sets than ones based on 3′UTR length alone (likelihood ratio test P < 10−7 for synthetic antibody and P < 10−11 for the anti-GFP, 5-fold; Figure 6 and Supplementary Table S15), indicating that the presence of these motifs explains Staufen’s binding preferences better than the strong bias in 3′UTR length noted earlier.
Having identified this core set of motifs through our analysis of 3′UTRs, we also asked whether there was enrichment for these motifs in the 5′UTRs or ORFs of Staufen-bound mRNAs. To do this, we asked whether the motif scores computed for the 5′UTR or ORF had significantly higher AUROCs than just the length of the corresponding region at the task of classifying transcripts according to Staufen binding, on either the synthetic antibody or anti-GFP 5-fold target sets (Supplementary Table S16). For both 5′UTR and ORF, none of the motifs was a better predictor than length. In addition, notably, the motif hits in the 3′UTR had consistently higher AUROCs than those in either the ORF or 5′UTR (e.g. AUROCs of 0.73–0.90 for those in the 3′UTR versus 0.48-0.63 for 5′UTR and ORF; see Supplementary Table S16).
Having identified several potential motifs that are enriched in the 3′UTRs of Staufen’s targets, we combined the Staufen-associated transcripts from the synthetic antibody and GFP 5-fold data sets to further refine our model of Staufen binding. This created a new set of Staufen target transcripts consisting of the union of the Staufen targets from the two data sets, and a new set of co-expressed non-targets consisting of the intersection of the non-targets from the two data sets. Repeating our regression analysis on these new sets, we found that the fit of the model containing only the 10 of 12 and 15 of 19 motifs (and 3′UTR length) was statistically indistinguishable from one that included all six motifs (and 3′UTR length) [P > 0.05, one-tailed likelihood ratio test (LRT)]. Furthermore, the model containing only the 10 of 12 and 15 of 19 motifs had a significantly better fit than one that only contained one of these two motifs (P < 0.05 for both, one-tailed LRT, Bonferroni-corrected). As such, for further analysis, we considered only 10 of 12 and 15 of 19.
Computational analysis of the properties of dsRNA stems bound by Staufen
The aforementioned analysis provides information for only one side of a potential dsRNA structure (i.e. the black strands illustrated in Figure 5B). As such, a single N of M designation describes a large number of potential secondary structures (for some examples, see the combinations of black and grey strands in Figure 5B). Thus, to further refine our analysis, we next sought to identify properties of the dsRNA stems that corresponded to the 10 of 12 and 15 of 19 motif hits. To do so, we used Sfold (47) to predict the secondary structure of a region of ∼300 nt centred on each of the 10 of 12 and 15 of 19 motif hits. Sfold outputs two results, both of which we considered in our analysis: a set of 1000 structures sampled from the ensemble of all possible secondary structures, and the ‘centroid’ structure, which is the single structure with the highest total agreement with all 1000 samples. Using the Sfold results, we then identified those centroids where the motif hit satisfied the following three critera, thus placing it in a dsRNA stem: (i) at least N of its M bases had to be paired, including the first and last bases; (ii) its ‘partner region’, which is the transcript sequence between the bases that pair with the first and last bases of the N of M hit, had to pair only with bases in the hit (i.e. contain no hairpins); (iii) the motif hit had to pair only with bases in its partner region (compare Figure 5B and C to see examples of structures that are filtered out, or retained, using these criteria). Based on these analyses, we found that a significantly higher percentage of motif hits were in dsRNA stems in the Staufen targets versus non-targets (Table 3, P < 10−9). We then removed hits that did not correspond to dsRNA stems and combined hits that were on either side of the same dsRNA stem into a single structure consisting of the motif hit and the sequence with which it pairs. We call these structures [12,10] and [19,15] where the first number indicates that at least one side of the dsRNA stem spans exactly 12 (or 19) nt, and the second number indicates that at least 10 (or 15) bases are paired on that side (e.g. see Figure 5C). The partner region could, in principle, span a different number of nucleotides and have fewer or more unpaired bases.
Table 3.
Sfold validation of 10 of 12 and 15 of 19 motif hits
| Motif hit | Staufen targets | Co-expressed non-targets | Hypergeometric P-value |
|---|---|---|---|
| 10 of 12 | |||
| No. of motif hits in dsRNA stem | 672 | 3272 | 1.83 × 10−10 |
| No. of motif hits | 954 | 5469 | |
| Percentage motif hits in dsRNA stem | 70.44 | 59.83 | |
| No. of genes with at least one hit in dsRNA stem | 45 | 572 | 4.41 × 10−11 |
| No. of genes with at least one hit | 55 | 1528 | |
| Percentage genes with at least one hit in dsRNA stem | 81.82 | 37.43 | |
| 15 of 19 | |||
| No. of motif hits in dsRNA stem | 487 | 1626 | 1.71 × 10−36 |
| No. of motif hits | 954 | 5469 | |
| Percentage motif hits in dsRNA stem | 51.05 | 29.73 | |
| No. of genes with at least one hit in dsRNA stem | 36 | 344 | 3.54 × 10−11 |
| No. of genes with at least one hit | 55 | 1528 | |
| Percentage genes with at least one hit in dsRNA stem | 65.45 | 22.51 |
On further analysis of the [12,10] and the [19,15] structures, we found clear differences between the Staufen targets and non-targets for both structures. Specifically, we assessed the presence of various structural features: (i) unpaired bases, which refer to those bases that do not have a corresponding partner base on the other strand of a stem, (ii) mismatches, which refer to two bases that are found across from one another in a secondary structure but are not canonical base pairs, (iii) internal loops, which are loops emanating from within the dsRNA stem that contain at least one mismatch and, possibly, some unpaired bases, and (iv) bulge loops, which are loops emanating from within one strand of the stem and that contain only one or more unpaired bases. These are diagrammed in Figure 5A.
Comparing Staufen targets with non-targets, our analysis revealed that the [12,10] and [19,15] structures in the targets had significantly fewer unpaired bases (Supplementary Figure S8) as well as a weak preference against mismatches. The bias against unpaired bases also manifested as a significant depletion for bulges in the target set (Supplementary Figure S8). In addition, there was a significant preference for smaller internal loops or bulges in the Staufen targets than the non-targets (Supplementary Figure S9). Taken together, these results suggest an overall model in which Staufen binds [12,10] and [19,15] dsRNA stems that have a small number of mismatches, zero or few unpaired bases and short internal loops.
Many transcripts in both the target and non-target sets contain multiple [12,10] and [19,15] structures; as such, we next considered the same criteria at the transcript level to determine features associated with the ‘best’ Staufen site in each transcript. We did this by assigning each transcript a mismatch, unpaired base, and loop count as well as loop length equal to the minimum of those values across all [12,10] and [19,15] structures in that transcript’s 3′UTR. This analysis also revealed a significantly lower number of unpaired bases in the ‘best’ structure in Staufen target transcripts compared with non-targets, which also manifested as an even stronger preference for balanced stems (i.e. those containing only paired or mismatched bases and no unpaired bases) (Figure 7). In particular, 82% of Staufen targets that had at least one [12,10] structure contained a balanced [12,10] structure (compared with 47% in non-targets). We use the designation [12,10,0] to describe these balanced structures where the last number refers to the maximum number of unpaired bases in the structure. The enrichment among the Staufen targets for transcripts containing balanced [19,15] structures (i.e. [19,15,0]) was even more striking (67 versus 12%). Because many more Staufen target transcripts have [19,15] structures than do non-targets, if we define a Staufen-bound transcript as one containing a [19,15] that is balanced (i.e. [19,15,0]), this rule would be satisfied by 44% (n = 24) of the target transcripts but only 3% of the non-target transcripts (n = 42).
Figure 7.

Characteristics of the stems bound by Staufen. We compared the 10 of 12 (A) and 15 of 19 (B) structures in the Staufen targets versus non-targets, by characterizing the structural features: (i) number of mismatches; (ii) number of unpaired bases; (iii) number of bulges; (iv) number of internal loops. If there was more than one structure in a transcript’s 3′UTR, the feature with the minimal score was used to represent the gene. The Wilcoxon rank sum test was used to assess the feature in Staufen targets (the solid line) versus non-targets (the dashed line). pos: targets; neg: non-targets.
We note that Staufen sites defined by previous structural, in vitro and/or in vivo studies are not nearly as predictive of its binding as the [12,10] and [19,15] structures: using the criteria described earlier in the text, only 31% of all targets and 4% of the non-targets contain a perfect 12 bp region of dsRNA (i.e. [12,12,0]), which was defined as an optimal structure for Staufen dsRBD3 binding (30), and none of the target transcripts and only two of the non-target transcripts (0.1%) contain a perfect 19 bp region of dsRNA (i.e. [19,19,0]) defined as an optimal site for human Staufen1 binding to ARF1’s 3′UTR (32).
That said, the [19,15,0] rule does not explain all instances of Staufen binding; in particular, the bicoid 3′UTR does not contain one of these high-confidence sites. However, we can slightly refine our model of the [12,10] and [19,15] structures to [12,10,2] and [19,15,4], respectively (Figure 5). A model including these latter structures is better at distinguishing Staufen targets from non-targets than the [12,10] and [19,15] structures. Although this model removes individual sites from the Staufen target set, all Staufen targets with a [12,10] or a [19,15] structure contain at least one [12,10,2] or [19,15,4] structure, respectively. On the other hand, this rule reduces the number of non-target transcripts with these structures by removing those that have a large number of unpaired bases in the partner region (e.g. one [19,15] structure in the non-target set had >100 unpaired bases). Note that all [19,15,0] structures are also [19,15,4] structures because the latter includes structures with three, two, one and zero unpaired bases. In total, 65% of Staufen targets and 18% of non-targets contain a [19,15,4] structure, whereas 82% of Staufen targets and 33% of non-targets contain a [12,10,2] structure.
Definition of Staufen-recognized structures (SRSs)
Having defined the structural characteristics of the dsRNA stems bound by Staufen, we next mapped these structures onto: (i) the 3′UTRs of the Staufen target transcripts; (ii) length-matched non-target transcript 3′UTRs; and (iii) a random subset of non-target 3′UTRs (Supplementary Figure S10). This led us to note a substantial overlap of [19,15,4] and [12,10,2] structures in the Staufen target set. As such, we next assessed whether the [12,10,2] structures are independently associated with Staufen binding or if they are simply features of more predictive [19,15,4] structures. To do so, we first assessed whether the presence of [12,10,2] in the absence of [19,15,4] was predictive of Staufen binding by comparing Staufen target and non-target transcripts that did not contain a [19,15,4] structure. We found that 47% (9 of 19) of such transcripts in the Staufen target set have a [12,10,2] structure, whereas only 22% (279 of 1245) of the non-targets have one (P = 0.02, Hypergeometric test). We then asked whether Staufen target transcripts were more likely to have a [19,15,4] structure that contained a [12,10,2] structure than non-targets. Here, we found a highly significant difference: 94% (34 of 36) of Staufen target transcripts with a [19,15,4] structure had one containing a [12,10,2] structure but only 61% (174 of 283) of such non-target transcripts did (P < 2 × 10−5, Hypergeometric test). We also noted a similar, but not statistically significant, preference for [19,15,0] structures containing a [12,10,0] structure in the Staufen targets: 91.7% (22 of 24) versus 69% (29 of 42). These results indicate that [12,10] structures are not only independent predictors of Staufen binding—although the statistical significance of this association is weak—but are also more likely to be found in Staufen targets as features of [19,15] structures.
Based on these results, we define three classes of SRSs: Type I is a [19,15,0] containing a [12,10,0], which we abbreviate as [19,15,0]&[12,10,0]; Type II is a [19,15,4] containing a [12,10,2], abbreviated [19,15,4]&[12,10,2]; and Type III is a [12,10,2]. An example of a Type II SRS is shown in Figure 5D.
Figure 8 individually compares the true- and false-positive rates for each of the aforementioned structures. If we classify transcripts based on which type of SRS they contain, we achieve an AUROC of 81.6% by training the whole data set (i.e. as described earlier in the text, the union of the Staufen targets from the synthetic and GFP 5-fold data sets, and the set of co-expressed non-targets consisting of the intersection of the non-targets from the two data sets), which is comparable with that achieved on held-out data by a logistic regression classifier that uses presence or absence of all the structures mentioned in the previous paragraph plus 3′UTR length as features (data not shown). Because we used the training set to define these structures and the SRSs, the AUROC is likely to over-estimate the classification performance we should expect on new data; however, we suspect that this over-estimate is small because (i) we used a simple classification model; (ii) the enrichment P-values we used to define these features were all highly significant; and (iii) we only made a small number of feature selection choices. We also note that [12,10,0] lies directly on the curve; therefore, it has no added classification power in our training set above the three types of SRSs, and, as such, we do not identify it as an additional SRS. [19,15,0] lies slightly to the left of the ROC curve formed by the three SRSs; therefore, it does have added classification power; however, the increase is small, and, as such, we omit it as a separate feature to ensure that the SRSs are nested, thereby forming the basis of our transcript scoring scheme (see ‘Materials and Methods’ section).
Figure 8.
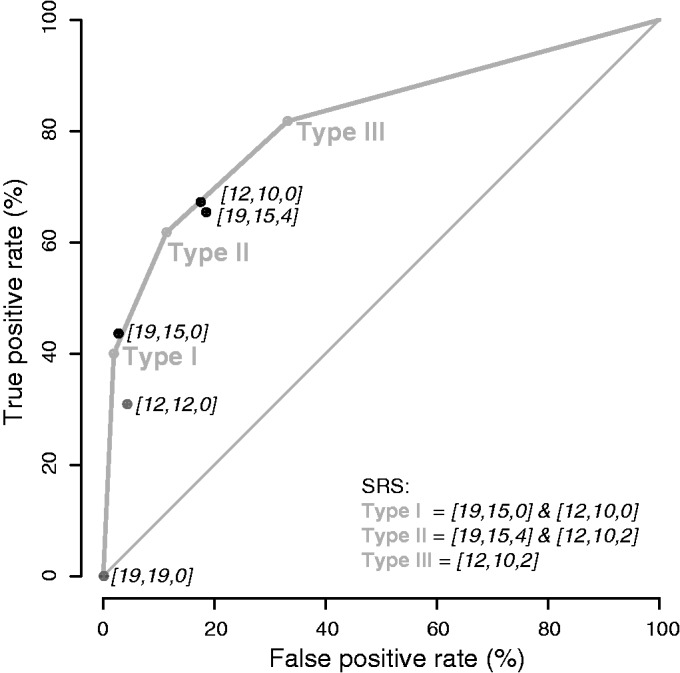
Classification of transcripts based on SRS type. ROC curve to assess how well the SRSs distinguish Staufen targets from non-targets (see ‘Materials and Methods’ section for detailed information). The figure also shows the true- and false-positive rate using the presence and absence of other structural elements, including [19,15,0], [19,15,4], [12,10,0], [12,12,0] and [19,19,0].
The crystal structures of dsRBDs from Drosophila Staufen or yeast Rnt1p endonuclease bound to dsRNA show interactions between the RBD and the hairpin loop (30,58). We, therefore, next asked whether there was any preference for the location of the three types of SRSs relative to hairpin loops. We scored transcripts based on the linear distance on the mRNA transcript between the two paired regions in the SRS and found that this minimum distance was significantly larger in target transcripts for Type I SRSs but shorter for Type II and Type III SRSs (Supplementary Figure S11). These results were, therefore, inconclusive with respect to the preferred location of SRSs relative to hairpin loops.
Mapping of SRSs in Drosophila and human mRNAs
We next mapped the locations of the three types of SRSs within the 3′UTRs of all Drosophila mRNAs (Supplementary Table S17). Figure 9 shows the locations of SRSs in the 3′UTRs of the Staufen target transcripts, length-matched non-target transcript 3′UTRs and a random subset of non-target 3′UTRs.
Figure 9.



Mapping of SRSs in the 3′UTRs of Staufen targets and non-targets. The three types of SRSs were mapped in the 3′UTRs of Staufen targets (A), length-matched non-targets (B) and a random subset of non-targets (C). The x-axis represents the 3′UTR in nucleotides, starting from the first nucleotide after the stop codon. Each 3′UTR is represented by a grey bar, within which the predicted SRS hits are represented by coloured bars (Type I: dark blue; Type II: light blue; Type III: red; Type III embedded in or overlapping with Type I: black; Type III embedded in or overlapping with Type II: magenta). For each SRS, the 5′-most nucleotide in the corresponding 15 of 19 or 10 of 12 motif is connected to the paired nucleotide in the partner arm by a line of the same colour as the SRS (Type I: dark blue, Type II: light blue, Type III: red).
We then asked whether the identified Drosophila SRSs predict human Staufen targets better than 3′UTR length alone and found that none had better AUROC than 3′UTR length for either Staufen1 or Staufen2 (Supplementary Table S18). These results, as well as the failure to predict a motif for Drosophila Staufen from the GFP 2-fold data set, may be a consequence of these lists containing a significant fraction of weakly associated targets or co-expressed transcripts that are not normally bound by Staufen but are scored as bound in these data sets due to overexpression of the tagged Staufen protein used in the RIP-Chip experiments. As such, those transcripts may not be enriched for SRSs (see ‘Discussion’ section).
Finally, to assess whether SRSs map to the experimentally determined in vivo Staufen-binding sites, we focused on Drosophila bicoid (13,31) and human ARF1 (32). In the bicoid 3′UTR, the SRSs mapped almost exclusively to the three experimentally determined Staufen-binding regions (Figure 10A): for Type II SRSs, the precision was 1.0, and for Type III SRSs, the precision was 0.94 (baseline precision = 0.47; there were no Type I SRSs). For human ARF1’s 3′UTR, again, the SRSs mapped almost exclusively to the two known Staufen1-binding regions (Figure 10B): for all three types of SRSs, the precision was 1.0 (baseline precision = 0.13). Taken together, these results provide strong evidence that the computationally identified structures correspond to bona fide, in vivo Staufen-binding sites.
Figure 10.
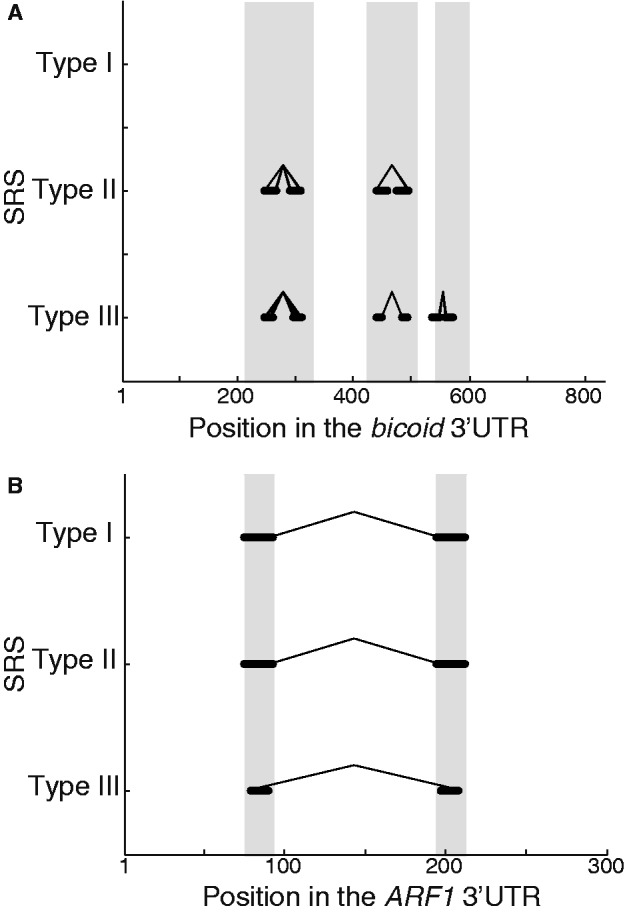
Predicted SRSs map with high precision to the known Staufen-binding regions in Drosophila bicoid and human ARF1 3′UTRs. Mapping of the predicted Drosophila SRSs to experimentally determined in vivo Staufen-binding regions in (A) Drosophila bicoid (12) and (B) human ARF1 (31) 3′UTRs. The grey shading in the background indicates the regions that are important for Staufen binding in vivo as defined in those studies. The x-axis presents the relevant region of the 3′UTR in nucleotides, starting from the first nucleotide after the stop codon. The black lines represent the entire span of each predicted SRS hit that is indicated on the y-axis, mapped onto the 3′UTR sequence. For each SRS, the 5′-most nucleotide in the corresponding 15 of 19 or 10 of 12 motif is connected to the paired nucleotide in the partner arm by a line.
DISCUSSION
Here, we have performed RIP-Chip to identify mRNA targets of Staufen in vivo in early Drosophila embryos, using a synthetic antibody to immunoprecipitate endogenous Staufen and an anti-GFP antibody to immunoprecipitate transgenic GFP-Staufen. In silico analyses of the functions and subcellular localization of these bound transcripts suggested novel roles for Staufen in early embryos. In addition, computational analyses identified dsRNA structures that are highly specific to Staufen’s in vivo targets.
Localization and functions of Staufen-associated transcripts
The localization patterns of the Staufen-associated transcripts that we have identified here are largely consistent with known localization of Staufen protein in early Drosophila embryos. We found strong enrichment for posteriorly localized mRNAs among Staufen’s targets, consistent with the fact that Staufen is concentrated at the posterior of early embryos (8). The 30 newly identified posterior-localized Staufen targets include mRNAs that encode additional post-transcriptional regulators, several of which are known to function in the germ plasm (nanos, arrest, lost, Imp and orb). Thus, Staufen may play a more general role in localization and regulation of germ-plasm transcripts than previously suspected.
Our gene set annotation enrichment analyses suggest that Staufen also plays a more general role in cell fate specification than previously thought, via regulation of mRNAs encoding intra- and inter-cellular signalling molecules, as well as transcriptional and post-transcriptional regulators. This more general role may not be limited to early embryos but might extend into the developing nervous system as well, as Staufen targets were enriched for transcripts that confer neuroblast and neuronal fates. Although Drosophila Staufen was previously known to localize prospero transcripts in the neuroblast lineage (14–17), a more general role for Drosophila Staufen in neuronal development and function is consistent with the previously reported role of mammalian Staufens in these processes (18–23).
Our gene set annotation enrichment analyses also suggest that Staufen may serve a more general role in regulating transcription in early embryos via the basal transcriptional machinery, additional homeodomain-encoding transcripts and several known epigenetic regulators. One of the new targets encodes Zelda (also known as Vielfaltig), an important regulator of zygotic genome activation during the maternal-to-zygotic transition (59–62). Thus, Staufen may regulate both spatial and temporal aspects of zygotic transcription.
Finally, our analyses suggest a potential role for Drosophila Staufen in regulation of alternative splicing and/or a preference for binding to alternatively spliced transcripts. Although Drosophila Staufen has not been observed to localize to nuclei, it contains at least one putative nuclear localization signal as predicted by NLStradamus (63) and NucPred (64) web tools. Furthermore, mammalian Staufen1 contains a nuclear localization signal, has been observed in nuclei (27,28,65) and has been shown to function in splicing regulation (26). Posterior localization of one of Drosophila Staufen’s targets, oskar, requires splicing of its first intron and deposition of the exon junction complex (9,66–71). This raises the possibility that Staufen’s role in posterior localization of its targets could, in part, be related to a function in splicing regulation.
The role of 3′UTR length in Staufen’s targets
We have shown that 3′UTR length is a major feature that distinguishes both the Drosophila and human Staufen targets from non-targets. Our analyses also indicated that the targets of single-stranded RBPs had long 3′UTRs relative to co-expressed but unbound transcripts. However, the fold-increase was less than for fly and human Staufen targets: 1.5–2.5-fold rather 3.0–4.5-fold. Thus, although Staufen targets have particularly long 3′UTRs, long 3′UTRs are a feature of the targets of both double-stranded and single-stranded RBPs. Indeed, long 3′UTRs may be a feature of post-transcriptionally regulated transcripts in general, particularly maternal mRNAs (72) and nervous-system isoforms of mRNAs (73). We speculate that there may be a correlation between the extent that a particular mRNA is post-transcriptionally regulated and the length of its 3′UTR. If so, an implication of our results is that Staufen targets have particularly long 3′UTRs because they undergo a great deal of such regulation.
An alternative explanation is that the 3′UTR length derives from a particular mechanism that is specific to this set of targets. For example, studies of human Staufen suggest that Staufen-mediated decay (SMD) is dependent on the presence of an upstream termination codon, and therefore Staufen binding sites that direct SMD must be located in the 3′UTR of targets (23). In addition, in yeast, C. elegans, Drosophila and mammals, transcripts with longer 3′UTRs show increased susceptibility to nonsense-mediated decay, which shares many mechanistic similarities with SMD (74–79). It is therefore possible that the longer 3′UTRs of Staufen targets may be important for SMD. There is not, however, currently any evidence for SMD in Drosophila embryos. If SMD were a major role of Drosophila Staufen, then a large fraction of its targets should be degraded and, although 19 to 25% of the Staufen targets we have identified are degraded in early embyros, this number is similar to the overall percentage of maternal mRNAs that is cleared (72,78).
Staufen-recognized secondary structures
Previous in vitro studies on the binding of dsRBD3 from Drosophila Staufen to artificial RNA substrates (30) and in vivo studies on the binding of mammalian Staufen1 to ARF1 mRNA (32) have identified 12 and 19 bp Watson–Crick (or G-U) paired stems, respectively, as binding sites for Staufen. In addition, studies of two other dsRBPs have shown that 16 bp stems can also act as binding sites for RBPs containing two or more dsRBDs in vitro (6,81). Our computational analysis of Drosophila Staufen’s in vivo targets has revealed enrichment for three types of SRSs in their 3′UTRs: Type I [19,15,0]&[12,10,0], Type II [19,15,4]&[12,10,2] and Type III [12,10,2]. Together, the Type I, II and III SRSs map to the previously identified Staufen-binding regions in Drosophila bicoid and human ARF1 mRNAs with a precision of ≥0.94.
To our knowledge, this is the first report that uses genome-wide in vivo binding data to define structural preferences—and thus specificity—for a double-stranded RBP. Although in vitro studies have suggested that mismatches or unpaired bases in dsRNA stems reduce the degree of dsRBD-RNA binding (6,30,82), all three SRSs predicted from our in vivo data allow one or more mismatches, and Type II and III SRSs also allow unpaired bases. Consistent with this finding, in Drosophila, the experimentally identified regions within the 3′UTR of bicoid RNA that are required for Staufen binding contain imperfect stems of the length and mismatch number predicted by our computational analysis (13). Moreover, in other cases, where the binding of dsRBDs to endogenous substrates has been studied, bulges or mismatches are often present. For example, mammalian ADAR2 binds a structure in GluR-2 mRNA referred to as the R/G stem-loop, which contains mismatches required for binding (83,84). In addition, mammalian PKR binds to a variety of cellular and viral RNAs that contain mismatches (28,85–87). Thus, a perfect Watson–Crick/G-U dsRNA helix is not a prerequisite for dsRBD binding; indeed, we argue that imperfect stems provide the major specificity of Staufen’s binding to its targets. Experimental analyses will be required to test this proposal. We note that our definition of SRSs permits non-canonical base pairs (or the rotation of unpaired bases) that lead to stable A-form helical stems in the absence of perfect canonical base pairing. Indeed, the strong preference for mismatches over unpaired bases supports this possibility.
How does Staufen recognize and bind stems of different lengths?
In addition to being imperfect stems, the SRSs we have identified are comprised of two major substructures of length 19 and 12, with the latter often contained within the former. One possibility to explain how Staufen binds structures with different lengths is that the shorter stems mediate the binding of a single dsRBD and the longer stems mediate the binding of two or more dsRBDs, either housed in the same dsRBP or in two simultaneously bound dsRBP molecules. Indeed, it has recently been shown that mammalian Staufen 1 and 2 can form both homo- and heterodimers in vitro as well as in tissue-culture cells (25,88). Consistent with the possibility that the longer stems mediate the binding of two or more dsRBDs, three of Staufen’s dsRBDs bind RNA in vitro (9) and structural studies of Xenopus Xlrbpa and mammalian ADAR2 have shown that the binding of a single dsRBD involves interactions with only one face of a dsRNA helix, thereby allowing a second dsRBD to bind a different face of the same helix (81,83,84).
A second, not necessarily mutually exclusive, possibility is that that the short Type III SRS represents the binding site for a dsRBD only in the context of a stem-loop structure, whereas the longer SRSs represent binding to stems with no adjacent hairpin loop. With respect to the former, in vitro studies of the binding of dsRBD3 of Drosophila Staufen to RNA stem loops have shown that a stem length of 12 bp with a loop is optimal for binding, and that, in this context, the dsRBD makes contacts with the loop as well as the stem (30). Similarly, the structure of the dsRBD from the yeast Rnt1p endonuclease bound to a small nucleolar RNA substrate shows binding to a 13 bp stem capped by a tetraloop, with the protein again making contacts with both stem and loop (58). With respect to the longer SRSs, the mammalian Staufen1-binding site within ARF1 mRNA appears to be a 19 bp stem with no adjacent hairpin loop (i.e. the ‘loop’ enclosed by the stem contains multiple hairpin loop structures), and mutations that shorten this stem reduce Staufen binding (32). That different dsRBDs in the same protein may have different binding preferences is supported by data from mammalian ADAR2: one of its dsRBDs prefers hairpin loops, whereas the other prefers duplexes that contain mismatches within internal loops (83).
Staufen levels as a determinant of target mRNA selection
Our data suggest that modest changes in the levels of Staufen might have a significant effect on its compendium of bound mRNAs. Thus, mechanisms that regulate the levels of Staufen could have biologically relevant effects on the processes that Staufen regulates. This has important implications for RIP experiments that use over-expression of tagged Staufen proteins, and likely other dsRBPs, in cell lines or in vivo, as they may lead to identification of spurious target mRNAs. It may be argued that Staufen and other dsRBPs are particularly prone to artefactual target mRNA identification because of their propensity to bind a range of double-stranded motifs rather than to specific sequences. However, given the low complexity and the redundancy within the recognition sites of many sequence-specific RBPs, it is possible that target site selection for many RBPs is strongly influenced by RBP levels.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Canadian Institutes of Health Research [MOP-14409 to H.D.L. and MOP-125894 to Q.D.M.]; University of Toronto Open Scholarships (to J.D.L., K.A. and X.L.). Funding for open access charge: Canadian Institutes of Health Research.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Daniel St Johnston (Cambridge, UK) for providing the GFP-Staufen Drosophila stock and the rabbit polyclonal anti-Staufen antibody, Luc DesGroseillers (Montreal, Canada) for providing the raw data from the human Staufen1 and 2 microarray experiments and Anna Soltyk (Canadian Drosophila Microarray Centre, University of Toronto, Mississauga) for help with the microarray experiments. Extensive use was made during this study of the following resources: Fly-FISH, FlyBase, the Bloomington Drosophila Stock Center, and the Berkeley Drosophila Genome Project. H.D.L. and C.A.S. conceived of the project and designed its overall goals; synthetic antibody RIP-Chip was carried out by J.D.L.; anti-GFP RIP-Chip and western blots by K.A.; RT-qPCR validation experiments by J.D.L. and K.A.; GO term analysis by J.D.L. and K.A.; subcellular localization analysis by J.D.L.; 3′UTR length analysis, motif finding and structural analysis by X.L. under the guidance of Q.D.M.; J.D.L., K.A. and X.L. wrote the first draft of the manuscript, which was revised by H.D.L. and Q.D.M. with input from the other authors. J.D.L. is co-supervised by H.D.L. and C.A.S.; K.A. by J.T.W. and H.D.L.; X.L. by Q.D.M. and H.D.L.
REFERENCES
- 1.Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat. Rev. Mol. Cell. Biol. 2007;8:479–490. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bycroft M, Grunert S, Murzin AG, Proctor M, St Johnston D. NMR solution structure of a dsRNA binding domain from Drosophila staufen protein reveals homology to the N-terminal domain of ribosomal protein S5. EMBO J. 1995;14:3563–3571. doi: 10.1002/j.1460-2075.1995.tb07362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kharrat A, Macias MJ, Gibson TJ, Nilges M, Pastore A. Structure of the dsRNA binding domain of E. coli RNase III. EMBO J. 1995;14:3572–3584. doi: 10.1002/j.1460-2075.1995.tb07363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nanduri S, Carpick BW, Yang Y, Williams BR, Qin J. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J. 1998;17:5458–5465. doi: 10.1093/emboj/17.18.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.St Johnston D, Brown NH, Gall JG, Jantsch M. A conserved double-stranded RNA-binding domain. Proc. Natl Acad. Sci. USA. 1992;89:10979–10983. doi: 10.1073/pnas.89.22.10979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bevilacqua PC, Cech TR. Minor-groove recognition of double-stranded RNA by the double-stranded RNA-binding domain from the RNA-activated protein kinase PKR. Biochemistry. 1996;35:9983–9994. doi: 10.1021/bi9607259. [DOI] [PubMed] [Google Scholar]
- 7.Schupbach T, Wieschaus E. Germline autonomy of maternal-effect mutations altering the embryonic body pattern of Drosophila. Dev. Biol. 1986;113:443–448. doi: 10.1016/0012-1606(86)90179-x. [DOI] [PubMed] [Google Scholar]
- 8.St Johnston D, Beuchle D, Nusslein-Volhard C. Staufen, a gene required to localize maternal RNAs in the Drosophila egg. Cell. 1991;66:51–63. doi: 10.1016/0092-8674(91)90138-o. [DOI] [PubMed] [Google Scholar]
- 9.Micklem DR, Adams J, Grunert S, St Johnston D. Distinct roles of two conserved Staufen domains in oskar mRNA localization and translation. EMBO J. 2000;19:1366–1377. doi: 10.1093/emboj/19.6.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim-Ha J, Smith JL, Macdonald PM. Oskar mRNA is localized to the posterior pole of the Drosophila oocyte. Cell. 1991;66:23–35. doi: 10.1016/0092-8674(91)90136-m. [DOI] [PubMed] [Google Scholar]
- 11.Ephrussi A, Dickinson LK, Lehmann R. Oskar organizes the germ plasm and directs localization of the posterior determinant nanos. Cell. 1991;66:37–50. doi: 10.1016/0092-8674(91)90137-n. [DOI] [PubMed] [Google Scholar]
- 12.St Johnston D, Driever W, Berleth T, Richstein S, Nusslein-Volhard C. Multiple steps in the localization of bicoid RNA to the anterior pole of the Drosophila oocyte. Development. 1989;107:13–19. doi: 10.1242/dev.107.Supplement.13. [DOI] [PubMed] [Google Scholar]
- 13.Ferrandon D, Elphick L, Nusslein-Volhard C, St Johnston D. Staufen protein associates with the 3′UTR of bicoid mRNA to form particles that move in a microtubule-dependent manner. Cell. 1994;79:1221–1232. doi: 10.1016/0092-8674(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 14.Broadus J, Doe CQ. Extrinsic cues, intrinsic cues and microfilaments regulate asymmetric protein localization in Drosophila neuroblasts. Curr. Biol. 1997;7:827–835. doi: 10.1016/s0960-9822(06)00370-8. [DOI] [PubMed] [Google Scholar]
- 15.Broadus J, Fuerstenberg S, Doe CQ. Staufen-dependent localization of prospero mRNA contributes to neuroblast daughter-cell fate. Nature. 1998;391:792–795. doi: 10.1038/35861. [DOI] [PubMed] [Google Scholar]
- 16.Schuldt AJ, Adams JH, Davidson CM, Micklem DR, Haseloff J, St Johnston D, Brand AH. Miranda mediates asymmetric protein and RNA localization in the developing nervous system. Genes Dev. 1998;12:1847–1857. doi: 10.1101/gad.12.12.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li P, Yang X, Wasser M, Cai Y, Chia W. Inscuteable and Staufen mediate asymmetric localization and segregation of prospero RNA during Drosophila neuroblast cell divisions. Cell. 1997;90:437–447. doi: 10.1016/s0092-8674(00)80504-8. [DOI] [PubMed] [Google Scholar]
- 18.Kiebler MA, Hemraj I, Verkade P, Kohrmann M, Fortes P, Marion RM, Ortin J, Dotti CG. The mammalian staufen protein localizes to the somatodendritic domain of cultured hippocampal neurons: implications for its involvement in mRNA transport. J. Neurosci. 1999;19:288–297. doi: 10.1523/JNEUROSCI.19-01-00288.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vessey JP, Macchi P, Stein JM, Mikl M, Hawker KN, Vogelsang P, Wieczorek K, Vendra G, Riefler J, Tubing F, et al. A loss of function allele for murine Staufen1 leads to impairment of dendritic Staufen1-RNP delivery and dendritic spine morphogenesis. Proc. Natl Acad. Sci. USA. 2008;105:16374–16379. doi: 10.1073/pnas.0804583105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goetze B, Tuebing F, Xie Y, Dorostkar MM, Thomas S, Pehl U, Boehm S, Macchi P, Kiebler MA. The brain-specific double-stranded RNA-binding protein Staufen2 is required for dendritic spine morphogenesis. J. Cell Biol. 2006;172:221–231. doi: 10.1083/jcb.200509035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duchaine TF, Hemraj I, Furic L, Deitinghoff A, Kiebler MA, DesGroseillers L. Staufen2 isoforms localize to the somatodendritic domain of neurons and interact with different organelles. J. Cell. Sci. 2002;115:3285–3295. doi: 10.1242/jcs.115.16.3285. [DOI] [PubMed] [Google Scholar]
- 22.Kusek G, Campbell M, Doyle F, Tenenbaum SA, Kiebler M, Temple S. Asymmetric segregation of the double-stranded RNA binding protein Staufen2 during mammalian neural stem cell divisions promotes lineage progression. Cell Stem Cell. 2012;11:505–516. doi: 10.1016/j.stem.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vessey JP, Amadei G, Burns SE, Kiebler MA, Kaplan DR, Miller FD. An asymmetrically localized Staufen2-dependent RNA complex regulates maintenance of mammalian neural stem cells. Cell Stem Cell. 2012;11:517–528. doi: 10.1016/j.stem.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 24.Kim YK, Furic L, Desgroseillers L, Maquat LE. Mammalian Staufen1 recruits Upf1 to specific mRNA 3'UTRs so as to elicit mRNA decay. Cell. 2005;120:195–208. doi: 10.1016/j.cell.2004.11.050. [DOI] [PubMed] [Google Scholar]
- 25.Park E, Gleghorn ML, Maquat LE. Staufen2 functions in Staufen1-mediated mRNA decay by binding to itself and its paralog and promoting UPF1 helicase but not ATPase activity. Proc. Natl Acad. Sci. USA. 2013;110:405–412. doi: 10.1073/pnas.1213508110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dugré-Brisson S, Elvira G, Boulay K, Chatel-Chaix L, Mouland AJ, DesGroseillers L. Interaction of Staufen1 with the 5' end of mRNA facilitates translation of these RNAs. Nucleic Acids Res. 2005;33:4797–4812. doi: 10.1093/nar/gki794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ravel-Chapuis A, Belanger G, Yadava RS, Mahadevan MS, DesGroseillers L, Cote J, Jasmin BJ. The RNA-binding protein Staufen1 is increased in DM1 skeletal muscle and promotes alternative pre-mRNA splicing. J. Cell. Biol. 2012;196:699–712. doi: 10.1083/jcb.201108113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elbarbary RA, Li W, Tian B, Maquat LE. STAU1 binding 3' UTR IRAlus complements nuclear retention to protect cells from PKR-mediated translational shutdown. Genes Dev. 2013;27:1495–1510. doi: 10.1101/gad.220962.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Legendre JB, Campbell ZT, Kroll-Conner P, Anderson P, Kimble J, Wickens M. RNA targets and specificity of Staufen, a double-stranded RNA-binding protein in C. elegans. J. Biol. Chem. 2012;288:2532–2545. doi: 10.1074/jbc.M112.397349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramos A, Grunert S, Adams J, Micklem DR, Proctor MR, Freund S, Bycroft M, St Johnston D, Varani G. RNA recognition by a Staufen double-stranded RNA-binding domain. EMBO J. 2000;19:997–1009. doi: 10.1093/emboj/19.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferrandon D, Koch I, Westhof E, Nusslein-Volhard C. RNA-RNA interaction is required for the formation of specific bicoid mRNA 3' UTR-STAUFEN ribonucleoprotein particles. EMBO J. 1997;16:1751–1758. doi: 10.1093/emboj/16.7.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim YK, Furic L, Parisien M, Major F, DesGroseillers L, Maquat LE. Staufen1 regulates diverse classes of mammalian transcripts. EMBO J. 2007;26:2670–2681. doi: 10.1038/sj.emboj.7601712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Furic L, Maher-Laporte M, DesGroseillers L. A genome-wide approach identifies distinct but overlapping subsets of cellular mRNAs associated with Staufen1- and Staufen2-containing ribonucleoprotein complexes. RNA. 2008;14:324–335. doi: 10.1261/rna.720308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3' UTRs via Alu elements. Nature. 2011;470:284–288. doi: 10.1038/nature09701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Gong C, Maquat LE. Control of myogenesis by rodent SINE-containing lncRNAs. Genes Dev. 2013;27:793–804. doi: 10.1101/gad.212639.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laver JD, Ancevicius K, Sollazzo P, Westwood JT, Sidhu SS, Lipshitz HD, Smibert CA. Synthetic antibodies as tools to probe RNA-binding protein function. Mol. Biosyst. 2012;8:1650–1657. doi: 10.1039/c2mb00007e. [DOI] [PubMed] [Google Scholar]
- 37.Lehmann R, Nusslein-Volhard C. The maternal gene nanos has a central role in posterior pattern formation of the Drosophila embryo. Development. 1991;112:679–691. doi: 10.1242/dev.112.3.679. [DOI] [PubMed] [Google Scholar]
- 38.Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- 39.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl Acad. Sci. USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, Quackenbush J. TM4 microarray software suite. Methods Enzymol. 2006;411:134–193. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- 41.Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 42.Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 44.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bernhart SH, Hofacker IL, Stadler PF. Local RNA base pairing probabilities in large sequences. Bioinformatics. 2006;22:614–615. doi: 10.1093/bioinformatics/btk014. [DOI] [PubMed] [Google Scholar]
- 46.Lange SJ, Maticzka D, Mohl M, Gagnon JN, Brown CM, Backofen R. Global or local? predicting secondary structure and accessibility in mRNAs. Nucleic Acids Res. 2012;40:5215–5226. doi: 10.1093/nar/gks181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ding Y, Lawrence CE. A statistical sampling algorithm for RNA secondary structure prediction. Nucleic Acids Res. 2003;31:7280–7301. doi: 10.1093/nar/gkg938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hulsen T, de Vlieg J, Alkema W. BioVenn - a web application for the comparison and visualization of biological lists using area-proportional venn diagrams. BMC Genomics. 2008;9:488. doi: 10.1186/1471-2164-9-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lécuyer E, Yoshida H, Parthasarathy N, Alm C, Babak T, Cerovina T, Hughes TR, Tomancak P, Krause HM. Global analysis of mRNA localization reveals a prominent role in organizing cellular architecture and function. Cell. 2007;131:174–187. doi: 10.1016/j.cell.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 50.Tomancak P, Beaton A, Weiszmann R, Kwan E, Shu S, Lewis SE, Richards S, Ashburner M, Hartenstein V, Celniker SE, et al. Systematic determination of patterns of gene expression during Drosophila embryogenesis. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-12-research0088. RESEARCH0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tomancak P, Berman BP, Beaton A, Weiszmann R, Kwan E, Hartenstein V, Celniker SE, Rubin GM. Global analysis of patterns of gene expression during Drosophila embryogenesis. Genome Biol. 2007;8:R145. doi: 10.1186/gb-2007-8-7-r145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al. The developmental transcriptome of Drosophila melanogaster. Nature. 2011;471:473–479. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gerber AP, Luschnig S, Krasnow MA, Brown PO, Herschlag D. Genome-wide identification of mRNAs associated with the translational regulator PUMILIO in Drosophila melanogaster. Proc. Natl Acad. Sci. USA. 2006;103:4487–4492. doi: 10.1073/pnas.0509260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lopez de Silanes I, Zhan M, Lal A, Yang X, Gorospe M. Identification of a target RNA motif for RNA-binding protein HuR. Proc. Natl Acad. Sci. USA. 2004;101:2987–2992. doi: 10.1073/pnas.0306453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gama-Carvalho M, Barbosa-Morais NL, Brodsky AS, Silver PA, Carmo-Fonseca M. Genome-wide identification of functionally distinct subsets of cellular mRNAs associated with two nucleocytoplasmic-shuttling mammalian splicing factors. Genome Biol. 2006;7:R113. doi: 10.1186/gb-2006-7-11-r113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morris AR, Mukherjee N, Keene JD. Ribonomic analysis of human Pum1 reveals cis-trans conservation across species despite evolution of diverse mRNA target sets. Mol. Cell. Biol. 2008;28:4093–4103. doi: 10.1128/MCB.00155-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li X, Quon G, Lipshitz HD, Morris Q. Predicting in vivo binding sites of RNA-binding proteins using mRNA secondary structure. RNA. 2010;16:1096–1107. doi: 10.1261/rna.2017210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu H, Henras A, Chanfreau G, Feigon J. Structural basis for recognition of the AGNN tetraloop RNA fold by the double-stranded RNA-binding domain of Rnt1p RNase III. Proc. Natl Acad. Sci. USA. 2004;101:8307–8312. doi: 10.1073/pnas.0402627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liang HL, Nien CY, Liu HY, Metzstein MM, Kirov N, Rushlow C. The zinc-finger protein Zelda is a key activator of the early zygotic genome in Drosophila. Nature. 2008;456:400–403. doi: 10.1038/nature07388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harrison MM, Botchan MR, Cline TW. Grainyhead and Zelda compete for binding to the promoters of the earliest-expressed Drosophila genes. Dev. Biol. 2010;345:248–255. doi: 10.1016/j.ydbio.2010.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harrison MM, Li XY, Kaplan T, Botchan MR, Eisen MB. Zelda binding in the early Drosophila melanogaster embryo marks regions subsequently activated at the maternal-to-zygotic transition. PLoS Genet. 2011;7:e1002266. doi: 10.1371/journal.pgen.1002266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nien CY, Liang HL, Butcher S, Sun Y, Fu S, Gocha T, Kirov N, Manak JR, Rushlow C. Temporal coordination of gene networks by Zelda in the early Drosophila embryo. PLoS Genet. 2011;7:e1002339. doi: 10.1371/journal.pgen.1002339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nguyen Ba AN, Pogoutse A, Provart N, Moses AM. NLStradamus: a simple Hidden Markov Model for nuclear localization signal prediction. BMC Bioinformatics. 2009;10:202. doi: 10.1186/1471-2105-10-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brameier M, Krings A, MacCallum RM. NucPred—predicting nuclear localization of proteins. Bioinformatics. 2007;23:1159–1160. doi: 10.1093/bioinformatics/btm066. [DOI] [PubMed] [Google Scholar]
- 65.Martel C, Macchi P, Furic L, Kiebler MA, Desgroseillers L. Staufen1 is imported into the nucleolus via a bipartite nuclear localization signal and several modulatory determinants. Biochem. J. 2006;393:245–254. doi: 10.1042/BJ20050694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Newmark PA, Boswell RE. The mago nashi locus encodes an essential product required for germ plasm assembly in Drosophila. Development. 1994;120:1303–1313. doi: 10.1242/dev.120.5.1303. [DOI] [PubMed] [Google Scholar]
- 67.Hachet O, Ephrussi A. Drosophila Y14 shuttles to the posterior of the oocyte and is required for oskar mRNA transport. Curr. Biol. 2001;11:1666–1674. doi: 10.1016/s0960-9822(01)00508-5. [DOI] [PubMed] [Google Scholar]
- 68.Mohr SE, Dillon ST, Boswell RE. The RNA-binding protein tsunagi interacts with Mago Nashi to establish polarity and localize oskar mRNA during Drosophila oogenesis. Genes Dev. 2001;15:2886–2899. doi: 10.1101/gad.927001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Palacios IM, Gatfield D, St Johnston D, Izaurralde E. An eIF4AIII-containing complex required for mRNA localization and nonsense-mediated mRNA decay. Nature. 2004;427:753–757. doi: 10.1038/nature02351. [DOI] [PubMed] [Google Scholar]
- 70.Hachet O, Ephrussi A. Splicing of oskar RNA in the nucleus is coupled to its cytoplasmic localization. Nature. 2004;428:959–963. doi: 10.1038/nature02521. [DOI] [PubMed] [Google Scholar]
- 71.Ghosh S, Marchand V, Gaspar I, Ephrussi A. Control of RNP motility and localization by a splicing-dependent structure in oskar mRNA. Nat. Struct. Mol. Biol. 2012;19:441–449. doi: 10.1038/nsmb.2257. [DOI] [PubMed] [Google Scholar]
- 72.Tadros W, Goldman AL, Babak T, Menzies F, Vardy L, Orr-Weaver T, Hughes TR, Westwood JT, Smibert CA, Lipshitz HD. SMAUG is a major regulator of maternal mRNA destabilization in Drosophila and its translation is activated by the PAN GU Kinase. Dev. Cell. 2007;12:143–155. doi: 10.1016/j.devcel.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 73.Hilgers V, Perry MW, Hendrix D, Stark A, Levine M, Haley B. Neural-specific elongation of 3’UTRs during Drosophila development. Proc. Natl. Acad. Sci. USA. 2011;108:15864–15869. doi: 10.1073/pnas.1112672108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pulak R, Anderson P. mRNA surveillance by the caenorhabditis elegans smg genes. Genes Dev. 1993;7:1885–1897. doi: 10.1101/gad.7.10.1885. [DOI] [PubMed] [Google Scholar]
- 75.Muhlrad D, Parker R. Aberrant mRNAs with extended 3' UTRs are substrates for rapid degradation by mRNA surveillance. RNA. 1999;5:1299–1307. doi: 10.1017/s1355838299990829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Amrani N, Ganesan R, Kervestin S, Mangus DA, Ghosh S, Jacobson A. A faux 3'-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature. 2004;432:112–118. doi: 10.1038/nature03060. [DOI] [PubMed] [Google Scholar]
- 77.Buhler M, Steiner S, Mohn F, Paillusson A, Muhlemann O. EJC-independent degradation of nonsense immunoglobulin-mu mRNA depends on 3' UTR length. Nat. Struct. Mol. Biol. 2006;13:462–464. doi: 10.1038/nsmb1081. [DOI] [PubMed] [Google Scholar]
- 78.Behm-Ansmant I, Gatfield D, Rehwinkel J, Hilgers V, Izaurralde E. A conserved role for cytoplasmic poly(A)-binding protein 1 (PABPC1) in nonsense-mediated mRNA decay. EMBO J. 2007;26:1591–1601. doi: 10.1038/sj.emboj.7601588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hansen KD, Lareau LF, Blanchette M, Green RE, Meng Q, Rehwinkel J, Gallusser FL, Izaurralde E, Rio DC, Dudoit S, et al. Genome-wide identification of alternative splice forms down-regulated by nonsense-mediated mRNA decay in Drosophila. PLoS Genet. 2009;5:e1000525. doi: 10.1371/journal.pgen.1000525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.De Renzis S, Elemento O, Tavazoie S, Wieschaus EF. Unmasking activation of the zygotic genome using chromosomal deletions in the Drosophila embryo. PLoS Biol. 2007;5:e117. doi: 10.1371/journal.pbio.0050117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ryter JM, Schultz SC. Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J. 1998;17:7505–7513. doi: 10.1093/emboj/17.24.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schmedt C, Green SR, Manche L, Taylor DR, Ma Y, Mathews MB. Functional characterization of the RNA-binding domain and motif of the double-stranded RNA-dependent protein kinase DAI (PKR) J. Mol. Biol. 1995;249:29–44. doi: 10.1006/jmbi.1995.0278. [DOI] [PubMed] [Google Scholar]
- 83.Stefl R, Xu M, Skrisovska L, Emeson RB, Allain FH. Structure and specific RNA binding of ADAR2 double-stranded RNA binding motifs. Structure. 2006;14:345–355. doi: 10.1016/j.str.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 84.Stefl R, Oberstrass FC, Hood JL, Jourdan M, Zimmermann M, Skrisovska L, Maris C, Peng L, Hofr C, Emeson RB, et al. The solution structure of the ADAR2 dsRBM-RNA complex reveals a sequence-specific readout of the minor groove. Cell. 2010;143:225–237. doi: 10.1016/j.cell.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Katze MG, DeCorato D, Safer B, Galabru J, Hovanessian AG. Adenovirus VAI RNA complexes with the 68 000 Mr protein kinase to regulate its autophosphorylation and activity. EMBO J. 1987;6:689–697. doi: 10.1002/j.1460-2075.1987.tb04809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Davis S, Watson JC. In vitro activation of the interferon-induced, double-stranded RNA-dependent protein kinase PKR by RNA from the 3' untranslated regions of human alpha-tropomyosin. Proc. Natl Acad. Sci. USA. 1996;93:508–513. doi: 10.1073/pnas.93.1.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Robertson HD, Manche L, Mathews MB. Paradoxical interactions between human delta hepatitis agent RNA and the cellular protein kinase PKR. J. Virol. 1996;70:5611–5617. doi: 10.1128/jvi.70.8.5611-5617.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gleghorn ML, Gong C, Kielkopf CL, Maquat LE. Staufen1 dimerizes through a conserved motif and a degenerate dsRNA-binding domain to promote mRNA decay. Nat. Struct. Mol. Biol. 2013;20:515–524. doi: 10.1038/nsmb.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.