

Key Points
Neutrophils are necessary and sufficient for mAb-induced therapy of subcutaneous syngeneic or xenograft tumors in mice.
Antitumor immunoglobulin G mAb therapy requires a Syk-dependent FcγR-induced killing of tumors by neutrophils.
Abstract
Tumor engraftment followed by monoclonal antibody (mAb) therapy targeting tumor antigens represents a gold standard for assessing the efficiency of mAbs to eliminate tumor cells. Mouse models have demonstrated that receptors for the Fc portion of immunoglobulin G (FcγRs) are critical determinants of mAb therapeutic efficacy, but the FcγR-expressing cell populations responsible remain elusive. We show that neutrophils are responsible for mAb-induced therapy of both subcutaneous syngeneic melanoma and human breast cancer xenografts. mAb-induced tumor reduction, abolished in neutropenic mice, could be restored in FcγR-deficient hosts upon transfer of FcγR+ neutrophils or upon human FcγRIIA/CD32A transgenic expression. Finally, conditional knockout mice unable to perform FcγR-mediated activation and phagocytosis specifically in neutrophils were resistant to mAb-induced therapy. Our work suggests that neutrophils are necessary and sufficient for mAb-induced therapy of subcutaneous tumors, and represent a new and critical focal point for optimizing mAb-induced immunotherapies that will impact on human cancer treatment.
Introduction
Murine tumor models are the main preclinical tools used to screen and optimize monoclonal antibodies (mAbs) for potential antitumor mAb-mediated therapy in the clinic. These models consist of implanting syngeneic mouse cancer cells into immunocompetent mice or xenogeneic human cancer cells into immunodeficient mice, followed by intravenous injections of potential therapeutic mAbs. Most antitumor therapeutic mAbs target an antigen expressed by the tumor and were designed to limit tumor growth by inducing cellular apoptosis or growth arrest.1 Several reports, however, indicate that the immune effector response is highly relevant to the efficacy of therapeutic mAbs in vivo in mouse models.2 Importantly, mice deficient for all activating FcγRs (FcRγ−/− mice) are not protected from the growth of glycoprotein 75 (gp75)–expressing syngeneic melanoma or of HER2-expressing breast cancer xenografts following anti-gp75 (TA99) or anti-HER2 (Trastuzumab) mAb treatment, respectively.3,4 Furthermore, polymorphisms in FcγR-encoding genes in patients (eg, FcγRIIIA/CD16A and FcγRIIA/CD32A) have been reported to impact mAb therapeutic efficacy.5,6
However, the FcγR-expressing cell populations responsible for the mAb-induced therapeutic activities on tumors have not been formally identified. In vitro, FcγR+ natural killer (NK) cells and various FcγR+ myeloid cells7-10 can all kill mAb-opsonized tumor cells. In vivo, however, it is unclear which of these cell types plays the dominant role in mAb-induced antitumor effects.
Study design
We used tumor cell lines expressing the enhanced firefly luciferase (luc2) to allow accurate, noninvasive assessment of tumor burden over time using bioluminescence acquisition.11,12 A subcutaneous injection of luc2-expressing syngeneic gp75+ B16-F10 (B16-luc2) melanoma into wild-type mice led to a localized tumor development (Figure 1A; supplemental Figure 1A, available on the Blood Web site). Recurrent injections of anti-gp75 mAb TA99 reduced bioluminescence to background level as early as 24 to 48 hours following the first injection and prevented reoccurrence of detectable tumors in wild-type mice (Figure 1A; supplemental Figure 1A) but not in FcRγ−/− mice (supplemental Figure 1B), as reported.3 Anti-gp75 mAb injections starting on day 0 or day 2, but not on day 7, post–tumor engraftment efficiently reduced the tumor burden (supplemental Figure 1C). The protective effect in this mAb therapy model can therefore be monitored using bioluminescence before appearance of detectable tumor masses, and mimics the clinical efficacy of antitumor mAbs on small or residual tumors and their relative inefficiency on larger tumors.13 The potential contribution of FcγR+ cell populations14 to antitumor mAb immunotherapy could therefore be investigated in the first days following mAb therapy (see supplemental Material and methods).
Figure 1.

Neutrophils are required for anti-gp75 mAb therapy of melanoma. (A-F) Indicated mice were injected subcutaneously with 5 × 104 B16-luc2 cells at day 0, intravenously with 200 μg of mAb TA99 or isotype Ctrl on days 0, 1, and 2, and intraperitoneally with d-luciferin immediately before total photon flux acquisition (photons per second). Indicated mice were also injected on days −1, 1, 3, 5, and 7 with (C) 200 μg/mouse clodronate-containing liposomes (Clodronate) or (E) 300 μg/mouse anti-Gr1 mAbs, or (D) on days 0, 1, and 2 with 2 × 106 WT B.M. cells (▲). (A-F) Data are represented as mean ± SEM (n.s.: P > .05; *P < .05; **P < .01; ***P < .001) and are representative of at least 2 independent experiments (n ≥ 4). B.M., bone marrow; Ctrl, control; KO, knockout; n.s., not significant; WT, wild type.
Results and discussion
NK cells did not detectably contribute to anti-gp75 immunotherapy, as demonstrated by NK-cell deficiency15 (Figure 1B) or depletion (supplemental Figure 1D). Similarly, monocytes/macrophages were not involved, as demonstrated by monocyte/macrophage depletion (Figure 1C; supplemental Figure 2A) or by their inhibition by gadolinium (data not shown). This latter result was unexpected in view of the critical role of macrophages reported in the depletion of B cells after anti-CD20 therapy,10,16 but may rely on the tissue localization of the target cells, that is, subcutaneous vs circulating, respectively. Finally, a role for mast cells, basophils, or eosinophils could be ruled out (supplemental Figure 2B-D). Mouse protocols were approved by the Animal Care and Use Committees of Paris, France.
As demonstrated previously,3 FcRγ−/− mice failed to respond to anti-gp75 treatment following tumor transfer (supplemental Figure 1B). Although bone marrow cell transfers from wild-type mice into FcRγ−/−RAG−/− mice restored anti-gp75 immunotherapy (Figure 1D), transfers on days 0 and 1 were not sufficient to protect mice from tumor outgrowth (supplemental Figure 3A). This result suggested that a short-lived bone marrow cell population may mediate the protection. Among short-lived bone marrow cells, neutrophils have been reported to have a lifespan of 12.5 hours in mice.17 Importantly, anti-Gr1–induced depletion of neutrophils abolished anti-gp75 immunotherapy (Figure 1E; supplemental Figure 3B-C). Although myeloid-derived suppressor cells (MDSCs) also express Gr1, it is unlikely that the depletion of MDSCs is contributing to the loss of the therapeutic activity of TA99 in this model, as B16 cells have been reported not to induce MDSCs.18 Because antibody-induced cell depletion might also affect other cell populations in this setting, we used a mouse model of neutropenia,19 induced by the absence of transcriptional repressor growth factor independence-1 (Gfi1)20 (supplemental Figure 3D). Whereas tumor growth was identical in Gfi1-deficient and Gfi1-sufficient (Gfi1+/−) mice, anti-gp75 immunotherapy was abolished in the absence of Gfi1 (Figure 1F; supplemental Figure 3E). Taken together, our results suggest that Gr1+ cells, deficient in Gfi1−/− mice, that is, neutrophils, are mandatory for antitumor mAb therapeutic efficacy.
We next extended our observations to the HER2/neu+ human breast cancer cell line BT474-M1 that has been used to assess the therapeutic activity of Trastuzumab.4 A subcutaneous injection of luc2-expressing BT474 cells (BT474-luc2; supplemental Figure 4A-B) in matrigel leads to a localized bioluminescent tumor mass in immunodeficient nude mice. Trastuzumab injections reduced bioluminescence to background level in 7 days and prevented detectable tumors from appearing in nude mice but not in FcRγ−/− nude mice (supplemental Figure 4C-D), in agreement with earlier findings.4 Anti-Gr1–mediated depletion of neutrophils abolished Trastuzumab immunotherapy (Figure 2A). Using suboptimal doses of anti-Gr1 resulted in a partial reduction of neutrophil numbers that correlated with a partial loss of the efficacy of Trastuzumab on tumor growth (supplemental Figure 4E). Of note, BT474 cells, like B16 cells, do not produce granulocyte macrophage–colony-stimulating factor and therefore do not induce MDSCs.18 Moreover, Gfi1-deficient nude mice were resistant to Trastuzumab treatment (Figure 2B). These data indicate that neutrophils are also mandatory for the antitumor effect of Trastuzumab on HER2-expressing breast cancer xenografts. To further demonstrate a role for myeloid cells, but not for NK cells in this model, we used transgenic mice expressing the human FcγRIIA/CD32A gene in neutrophils and other myeloid cells, but not NK cells.21,22 Expression of this transgene restored Trastuzumab efficacy in FcRγ−/− nude mice (Figure 2C; supplemental Figure 4F).
Figure 2.
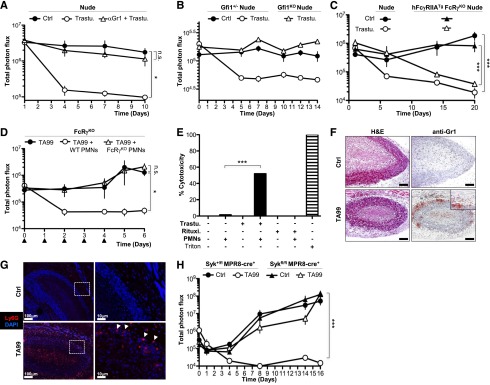
Trastuzumab efficacy on HER2+ xenografts also relies on neutrophils, and FcγRs and Syk are required in neutrophils for mAb-induced antitumor activity. (A-C) Indicated mice (n ≥ 4) were injected subcutaneously with 5 × 106 BT474-luc2 cells in matrigel at day 0, intravenously with 100 μg of Trastuzumab or isotype Ctrl weekly starting day 1, and total photon flux was acquired (photons per second). Mice were also injected (A) on days −1, 1, 3, 5, and 7 with anti-Gr1 mAbs. (B) Data are compiled from 2 identical experiments. (Nota bene: Anti-Gr1 mAb-treated nude mice and littermates, Gfi1−/− nude, and Gfi1+/− nude littermates were kept under sulfamethoxypyridazin plus trimethoprim.) (D) FcRγ−/− mice (n ≥ 4) were injected with B16-luc2 cells and mAb TA99 as in Figure 1, daily with 2 × 106 neutrophils purified from indicated mice (▲: neutrophil injection). (E) Ex vivo cytotoxicity of human neutrophils (PMNs) on opsonized-BT474-luc2 cells at a 50:1 effector:target ratio. (Note: BT474 cells express HER2 but not CD20; thus anti-CD20 Rituximab [Rituxi.] represents a negative control. Triton lysis of BT474-luc2 is used as a positive control. Mean of triplicates is represented.) (F) H&E staining or anti-Gr1 immunolabeling, or (G) DAPI staining and anti-Ly6G immunolabeling of sections of 7-day-old B16-luc2 tumors 24 hours after mAb TA99 injection. (F) Original magnification, ×10 (scale bar = 100 μm). (H) Mice (n ≥ 4) were injected with B16-luc2 cells, mAb TA99, and analyzed as in Figure 1. (A-E,H) Data are represented as mean ± SEM (not significant: P > .05; *P < .05; ***P < .001) and are representative from at least 2 independent experiments. Ctrl, control; DAPI, 4′6 diamidino-2-phenylindole; H&E, hematoxylin and eosin; PMN, neutrophils; Trastu, Trastuzumab.
We next investigated whether neutrophils were sufficient to overcome a host environment resistant to mAb therapy. Daily transfers of purified neutrophils from wild-type mice, but not from FcRγ−/− mice, into recipient FcRγ−/− mice restored anti-gp75 immunotherapy (Figure 2D; supplemental Figure 5A). Thus activating immunoglobulin G (IgG) receptors are only required on neutrophils to allow mAb-mediated therapy. Neutrophils may thus be responsible by themselves for mAb-induced tumor reduction. Purified human blood neutrophils (supplemental Figure 5B) could, indeed, induce the killing of BT474-luc2 cells only in the presence of Trastuzumab (Figure 2E) suggesting a requirement for physical interaction between neutrophils and opsonized target cells in vivo.7 Histologic analysis revealed foci of Gr1+ cells with a neutrophil morphology in the tumor outer rim only after anti-gp75 mAb injection (Figure 2F), whereas similar numbers of CD3+, CD45R+, or F4/80+ cells were present in either the presence or absence of therapy (supplemental Figure 5C-D). These foci contained Ly6G+ cells, indicating that these were neutrophils (Figure 2G).
Finally, we investigated by which mechanism neutrophils contribute to these models of anticancer immunotherapy. Neither a deficiency in cytokines (tumor necrosis factor–α or interferon-γ), in proteases (elastase or myeloperoxidase), in phospholipase-A2–dependent mediators, nor in reactive oxygen species (gp47phox or gp91phox-NADPH oxidase complex) affected anti-gp75 immunotherapy, nor did inhibition of metalloproteases or blocking neutrophil-chemoattractant chemokine CXCL1 (supplemental Figure 6, data not shown). To investigate whether neutrophils required FcγR-mediated activation to contribute to tumor reduction in vivo, we used mice with a neutrophil-specific deficiency in the Syk kinase, that is, Sykfl/fl MPR8-cre+ mice.23 Syk has indeed been reported to be necessary for FcγR-dependent functions, including cell activation,24 antibody-dependent cell-mediated cytotoxicity,25 and phagocytosis,26 without impairing neutrophil migration to sites of antibody-induced inflammation.27 Importantly, Sykfl/fl MPR8-cre+ mice were resistant to anti-gp75 immunotherapy (Figure 2H), demonstrating an essential role for Syk-dependent FcγR-induced neutrophil antitumor activity.
Our work provides a mechanistic basis for the observed reduction in tumor burden following antitumor mAb injection in both syngeneic and xenograft mouse models of cancer immunotherapy. The selective requirement and the sufficiency of neutrophils to mediate IgG–induced antitumor activities we reveal may also extend to emerging models of IgA mAb-antitumor therapy, which have been proposed to rely on complement and on IgA receptor (CD89)–expressing neutrophils.28 Although significant differences between mouse and human neutrophils including the activating IgG receptors they express have been reported,14,17 the principles that have emerged from these mouse studies are likely to apply to human immunotherapy protocols. Polymorphisms of FcγRIIA/CD32A expressed by human neutrophils have indeed been reported to impact mAb therapeutic efficacy.6 Antibody therapy is, however, usually combined with chemotherapy that strongly reduces neutrophil numbers. If human neutrophils mediate the therapeutic effect of antitumor antibody in the clinic, chemotherapy may thus reduce the efficiency of antitumor mAbs by depleting their effector cell population.
Supplementary Material
Acknowledgments
The authors are thankful to our colleagues at Institut Pasteur (Paris, France): P. Bousso (Dynamics of Immune Responses Unit) for discussions and advice, M.-A. Nicola (Plate-Forme d'Imagerie Dynamique) for help with bioluminescence experiments, X. Montagutelli, Q. Mille, D. Montean, and G. Labas for help with mouse colony management (Central Animal Facility), C. Detchepare and C. Nizak for administrative help (Laboratoire Anticorps en Thérapie et Pathologie), and at University of California, San Francisco (San Francisco, CA): Y. Hu for animal husbandry. The authors are grateful to their colleagues for providing mice or reagents: R. Coffman (DNAX, Palo Alto, CA), B. Hann (University of California, San Francisco), J. J. Lee (Mayo Clinic, Scottsdale, AZ), H. Karasuyama (Tokyo Medical and Dental University Graduate School, Tokyo, Japan), M.P. Reilly (Jefferson Medical College, Philadelphia, PA), and M. Tarik (Montreal University, Montreal, QC, Canada).
This work was supported by the Institut Pasteur and the Institut National de la Santé et de la Recherche Médicale; by Agence Nationale de la Recherche (grant ANR-09-GENO-014-01), Fondation ARC pour la Recherche sur le Cancer and Ligue Nationale contre le Cancer (Comité de Paris) grants (P.B.); through an “Equipe Labellisée Ligue Contre le Cancer” grant (J.P.D.S.); and by National Institutes of Health, National Institute of Allergy and Infectious Diseases grants AI065495 and AI068150 (C.A.L.).
M.A. is a scholar of the Pasteur Paris University International Doctoral Program.
Footnotes
The online version of this article contains a data supplement.
There is an Inside Blood commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: M.A. performed all experiments with help from D.A.M. and F.J.; B.I. produced reagents and genotyped mice; L.F. performed histological analyses; J.P.D.S. and C.A.L. discussed results, provided ideas and mice, and critically read the manuscript; and P.B. conceived and funded the study and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for M.A. is Département d’Immunologie, Unité de Dynamique des Réponses Immunes, Institut Pasteur, 75015 Paris, France.
Correspondence: Pierre Bruhns, Laboratoire Anticorps en Thérapie et Pathologie, Département d’Immunologie, Institut Pasteur, 25 rue du Docteur Roux, 75015 Paris, France; e-mail: bruhns@pasteur.fr.
References
- 1.Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10(5):317–327. doi: 10.1038/nri2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clynes R. Antitumor antibodies in the treatment of cancer: Fc receptors link opsonic antibody with cellular immunity. Hematol Oncol Clin North Am. 2006;20(3):585–612. doi: 10.1016/j.hoc.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 3.Clynes R, Takechi Y, Moroi Y, Houghton A, Ravetch JV. Fc receptors are required in passive and active immunity to melanoma. Proc Natl Acad Sci U S A. 1998;95(2):652–656. doi: 10.1073/pnas.95.2.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6(4):443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 5.Cartron G, Dacheux L, Salles G, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99(3):754–758. doi: 10.1182/blood.v99.3.754. [DOI] [PubMed] [Google Scholar]
- 6.Musolino A, Naldi N, Bortesi B, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol. 2008;26(11):1789–1796. doi: 10.1200/JCO.2007.14.8957. [DOI] [PubMed] [Google Scholar]
- 7.Gale RP, Zighelboim J. Polymorphonuclear leukocytes in antibody-dependent cellular cytotoxicity. J Immunol. 1975;114(3):1047–1051. [PubMed] [Google Scholar]
- 8.Tepper RI, Coffman RL, Leder P. An eosinophil-dependent mechanism for the antitumor effect of interleukin-4. Science. 1992;257(5069):548–551. doi: 10.1126/science.1636093. [DOI] [PubMed] [Google Scholar]
- 9.Gould HJ, Mackay GA, Karagiannis SN, et al. Comparison of IgE and IgG antibody-dependent cytotoxicity in vitro and in a SCID mouse xenograft model of ovarian carcinoma. Eur J Immunol. 1999;29(11):3527–3537. doi: 10.1002/(SICI)1521-4141(199911)29:11<3527::AID-IMMU3527>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 10.Uchida J, Hamaguchi Y, Oliver JA, et al. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J Exp Med. 2004;199(12):1659–1669. doi: 10.1084/jem.20040119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tiffen JC, Bailey CG, Ng C, Rasko JE, Holst J. Luciferase expression and bioluminescence does not affect tumor cell growth in vitro or in vivo. Mol Cancer. 2010;9:299. doi: 10.1186/1476-4598-9-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albanesi M, Mancardi DA, Macdonald LE, et al. Cutting edge: FcγRIII (CD16) and FcγRI (CD64) are responsible for anti-glycoprotein 75 monoclonal antibody TA99 therapy for experimental metastatic B16 melanoma [published correction appears in J Immunol. 20131;190(3):1381]. J Immunol. 2012;189(12):5513–5517. doi: 10.4049/jimmunol.1201511. [DOI] [PubMed] [Google Scholar]
- 13.Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157(2):220–233. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood. 2012;119(24):5640–5649. doi: 10.1182/blood-2012-01-380121. [DOI] [PubMed] [Google Scholar]
- 15.DiSanto JP, Müller W, Guy-Grand D, Fischer A, Rajewsky K. Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor gamma chain. Proc Natl Acad Sci U S A. 1995;92(2):377–381. doi: 10.1073/pnas.92.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minard-Colin V, Xiu Y, Poe JC, et al. Lymphoma depletion during CD20 immunotherapy in mice is mediated by macrophage FcgammaRI, FcgammaRIII, and FcgammaRIV. Blood. 2008;112(4):1205–1213. doi: 10.1182/blood-2008-01-135160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pillay J, den Braber I, Vrisekoop N, et al. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood. 2010;116(4):625–627. doi: 10.1182/blood-2010-01-259028. [DOI] [PubMed] [Google Scholar]
- 18.Bronte V, Chappell DB, Apolloni E, et al. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol. 1999;162(10):5728–5737. [PMC free article] [PubMed] [Google Scholar]
- 19.Möröy T. The zinc finger transcription factor growth factor independence 1 (Gfi1). Int J Biochem Cell Biol. 2005;37(3):541–546. doi: 10.1016/j.biocel.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 20.Hock H, Hamblen MJ, Rooke HM, et al. Intrinsic requirement for zinc finger transcription factor Gfi-1 in neutrophil differentiation. Immunity. 2003;18(1):109–120. doi: 10.1016/s1074-7613(02)00501-0. [DOI] [PubMed] [Google Scholar]
- 21.McKenzie SE, Taylor SM, Malladi P, et al. The role of the human Fc receptor Fc gamma RIIA in the immune clearance of platelets: a transgenic mouse model. J Immunol. 1999;162(7):4311–4318. [PubMed] [Google Scholar]
- 22.Jönsson F, Mancardi DA, Zhao W, et al. Human FcγRIIA induces anaphylactic and allergic reactions. Blood. 2012;119(11):2533–2544. doi: 10.1182/blood-2011-07-367334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Ziffle JA, Lowell CA. Neutrophil-specific deletion of Syk kinase results in reduced host defense to bacterial infection. Blood. 2009;114(23):4871–4882. doi: 10.1182/blood-2009-05-220806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest. 2002;109(3):363–371. doi: 10.1172/JCI13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vivier E, da Silva AJ, Ackerly M, Levine H, Rudd CE, Anderson P. Association of a 70-kDa tyrosine phosphoprotein with the CD16: zeta: gamma complex expressed in human natural killer cells. Eur J Immunol. 1993;23(8):1872–1876. doi: 10.1002/eji.1830230821. [DOI] [PubMed] [Google Scholar]
- 26.Greenberg S, Grinstein S. Phagocytosis and innate immunity. Curr Opin Immunol. 2002;14(1):136–145. doi: 10.1016/s0952-7915(01)00309-0. [DOI] [PubMed] [Google Scholar]
- 27.Elliott ER, Van Ziffle JA, Scapini P, Sullivan BM, Locksley RM, Lowell CA. Deletion of Syk in neutrophils prevents immune complex arthritis. J Immunol. 2011;187(8):4319–4330. doi: 10.4049/jimmunol.1100341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Egmond M, Bakema JE. Neutrophils as effector cells for antibody-based immunotherapy of cancer. Semin Cancer Biol. 2013;23(3):190–199. doi: 10.1016/j.semcancer.2012.12.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.