

Background: The regulation of the core clock gene Bmal1 is not fully understood despite many studies.
Results: NF-Y cooperates with Sp1 to activate the transcription of Bmal1 by binding to the CCAAT-boxes in the Bmal1 promoter.
Conclusion: NF-Y/Sp1 and Rev-Erbα serve as positive and negative regulators of Bmal1 expression, respectively.
Significance: Elucidation of Bmal1 regulation provides a better understanding of its biological functions and the circadian clock overall.
Keywords: Circadian Clock, Clock Genes, Promoters, Sp1, Transcription Coactivators, Bmal1, NF-Y, Rev-Erbα
Abstract
The circadian clock enables organisms to adjust to daily environmental changes and synchronize multiple molecular, biochemical, physiological, and behavioral processes accordingly. In mammalian clock work, Bmal1 is the most important core clock gene, which works with another core clock gene Clock to drive the expression of other clock genes and clock-controlled genes. However, the regulation of Bmal1 has not been fully understood. This work was aimed at identifying the positive regulator(s) of Bmal1 transcription. A series of 5′ deletion reporter constructs was generated, and binding site mutations of mouse Bmal1 promoter fragments were cloned into pGL3-basic and pGL3(R2.1)-basic plasmids and transfected into NIH 3T3 cells. Luciferase activity was either measured 48 h after transfection or recorded for 4 days after serum shock. DNA affinity precipitation assay was used to detect the transcription factors binding to Bmal1 promoter. Small interfering RNA against nuclear factor Y, subunit A (NF-YA) and dominant negative NF-YA were employed to study the role of NF-Y in Bmal1 transcription regulation. Deletion and mutation analyses identified two clusters of CCAAT/GC-boxes at the proximal region of Bmal1 promoter as the activating cis-elements. Bmal1 promoter activity was up-regulated by NF-Y and/or Sp1 and repressed by dominant negative NF-YA or siRNA against NF-YA. The activation of Bmal1 promoter activity by NF-Y and Sp1 was inhibited by Rev-Erbα. DNA affinity precipitation assay showed that NF-Y and Sp1 bound to the two CCAAT/GC clusters of Bmal1 promoter. These results indicate that NF-Y is a functional activator of Bmal1 transcription and it cooperates with Sp1 and Rev-Erbα to generate the daily cycle of Bmal1 expression.
Introduction
The circadian rhythm is the ∼24-h cycle that enables organisms to adjust to daily environmental changes and synchronize multiple molecular, biochemical, physiological, and behavioral processes accordingly (1). The mammalian master pacemaker is located within the suprachiasmatic nucleus of the anterior hypothalamus (2). Recent studies have shown that endogenous oscillators also exist in peripheral tissues as well as in embryos and isolated cells, suggesting the existence of autonomous circadian clocks in each cell of the organisms. Peripheral oscillators are generally under the control of the suprachiasmatic nucleus master pacemaker, but they might be able to reset by alternative means such as restricted feeding (2).
In mammals, eight core clock genes have been identified. The clock and bmal1 (brain-muscle Arnt-like protein1) genes encode basic helix-loop-helix (bHLH)-PAS (Per-Arnt-Sim domain) transcription factors. Three period genes (per1–3) encode PAS domain proteins. Casein kinase Iϵ (CKIϵ) and two cryptochrome genes (cry1 and cry2) are the other core clock genes (2). The levels of transcripts and proteins of all those genes except for clock and casein kinase Iϵ show robust circadian rhythm with an approximate 24-h period.
The circadian rhythms are sustained by interlocking transcriptional-translational feedback loops. The two transcription activators, Clock and Bmal1, form heterodimers and bind to the E-boxes (consensus sequence as CACGTG) to drive the transcription of their target genes, including per1–3 and cry1 and cry2. The Per and Cry proteins dimerize in the cytoplasm and translocate into the nucleus to repress their own transcription by inhibiting the activity of Clock-Bmal1 complex. The activity and stability of Per and Cry proteins are regulated by casein kinase I and glycogen synthase kinase-3β (GSK-3β) (3). On the other hand, Bmal1 expression has been shown to be repressed by orphan nuclear receptor Rev-Erbα (4) and activated by RORα4 (5), which in turn are under circadian control. However, mutating RORα did not show either drastic decrease of Bmal1 expression or disruption of Per2 expression rhythm (5). Using small interfering RNA knockdown, RORα also had only minimal effects on Bmal1 expression in mouse embryonic fibroblasts (6). It is of great interest to identify the dominant activator of Bmal1 transcription because high level Bmal1 expression is required for generating oscillation of both the positive and the negative loops.
The mammalian CCAAT-box-binding factor nuclear factor Y (NF-Y) is an evolutionarily conserved transcription factor presented from yeast to human (7). A functional NF-Y factor consists of three different subunits, NF-YA, NF-YB, and NF-YC. Both the NF-YB and the NF-YC subunits contain a histone-fold motif and interact with each other to form a NF-YB/NF-YC heterodimer. The B/C complex then interacts with the NF-YA subunit to form the NF-Y heterotrimeric transcription factor. All three subunits are required for the binding of NF-Y complex to DNA and NF-Y-directed transcription (7).
The CCAAT motif (Y-box) is present in promoters of many mammalian genes spanning from developmentally controlled and tissue-specific, housekeeping and inducible, to cell cycle-regulated. The position of the CCAAT-box in promoters is strongly biased, and it is typically found as a single copy element in either forward or reverse direction between −30 and −150 bp from major transcription start sites (8). Bmal1 promoters from mouse to human are highly conserved at the proximal region where multiple copies of CCAAT-boxes are located upstream of the transcription start site. Although NF-Y was implicated as a regulator of the circadian clock (9), there has been no experimental evidence to confirm its role in the transcriptional regulation of clock genes. Here we report the identification of NF-Y as the major activator of Bmal1 transcription.
MATERIALS AND METHODS
Cell Culture, Transient Transfection, and Luciferase Assays
Mouse embryonic fibroblast NIH 3T3 cells (ATCC, Manassas, VA) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% bovine serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin (all from Invitrogen, Shanghai, China) at 37 °C in a humidified atmosphere (5% CO2). On the day before transfection, NIH 3T3 cells were seeded into 12-well plates to have a confluence about 30–50% on the day of transfection. Reporter constructs and expression vectors were transfected using Lipofectamine Plus (Invitrogen) according to manufacturer's instructions. 50 μg of pCMV-lacZ (Promega, Madison, WI) was transfected as the internal control. The total DNA amount was kept at 1.0 mg/well. Corresponding empty vectors were used to make up the differences among treatments. Luciferase activity was measured 48 h later using a microtiter plate luminometer (Dynex, Chantilly, VA) and luciferase assay kit (Promega). All experiments were done by triplicates and performed at least three times independently. Results were shown as relative luciferase activity and expressed as mean ± S.E.
Plasmids
The mouse Bmal1 promoter luciferase reporter constructs were generated by PCR (see Table 1 for primer sequences) and digested with MluI and XhoI and then ligated into pGL3-basic (Promega) predigested with the same enzymes. For bioluminescence recording, the same MluI/XhoI promoter fragments were transferred into pGL3-Basic(R2.1) (Promega). The mutation of the NF-Y-, Sp1-, or RORα-binding site was done by site-directed mutagenesis with the primers listed in Table 1. Heterologous promoter reporter vectors were generated by cloning custom-made oligonucleotides corresponding to either the proximal CCAAT/GC cluster or the RORα-binding element of murine Bmal1 promoter (refer to Table 1 for primer sequences) into BamHI/XhoI sites of enhancerless luciferase reporter vector pT81. All the constructs were verified by DNA sequencing. NF-Y expression constructs (Δ4NF-YA, Δ4NF-YB, and Δ4NF-YC) were gifts from Dr. Mantovani (Universita di Milano, Italy). Dominant negative NF-YA (YA m29) was generated by site-directed mutagenesis as described previously (10). pCMV-Bmal1 and mPer1 promoter reporter constructs have been described previously (11).
TABLE 1.
Sequences of primers used in this study
m, mutant; D, Bmal1 promoter deletion construct 5′ primer; Bm_luc, Bmal1 promoter deletion construct 3′ primer; pT81, enhancer-less reporter vector.
| D318-MluI | GGTACGCGTAAAGTAGCAGGTAAACCAGCCCT |
| D275-MluI | GGTACGCGTAGGCTTTCCTATCGGTCACTCGAT |
| D165-MluI | GGTACGCGTGAAGAGGCAGGTATCCGGGCTG |
| D107-MluI | GGTACGCGTGGGCACAGCGATTGGTGGGC |
| D77-MluI | GGTACGCGTGCCTGGGCCGGCGGGGAGCGG |
| D52-MluI | GGTACGCGTGTCGGAAAGTAGGTTAGTGGTG |
| Bm_luc-XhoI | GGTCTCGAGTGACCTACTTTCTGCCTTCCCT |
| mSp1–1F | CATTGGTGGTATGGGGAAGGG |
| mSp1–1R | CCCTTCCCCATACCACCAATG |
| mSp1–2F | CGATTGGTGGTATGGGGTATGGGCCTGGGC |
| mSp1–2R | GCCCAGGCCCATACCCCATACCACCAATCG |
| mY-1F | CTCCTCCATACGGGGCGGG |
| mY-1R | CCCGCCCCGTATGGAGGAG |
| mY-2F | GCACAGCGATCAGTGGGCGGG |
| mY-2R | CCCGCCCACTGATCGCTGTGC |
| mRORE-1F | AGCGGATTGGTCGGAACGCCTTCTAGTGGTGCGACATTTAG |
| mRORE-1R | CTAAATGTCGCACCACTAGAAGGCGTTCCGACCAATCCGCT |
| mRORE-2F | CATTTAGGGAAGGCAGACCATCCTCCAGGGACGGAGGTGCC |
| mRORE-2R | GGCACCTCCGTCCCTGGAGGATGGTCTGCCTTCCCTAAATG |
| NF-YAm29_F | GCACGGAAGGCTGCGGCAGGGGGCCGC |
| NF-YAm29_R | GCGGCCCCCTGCCGCAGCCTTCCGTGC |
| siYA-1S | GUACCUGUGUCUGGAACACTT |
| siYA-1R | GUGUUCCAGACACAGGUACTT |
| siYA-2S | CCAGCAGAUCAUCAUCCAATT |
| siYA-2R | UUGGAUGAUGAUCUGCUGGTT |
| pT81-C2F | CAGCGATTGGTGGGCGGGGGGCCGGGCCTGGG |
| pT81-C2R | CCCAGGCCCGGCCCCCCGCCCACCAATCGCTG |
| pT81-ROREF | GTCGGAAAGTAGGTTAGTGGTGCGACATTTAGGGAAGGCAGAAAGTAGGTCAGGGA |
| pT81-RORER | TCCCTGACCTACTTTCTGCCTTCCCTAAATGTCGCACCACTAACCTACTTTCCGAC |
Real-time Recording of the Circadian Expression of Bmal1
NIH 3T3 cells were seeded in 35-mm plates with a density of greater than 95% confluence and transfected the next day with the indicated DNA constructs by Lipofectamine Plus. 24 h after transfection, cells were treated with 50% horse serum for 2 h and replaced with culture medium without serum. Luciferin was added at a final concentration of 1 μm. Light emission was measured and integrated for 1 min at intervals of 15 min by an LM-2400 photon detection unit (Hamamatsu, Japan).
Preparation of Nuclear Extract and DNA Affinity Precipitation Assays (DAPA)
Nuclear extracts from 24 h post-serum-shocked NIH 3T3 cells were prepared as described previously (19). DAPA have been described previously (19). The sequences for the oligonucleotides used in DAPA were as follows (with mutated nucleotides in lowercase): 1-WT, 5′-CTC CAT TGG TGG GCG GGG AAG G; 1-NF-Y, 5′-CTC CAA ACG TGG GCG GGG AGG G; 1-Sp1, 5′-CTC CAT TGG TGG TAT GGG AAG G; 1-Mut, 5′-CTC CAA ACG TGG TAT GGG AAG G; 2-WT, 5′-GCG ATT GGT GGG CGG GAG GCC GGG CCT; 2-NF-Y, 5′-GCG AAA CGT GGG CGG GAG GCC GGG CCT; 2-Sp1, 5′-GCG ATT GGT GGT ATG GAG TAT GGG CCT; and 2-Mut, 5′-GCG AAA CGT GGT ATG GAG TAT GGG CCT. 5′-Biotinylated double-stranded oligonucleotides corresponding to wild type or mutant (CCAAT- and GC-box mutated individually or combined) sequences of either distal or proximal clusters of Y-box/GC-box were incubated with NIH 3T3 nuclear extract. The protein-DNA complexes were pulled down by TetralinkTM avidin resin and resolved on PAGE gel followed by immunoblotting with antibodies against NF-YA (G-2) and Sp1 (PEP2) (Santa Cruz Biotechnology, Inc. Santa Cruz, CA).
Small Interfering RNA (siRNA) against NF-YA
siRNAs were designed using the Invitrogen RNAi designing tool. Two pairs of RNA oligonucleotides targeting different regions of NF-YA and a control pair (enhanced green fluorescence protein) were custom-synthesized by Sangon (Shanghai, China). Double-stranded siRNA was transfected into NIH 3T3 cells using Lipofectamine 2000 (Invitrogen, Shanghai, China) according to the manufacturer's instructions. The protein or mRNA level was measured 48 h after transfection by immunoblotting with specified antibodies or quantitative real-time PCR using the ABI Prism 7000 detection system (Applied Biosystems, Foster City, CA).
RESULTS
Identification of Promoter Region Conferring Bmal1 Activation
It has been shown that Bmal1 expression is negatively regulated by Rev-Erbα, which drives the circadian oscillation of Bmal1 expression and forms the stabilizing loop in the positive limb of the circadian clock. However, the identity of the activator of Bmal1 transcription has been elusive. We employed in vitro real-time expression monitoring systems to identify the cis-element in the mouse Bmal1 promoter driving its transcription. A series of 5′ deletion reporter constructs was generated using a destabilized luciferase reporter vector to enhance the oscillation resolution. The reporter constructs were transfected into NIH 3T3 cells individually, and their bioluminescence was recorded. The expression level and circadian rhythm of Bmal1 were gradually reduced from −275 to −107 deletion constructs (Fig. 1, A and B). Further truncating Bmal1 promoter to −77, the promoter activity was further diminished (Fig. 1A) and circadian oscillation was almost flattened (Fig. 1B), indicating that the cis-element(s) critical for activation of Bmal1 transcription were localized within Bmal1 promoter regions between −275 to −77. As all promoter reporter constructs had intact transcription start site and both ROR-binding sites, the loss of transcriptional activity and rhythmicity indicated the requirement of (an) additional cis-element(s) within the promoter for the up-regulated expression of the Bmal1 gene.
FIGURE 1.

Identification of promoter element(s) responsible for mouse Bmal1 gene activation and rhythmic expression. A, 5′ deletion Bmal1 reporter constructs were transfected into NIH 3T3 cells. Cells were treated with 50% horse serum for 2 h on the next day followed by culturing in regular DMEM without serum and 1 μm luciferin (Luc) and placed in LM-2400 for bioluminescence recording. The highest activity recording of each construct was set as 1, respectively. B, data were normalized to the average luciferase activity produced over the duration of the recording and are plotted as the difference from the centered 24-h moving average.
NF-Y Activates Bmal1 Transcription through the Inverted CCAAT-boxes
The proximal regions of human and mouse Bmal1 promoter are highly conserved, and they include several inverted CCAAT motifs (Y-box), two clusters of Y-box and GC-boxes, as well as two ROR-binding sites (Fig. 2A). To determine whether NF-Y is truly an activator of Bmal1 gene, we first co-transfected mouse Bmal1 luciferase reporter gene with NF-Y expression constructs in transient transfection assay. Co-transfection of NF-Y yielded 4–10-fold increase of mouse Bmal1 promoter activity (Fig. 2B). Co-transfection of the dominant negative form of NF-YA expression vector (Δ4NF-YAm29) with Bmal1-luc strongly inhibited Bmal1 promoter activity (Fig. 2C) but maintained detectable oscillation (Fig. 2D), implicating the involvement of NF-Y in activating Bmal1 expression. Two lines of experiments were carried out to further dissect the sequence requirements of mouse Bmal1 promoter for NF-Y activation. First, a series of 5′-truncated promoter reporter genes were co-transfected with or without NF-Y constructs to locate the region conferring significant activation of mouse Bmal1 promoter. As deleting from −255 to −165 then to −107 caused gradual decreases of activation of Bmal1 expression, further deletion from −107 to −77 resulted in significant loss of activation (Fig. 2E). Next, we characterized the possible roles of two clusters of Y-box/GC-box in mediating NF-Y activation of Bmal1 expression. The Y-boxes and GC-boxes were mutated individually or in combination within each cluster. Mutating the GC-box of distal cluster (C1) did not have significant effect on either promoter activity or activation by NF-Y. Mutation of the Y-box of C1 or GC-boxes of proximal cluster (C2) had a moderate effect on NF-Y activation of Bmal1 expression. However, mutation of proximal Y-box caused more than 50% decrease of such activation (Fig. 2F). Furthermore, deletion of a small fragment from −118 to −77, which contained the proximal Y-box/GC-box cluster, from the −318 construct almost totally abolished the activation of Bmal1 expression by NF-Y (last lane of Fig. 2E). Alternatively, when the proximal cluster of Y-box/GC-boxes (C2) was cloned into an enhancerless luciferase reporter vector pT81, it was specifically activated by NF-Y, whereas pT81-RORE did not show any activation by NF-Y when compared with empty pT81 vector (Fig. 2G).
FIGURE 2.

NF-Y is the functional transcription activator of Bmal1 gene. A, alignment of human (hBmal) and mouse (mBmal) Bmal1 promoter proximal region. The important and highly conserved transcription factor-binding sites are shown in boldface and underlined. B, Bmal1 luciferase (Bm-luc) reporter gene (d318) was co-transfected with NF-Y or empty pSG5 vectors into NIH 3T3 cells. Luciferase activity was measured 48 h later. C, Bmal1 luciferase reporter gene (d318) was co-transfected with dominant negative NF-YA or empty pSG5 vectors into NIH 3T3 cells. Luciferase activity was measured 48 h later. The remaining luciferase activity was shown. D, NIH 3T3 cells were transfected with Bmal1 promoter construct in combination with control vector or pSG5-YAm29 and treated the same way as in Fig. 1. E, 5′-truncated Bmal1 promoter constructs were co-transfected with NF-Y or empty pSG5 vector. The ratio of luciferase activity of each reporter with and without NF-Y was shown. F, Bmal1 promoter (d318) reporter constructs with mutated different CCAAT-box or/and Sp1 site were co-transfected with either pSG5 or NF-Y expression constructs. The relative luciferase activity (left panel) and -fold of activation by NF-Y (right panel) were shown. G, pT81-carrying the proximal CCAAT/GC-box/GC-box (C2) and RORα-binding sites of mouse Bmal1 promoter were co-transfected with NF-Y, Sp1, and/or Rev-Erbα. The luciferase activity relative to β-galactosidase activity was shown. Error bars in B, C, E, F, and G indicate mean ± S.E.
NF-Y and Sp1 Bind to Two Y-box/GC-box Clusters
To test whether NF-Y actually binds to those two CCAAT-boxes, DAPA were performed. Wild type oligonucleotides of both distal (C1) and proximal (C2) elements pulled down NF-Y and Sp1, whereas mutating Y-box and GC-box simultaneously abolished their ability to bind either protein (Fig. 3, A and B). Individual mutation of Y-boxes reduced both NF-Y and Sp1 binding, whereas mutation of GC-boxes only reduced Sp1 binding capability but did not have any effect on NF-Y binding (Fig. 3, A and B). To confirm the specificity of such binding, we then performed competitive DAPA. Each wild type biotinylated oligonucleotide was incubated with NIH 3T3 cell nuclear extracts alone or with 10 times the amount of nonbiotinylated oligonucleotide (wild type or mutants) added. Wild type oligonucleotide competed away NF-Y and Sp1 binding to its own sequence as well as the other cluster of Y-box/GC-box (Fig. 3, C and D). The oligomers with mutated Y-box competed away Sp1 binding but not NF-Y binding, whereas mutation at the GC-box abrogated its ability to compete for Sp1 but not NF-Y binding (Fig. 3, C and D).
FIGURE 3.
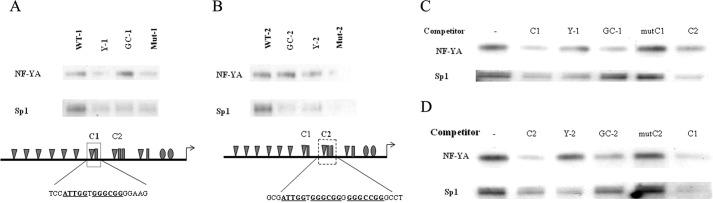
NF-Y and Sp1 bind to two CCAAT/Sp1 clusters. A and B, 5′-biotin-conjugated oligonucleotides corresponding to either one of the two CCAAT/Sp1 clusters and their mutants (CCAAT or Sp1 site or both mutated; indicated as Mut-1 and Mut-2) were incubated with NIH 3T3 cell nuclear extracts and precipitated with TetralinkTM avidin resin. The DNA-protein complexes were resolved in SDS-PAGE gel and immunoblotted with specified antibodies. C and D, 5′-biotin-conjugated wild type oligonucleotides preincubated with 20-fold indicated unconjugated oligonucleotides for 5 min before the procedure in A and B. mutC1 and mutC2 indicate mutant 1 and mutant 2.
NF-Y Activation of Bmal1 Expression Is Inhibited by Rev-Erbα
As Rev-Erbα has been shown to be a negative regulator of Bmal1 transcription to generate the oscillation of Bmal1 expression, we next tested whether NF-Y activation of Bmal1 was repressed by Rev-Erbα. In a transient transfection luciferase assay, we co-transfected Bmal1 promoter reporter with NF-Y and an increasing amount of Rev-Erbα expression construct. The activation of Bmal1 promoter activity by NF-Y was dose-dependently repressed by Rev-Erbα (Fig. 4A). Furthermore, Bmal1 promoter was activated by NF-Y and Sp1 synergistically, which was repressed by Rev-Erbα (Fig. 4B).
FIGURE 4.
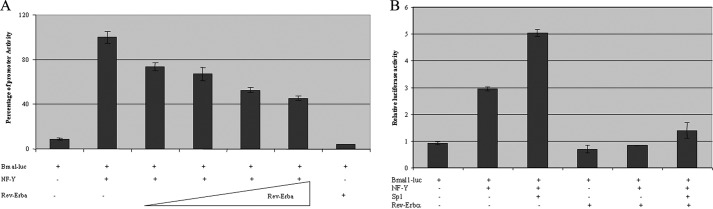
The activation of Bmal1 expression by NF-Y is repressed by Rev-Erbα. A, Bmal1 luciferase reporter construct was co-transfected with a constant amount of NF-Y expression vectors and an increased amount of Rev-Erbα expression construct. B, Bmal1 luciferase reporter construct was co-transfected with NF-Y, Sp1, and Rev-Erbα mammalian expression vectors alone or in combination. Error bars indicate mean ± S.E.
The Elements in Bmal1 Promoter Binding Positive and Negative Regulators Are Separable
It has been shown that two ROR-binding sites are required for the repression and rhythmic expression of Bmal1 gene. On the other hand, although it was suggested that RORα bound to RORE to activate Bmal1 transcription, we identified the sequence between −165 and −77 (especially from −107 to −77) of Bmal1 promoter as the more important cis-elements for positive regulation of Bmal1 expression. To determine the role of RORE and the cis-element located within −107 to −77 in the transcriptional activation of Bmal1, we made two reporter constructs to specifically address this issue. The first construct had both RORα-binding sites mutated in the −275 construct (mRORE), and the second one had wild type RORα-binding sites but a deletion of the sequence from −118 to −77 (d118–77). These two reporter constructs showed completely opposite behavior. Although mRORE had high basal Bmal1 expression, d188–77 showed minimal promoter activity (Fig. 5A). Consequently, in serum-shocked NIH 3T3 cells, mRORE showed constantly high level expression, whereas d118–77 had extremely low level expression and without oscillation (Fig. 5B). Furthermore, mutating the Y-box within the region between −118 and −77 caused significant decrease of Bmal1 promoter activity in the presence of NF-Y but had no effect on the repression by Rev-Erbα in the presence or absence of NF-Y. On the contrary, disrupting the RORα-binding sites abolished the ability of Rev-Erbα to inhibit Bmal1 expression at basal or NF-Y-activated state (Fig. 5C).
FIGURE 5.
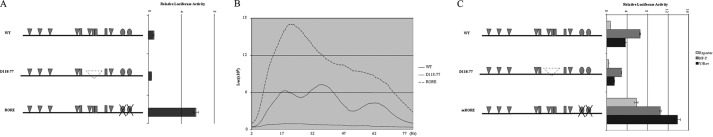
The cis-elements responsible for Bmal1 transcription activation and repression are separated. A, The d275 Bmal1-luc construct (WT), with a deletion from −77 to −118 (D118–77), or with both RORα-binding sites mutated (mRORE) was transfected into NIH 3T3 cells. Luciferase activity was measured 48 h later. B, WT, D118–77, or mRORE was transfected into NIH 3T3 cells and treated as in Fig. 1. C, WT, D118–77, or mRORE was co-transfected with NF-Y and/or Rev-Erbα into NIH 3T3 cells. Luciferase (Luc) activity was measured 48 h later. Error bars in A and C indicate mean ± S.E.
Endogenous Expression of Bmal1 and Its Target Gene Is Reduced by Knocking Down NF-Y Activity
The ability of NF-Y to regulate Bmal1 expression in vivo was tested using siRNA against NF-YA. Two siRNAs targeting different regions of mouse NF-YA were transfected into NIH 3T3 cells, and protein levels were measured 48 h after transfection. One siRNA (siYA-2) drastically knocked down the expression level of NF-YA, whereas the other one (siYA-1) did not have any effect on NF-YA level (Fig. 6A). Consequently, the Bmal1 level in siYA-2 treated cells was also decreased significantly, but that in siYA-1-treated cells did not change (Fig. 6, A and B). When siRNAs targeting NF-YA were co-transfected with Bmal1 or Per1 promoter luciferase reporter, siYA-2 significantly knocked down both Bmal1 and Per1 promoter activity, whereas siYA-1 did not have any effect (Fig. 6, C and D). Overexpression of Bmal1 restored Per1 promoter activity in siYA-2-treated cells to control level (Fig. 6D).
FIGURE 6.
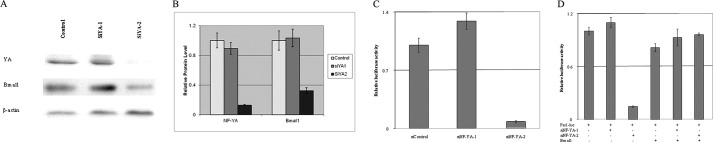
NF-Y activates Bmal1 expression and regulates Per1 expression through Bmal1. A, knocking down NF-YA (YA) level resulted in the down-regulation of Bmal1 expression. Small interfering RNA targeting either NF-YA (SiYA-1 and SiYA-2) or control (enhanced green fluorescent protein) was transfected into NIH 3T3 cells. 48 h after transfection, total cell lysates were subjected to immunoblotting assay to measure the expression levels of NF-YA, Bmal1, and β-actin. B, quantification of the bands in A. The ratio of NF-YA and β-actin or Bmal1 and β-actin from control siRNA-treated sample was set as 1. C and D, Bmal1 and Per1 promoter activity was inhibited by siRNA targeting NF-YA (siNF-YA-1 and siNF-YA-2). Bmal1 (C) or Per1 (D) luciferase (Perl-luc) construct was co-transfected with control(siControl) or NF-YA siRNA. Luciferase activity was measured 48 h later. Error bars in B–D indicate mean ± S.E.
DISCUSSION
Our results demonstrated that heterotrimeric transcription factor NF-Y was a functional activator of Bmal1 transcription. First, NF-Y activated, whereas dominant negative NF-YA or small interfering RNA against NF-YA inhibited Bmal1 promoter activity in transient luciferase assays. Second, NF-Y (and Sp1) bound to the Bmal1 promoter in vivo and in vitro. Third, RNAi against NF-YA significantly decreased Bmal1 expression level. Fourth, deletion of CCAAT/GC elements reduced both expression level and oscillation amplitude of Bmal1.
It has been shown that Bmal1 expression is inhibited by another clock-controlled gene, Rev-Erbα, which drives the circadian oscillation of Bmal1 expression (4). Although it is speculated that RORα acts as the activator of Bmal1 transcription, mice with mutated RORα (and embryonic fibroblasts originating from such mice) or cells with decreased RORα level by RNA interference had Bmal1 expression level and rhythm comparable with that of wild type. Moreover, other results (12) and our results showed that RORα-binding sites were not sufficient to drive rhythmic Bmal1 expression as the promoter reporter constructs containing intact transcription start site and both RORα-binding sites had extremely low promoter activity and flattened oscillation, which was different from the conclusion drawn by Ueda et al. (13) when they fused a DNA fragment containing RORα-binding sites of Bmal1 promoter with SV40 promoter. This approach had two major flaws. First, it completely changed the configuration around RORα-binding sites in the promoter and the distance of RORα-binding sites from the transcription start site. Second, the SV40 promoter masked the need for the activating elements of the Bmal1 promoter. Moreover, mutating the RORα-binding sites resulted in significantly high promoter activity and dampened rhythm at peak level of the Bmal1 gene, indicating that the RORα-binding sites of Bmal1 promoter served primarily as negative-regulating elements for Rev-Erbα binding and that the role of RORα activation in Bmal1 expression was marginal.
Deleting the proximal cluster or mutating the Y-box and two GC-boxes of the cluster greatly reduced Bmal1 level and flattened the oscillation of Bmal1 expression at the trough level, indicating that the activation of Bmal1 transcription was mainly mediated through the proximal Y-box/GC-box cluster. As NF-Y and Sp1 cooperatively regulate many genes (14–18), it was not surprising to see that Bmal1 transcription was up-regulated by NF-Y and Sp1 through a tandem of one Y-box and two GC-boxes. NF-Y bound to CCAAT-box of various promoters and recruited RNA polymerase II and general transcription factors onto those promoters (7). NF-Y has been shown to regulate the transcription of genes involved in development (14), cancer (14, 15), cell growth (16), and cell cycle control (17). A previous study (9) implicated a possible role for NF-Y in the regulation of the circadian clock, but we for the first time provided direct evidences that NF-Y activated Bmal1 transcription by binding to the CCAAT-boxes in Bmal1 promoter.
Based on previously published data and our results (4–6, 12), we propose a new model for the clock work and Bmal1 regulation (Fig. 7). Unlike other clock genes, the oscillation of Bmal1 expression is solely achieved by the oscillation of negative regulator Rev-Erbα. The high basal level of Bmal1 expression is driven by NF-Y/Sp1. Bmal1 and Clock activate the transcription of per1–3, cry1, cry2, Rev-Erbα, and other clock-controlled genes, which is repressed by Per/Cry, whereas the transcription of Bmal1 is repressed by Rev-Erbα.
FIGURE 7.

A proposed model of Bmal1 regulation and clock work. NF-Y/Sp1 and other factors drive Bmal1 transcription, which is repressed by Rev-Erbα. The oscillation of Rev-Erbα results in the anti-phased cyclic expression of Bmal1 (refer to “Results” for details).
Acknowledgment
We thank Dr. Mantovani (Universita di Milano, Italy) for providing NF-Y constructs.
This article was selected as a Paper of the Week.
- RORα
- retinoic acid receptor-related orphan receptor α
- RORE
- RORα response element
- mRORE
- mutated RORE
- NF-Y
- nuclear factor Y
- DAPA
- DNA affinity precipitation assays.
REFERENCES
- 1. Kondratova A. A., Kondratov R. V. (2012) The circadian clock and pathology of the ageing brain. Nat. Rev. Neurosci. 13, 325–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Reppert S. M., Weaver D. R. (2002) Coordination of circadian timing in mammals. Nature 418, 935–941 [DOI] [PubMed] [Google Scholar]
- 3. Buhr E. D., Takahashi J. S. (2013) Molecular components of the mammalian circadian clock. Handb. Exp. Pharmacol. 217, 3–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Preitner N., Damiola F., Lopez-Molina L., Zakany J., Duboule D., Albrecht U., Schibler U. (2002) The orphan nuclear receptor REV-ERBα controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 110, 251–260 [DOI] [PubMed] [Google Scholar]
- 5. Sato T. K., Panda S., Miraglia L. J., Reyes T. M., Rudic R. D., McNamara P., Naik K. A., FitzGerald G. A., Kay S. A., Hogenesch J. B. (2004) A functional genomics strategy reveals Rora as a component of the mammalian circadian clock. Neuron 43, 527–537 [DOI] [PubMed] [Google Scholar]
- 6. Akashi M., Takumi T. (2005) The orphan nuclear receptor RORα regulates circadian transcription of the mammalian core-clock Bmal1. Nat. Struct. Mol. Biol. 12, 441–448 [DOI] [PubMed] [Google Scholar]
- 7. Dolfini D., Gatta R., Mantovani R. (2012) NF-Y and the transcriptional activation of CCAAT promoters. Crit. Rev. Biochem. Mol. Biol. 47, 29–49 [DOI] [PubMed] [Google Scholar]
- 8. Dolfini D., Zambelli F., Pavesi G., Mantovani R. (2009) A perspective of promoter architecture from the CCAAT box. Cell Cycle 8, 4127–4137 [DOI] [PubMed] [Google Scholar]
- 9. Bozek K., Relógio A., Kielbasa S. M., Heine M., Dame C., Kramer A., Herzel H. (2009) Regulation of clock-controlled genes in mammals. PLoS One 4, e4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mantovani R., Li X. Y., Pessara U., Hooft van Huisjduijnen R., Benoist C., Mathis D. (1994) Dominant negative analogs of NF-YA. J. Biol. Chem. 269, 20340–20346 [PubMed] [Google Scholar]
- 11. Lee Y., Lee J., Kwon I., Nakajima Y., Ohmiya Y., Son G. H., Lee K. H., Kim K. (2010) Coactivation of the CLOCK-BMAL1 complex by CBP mediates resetting of the circadian clock. J. Cell Sci. 123, 3547–3557 [DOI] [PubMed] [Google Scholar]
- 12. Onishi Y., Hanai S., Ohno T., Hara Y., Ishida N. (2008) Rhythmic SAF-A binding underlies circadian transcription of the Bmal1 gene. Mol. Cell Biol. 28, 3477–3488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ueda H. R., Chen W., Adachi A., Wakamatsu H., Hayashi S., Takasugi T., Nagano M., Nakahama K., Suzuki Y., Sugano S., Iino M., Shigeyoshi Y., Hashimoto S. (2002) A transcription factor response element for gene expression during circadian night. Nature 418, 534–539 [DOI] [PubMed] [Google Scholar]
- 14. Smith J., Mowla S., Prince S. (2011) Basal transcription of the human TBX3 gene, a key developmental regulator which is overexpressed in several cancers, requires functional NF-Y and Sp1 sites. Gene 486, 41–46 [DOI] [PubMed] [Google Scholar]
- 15. Lützner N., De-Castro Arce J., Rösl F. (2012) Gene expression of the tumour suppressor LKB1 is mediated by Sp1, NF-Y and FOXO transcription factors. PLoS One 7, e32590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Armengol S., Arretxe E., Rodríguez L., Ochoa B., Chico Y., Martínez M. J. (2013) NF-κB, Sp1 and NF-Y as transcriptional regulators of human SND1 gene. Biochimie 95, 735–742 [DOI] [PubMed] [Google Scholar]
- 17. Dalvai M., Mondesert O., Bourdon J. C., Ducommun B., Dozier C. (2011) Cdc25B is negatively regulated by p53 through Sp1 and NF-Y transcription factors. Oncogene 30, 2282–2288 [DOI] [PubMed] [Google Scholar]
- 18. Wu Y. F., Matsuo N., Sumiyoshi H., Yoshioka H. (2010) The Sp1 and CBF/NF-Y transcription factors cooperatively regulate the mouse pro-α3(V) collagen gene (Col5a3) in osteoblastic cells. Acta Med. Okayama. 64, 95–108 [DOI] [PubMed] [Google Scholar]
- 19. Lei S., Dubeykovskiy A., Chakladar A., Wojtukiewicz L., Wang T. C. (2004) The murine gastrin promoter is synergistically activated by transforming growth factor-β/Smad and Wnt signaling pathways. J. Biol. Chem. 279, 42492–42502 [DOI] [PubMed] [Google Scholar]