

Background: Protein phosphatase 2A (PP2A) controls most cell signaling, but the current understanding of its many isoforms, e.g. as tumor suppressors, is limited.
Results: Differentiation was controlled by the small fraction of PP2A using the Aβ scaffold.
Conclusion: PP2A Aβ regulates Akt.
Significance: Given the extreme importance of Akt in cancer, these findings help explain why Aβ is a tumor suppressor.
Keywords: Akt, Differentiation, PI 3-Kinase (PI3K), PP2A, Protein Kinases, Protein Phosphatase, SV40, Polyomavirus
Abstract
Protein phosphatase 2A (PP2A) regulates almost all cell signaling pathways. It consists of a scaffolding A subunit to which a catalytic C subunit and one of many regulatory B subunits bind. Of the more than 80 PP2A isoforms, 10% use Aβ as a scaffold. This study demonstrates the isoform-specific function of the A scaffold subunits. Polyomaviruses have shown the importance of phosphotyrosine, PI3K, and p53 in transformation. Comparisons of polyoma and SV40 small T antigens implicate Aβ in the control of differentiation. Knockdown of Aβ enhanced differentiation. Akt signaling regulated differentiation; its activation or inhibition promoted or blocked it, respectively. Aβ bound Akt. Enhancement of PP2A Aβ/Akt interaction by polyoma small T antigen increased turnover of Akt Ser-473 phosphorylation. Conversely, knockdown of Aβ promoted Akt activity and reduced turnover of phosphate at Ser-473 of Akt. These data provide new insight into the regulation of Akt, a protein of extreme importance in cancer. Furthermore, our results suggest that the role for Aβ in differentiation and perhaps tumor suppression may lie partly in its ability to negatively regulate Akt.
Introduction
Protein phosphatase 2A (PP2A)2 accounts for 30–50% of the total cellular phosphoserine/threonine phosphatase activity (1–4). Regulating at least 50 protein kinases (5), PP2A is involved in most cell processes. PP2A is a heterotrimeric ABC complex. There are two isoforms of the scaffold A subunits, a major Aα and minor (∼10% (6)) Aβ isoform. There are also two forms of the catalytic C subunit and four B subunit families, each with several members. B subunits target PP2A to particular locations or substrates (7, 8). PP2A is being increasingly connected to cancer (9–11). For example, decreasing the activity of PP2A toward Myc has been associated with head and neck and colon cancers (12). In this study, we focused on the forms of PP2A that use Aβ as a scaffold. Aβ acts as a tumor suppressor, and mutations in it are associated with lung and colon cancers (13, 14).
Polyomaviruses have identified signal regulators important for transformation, such as PI3K and p53, each of which were identified via their interactions with polyomavirus early proteins, the so called T (tumor) antigens. Polyomavirus small T antigens (STs) bind to the PP2A A and C subunits, displacing or preventing B subunits from binding (15, 16). Murine polyoma ST antigen (PyST) causes apoptosis (17) or inhibits differentiation through association with PP2A (18). Although simian virus SV40 ST antigen (SVST) also binds PP2A, it does not do either of these things. Determining why two homologous proteins that both bind PP2A have very different outcomes provides an opportunity to understand the regulation of PP2A. The comparison of PyST and SVST focused attention on specific PP2A A isoforms. PyST, but not SVST, binds to the Aβ isoform of PP2A (18). Although mutations in Aβ are associated with cancer (13, 14), there is relatively little information about Aβ-specific functions. However, it is known to regulate the Ral family of small GTPases (19).
PI3K signaling is deregulated in the majority of human tumors. Akt kinases are the most important PI3K effector molecules. Studies of both transformation (11, 20, 21) and cell death (22) have focused attention on Akt. Translocated to membranes by stimulation of PI3K signaling, Akt is activated by phosphorylation at Thr-308 by PDK1 (PI3K-dependent protein kinase 1) (23) and at Ser-473 by the mTORC2 complex (24). It has been clear for a long time that PP2A can affect Akt. PP2A dephosphorylates and reduces Akt kinase activity in vitro, whereas treatment of cells with okadaic acid, a PP2A inhibitor, causes Akt activation (22). Both STs affect Akt but in different ways. SVST signaling through Akt contributes to transformation (21). Depending on cell conditions, PyST can kill or protect 3T3 cells in a PP2A/Akt-dependent manner (17).
Akt activity has been implicated as a necessary component in preadipocyte (25, 26) and myoblast (27–29) differentiation. Both Akt1 and Akt2 play a role in myoblast differentiation (28, 30, 31), and knockdown of either blocks myoblast differentiation (30, 31). Conversely, Akt2 expression is increased during the differentiation process (28). Akt may have multiple functions during differentiation. It may reduce cyclin D levels and thereby proliferation (32). Akt promotes transcription of peroxisome proliferator-activated receptor-γ (PPARγ) (25) and myogenin (29), two transcription factors required for fat and muscle cell differentiation, respectively.
In this study, we explored the connections among PP2A, Akt, and the viral STs and their effects on differentiation and established that PP2A Aβ complexes control differentiation through regulation of Akt. Given the extreme importance of Akt in cancer, it is likely that our findings may help explain why Aβ is a tumor suppressor.
EXPERIMENTAL PROCEDURES
Transient Transfection
Fresh DMEM with 10% FBS and supplements was added to 60–80% confluent cells on 10-cm plates for 4 h. 3 μg of PEI and at least 7 μg of the plasmid DNA were mixed in 1 ml of Opti-MEM I. The mixture was incubated for 5 min and slowly distributed into the medium. The medium was changed the next day. 48 h was usually needed for optimal expression.
Retroviral Infection and Cell Line Derivation
Phoenix-AMPHO cells were transfected with 10 μg of pBABE retrovirus constructs. 48 h later, the supernatants were removed and replaced with fresh medium. The filtered viral supernatant mixed with fresh medium at a 1:1 ratio was placed on target cells with 2 μg/ml Polybrene (Sigma AL-118) for 4 h twice. The next day, the process was repeated twice. Cells were selected the following day with either puromycin (2 μg/ml) or G418 (500 μg/ml).
Cell Lysates and Western Blotting
Harvested cells washed with PBS were suspended in T antigen extraction buffer (137 mm NaCl, 20 mm Tris, 0.92 mm Ca2+, 0.49 mm Mg2+, 1% Nonidet P-40, and 10% glycerol adjusted to pH 7) and incubated for 30 min at 4 °C. The cleared supernatant was boiled in SDS loading buffer.
Adipocyte Differentiation and Oil Red O Staining
3T3-L1 cell lines were allowed to grow to confluence in DMEM containing 10% FBS. 2 days later, this was replaced with induction medium (10% FBS in DMEM supplemented with 167 nm insulin, 1 μm dexamethasone, 0.5 mm isobutylmethylxanthine, and 5 μm troglitazone) for 2 days. The cells were stimulated for 1 day with DMEM containing 10% FBS and 167 nm insulin. After this, the cells were allowed to fully differentiate for 8 days with fresh medium supplied every 2 days. For Oil Red O staining, the differentiated cells were fixed in 10% formalin for 10 min at room temperature and replaced with 10% formalin for 1 h. The plates were washed with 60% isopropyl alcohol, dried, and stained with 3.5% Oil Red O in 60% isopropyl alcohol for 10 min at room temperature. The plates were subsequently washed four times with water and dried, and images were taken at ×10 magnification.
Myoblast Differentiation
C2C12 cells were grown to confluence in DMEM containing 10% FBS. The medium was then replaced with DMEM containing 2% horse serum every 2 days. Cell lysates were collected 4∼6 days post-confluence.
Bone Differentiation
To initiate bone differentiation, C2C12 cells were grown to confluence in DMEM with 10% FBS. The growth medium supplemented with 300 ng/ml bone morphogenetic protein (R&D Systems 355-BM-010) was changed every 2 days. Bone differentiation was analyzed with an alkaline phosphatase activity detection kit from (Sigma 85L3R).
Knockdown
The sequence and use of shRNA4 of PP2A Aβ were described previously (18). For siRNA knockdown, four PP2A Aβ siRNAs were obtained (Qiagen GS73699). Immediately after cells were resuspended on 6-cm plates with growth medium, 250 μl of Opti-MEM I preincubated for 10 min with 10 nmol of each siRNA and 2.5 μl of HiFect transfection reagent (Qiagen 301802) was added to the cells. The medium was changed after 24 h. After 72 h, the cells were assayed for the level of knockdown. The levels of Aβ compared with GAPDH using real-time PCR described previously showed the extent of knockdown (18). Puromycin resistance gene expression measured by real-time PCR was used to control the levels of lentiviral infection (18).
Immunoprecipitation
T antigen extraction buffer lysates from transfected 293T cells were added to antibody premixed with either protein A beads (GenScript L00210) or protein G beads (GenScript L00209) for 1–2 h at 4 °C. Each of the samples was subsequently washed four times with T antigen extraction buffer, boiled in loading buffer, and analyzed by Western blotting.
Antibodies and Reagents
Antibody MF20 to myosin heavy chain was from the Developmental Studies Hybridoma Bank. Monoclonal antibodies PAB419 and PN116 recognize the N termini of SVST and PyST, respectively (18). The following antibodies were from Cell Signaling Technologies: anti-Akt (9272), anti-phospho-Ser (pSer)-473 Akt (XP 4060), anti-phospho-Thr (pThr)-246 PRAS40 (2997), anti-PRAS40 (2691), anti-pSer-21/9 GSK3α/β (9331), anti-GSK3β (9315), anti-PPARγ (2443), anti-mTOR (2972), anti-pThr-2481 mTOR (2974), anti-pThr-389 p70 S6 kinase (9206), and anti-Akt substrate (9614). Anti-myogenin antibody (sc-12732) was from Santa Cruz Biotechnology, antibody HA11 (MMS-101P) was from Covance, anti-Glu-Glu epitope tag antibody (AB3788) was from Millipore, and anti-actin antibody (A2066) was from Sigma. MK2206 was a gift from Phillip Tsichlis. Torin2 (4248) was from Tocris Bioscience, and rapamycin (553210) was obtained from Calbiochem.
RESULTS
Comparisons of PyST and SVST, Two Polyomavirus Small T Antigens, Indicate a Role for PP2A Aβ in Regulating Differentiation
Previous work, including our own, indicated that PyST can interfere with differentiation (18, 33). Fig. 1 shows this in three systems: differentiation of 3T3-L1 cells into adipocytes and differentiation of C2C12 cells into myocytes or osteoblasts. 3T3-L1 preadipocytes were infected with retrovirus expressing wild-type PyST, PP2A binding-deficient mutant W157S, SVST, or GFP. Selected pools were tested for the ability to differentiate as measured by the formation of lipid droplets. Compared with controls, wild-type PyST, but not the PP2A-defective W157S allele, blocked differentiation (Fig. 1A). When C2C12 cells are treated with BMP-2 (bone morphogenetic protein 2), they differentiate into osteoblasts and express alkaline phosphatase activity. Lack of alkaline phosphatase activity showed that PyST blocked this differentiation pathway (Fig. 1B). C2C12 cells can also be differentiated into myocytes. Myosin heavy chain (MHC) expression is an indication that myoblast differentiation has occurred. Lack of MHC expression demonstrated that PyST blocked C2C12 cell differentiation into myocytes (Fig. 1C).
FIGURE 1.
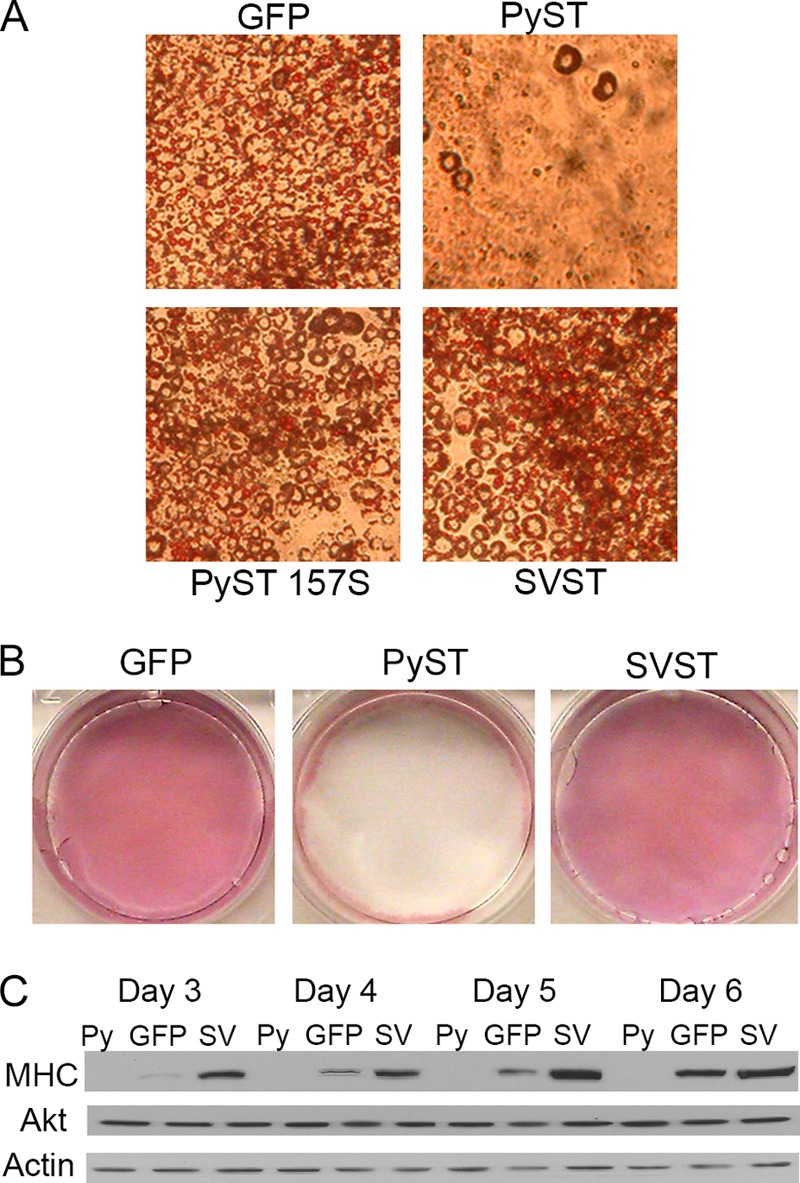
PyST, but not SVST, blocks differentiation in several systems. A, 3T3-L1 cell lines expressing PyST, the PP2A-binding defective PyST W157S, SVST, or a GFP control were differentiated to adipocytes according to protocol. The cells were fixed and stained with Oil Red O. Images were then taken at ×10 magnification. B, C2C12 cell lines expressing PyST, SVST, or a GFP control were differentiated for 7 days using the bone differentiation protocol. The cells were then stained for alkaline phosphatase activity. C, PyST-expressing (Py), GFP-expressing, and SVST-expressing (SV) cells were placed under differentiation conditions. Cell extracts collected at days 3–6 were analyzed for MHC with respect to loading controls (total Akt and actin).
SVST had a different phenotype. It did not interfere with adipocyte or osteoblast differentiation. SVST cells showed earlier and increased expression of MHC (Fig. 1C). This indicated that SVST was promoting differentiation. PyST, but not SVST, binds PP2A Aβ (18), so we wondered if Aβ was involved. Examination of the SVST-PP2A Aα structure (34) showed that residues in the Aα/SVST interface were identical in Aβ. Nonetheless, mutation of SVST Lys-130 and Asp-131, two residues close to PP2A A, resulted in a mutant (SVST K130A/D131A) that could bind to Aβ as well as Aα (Fig. 2A). Although clearly not as effective as PyST, SVST K130A/D131A suppressed differentiation as measured by MHC expression rather than activating differentiation as wild-type SVST did (Fig. 2B). Taken together, the ST results suggest a connection between Aβ and differentiation.
FIGURE 2.
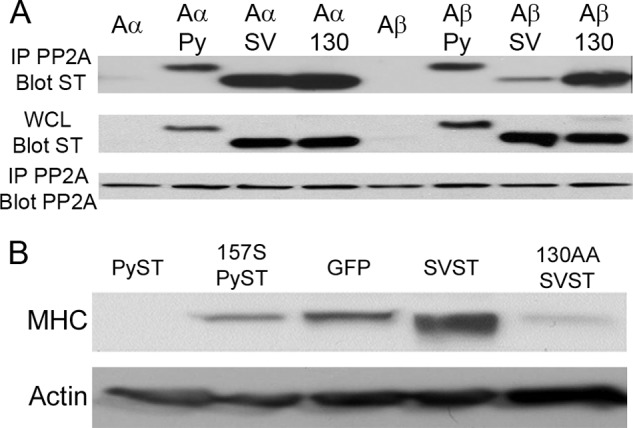
SVST interaction with PP2A Aβ blocks differentiation. A, Glu-Glu epitope-tagged PP2A Aα or Aβ was transfected with PyST (Py), SVST (SV), and SVST K130A/D131A (130) into NIH-3T3 cells. 48 h post-transfection, whole cell lysates (WCL) were collected, and immunoprecipitation (IP) was performed using anti-Glu-Glu epitope tag antibody. The whole cell lysate and immunoprecipitated fractions were analyzed for ST and PP2A A interaction by Western blotting for STs using antibody PN116 for PyST, antibody PAB419 for SVST, and anti-Glu-Glu epitope tag antibody for PP2A A. B, C2C12 cell lines expressing PyST, PyST W157S (157S), SVST, SVST K130A/D131A (130AA; an SVST mutant that binds Aβ), or a GFP control were differentiated for 4 days. Cell extracts were collected and analyzed for MHC and β-actin as a loading control.
Knockdown of PP2A Aβ Enhances Differentiation
Because the STs interfered with differentiation in an Aβ-dependent fashion, we directly examined the Aβ role in differentiation. We used two different siRNAs to achieve knockdown (Fig. 3A). Each achieved a moderate (∼60%) knockdown and stimulated differentiation as measured by MHC expression. As further confirmation, lentiviral infection of an shRNA targeting a different Aβ sequence also enhanced MHC expression (Fig. 3B). These results suggest that Aβ restricts the activity of a protein(s) important for differentiation. The simplest interpretation of the difference in the outcome between Aβ knockdown and PyST binding to Aβ would be that PyST enhances Aβ activity toward this target.
FIGURE 3.
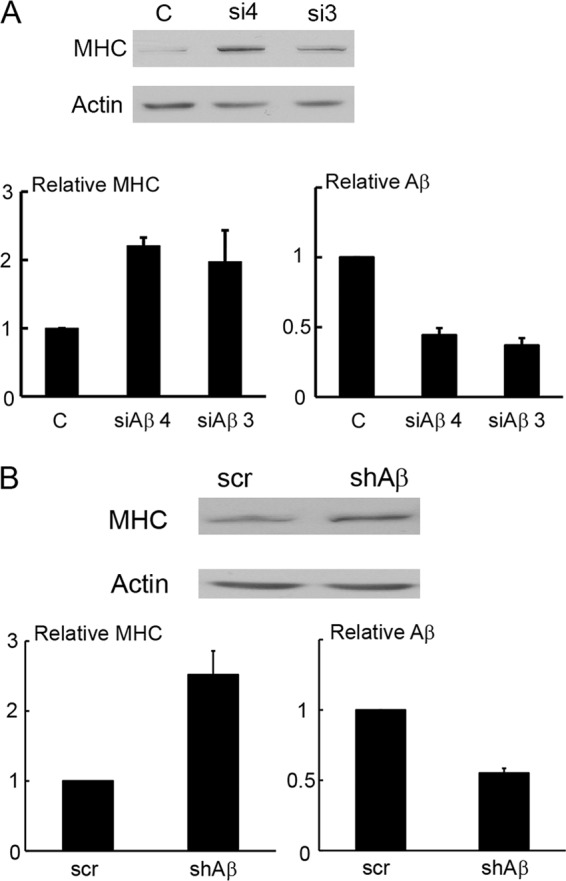
Knockdown of PP2A Aβ enhances differentiation. A, C2C12 cells were transfected with two different siRNAs to Aβ (si4 and si3) or a control (C). Cells were then differentiated for 3 days and analyzed by Western blotting for MHC and β-actin as a control (upper). Results from three experiments were quantitated (lower left). RT-PCR was used to determine the relative expression of Aβ using GAPDH as a control (lower right). B, C2C12 cells were infected with lentivirus to Aβ as well as a pLKO scrambled control (scr). 48 h post-infection, the relative expression of Aβ was assayed by RT-PCR with GAPDH as a control (lower right). The cells were then differentiated for 3 days and analyzed by Western blotting for MHC and β-actin as a control (upper). Results from four experiments were quantitated (lower left). shAβ, Aβ shRNA.
Akt Activity Controls Differentiation of C2C12 and 3T3-L1 Cells
Akt activity has been implicated in differentiation of preadipocytes and myoblasts (25–31). As expected, Akt was activated during differentiation as seen by increased phosphorylation at Ser-473 (Fig. 4A). We altered the specific Akt activity to confirm the importance of Akt in our systems. Retroviral expression of activated Akt2 (myristoylated Akt2) in C2C12 cells promoted differentiation, as shown by elongated morphology and by expression of myogenin and MHC (Fig. 4, B and C). Alternatively, inhibition of Akt with the specific allosteric inhibitor MK2206 or inhibition of mTORC2, the activator of Akt, with Torin2 blocked differentiation (Fig. 4D). Fig. 4 (E and F) shows that activated Akt promoted differentiation of 3T3-L1 cells into adipocytes in the absence of insulin, as measured by lipid staining or by expression of PPARγ, the primary driver of adipocyte differentiation. Inhibition of Akt with MK2206 blocked differentiation (Fig. 4, G and H).
FIGURE 4.
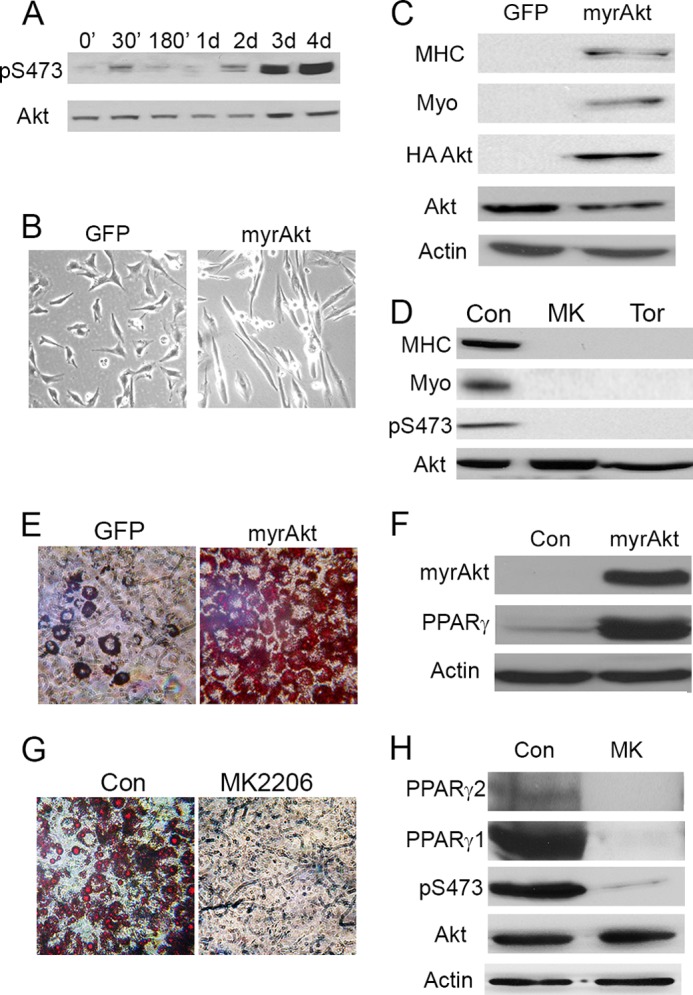
Akt activity controls differentiation of C2C12 and 3T3-L1 cells. A, C2C12 cells were differentiated for up to 4 days. Cell extracts were collected at the indicated times and analyzed by Western blotting for Akt Ser-473 phosphorylation (pS473) using total Akt as a control. d, days. B, naïve C2C12 cells were infected with retrovirus to express myristoylated Akt2 (myrAkt). The cell lines were selected with puromycin and allowed to grow for ∼10 days. Images of the cells were taken at ∼8–10 days post-selection. C, lysates of cells from B were collected and analyzed by Western blotting for MHC, myogenin (myo), HA-myristoylated Akt2, total Akt, and β-actin as a control. D, cell extracts were collected from C2C12 cells differentiated for 4 days in the presence of 2 μm MK2206 (MK) or 250 nm Torin2 (Tor) and then analyzed by Western blotting for myogenin, MHC, phosphorylation of Akt at Ser-473, and total Akt as a control. E, 3T3-L1 cell lines expressing either myristoylated Akt or a GFP control were grown to confluence for 2 days. The cells were then differentiated to adipocytes in insulin-free differentiation medium. The cells were fixed and stained with Oil Red O. Images were taken at ×10 magnification. F, cells from E were collected at day 3 after induction. Western blots were used to analyze HA-myristoylated Akt expression, PPARγ, total Akt, and β-actin as a control. G, 3T3-L1 cells were grown to confluence for 2 days and allowed to differentiate to adipocytes in full induction medium with either 2 μm MK2206 or a control (Con). The cells were fixed and stained with Oil Red O. H, lysates of cells from G were collected at day 3 post-induction and analyzed for Akt Ser-473 phosphorylation, total Akt, PPARγ1, PPARγ2, and actin.
Action of Both SVST and PyST in Differentiation Is Connected to Akt
Given the role of Akt in differentiation, it became important to determine how the STs regulate Akt activity during differentiation. The phosphorylation of Akt at Thr-308 and Ser-473 was determined (Fig. 5A). Although SVST promoted phosphorylation at both sites, PyST had an inhibitory effect. Both Akt1 and Akt2 were affected by the STs. Interestingly, Fig. 5B shows that, although wild-type SVST promoted Akt phosphorylation, the Aβ-binding SVST mutant K130A/D131A had a suppressive effect on Akt phosphorylation at both Thr-308 and Ser-473. Probing blots of total cell proteins with an antibody to Akt consensus phosphorylation sites (RXRXX(S/T)) showed that SVST increased all bands. This confirmed that Akt activity was enhanced by SVST (Fig. 5C). When PyST was present, many (but not all) Akt substrates showed reduced phosphorylation, reflecting reduced activity (Fig. 5C). The direct substrates of Akt, PRAS40 at Thr-246 and TSC2 at Thr-939, were suppressed, whereas PyST also attenuated phosphorylation of p70 S6 kinase at Thr-389, a site indicative of mTORC1 activity. PyST did not affect GSK3 phosphorylation. This pattern of differential effects has been noted previously in cell death experiments (17). Unlike PyST, SVST had a general enhancing effect on the same substrates. The results showed that regulation of differentiation by STs had a strong correlation with their effects on Akt activity. We further demonstrated that the ability of the STs to regulate differentiation was also highly dependent on Akt activity. Akt inhibitor MK2206 blocked enhanced differentiation in SVST cells in a dose-dependent manner (Fig. 5D). Expression of activated Akt overcame the PyST block of differentiation (Fig. 5E).
FIGURE 5.

Both SVST and PyST regulate Akt activity during differentiation. A, SVST- and GFP-expressing C2C12 cells were differentiated for 2 days, whereas PyST and another GFP cell line were differentiated for 4 days. Cell lysates were collected and blotted for phosphorylation of Akt at Thr-308 (Akt pT308) and Ser-473 (Akt pS473), Akt1 at Ser-473 (Akt1 pS473), Akt2 at Ser-474 (Akt2 pS474), and total Akt using β-actin as a control. B, the indicated C2C12 cell lines were differentiated for 2 days, and the lysate was collected and analyzed for Akt phosphorylation at Thr-308 and Ser-473 using Akt and actin as loading controls. C, cell lysates were collected from C2C12 cells expressing SVST or GFP differentiated for 2 days and PyST cells differentiated for 4 days, followed by Western blotting for phosphorylation of Akt substrates. Brackets indicates regions in which PyST reduced Akt substrate phosphorylation. The lysates were then analyzed for relative phosphorylation of PRAS40 at Thr-246 (pT246), TSC2 at Thr-939 (pT939), GSK3α/3β at Ser-9/Ser-21 (pS9/S21), p70 S6 kinase at Thr-389 (p70S6 pT389), and Akt at Ser-473. Total Akt and β-actin were used as protein controls. D, SVST-expressing C2C12 cells were differentiated in the presence of the indicated amounts of MK2206. Cell lysates were collected 3 days post-induction and analyzed for MHC, total Akt, and Akt Ser-473 phosphorylation. E, PyST- or GFP-expressing C2C12 cell lines were infected with myristoylated Akt2 (myrAkt) and selected with neomycin. Cell extracts were collected and analyzed for MHC, myogenin, and total Akt. PyST- and GFP-expressing cells differentiated for 5 days were used as controls.
PyST Alters the Turnover of Phosphate at Akt Ser-473
The simplest explanation for why PyST reduces the level of Akt phosphorylated at Ser-473 is that PyST association with PP2A increases the rate of Akt dephosphorylation. To measure phosphatase activity toward Akt pSer-473, loss of Akt Ser-473 phosphate was measured after blocking de novo phosphorylation by its principle kinase, mTORC2 (24). mTORC2 activity can be blocked by Torin2 (35). The addition of Torin2 prevented further phosphorylation at Ser-473 (Fig. 6, A and B); this allowed measurement of the rate of dephosphorylation at Akt Ser-473 in the presence of STs. Expression of PyST caused a higher rate of turnover at pSer-473 compared with controls (Fig. 6A). Quantification by ImageJ demonstrated that the half-life of Akt Ser-473 phosphorylation was <1 min in PyST cells, whereas in control cells, it was ∼6 min. Considering that some time must be required for Torin2 to enter and inhibit mTORC2, this experiment probably underestimated the difference caused by PyST. Turnover experiments examining phosphorylation at Thr-308 showed that it was similarly dephosphorylated in the timescale of our experiments (data not shown), even though Torin2 does not directly block PDK1, the kinase of Akt Thr-308.
FIGURE 6.
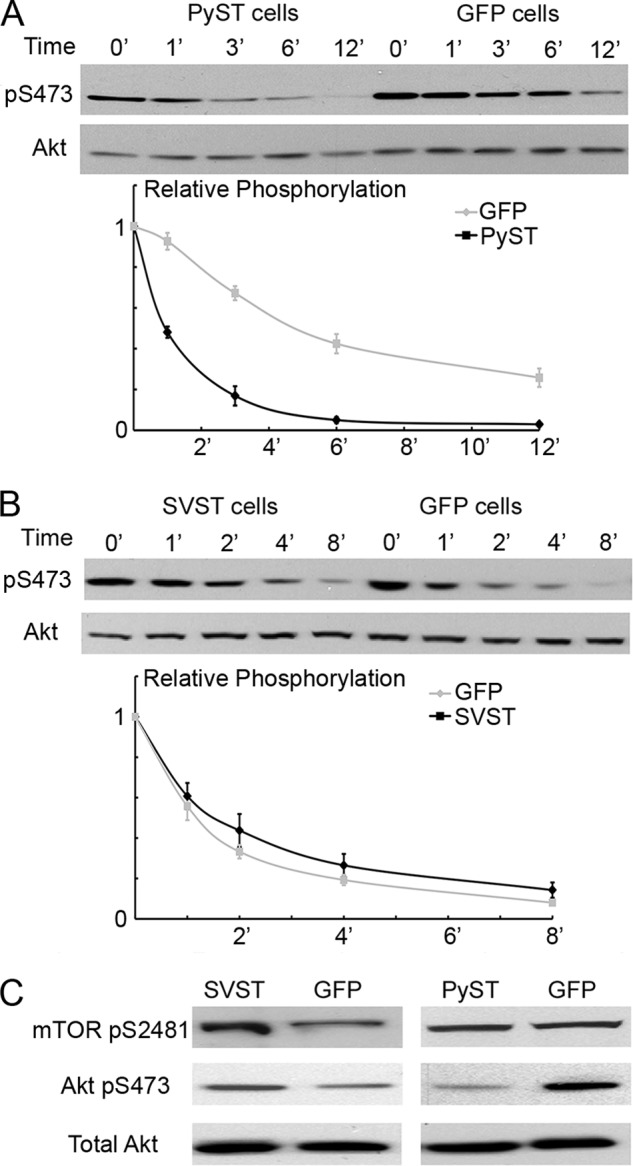
PyST alters the turnover of Akt phosphate at Ser-473 by enhancing Akt and PP2A interaction. A, cells were stimulated with fresh medium containing 10% FBS for 15 min prior to the experiment. At time 0, Torin2 was added to the cells, and cell extracts were collected at the indicated times. Akt Ser-473 phosphorylation was determined by Western blotting. Results were quantified by ImageJ, and the intensity of Akt Ser-473 phosphorylation (pS473) was normalized to total Akt. The relative phosphorylation was then normalized again to 100% with respect to time 0. The average of five experiments was plotted. Similar differences were seen in C2C12 cells. B, SVST- or GFP-expressing C2C12 cells were stimulated with medium containing 10% FBS for 15 min before Torin2 was added to the cells. A similar analysis was performed as described for A to determine turnover of the phosphorylation at Akt Ser-473. The average results from four experiments were plotted to visualize the rate of dephosphorylation. C, C2C12 cell lines expressing SVST and PyST were differentiated for 1.5 (left) and 4 (right) days. Cell extracts were collected and analyzed by Western blotting for phosphorylation of mTOR at Ser-2481 (pS2481), Akt at Ser-473, and total Akt.
Unlike PyST, SVST had little or no effect on pSer-473 turnover after Torin2 treatment (Fig. 6B). This might seem surprising because SVST causes increased Ser-473 phosphorylation. However, the level of phosphorylation at Akt Ser-473 could be affected by either phosphatase or kinase activity. SVST may be activating mTORC2 kinase because it increased phosphorylation at mTOR Ser-2481 (Fig. 6C). This might account for the observed increase in Ser-473 phosphorylation.
PP2A Aβ Regulates Akt
Because the PyST/Aβ interaction correlates with enhanced Akt pSer-473 turnover, we investigated the connection between Aβ and Akt. Fig. 7A shows that both PP2A A isoforms associated with Akt, but Aβ was significantly better than Aα. PyST enhanced the interactions of Akt with PP2A, so the most complex formation was observed with PyST and PP2A Aβ. This would account for the loss of pSer-473 in the turnover experiments. Fig. 7B shows that PyST associated with Akt. This, along with the enhancement of PP2A/Akt binding by PyST, suggests that the three might associate together. SVST (and SVST K130A/D131A) seemed to have caused a decrease in the PP2A/Akt interactions (Fig. 7A). This was unexpected based on the turnover experiments of Fig. 6 and may relate to the fact that the binding experiments were done as transfections.
FIGURE 7.

PP2A Aβ negatively regulates Akt activation at Ser-473. A, in 293T cells, Glu-Glu epitope-tagged PP2A Aα or Aβ was cotransfected along with Myc-tagged Akt, PyST (Py), and SVST (SV) as indicated. Immunoprecipitation (IP) was performed using rabbit anti-Glu-Glu epitope tag antibody. The whole cell lysate (WCL) and immunoprecipitates were analyzed for PP2A A/Akt interaction by Western blotting for PP2A with mouse anti-Glu-Glu epitope tag antibody and for Akt with a mouse anti-Myc antibody. B, in 293T cells, Myc-tagged Akt was cotransfected along with PyST as indicated. Immunoprecipitation was performed using rabbit anti-Myc antibody. The whole cell lysate and immunoprecipitated fractions were analyzed for Akt and PyST interaction by Western blotting for PyST with antibody PN116 and for Akt with mouse anti-Myc antibody. C, Aβ in C2C12 cells was knocked down (50%) by siRNA. 24 h after the initial transfection, the cells were differentiated for 3 days. Lysates were analyzed for phosphorylation of Akt at Ser-473 (pS473), total Akt, and β-actin by Western blotting. C, control siRNA; si4, Aβ siRNA4. D, 3T3-L1 cells were infected with lentivirus expressing shRNA to Aβ or scrambled control (scr). The cell lysates were collected and analyzed for Akt Ser-473 phosphorylation 2 days post-infection (upper). Cells were also analyzed for the extent of Aβ knockdown (KD) by RT-PCR using GAPDH as a control (lower). The amount of infection was controlled by the puromycin-resistant gene (PAC). E, wild-type HEK and HEK cells with Aβ knockdown (95% knockdown by RT-PCR; kindly provided by William Hahn) were analyzed for phosphorylation of Akt at Thr-308 (pT308) and at Ser-473, total Akt, and Akt phospho-substrate phosphorylation via Western blotting using β-actin as a control. F, HEK cells from E were stimulated for 15 min with fresh medium containing 10% FBS. Torin2 was added to the cells to analyze the rate of Akt Ser-473 turnover as described for Fig. 6 (A and B). The average of three experiments was plotted after quantitation of relative Akt Ser-473 phosphorylation with respect to total Akt.
Because Akt binds Aβ even in the absence of PyST, the possibility that Aβ controls Akt activity was tested by knockdown experiments. A transient 50% knockdown of Aβ with siRNA increased the levels of phosphorylation at Ser-473 (Fig. 7C). Furthermore, lentiviral infection of a pLKO vector expressing an Aβ shRNA directed toward a different Aβ sequence decreased mRNA levels by 70% and resulted in greater Ser-473 phosphorylation compared with a scrambled control (Fig. 7D). Finally, HEK cells with stable Aβ knockdown were obtained from William Hahn (19). These cells have an ∼95% knockdown of Aβ. Substantially increased phosphorylation of Akt at Ser-473 was seen, along with increased phosphorylation of a variety of substrates measured with anti-Akt substrate antibody in these cells (Fig. 7E). These three different kinds of knockdown experiments all indicated that Aβ is important for suppression of Akt phosphorylation at Ser-473. In principle, Aβ could control activation of Akt indirectly, or it could directly control dephosphorylation at Ser-473. We again measured Ser-473 phosphorylation as a function of time after blocking the kinase mTORC2 with Torin2. These turnover experiments showed that the phosphorylated form of Akt was much more stable in the absence of Aβ (Fig. 7F). This supports the idea that PP2A Aβ plays an important role in deactivating Akt.
Aβ Is Reduced during Differentiation
One final issue is whether effects on Aβ contribute to the activation of Akt seen in differentiation (Fig. 4A). Fig. 8A shows that the expression of Aβ was down-regulated relatively early in differentiation; this coincided with increased Akt phosphorylation at Ser-473. Additionally, turnover experiments demonstrated that upon blocking mTORC2 activity with Torin2, dephosphorylation of Akt pSer-473 was significantly slower in differentiated cells relative to growing cells (Fig. 8B). These results emphasize that phosphatases, as well as kinases, can be controlled to regulate levels of protein phosphorylation.
FIGURE 8.
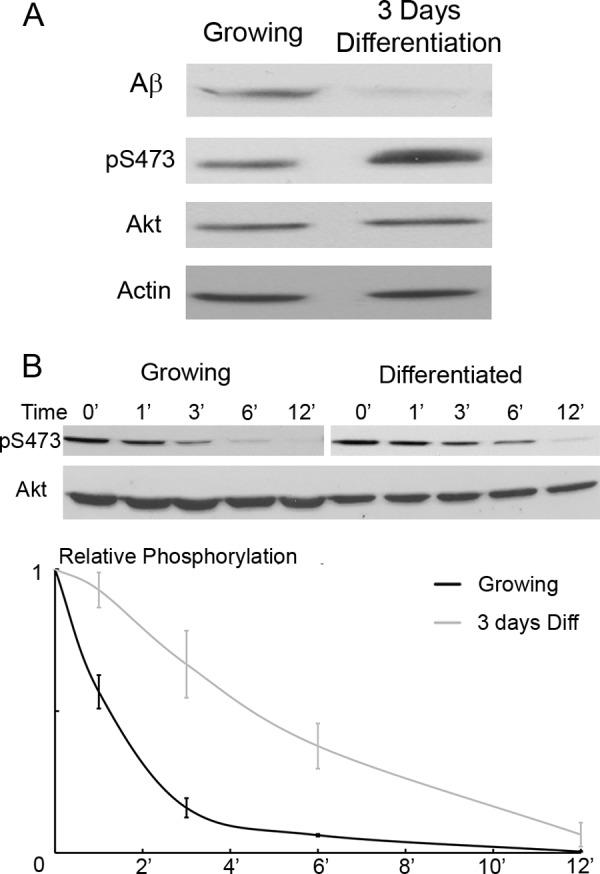
Aβ and Akt dephosphorylation is attenuated during differentiation. A, C2C12 cells were differentiated (Diff) for 3 days. The cell extracts were collected and analyzed for expression of Aβ, Akt phosphorylation at Ser-473 (pS473), total Akt, and β-actin by Western blotting. The undifferentiated control cells were 80% confluent. B, cells from A were analyzed for the rate of Akt Ser-473 turnover as described for Fig. 6 (A and B) and Fig. 7F. These cells were not stimulated before the addition of Torin2. The average of three experiments was plotted after quantitation of relative Akt Ser-473 phosphorylation with respect to total Akt.
DISCUSSION
The PP2A A subunit is generally seen as the scaffolding subunit for the PP2A holoenzyme. Aα is the predominant isoform and represents >90% of total PP2A A (6). Although quantitatively a minor species, Aβ clearly has important functions. Aβ knockdown cells show activated RalA and are tumorigenic (19). Inactivating mutations of Aβ are seen in colon, neck, and breast cancers (13, 14, 36). Small DNA tumor viruses clearly discern differences between the two A isoforms. Murine PyST evolved to bind both PP2A Aα and Aβ, whereas SVST, a monkey polyomavirus, strongly prefers Aα. In this work, we have demonstrated the functional importance of the discrimination between A isoforms.
Here, we have shown that the PP2A Aβ subunit is a negative regulator of Akt activity. Knocking down PP2A Aβ by siRNA or shRNA in three different cell systems greatly enhanced phosphorylation at Akt Ser-473 (Fig. 7. C–E). The turnover rate of Akt Ser-473 was reduced drastically in Aβ knockdown cells (Fig. 7F), suggesting that Aβ isoforms make a major contribution to Akt dephosphorylation. Consistent with this idea, PP2A Aβ could be found in association with Akt in co-immunoprecipitation experiments (Fig. 7A). This could be a direct interaction. However, because Akt is known to associate with a number of scaffolds (11, 37, 38), one of those may mediate the interaction. Aα could also be detected in complex with Akt, albeit at much lower levels (Fig. 7A). Okadaic acid completely blocked turnover of phosphate at Akt Ser-473 in the time frame of our experiments.3 Because this was a more drastic effect than seen with Aβ knockdown, Aα may contribute to a lower extent to Akt Ser-473 dephosphorylation. The okadaic acid result also suggests that PHLLP phosphatases, which have been implicated in Akt regulation (see Ref. 39 for a review), are unlikely to be making a major contribution here.
There are several possibilities that might explain differences in Aα and Aβ function, even though they are 87% identical in sequence (40). Aα and Aβ could interact with different B subunits. B subunits are thought to regulate PP2A function. Knockdown or mutation of particular B subunits leads to hyperactivation of specific substrates (7, 8). In fact, there is evidence that B55α targets PP2A to dephosphorylate Akt at Thr-308 (7). This observation with B55α is probably not connected to Aβ because Aβ is reported not to bind B55α (6). However, interactions with some of the other ∼20 B subunits could still be involved. How differences in B subunit binding could be achieved is not entirely clear. B subunits are reported to interact with HEAT repeats 1–10 (6). Fig. 9 shows that, although there are sequence differences between Aα and Aβ in that portion of the molecule, the structure (41) indicates that those differences are largely absent from the side of the molecule that binds the B subunit (Fig. 9). Perhaps shifts in A scaffold structure adjust the spatial relationship between the B and C subunits in a way that alters substrate specificity. Alternatively, the genetic differences seen between Aα and Aβ could possibly allow the PP2A holoenzyme to reach particular subcellular locations or bind molecular scaffolds. Fig. 9 shows structural differences at the ends of the molecule and some patches in the middle portion that could certainly be responsible for particular substrate or scaffold interactions. Future genetic experiments will be needed to test this directly.
FIGURE 9.

Comparison of PP2A Aα and Aβ. Upper, this is a model of Aα from Ref. 41, with amino residues marked in colors that are different in Aβ. Olive, conservative substitutions; magenta, charge changes; orange, polar substitutions for charged; yellow, proline additions or subtractions; pink, polar for polar; blue, polar for non-polar; cyan, non-polar for polar. In the left view, the A surface interacting with B is shown. The N terminus of A is on the left. The center view is rotated 180° to show the backside of the structure, and the N terminus is now on the right. The right view shows the same orientation with the B (red) and C (white) subunits present. Lower, the sequence of human Aβ (GenBankTM AAC63525.1) is compared with Aα (NM_014225.5) by NCBI BLAST. HEAT repeats 1–15 of the A subunits are aligned as in Ref. 6. This comparison shows identical residues in black, conserved residues in green, and more pronounced differences in red. Ruediger et al. (48) have shown that SVST associates with HEAT repeats 3–6, whereas PyST associates with HEAT repeats 2–8.
The results presented here confirm and extend earlier results on the connection between Akt and differentiation. They also suggest mechanisms by which the STs affect differentiation. Blocking Akt activity with either PyST or the Akt inhibitor MK2206 blocked differentiation (Fig. 1, A–C, and Fig. 4, D, G, and H). Conversely, activation of Akt by expression of constitutively activated forms (Fig. 4, B and C) or SVST (Fig. 1C and Fig. 5, A–C) promoted differentiation. Activated Akt could also reverse the block caused by PyST (Fig. 5E), whereas MK2206 titrated down SVST promotion of differentiation (Fig. 5D). The results presented here apply to C2C12 cell differentiation to myocytes or 3T3-L1 cell differentiation to adipocytes. PyST reduced phosphorylation of both Akt1 and Akt2 during differentiation (Fig. 5A). It did this by enhancing dephosphorylation at Akt Ser-473 by increasing the interaction of PP2A and Akt (Fig. 7, A and B). From this point of view, by binding Aβ, PyST functions as a viral PP2A B regulatory subunit, enhancing Aβ activity toward its Akt target. Alternatively, there is evidence that expression of SVST changes the spectrum of B subunits in cells as part of its transforming activity (42, 43). It is possible that PyST has similar indirect effects. Finally, there are other PyST interactions, such as TAZ (44) and lipin (18), that could also affect differentiation.
The biological significance of the effect of Αβ on Akt is likely to be far-reaching. Blotting with anti-Akt substrate antibody showed that many cell proteins have enhanced phosphorylation when Aβ is knocked down (Fig. 7E). Given the significant role of Akt in promoting differentiation (Fig. 4), it was no surprise that knockdown of Aβ with either siRNA or shRNA activated Akt (Fig. 7, C and D) and enhanced differentiation (Fig. 3). Perturbation of Aβ could affect other Akt-dependent cell processes, such as transformation.
Although most attention has been focused on activation of Akt by phosphorylation downstream of PI3K, this work suggests that regulation of PP2A activity that controls dephosphorylation could also be important. Fig. 8 shows a reduction of Aβ associated with Akt activation and differentiation. PP2A can also be regulated by modifications, such as methylation (45) and phosphorylation (46). It can also be affected by binding proteins, such as type 2A-interacting protein (47) and α4 (37). Akt can associate with a number of molecular scaffolds (11, 38), and at least the one with RACK1 also binds PP2A (7). It is possible that PyST enhances recruitment to one of these, resulting in suppression of Akt signaling and subsequent inhibition of differentiation (Fig. 1). Not surprisingly, SVST, which activated Akt, did not bring Akt and PP2A together.
In this work, we delved into the mechanisms of Akt regulation in differentiation by comparing two distinct regulators of Akt, PyST and SVST. However, the most important outcome of our study is the realization that Aβ is a negative regulator of Akt in the absence of viral proteins. The clear implication of this work is that at least one role of Aβ as a tumor suppressor may lie in its ability to regulate this all-important oncogenic kinase.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 CA34722 and P01 CA50661.
J. H. Hwang and B. S. Schaffhausen, unpublished data.
- PP2A
- protein phosphatase 2A
- ST
- small T antigen
- PyST
- polyoma ST
- SVST
- simian virus SV40 ST
- PPARγ
- peroxisome proliferator-activated receptor-γ
- pSer
- phospho-Ser
- pThr
- phospho-Thr
- MHC
- myosin heavy chain.
REFERENCES
- 1. Lechward K., Awotunde O. S., Swiatek W., Muszyńska G. (2001) Protein phosphatase 2A: variety of forms and diversity of functions. Acta Biochim. Pol. 48, 921–933 [PubMed] [Google Scholar]
- 2. Janssens V., Goris J. (2001) Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 353, 417–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Janssens V., Goris J., Van Hoof C. (2005) PP2A: the expected tumor suppressor. Curr. Opin. Genet. Dev. 15, 34–41 [DOI] [PubMed] [Google Scholar]
- 4. Shi Y. (2009) Serine/threonine phosphatases: mechanism through structure. Cell 139, 468–484 [DOI] [PubMed] [Google Scholar]
- 5. Millward T. A., Zolnierowicz S., Hemmings B. A. (1999) Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem. Sci. 24, 186–191 [DOI] [PubMed] [Google Scholar]
- 6. Zhou J., Pham H. T., Ruediger R., Walter G. (2003) Characterization of the Aα and Aβ subunit isoforms of protein phosphatase 2A: differences in expression, subunit interaction, and evolution. Biochem. J. 369, 387–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kuo Y. C., Huang K. Y., Yang C. H., Yang Y. S., Lee W. Y., Chiang C. W. (2008) Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55α regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J. Biol. Chem. 283, 1882–1892 [DOI] [PubMed] [Google Scholar]
- 8. Li X., Yost H. J., Virshup D. M., Seeling J. M. (2001) Protein phosphatase 2A and its B56 regulatory subunit inhibit Wnt signaling in Xenopus. EMBO J. 20, 4122–4131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Westermarck J., Hahn W. C. (2008) Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol. Med. 14, 152–160 [DOI] [PubMed] [Google Scholar]
- 10. Janssens V., Rebollo A. (2012) The role and therapeutic potential of Ser/Thr phosphatase PP2A in apoptotic signalling networks in human cancer cells. Curr. Mol. Med. 12, 268–287 [DOI] [PubMed] [Google Scholar]
- 11. Hao Y., Wong R., Feig L. A. (2008) RalGDS couples growth factor signaling to Akt activation. Mol. Cell. Biol. 28, 2851–2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Junttila M. R., Puustinen P., Niemelä M., Ahola R., Arnold H., Böttzauw T., Ala-aho R., Nielsen C., Ivaska J., Taya Y., Lu S. L., Lin S., Chan E. K., Wang X. J., Grènman R., Kast J., Kallunki T., Sears R., Kähäri V. M., Westermarck J. (2007) CIP2A inhibits PP2A in human malignancies. Cell 130, 51–62 [DOI] [PubMed] [Google Scholar]
- 13. Wang S. S., Esplin E. D., Li J. L., Huang L., Gazdar A., Minna J., Evans G. A. (1998) Alterations of the PPP2R1B gene in human lung and colon cancer. Science 282, 284–287 [DOI] [PubMed] [Google Scholar]
- 14. Ruediger R., Pham H. T., Walter G. (2001) Alterations in protein phosphatase 2A subunit interaction in human carcinomas of the lung and colon with mutations in the Aβ subunit gene. Oncogene 20, 1892–1899 [DOI] [PubMed] [Google Scholar]
- 15. Pallas D. C., Shahrik L. K., Martin B. L., Jaspers S., Miller T. B., Brautigan D. L., Roberts T. M. (1990) Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell 60, 167–176 [DOI] [PubMed] [Google Scholar]
- 16. Walter G., Ruediger R., Slaughter C., Mumby M. (1990) Association of protein phosphatase 2A with polyoma virus medium tumor antigen. Proc. Natl. Acad. Sci. U.S.A. 87, 2521–2525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Andrabi S., Gjoerup O. V., Kean J. A., Roberts T. M., Schaffhausen B. (2007) Protein phosphatase 2A regulates life and death decisions via Akt in a context-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 104, 19011–19016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Andrabi S., Hwang J. H., Choe J. K., Roberts T. M., Schaffhausen B. S. (2011) Comparisons between murine polyomavirus and simian virus 40 show significant differences in small T antigen function. J. Virol. 85, 10649–10658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sablina A. A., Chen W., Arroyo J. D., Corral L., Hector M., Bulmer S. E., DeCaprio J. A., Hahn W. C. (2007) The tumor suppressor PP2A Aβ regulates the RalA GTPase. Cell 129, 969–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yuan H., Veldman T., Rundell K., Schlegel R. (2002) Simian virus 40 small tumor antigen activates AKT and telomerase and induces anchorage-independent growth of human epithelial cells. J. Virol. 76, 10685–10691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao J. J., Gjoerup O. V., Subramanian R. R., Cheng Y., Chen W., Roberts T. M., Hahn W. C. (2003) Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell 3, 483–495 [DOI] [PubMed] [Google Scholar]
- 22. Andjelković M., Jakubowicz T., Cron P., Ming X. F., Han J. W., Hemmings B. A. (1996) Activation and phosphorylation of a pleckstrin homology domain containing protein kinase (RAC-PK/PKB) promoted by serum and protein phosphatase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 93, 5699–5704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alessi D. R., James S. R., Downes C. P., Holmes A. B., Gaffney P. R., Reese C. B., Cohen P. (1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 7, 261–269 [DOI] [PubMed] [Google Scholar]
- 24. Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 25. Kim S. P., Ha J. M., Yun S. J., Kim E. K., Chung S. W., Hong K. W., Kim C. D., Bae S. S. (2010) Transcriptional activation of peroxisome proliferator-activated receptor-γ requires activation of both protein kinase A and Akt during adipocyte differentiation. Biochem. Biophys. Res. Commun. 399, 55–59 [DOI] [PubMed] [Google Scholar]
- 26. Naiki T., Saijou E., Miyaoka Y., Sekine K., Miyajima A. (2007) TRB2, a mouse Tribbles ortholog, suppresses adipocyte differentiation by inhibiting AKT and C/EBPβ. J. Biol. Chem. 282, 24075–24082 [DOI] [PubMed] [Google Scholar]
- 27. Wilson E. M., Tureckova J., Rotwein P. (2004) Permissive roles of phosphatidylinositol 3-kinase and Akt in skeletal myocyte maturation. Mol. Biol. Cell 15, 497–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gonzalez I., Tripathi G., Carter E. J., Cobb L. J., Salih D. A., Lovett F. A., Holding C., Pell J. M. (2004) Akt2, a novel functional link between p38 mitogen-activated protein kinase and phosphatidylinositol 3-kinase pathways in myogenesis. Mol. Cell. Biol. 24, 3607–3622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu Q., Wu Z. (2000) The insulin-like growth factor-phosphatidylinositol 3-kinase-Akt signaling pathway regulates myogenin expression in normal myogenic cells but not in rhabdomyosarcoma-derived RD cells. J. Biol. Chem. 275, 36750–36757 [DOI] [PubMed] [Google Scholar]
- 30. Héron-Milhavet L., Mamaeva D., Rochat A., Lamb N. J., Fernandez A. (2008) Akt2 is implicated in skeletal muscle differentiation and specifically binds Prohibitin2/REA. J. Cell Physiol. 214, 158–165 [DOI] [PubMed] [Google Scholar]
- 31. Rotwein P., Wilson E. M. (2009) Distinct actions of Akt1 and Akt2 in skeletal muscle differentiation. J. Cell Physiol. 219, 503–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gherzi R., Trabucchi M., Ponassi M., Gallouzi I. E., Rosenfeld M. G., Briata P. (2010) Akt2-mediated phosphorylation of Pitx2 controls Ccnd1 mRNA decay during muscle cell differentiation. Cell Death Differ. 17, 975–983 [DOI] [PubMed] [Google Scholar]
- 33. Yen A., Placanica L., Bloom S., Varvayanis S. (2001) Polyomavirus small t antigen prevents retinoic acid-induced retinoblastoma protein hypophosphorylation and redirects retinoic acid-induced G0 arrest and differentiation to apoptosis. J. Virol. 75, 5302–5314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cho U. S., Morrone S., Sablina A. A., Arroyo J. D., Hahn W. C., Xu W. (2007) Structural basis of PP2A inhibition by small t antigen. PLoS Biol. 5, e202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu Q., Wang J., Kang S. A., Thoreen C. C., Hur W., Ahmed T., Sabatini D. M., Gray N. S. (2011) Discovery of 9-(6-aminopyridin-3-yl)-1-(3-(trifluoromethyl)phenyl)benzo[h][1,6]naphthyridin-2(1H)-one (Torin2) as a potent, selective, and orally available mammalian target of rapamycin (mTOR) inhibitor for treatment of cancer. J. Med. Chem. 54, 1473–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Esplin E. D., Ramos P., Martinez B., Tomlinson G. E., Mumby M. C., Evans G. A. (2006) The glycine 90 to aspartate alteration in the Aβ subunit of PP2A (PPP2R1B) associates with breast cancer and causes a deficit in protein function. Genes Chromosomes Cancer 45, 182–190 [DOI] [PubMed] [Google Scholar]
- 37. Chen J., Peterson R. T., Schreiber S. L. (1998) α4 associates with protein phosphatases 2A, 4, and 6. Biochem. Biophys. Res. Commun. 247, 827–832 [DOI] [PubMed] [Google Scholar]
- 38. Nazarewicz R. R., Salazar G., Patrushev N., San Martin A., Hilenski L., Xiong S., Alexander R. W. (2011) Early endosomal antigen 1 (EEA1) is an obligate scaffold for angiotensin II-induced, PKC-α-dependent Akt activation in endosomes. J. Biol. Chem. 286, 2886–2895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brognard J., Newton A. C. (2008) PHLiPPing the switch on Akt and protein kinase C signaling. Trends Endocrinol. Metab. 19, 223–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hemmings B. A., Adams-Pearson C., Maurer F., Müller P., Goris J., Merlevede W., Hofsteenge J., Stone S. R. (1990) α- and β-forms of the 65-kDa subunit of protein phosphatase 2A have a similar 39 amino acid repeating structure. Biochemistry 29, 3166–3173 [DOI] [PubMed] [Google Scholar]
- 41. Xu Y., Chen Y., Zhang P., Jeffrey P. D., Shi Y. (2008) Structure of a protein phosphatase 2A holoenzyme: insights into B55-mediated Tau dephosphorylation. Mol. Cell 31, 873–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen W., Possemato R., Campbell K. T., Plattner C. A., Pallas D. C., Hahn W. C. (2004) Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell 5, 127–136 [DOI] [PubMed] [Google Scholar]
- 43. Moreno C. S., Ramachandran S., Ashby D. G., Laycock N., Plattner C. A., Chen W., Hahn W. C., Pallas D. C. (2004) Signaling and transcriptional changes critical for transformation of human cells by simian virus 40 small tumor antigen or protein phosphatase 2A B56γ knockdown. Cancer Res. 64, 6978–6988 [DOI] [PubMed] [Google Scholar]
- 44. Tian Y., Li D., Dahl J., You J., Benjamin T. (2004) Identification of TAZ as a binding partner of the polyomavirus T antigens. J. Virol. 78, 12657–12664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tolstykh T., Lee J., Vafai S., Stock J. B. (2000) Carboxyl methylation regulates phosphoprotein phosphatase 2A by controlling the association of regulatory B subunits. EMBO J. 19, 5682–5691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brautigan D. L. (1995) Flicking the switches: phosphorylation of serine/threonine protein phosphatases. Semin. Cancer Biol. 6, 211–217 [DOI] [PubMed] [Google Scholar]
- 47. McConnell J. L., Gomez R. J., McCorvey L. R., Law B. K., Wadzinski B. E. (2007) Identification of a PP2A-interacting protein that functions as a negative regulator of phosphatase activity in the ATM/ATR signaling pathway. Oncogene 26, 6021–6030 [DOI] [PubMed] [Google Scholar]
- 48. Ruediger R., Roeckel D., Fait J., Bergqvist A., Magnusson G., Walter G. (1992) Identification of binding sites on the regulatory A subunit of protein phosphatase 2A for the catalytic C subunit and for tumor antigens of simian virus 40 and polyomavirus. Mol. Cell. Biol. 12, 4872–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]