

Abstract
Cell-lineage–specific transcripts are essential for differentiated tissue function, implicated in hereditary organ failure, and mediate acquired chronic diseases. However, experimental identification of cell-lineage–specific genes in a genome-scale manner is infeasible for most solid human tissues. We developed the first genome-scale method to identify genes with cell-lineage–specific expression, even in lineages not separable by experimental microdissection. Our machine-learning–based approach leverages high-throughput data from tissue homogenates in a novel iterative statistical framework. We applied this method to chronic kidney disease and identified transcripts specific to podocytes, key cells in the glomerular filter responsible for hereditary and most acquired glomerular kidney disease. In a systematic evaluation of our predictions by immunohistochemistry, our in silico approach was significantly more accurate (65% accuracy in human) than predictions based on direct measurement of in vivo fluorescence-tagged murine podocytes (23%). Our method identified genes implicated as causal in hereditary glomerular disease and involved in molecular pathways of acquired and chronic renal diseases. Furthermore, based on expression analysis of human kidney disease biopsies, we demonstrated that expression of the podocyte genes identified by our approach is significantly related to the degree of renal impairment in patients. Our approach is broadly applicable to define lineage specificity in both cell physiology and human disease contexts. We provide a user-friendly website that enables researchers to apply this method to any cell-lineage or tissue of interest. Identified cell-lineage–specific transcripts are expected to play essential tissue-specific roles in organogenesis and disease and can provide starting points for the development of organ-specific diagnostics and therapies.
Cell-lineage differentiation plays a defining role in biology. Impairment of differentiated cell functions is responsible for the organ-specific manifestation of acquired chronic degenerating diseases, including Alzheimer's disease, diabetes, and chronic kidney disease (CKD). Defining lineage-specific cellular function in human physiology and disease remains challenging, as it is frequently impossible to physically isolate a specific cell lineage from the heterogeneous lineages that make up many solid human tissues. This inability to obtain a pure cell preparation from human tissue in vivo and to identify the functional context of cell lineages on a genome-scale is a significant barrier to developing an understanding of molecular interactions in complex tissues and diseases.
Here we develop a computational approach (Fig. 1) that identifies genes specifically expressed in a cell lineage from high-throughput expression data of complex solid tissue biopsies. This problem is of significant biological and clinical relevance, however, obtaining a pure ex vivo cell population of sufficient size from the lineage of interest for direct assay is often technically infeasible, particularly when the lineage of interest is a component of solid tissues. This challenge imposes severe limitations on researchers' ability to account for the cell-lineage–specific expression and function of most human genes. This problem is distinct from the task of identifying the fractional composition of a heterogeneous sample (e.g., whole blood), and methods to address such problems require whole-genome expression measurements for each underlying cell type, which are unavailable for most solid human cell lineages (Shen-Orr et al. 2010). Our iterative machine-learning–based approach leverages heterogeneous expression data from human tissue homogenates. We term this approach “in silico nanodissection” because it is a computational approach that can analyze lineages that are not separable by experimental microdissection.
Figure 1.
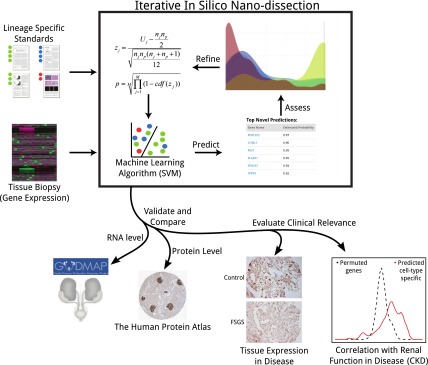
Schematic overview of the in silico nanodissection workflow, an iterative approach for cell-lineage–specific gene prediction, validation, and functional analysis. Expert-curated literature annotations are iteratively combined with gene-expression data to predict genes specific to a cell lineage. These predictions are assessed, and the standards are refined. Validation of podocyte specificity of our predictions used publicly available resources followed by evaluation of intrarenal mRNA and protein expression analysis in correlation with clinical phenotypes to define regulation of predicted gene sets in human disease.
Chronic diseases like diabetes and hypertension cause morbidity and mortality via alteration of differentiated organ function in a wide range of tissues. Here we use kidney disease as a proof of concept application, focusing on the podocytes, the highly differentiated glomerular epithelial cells responsible for most hereditary and acquired glomerular disease (Gerstein 2001; Kim et al. 2003; Roselli et al. 2004; Groop et al. 2009; D'Agati et al. 2011; Niewold 2011). As with most other differentiated cell lineages in solid tissues, discovering human podocyte-specific genes on a whole-genome scale has remained infeasible due to the challenge of obtaining pure ex vivo populations of sufficient size for high-throughput evaluation, making this important cell lineage an ideal proof of concept application for nanodissection. Restricted gene expression is defined in this study as podocyte “specific” within the renal context if it shows gene expression limited to podocytes within the kidney, and “podocyte specific within the renal glomerulus” if it is expressed only in podocytes within the glomeruli but detectable in other extraglomerular cell lineages in the kidney. Previous high-throughput strategies have relied on mouse (Endlich et al. 2002; Brunskill et al. 2011; Jain et al. 2011) or human (Saleem et al. 2008) immortalized glomerular visceral epithelial cells, but in vitro culture leads to rapid loss of both lineage-specific phenotypes and lineage-specific gene expression. Whole-tissue–based molecular profiles of renal disease are attainable (Henger et al. 2004; Higgins et al. 2004; Schmid et al. 2006; Bennett et al. 2007; Ju et al. 2009; Hodgin et al. 2010; Lindenmeyer et al. 2010; Woroniecka et al. 2011) since human renal tissue is routinely obtained by diagnostic fine needle biopsy, but whole-tissue expression profiles have not previously been capable of identifying gene expression at the cell-lineage level. This difficulty is not unique to renal disease. Similar challenges exist for other clinically important lineages, e.g., neuronal cell-lineage–specific markers in neurodegenerative diseases like Alzheimer's disease or multiple sclerosis. Employing a computational approach to identify cell-lineage–specific molecules for noninvasive monitoring of neuronal functional status would help to address one of the key challenges pursued in the study of such diseases (Reddy et al. 2011).
When applied to renal gene expression data sets, our nanodissection method predicts 136 genes not previously known to be podocyte specific. Through systematic immunohistochemistry-based evaluation, we show that our iterative in silico method significantly outperforms experimental strategies using fluorescence-activated cell sorting (FACS) separated GFP-tagged murine cells for identification of cell-lineage–enriched transcripts. We further demonstrate that expression of the nanodissection predicted podocyte-specific genes significantly correlates with kidney function, as measured by the glomerular filtration rate (GFR), in patients with CKD. The nanodissection method also predicts the most recently identified gene responsible for hereditary nephrotic syndrome (Mele et al. 2011). These findings reinforce the concept that defining cell-lineage–specific genes can provide important insights into the pathogenesis of and targeted therapies for degenerative human disease of the kidney, central nervous system, and other highly differentiated tissues. Our approach is freely available in a user-friendly website that allows researchers to easily explore any cell-lineage or tissue of interest (http://nano.princeton.edu) and through an open-source C++ library (http://libsleipnir.bitbucket.org).
Results
In silico nanodissection approach discovers cell-lineage–specific genes
To discover cell-lineage–specific genes, we developed in silico nanodissection, an iterative computational approach that predicts cell-lineage–specific expression of human genes using high-throughput genomic expression data derived from tissue homogenates. This method uses an iterative machine learning framework that makes robust predictions, even when only limited prior knowledge about cell-lineage–specific markers is available (for detailed description of the approach, see Methods). Intuitively, our approach discovers patterns of coexpression of the cell-lineage markers in whole-tissue homogenates from a variety of genetic backgrounds, physiological and pathophysiological states. The approach leverages human curated markers of the cell lineage of interest (podocyte in this case) (Supplemental Tables 1, 2) to identify the genetic or pathophysiological perturbations in which the expression patterns of these markers are predictive of their cell-lineage specificity. These patterns of informative conditions are identified from comprehensive transcriptional data sets derived from tissue homogenates, often represent only a small fraction of all data, and are likely reflective of the markers' biological functions. The condition-specific patterns are then used to identify additional cell-lineage–specific genes. Our approach uses an iterative algorithm to refine the weighting of informative perturbations in a manner robust to the limited availability of curated markers (gold standards). Each gold standard provides differential specificity (e.g., those based on double immunofluorescence, immunohistochemical (IHC) staining, or RNA abundance) and quality. With our strategy, standards are assessed without the need for genome-scale measurements from a pure sample of the cell lineage of interest. Our method is robust to variable standard quality, and the machine learning component of our approach by itself is not sufficient for this robustness (Supplemental Fig. 5).
We applied this nanodissection strategy to a data set of 452 microarray measurements for microdissected human kidney biopsies and predicted 136 genes with novel podocyte-specific expression in the renal context (Supplemental Table 3). These represented all non–gold-standard genes among the top 150 predictions. The selected genes are the set with the maximum F-measure (Supplemental Methods) as assessed by cross-validation where precision was weighted five times as much as recall and resulted in a number of genes practical for systematic verification and validation. In silico nanodissection separated known podocyte genes from genes specific to the other glomerular cell lineages and tubular cells (Fig. 2), while a simple correlation-based approach failed to do so (Supplemental Fig. 1; Supplemental Methods).
Figure 2.
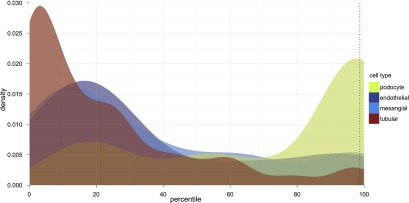
In silico nanodissection. Distribution of cell-type–specific prediction by percentile, estimated using a Gaussian kernel. Genes are ordered on the x-axis from worst (zero percentile) to best (100th percentile). The dotted line shows in silico nanodissection cutoff for the top 136 genes. Nanodissection successfully separates (area under the curve [AUC] 0.83) podocyte-specific genes (green) from genes specific to other renal cell lineages (glomerular endothelial in dark blue, glomerular mesangial in light blue, and tubular in red).
The applicability of our nanodissection strategy is not limited to the podocyte: It can accurately separate genes from tissues as diverse as skin (skin fibroblast genes from melanocyte and keratinocyte genes) and neuronal tissue (astrocyte genes from other glial cell–specific genes) from publicly available expression data for the corresponding tissue homogenates (Supplemental Figs. 2, 3).
Confirmation of the specificity of in silico podocyte predictions
In addition to the systematic evaluation by IHC staining (below), we found nanodissection-identified genes that were previously reported to have podocyte-specific expression patterns, such as PLA2R1 (Beck et al. 2009) and GJA1 (Supplemental Tables 3, 4; Yaoita et al. 2002; Sawai et al. 2006), but that were withheld during the expert curation. In addition to recapitulating past literature, this strategy predicted concurrent discoveries. While this manuscript was being prepared, two genes predicted by nanodissection, myosin IE (MYO1E) and PDZ and LIM domain 2 (PDLIM2), were shown to display podocyte-specific expression and play a role in renal function and hereditary and acquired glomerular disease (Mele et al. 2011; Sistani et al. 2011). MYO1E mutations were shown to cause childhood-onset, glucocorticoid-resistant focal segmental glomerulosclerosis (FSGS) (Ingelfinger 2011; Mele et al. 2011), and PDLIM2 exhibited a reduced expression in patients with minimal change disease (MCD) and membranous nephropathy (MN) (Sistani et al. 2011).
We used high-throughput IHC stainings from the Human Protein Atlas (HPA) (http://www.proteinatlas.org) to systematically validate the podocyte-specific expression of genes identified by nanodissection. Although genes with staining data available in the HPA were not annotated to the level of cell-lineage localization, intraglomerular cell types were identified based on their localization pattern inside of the glomerular tuft by three investigators with expertise in renal histopathology independently in a blinded manner (see Methods). This enabled us to systematically evaluate our predictions by IHC and to compare the performance of in silico nanodissection with experimental predictions from in vivo fluorescence-tagged murine podocytes (Fig. 3).
Figure 3.

Evaluation of podocyte-specific genes based on qualified Human Protein Atlas (HPA) staining images. (A) HPA images demonstrate podocyte-specific pattern of positive standard markers and predicted genes. Staining pattern of positive standard markers: podocyte-specific in kidney (I): nephrin (NPHS1) and podocyte-specific in glomerulus: (II) SYNPO and (III) CD2AP. Exemplary staining patterns for de novo nanodissection predicted proteins (IV–IX): (IV) FGF1; (V) ARHGAP28; (VI) PRKAR2B; (VII) PCOLCE2; (VIII) GJA1; and (IX) ZDHHC6. (B) HPA-based distribution intrarenal protein staining pattern in random gene set, nanodissection-identified gene set, and the murine experimental approach-derived gene set: The in silico nanodissection approach (65%) significantly outperforms a random set of genes (12%) and the ex vivo murine experimental approach (23%) for identifying podocyte-specific genes. Gray bars show the proportion of genes with exclusively podoctye-specific staining within the kidney, and black bars show the proportion of genes with exclusively podocyte-specific staining within the glomerulus.
As the first step of the validation strategy, a blinded evaluation of the localization of predicted podocyte proteins in IHC staining images from HPA was performed for predicted podocyte-specific transcripts and an equivalently sized set of randomly selected genes (included as control). Of the predicted 136 podocyte-specific proteins in HPA, 31 were found to have sufficient expression pattern for evaluation at the time of our study (using HPA version 7.0-2010.11.15). Of these 31 proteins, 20 (65%) were found to be podocyte-specific within the renal context, with staining for seven (23%) exclusively attributed to the podocyte within kidney tissue and an additional 13 proteins (42%) stained exclusively to the podocyte within the glomerulus. The other 11 proteins (35%) were stained to other renal cells (Fig. 3B; Supplemental Table 4). The nanodissection-identified gene set significantly (Fisher's exact p = 3.256 × 10−6) outperformed the control (background) genes, of which one (2%) showed podocyte-specific staining within the kidney and five (10%) showed podocyte-specific staining within the glomerulus (Fig. 3B). Forty-two (88%) background genes were stained to other renal cells.
In contrast to humans, where podocyte-specific microdissection is technically infeasible, murine transgenic model systems have been developed by the GenitoUrinary Development Molecular Anatomy Project (GUDMAP) consortium specifically to define cell-lineage–specific genes (McMahon et al. 2008). Lineage tracing was established by GFP expression using cell-type–specific promoters, followed by FACS and genome-wide expression profiling of GFP-positive single-cell suspension (Brunskill et al. 2011). Using the podocyte, mesangial, and endothelial cell-lineage–specific GUDMAP expression data sets (Supplemental Table 5), we subtracted endothelial and mesangial gene expression profiles from the transcriptome obtained from the podocyte preparation and identified 102 podocyte-specific transcripts (Methods; Supplemental Table 6; McMahon et al. 2008). These transcripts underwent the same cell-lineage–specific evaluation in HPA as the in silico nanodissected human transcripts. Thirty of the 102 murine experimental approach–derived podocyte-specific transcripts had staining patterns identifiable in HPA, of which staining for two was exclusively attributed to the podocyte within human kidney tissue and five proteins were stained to the podocyte within the glomerulus (Fig. 3B). Thus the in silico nanodissection approach exhibited a significantly higher accuracy (Fisher's exact p = 0.0059) than the murine experimental strategy for discovering transcripts with podocyte-specific expression (nanodissection's 65% vs. murine experimental approach's 23% of predictions confirmed as podocyte-specific). The in silico prediction accuracy of cell-lineage enrichment using human tissue homogenate exceeded that obtained from in vivo fluorescence-tagged and sorted cells in a murine model system.
Disease-specific regulation of the nanodissection gene sets
To test the hypothesis that the discovered podocyte-specific genes were associated with human renal disease, the transcript with the highest podocyte-specific score and positive HPA validation, procollagen C-endopeptidase enhancer 2 (PCOLCE2), was selected for further characterization. PCOLCE2 protein modulates binding of procollagen C-proteases to collagen in a BMP1-dependent and cell-lineage–restricted manner (Steiglitz et al. 2002), a process with significant relevance for the development and function of the glomerular basement membrane (Tanaka et al. 2010). We investigated the disease-specific transcriptional regulation of PCOLCE2. The steady-state mRNA level of PCOLCE2 in glomeruli from human renal biopsies was significantly repressed in patients with FSGS (n = 17), a glomerular disease with podocyte damage and end-stage renal disease (ESRD), compared with controls (n = 39, p < 0.05) (Fig. 4A). In contrast, in glomeruli from patients with MCD (n = 12), a proteinuric disease without progression to ESRD, PCOLCE2 transcript levels were not significantly altered. In a cohort of CKD patients with heterogeneous glomerular pathophysiology (n = 139), loss of PCOLCE2 glomerular gene expression was significantly correlated with loss of renal function (r = 0.32, p = 1.17 × 10−4).
Figure 4.

Regulation of predicted podocyte-specific gene set in human disease. (A) Box-and-whisker plot of glomerular mRNA expression of PCOLCE2 in biopsies from living donor controls (LD, n = 35), minimal change disease (MCD) patients (n = 12), and focal segmental glomerulosclerosis (FSGS) patients (n = 19). Asterisk denotes a significant differential expression (p < 0.05). (B) IHC staining of PCOLCE2 on kidney biopsies from controls (I) and FSGS patients (II). In comparison with control kidneys, PCOLCE2 signal disappears in FSGS patients. Images shown are the representative images in the glomerulus of controls (n = 5) and FSGS patients (n = 8). (C) Density plot of the association (Pearson correlation, x-axis) of the 136 predicted podocyte-specific genes (red) with renal function as quantified by GFR value, compared with density plot of repeatedly (100 times) randomized gene expression–GFR associations (black). The randomized set shows a distribution centered on zero (meaning no correlation with GFR), whereas the podocyte-specific genes show a skewed distribution toward positive correlation, indicating reduced gene expression is associated with impaired renal function. Correlation with GFR of the 136 transcripts across all renal diseases analyzed was significantly enriched compared with the permuted sample (p < 0.01). Black line indicates the correlation of PCOLCE2 mRNA level with GFR.
Disease-specific PCOLCE2 regulation was further validated in human kidneys affected by glomerular disease using IHC staining in an independent biopsy cohort. In concordance to the IHC staining patterns reported in HPA (Fig. 3A, VII), the podocyte-specific localization of PCOLCE2 protein was confirmed. In contrast to the nuclear and perinuclear PCOLCE2 signal seen in IHC in glomeruli of five healthy kidneys (Fig. 4B, I), PCOLCE2 staining was not detectable in glomeruli from eight FSGS patients (Fig. 4B, II), demonstrating the ability of the nanodissection strategy to detect genes with both cell-lineage–specific expression and disease-specific alteration in glomerular failure.
Glomerular disease stratification by the de novo predicted podocyte-specific transcripts
Podocyte damage leads to progressive loss of kidney function and the need for dialysis and renal transplantation. To test the association with glomerular function, the glomerular regulation of the podocyte gene set discovered by nanodissection was compared between 39 controls and 17 patients with FSGS, a renal disease caused by severe podocyte damage (Kriz et al. 1994; Pavenstädt 2000) and the leading cause of glomerular failure in children. Using significance analysis of microarrays (SAM) (Tusher et al. 2001), 60 of the 136 genes identified by nanodissection were significantly repressed in glomeruli from FSGS patients versus controls (q-value < 0.05).
Next, the regulation of predicted transcripts in chronic renal disease was evaluated in a cohort of 139 patients with glomerular diseases, including FSGS, diabetic nephropathy (DN), IgA nephropathy (IgAN), MN, lupus nephritis (LN), and MCD (Supplemental Table 7). Steady-state mRNA expression measurements of nanodissection predicted podocyte-specific transcripts were correlated with GFR at the time of biopsy, currently the best overall index of kidney function used to classify the stages of CKD patients by the Kidney Disease Outcomes Quality Initiative (KDOQI). Expression of the set of 136 de novo predicted podocyte-specific genes was significantly (p < 0.01) more correlated with GFR than observed in permuted gene–GFR associations (Fig. 4C). These results demonstrate the potential for predicted podocyte-specific genes to be candidate markers for disease progression. This finding has significant clinical utility, as the cell-lineage–specific and disease-associated genes can provide superior specificity for biomarker testing in heterogeneous biofluids like urine or blood compared with ubiquitously expressed disease markers. The GFR correlation of the podocyte-specific gene set predicted by nanodissection supports the tight link of podocyte differentiation and function with renal impairment irrespective of initiation of renal disease by genetic or environmental causes.
Application of nanodissection to nonpodocyte lineages
Mesangial cells are one of the three major cell types in kidney glomeruli, and mesangial expansion is a hallmark of DN. We investigated the expression profile of the top 52 mesangial cell–specific genes predicted by nanodissection (cutoff based on the same F-measure criterion) (Supplemental Fig. 6) in an independent DN data set (data include glomerular gene expression profile of 13 healthy donors and nine patients with DN) (Woroniecka et al. 2011). In this data set, 50 of the 52 predicted mesangial cell–specific genes showed robust expression in the microdissected glomeruli. Forty-four percent of these genes (22 out of 50) exhibited increased steady-state mRNA levels in patients with DN compared with controls (SAM analysis q < 0.01); no transcript showed significantly reduced mRNA levels. This demonstrates both nanodissection's applicability to other renal cell lineages, as well as its ability to identify lineage-specific genes with increased mRNA levels.
We further evaluated nanodissection on nonrenal cell lineages. For this analysis, we used tissue annotations from the Human Protein Reference Database (HPRD) (Keshava Prasad et al. 2009) as gold standards for tissue-specific expression. For example, we have applied nanodissection to identify genes specifically expressed in skin fibroblasts (density estimate cross-validation–based evaluation) (Supplemental Fig. 3). We evaluated genes above the maximum F-measure criterion for significant enrichment in disease annotations from Online Mendelian Inheritance in Man (OMIM). The genes above the maximum F-measure criterion showed significant enrichment of genes involved in two collagen disorders with known fibroblast expression: Ehlers-Danlos syndrome and Osteogenesis imperfecta. Six of the 10 genes associated with Ehlers-Danlos syndrome were above this threshold (false discovery rate [FDR]-corrected; q = 0.00038), as were six of the eight genes associated with Osteogenesis imperfecta (FDR-corrected; q = 0.00011). None of these genes were included in the fibroblast gold standard from HPRD used by nanodissection; all were new predictions of fibroblast-specific genes. This demonstrates both the potential of nanodissection to identify cell-lineage–specific genes as well as the potential for those genes to be associated with cell-lineage–specific diseases.
Discussion
Organ-specific transcriptional programs define the final stages in tissue development and the mature function of metazoan organisms. Alterations in the functions of genes with cell-lineage–restricted expression patterns are widely believed to lead to tissue-specific disease manifestations. Furthermore, inherited diseases are frequently caused by mutations in genes with restricted expression patterns (Winter et al. 2004; D'Agati 2008; Cai and Petrov 2010). Mutations in such genes often do not cause early embryonic lethality but rather manifest disease at the time when the function of these genes becomes critical for a specific tissue and subsequently for organismal survival (D'Agati 2008). In acquired disease like diabetes or hypertension, the vulnerability of a specific organ to the systemic disease is defined by the expression of tissue-specific genes (Doublier et al. 2003; Koop et al. 2003; Woroniecka et al. 2011). Defining cell-lineage–specific transcripts therefore has immediate clinical implications for such cell lineages. However, a major challenge to define a specific cellular transcriptome has been the inability to obtain pure cell preparation from human tissue in vivo (e.g., as recently summarized by Lindenmeyer et al. [2010] for renal cell lineages).
To identify cell-lineage–specific transcripts on a genome-wide scale even when direct experimental assays are infeasible, as is the case for most solid human tissues, we developed in silico nanodissection. This iterative machine-learning–based approach robustly leverages existing knowledge about the cell lineage of interest to identify transcripts with similar behavior in heterogeneous transcriptional data sets of tissue homogenates. In silico nanodissection does not require expression data of pure genome-wide profiles from the cell lineage of interest and is robust to small numbers and varying specificity of available cell-lineage markers. Although our strategy uses support vector machines (SVM) as the machine learning component of the nanodissection method (Fig. 1), in principal any machine learning approach that leverages positive and negative examples for training can be integrated in place of the SVM.
This study represents the first high-throughput approach for identification of cell-lineage–specific genes for any cell lineage from in vivo human data. The approach is general—we found that our predictions remained robust (significantly overrepresented by podocyte-specific genes) even when the directly targeted expression data (renal glomeruli) constitutes only 5% of the total data sets (the rest being diverse human expression data from the Gene Expression Omnibus [GEO]). In silico nanodissection applied to public gene expression data from tissue homogenates (implementation available through our nano.princeton.edu website) is also capable of accurately separating cell-lineage–specific genes in skin (skin fibroblast genes from melanocyte and keratinocyte genes) and neuronal tissue (astrocyte genes from other glial cell–specific genes). Furthermore, this approach is effective even with a limited number of positive marker genes or in situations when some provided lineage-specific genes are inaccurate/irrelevant to the cell lineage (Supplemental Figs. 4, 5). This makes in silico nanodissection a promising approach to identify cell-lineage–specific genes that might be potentially associated with other acquired or inherited diseases, for which targeted data may not be available.
Cell-type–specific markers predicted by nanodissection could be used to extend the applicability of methods that define the fractional composition of a mixture. These methods are then capable of deconvoluting an expression signal to perform tests of gene expression within individual lineages. While some approaches require knowledge of the mixture's fractional composition, which limits their application to cell types measurable in cell sorter experiments (Shen-Orr et al. 2010), others can start with either pure ex vivo samples of each mixture component to estimate the sample composition or a high quality set of known markers (Kuhn et al. 2011). These requirements currently limit the applicability of these methods, as they require information not available in studies of most complex solid human tissues. Markers identified by nanodissection provide a promising starting point for the application of such deconvolution approaches to many more human cell lineages, including those not amenable to experimental microdissections.
Nanodissection is the first genome-scale method that identifies cell-lineage–specific genes important for renal disease in humans. In addition to the de novo identified podocyte-specific genes, genes that have been shown by genetic or functional studies to be causally involved in hereditary glomerular diseases and chronic progressive renal failure, including DACH1 (nanodissection rank 184) (Köttgen et al. 2010), APOL1 (rank 185) (Genovese et al. 2010; Kopp et al. 2011), VEGFA (rank 247) (Eremina et al. 2008; Köttgen et al. 2010), and MYH9 (rank 260) (Kao et al. 2008; Kopp et al. 2008), were ranked highly by our method. During the preparation of this manuscript, MYO1E (rank 75) was reported to be associated with autosomal-recessive, glucocorticoid-resistant nephrotic syndrome (Mele et al. 2011). MYO1E was found to exhibit, as predicted by our study, podocyte-specific expression and appears to interact with other cell-lineage–specific genes in podocyte cytoskeletal dynamics. PDLIM2 (rank 121), another gene identified by nanodissection, was recently reported to show podocyte-specific expression and repression in acquired glomerular disease (Mele et al. 2011; Sistani et al. 2011). Interestingly, neither of these genes is present in the list of 102 genes (Supplemental Table 6) identified as podocyte-specific using the murine ex vivo cell-lineage separation in the GUDMAP data sets. Brunskill et al. (2011) recently generated a transcriptional data set (144 genes) regulated during murine podocyte development and enriched in adult podocytes in comparison with the renal cortex. Analysis in HPA of the human orthologs exhibited a similar enrichment to the GUDMAP ex vivo cell-lineage data set used in Figure 3 (two podocyte-specific in kidney [5%] and 13 podocyte-specific in glomeruli [30%]), but the murine data did not reach the specificity of the in silico nanodissection approach (65%).
Cell-lineage–specific strategies capable of identifying genes associated with a disease provide additional value compared with unbiased genome-wide approaches that identify genes with expression correlation to a specific phenotype (like GFR). The latter captures a different pool of transcripts: Abundantly expressed non-cell-lineage–specific genes constitute the majority of the transcripts correlated with renal function, but these transcripts do not necessarily perform cell-lineage–specific functions and may not be associated with hereditary disease. For example, expression levels of MYO1E and APOL1 are not strongly correlated to GFR (ranked 1236 and 7430, respectively, by GFR-expression correlation), yet these two genes were identified by nanodissection to be cell-type–specific and have been shown experimentally (independent of and parallel to our work) to cause hereditary glomerular disease (Genovese et al. 2010; Kopp et al. 2011; Mele et al. 2011) Furthermore, a systematic analysis focusing on literature-curated hereditary FSGS-associated genes that do not overlap with our podocyte gold standard (see Supplemental Methods) also demonstrates that a genome-wide assessment of GFR-transcript expression correlation alone could not identify genes associated with this hereditary renal disease (Supplemental Fig. 8A). In contrast, nanodissection can identify genes associated with disease, with FSGS genes receiving significantly higher podocyte nanodissection scores than those without known FSGS association (Supplemental Fig. 8B). Thus, nanodissection's ability to identify cell-lineage specificity is important for identifying genes potentially associated with such diseases and clinical phenotypes. Beyond simply addressing issues of statistical power, methods that consider cell-lineage specificity provide additional utility because they address targeted biological questions that are tightly coupled to the disease etiology.
Our findings have significant potential for clinical utility. In the study of hereditary diseases, next-generation exome sequencing technologies are now widely applied across hereditary diseases and are capable of identifying putative causal genetic variants in very small pedigrees. However, these studies often result in multiple candidate genes in need of further prioritization. As hereditary diseases are often caused by cell-lineage–specific transcripts (see above and Hinkes et al. 2006), the systematic scoring system for cell-lineage–specific enrichment provided by the nanodissection approach can become a crucial tool to prioritize candidate genes for further validation using their cell-lineage enrichment scores. Vice versa, several hundreds of tissue specific genes identified by nanodissection can be screened comprehensively in families with a hereditary disease of the organ of interest using targeted exon sequencing strategies as currently is pursued by our group in a rare disease cohort (Halbritter et al. 2012; Gadegbeku et al. 2013).
For acquired chronic disease, the search is still ongoing to define specific and robust biomarkers of differentiated organ function. Unbiased molecular screening approaches have been largely disappointing in this context. Our data strongly support the close association of cell-lineage–specific transcripts and loss of end organ function in complex, chronic human kidney disease. Proteins encoded by these genes may be detected in plasma and urine and provide a noninvasive means to measure organ function in a cell-lineage–specific manner. In contrast to ubiquitously expressed molecules involved in fibrosis and inflammation, which are currently the most common source of candidate biomarkers for chronic diseases, cell-lineage–specific biomarkers are less likely to be confounded by extrarenal processes and should provide superior diagnostic specificity (Fukuda et al. 2012). This has been demonstrated in the context of podocyte failure in model systems and human disease (Sato et al. 2009), and nanodissection provides an opportunity to expand the scope of podocyte-specific transcripts analyzed in a complex mixture of urinary cells by these approaches. Finally, functional studies of the cell-lineage–specific genes identified in human disease tissue offer the opportunity to develop a targeted therapeutic approach for chronic disease. Targeting a disease-specific molecular mechanism selectively in the tissue manifesting the disease has the potential to significantly increase efficacy while reducing off-target effects.
In summary, nanodissection is a novel computational approach for defining the specificity of cell types at the transcriptional level. As demonstrated for glomerular disease, but applicable across all organs with large-scale transcriptional data sets available, nanodissection can reveal novel transcripts with essential tissue-specific function in organogenesis and hereditary human disease. In chronic progressive diseases, the nanodissection-identified transcripts can serve as highly specific markers of disease stages and provide a starting point for the development of organ-specific targeted therapies. While we have shown that HPRD annotations can guide successful nanodissection analyses, we believe the method is most powerful when combined with high-quality user constructed standards, which can be easily accomplished using our nanodissection web server. Nanodissection can be performed on user-curated, tissue-specific gene expression compendia via the user-friendly nanodissection web server at http://nano.princeton.edu. This web server includes 452 microarrays from microdissected kidney biopsy samples from this study, as well as 7539 samples across 28 diverse human tissue collections manually curated from the Gene Expression Omnibus (GEO). Nanodissection of investigator-specific, gene-expression data sets can be performed with the Sleipnir library for functional genomics (version 3 or higher) available for Windows, OS X, and Linux systems from http://libsleipnir.bitbucket.org/ (Huttenhower et al. 2008).
Methods
Patient characteristics
Human renal biopsy specimens were procured through an international multicenter study, the European Renal cDNA Bank-Kroener-Fresenius biopsy bank. Biopsies were obtained from patients after informed consent and with approval of the local ethics committees. All biopsies were stratified by the reference pathologist of the ERCB according to their histological diagnoses. Histology reports, clinical data, and gene expression information were stored in a de-identified manner. A total of 452 microarrays from kidney biopsies were used for nanodissection, of which 139 patients were used for kidney function correlation analysis. Demographic data of these 139 patients are provided in the Supplemental Table 7.
Microdissected human kidney biopsy data
Microdissection into glomerular and tubule-interstitial compartments and Affymetrix-based gene expression profiling were performed according to the method previously reported (Ju et al. 2009). Affymetrix GeneChip Human Genome U133A 2.0 and U133 Plus 2.0 Array were used in this study. For this analysis, we restricted ourselves to only the probesets present on both platforms. Normalized data files are uploaded on the GEO (Edgar et al. 2002) website and accessible under reference numbers GSE32591 (Berthier et al. 2012), GSE35488 (Reich et al. 2010), GSE37455 (Berthier et al. 2012), and GSE37460 (Berthier et al. 2012). For simplicity, we use “in vivo” to refer to these assays of genes measuring gene expression in human biopsies of complex tissues.
In silico nanodissection for the prediction of cell-lineage–specific gene expression
Our approach uses machine learning within a novel iterative framework to predict genes with cell-lineage–specific expression on the whole-genome scale based on gene expression data from tissue homogenates. This problem is especially challenging because, in order to work for cell lineages that are infeasible to microdissect experimentally such as the podocytes, our approach must function without example expression profiles of the lineage of interest.
Intuitively, our method leverages patterns of expression of cell-lineage–specific genes that it discovers from whole-genome expression compendia not resolved to the cell lineage of interest. These patterns are specific for each cell lineage and generally only found in a small subset of experimental conditions, which may include genetic, physiological, pathophysiological, environmental, or experimental states/perturbation (e.g., biopsy specimens from different patients). To discover these cell-lineage–specific expression patterns as well as the subsets of conditions that are informative for a given cell lineage, our approach uses a machine learning approach in an iterative probabilistic framework to combine an expert-provided standard of known cell-lineage–specific genes (positives) as well as example genes that are expressed in other cell lineages (negatives). However, most solid-tissue cell lineages cannot be studied experimentally in high-throughput, and thus only few cell-lineage–specific genes are often known with high accuracy (e.g., from IHC). The additional challenge here is that these standards are often limited in size (especially for cell lineages not amenable to experimental micro dissection) and can be of varying specificity (e.g., specific to cell lineage within the immediate structure or whole organ or defined by different experimental approaches).
Because it is experimentally infeasible to obtain pure example expression profiles for cell lineages from solid human tissues, our method must perform well even while available standards are often very limited in size and can be of highly varying specificity. This paucity of high-quality standards and the need to effectively leverage lower-quality or less specific examples severely limits the direct application of traditional machine learning approaches (e.g., SVM performance outside of the iterative framework is shown in Supplemental Fig. 5).
To address these challenges, we developed an iterative classification approach that continually refines both the predictive cell-lineage–specific patterns and informative conditions based on statistical scoring and refinement (through informative subset selection) of the provided standard. This iterative approach allows the user to provide tiered standards, i.e., the investigator identifies only the relative specificity of evidence tiers (i.e., low-throughput high specificity approaches are more reliable as compared to high-throughput experimental platforms with lower specificity). The in silico nanodissection method is then able to make high-accuracy predictions of cell-lineage–specific genes on the whole-genome scale and, within the tiered standard constraint, is robust to variable specificity of example cell-lineage–specific genes. The iterative strategy is necessary to allow investigators to add standards of questionable quality without dramatically compromising the quality of cell-lineage predictions. A linear SVM without this iterative approach fails when standards of lower quality are added to high-quality standards (Supplemental Fig. 5).
The researcher defines standards within tiers. Tiers represent levels of specificity (i.e., in descending order: double immunofluorescence, annotated in literature curated database, high-throughput protein expression). For each tier, nanodissection calculates the sum of the ranks of genes from the classifier (for the case of SVM, this is the ranked distance from the SVM hyperplane) for each positive example, 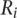 , (here podocyte genes) against each of
, (here podocyte genes) against each of 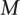 negative standards,
negative standards, 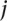 , (e.g., glomerular, mesangial, tubular) as
, (e.g., glomerular, mesangial, tubular) as 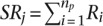 , where
, where 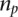 represented the number of positives and ranks were calculated from only the positive examples and the negative examples from standard
represented the number of positives and ranks were calculated from only the positive examples and the negative examples from standard 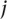 . It then computes a test statistic for this individual separation,
. It then computes a test statistic for this individual separation, 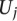 for each negative standard as
for each negative standard as 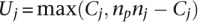 , where
, where
This is normalized by converting it to a z-score by using the mean and standard deviation through
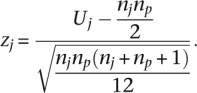 |
The scores for the individual separations are then combined to provide a final score for this tier of standards
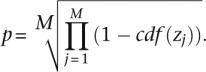 |
Nanodissection automatically selects the standards resulting in the lowest 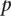 (which ranges from zero to one), i.e., that which corresponds to a better separation of positives from each negative standard.
(which ranges from zero to one), i.e., that which corresponds to a better separation of positives from each negative standard.
In certain cases, an additional (and optional) external validation gene set may be available. Because nanodissection can be applied where experimental microdissection was insufficient, these standards may represent both positives and negatives (e.g., in this case where additional microarray measurements of the renal glomerulus were available as validation). We termed genes in this standard as “high-throughput-validating” genes and other genes as “nonvalidating” genes. Nanodissection can use this validation set to identify the set of standards providing the best separation of validating genes by calculating 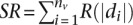 , where
, where 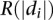 is the rank of the absolute value of the distance to the hyperplane of the validating gene
is the rank of the absolute value of the distance to the hyperplane of the validating gene 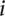 in a list containing the
in a list containing the 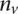 validating genes and the
validating genes and the 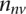 nonvalidating genes. It then calculates
nonvalidating genes. It then calculates 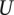 as
as 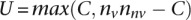 , where
, where
which is then converted to a z-score
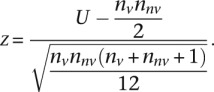 |
Finally, 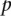 for validating versus nonvalidating is calculated as
for validating versus nonvalidating is calculated as 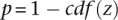 . Selecting the standard tier that provided the lowest p results in the standard where validating genes were most extreme (i.e., best separated from each other). Our results demonstrate that this approach enables us to use a non-cell-lineage–specific validation (i.e., glomerular) gene set to grade our separation of putative cell-lineage (podocyte) –specific genes by selecting that standard that leads to example genes on the extremes (in our example, this has potential podocytes at the top of the list and potential nonpodocyte glomerular genes at the bottom). In the case where there exists a validation standard of high-quality specific to our cell lineage of interest, we instead use
. Selecting the standard tier that provided the lowest p results in the standard where validating genes were most extreme (i.e., best separated from each other). Our results demonstrate that this approach enables us to use a non-cell-lineage–specific validation (i.e., glomerular) gene set to grade our separation of putative cell-lineage (podocyte) –specific genes by selecting that standard that leads to example genes on the extremes (in our example, this has potential podocytes at the top of the list and potential nonpodocyte glomerular genes at the bottom). In the case where there exists a validation standard of high-quality specific to our cell lineage of interest, we instead use 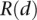 directly instead of
directly instead of 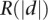 . In that case, this value would represent the one-sided Wilcoxon rank-sum p-value for a comparison of validating and nonvalidating genes. Because this iterative nanodissection approach relies on genome-scale data obtained from the surrounding compartment and because this evaluation was used to identify the optimum standards, this
. In that case, this value would represent the one-sided Wilcoxon rank-sum p-value for a comparison of validating and nonvalidating genes. Because this iterative nanodissection approach relies on genome-scale data obtained from the surrounding compartment and because this evaluation was used to identify the optimum standards, this 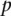 provides a quality measure for the resulting standard. Thus nanodissection allows us to obtain cell-lineage–specific signal from in vivo human data.
provides a quality measure for the resulting standard. Thus nanodissection allows us to obtain cell-lineage–specific signal from in vivo human data.
The nanodissection algorithm therefore proceeds as follows (for pseudocode, see Supplemental Fig. 7 ). Given user-supplied standards in tiers of increasing specificity, for each standard-level, k, combine standards of that level with all standards of higher specificity levels. Apply the selected classification algorithm (here we applied SVM from the SVMperf package [Joachims 2006] using the Sleipnir library [Huttenhower et al. 2008]) and generate a ranked list of predictions. Score the predictions for k as described above to calculate p for the kth level of specificity. Select the level of specificity providing the lowest p.
In this work, standards were obtained from expert literature review. The positive podocyte-specific standard genes were required to have at least one of the following levels of evidence: immunofluorescence staining, in situ hybridization, or electron microscopy image of immuno-gold staining of podocytes in vivo. Two levels of specificity were evaluated. The most stringent level contained genes specifically expressed only in podocytes and no other cell types in the human kidney, referred to as podocyte-specific in kidney (as an example, see nephrin staining pattern in Fig. 3A, I). The less stringent level contained all of the above, as well as genes expressed in podocytes and no other cell types in glomeruli, but did contain genes detected in extraglomerular cells of the kidney (synaptopodin [SYNPO] and CD2AP staining in Fig. 3A, II and III). For the majority of selected genes, evidence for disease association in human glomerular failure or murine model systems was also available. Application of nanodissection resulted in the use of both tiers of standards, which corresponded to a total of 46 genes that were both podocyte-specific and present in the gene expression data set.
Gene expression data extraction from the GUDMAP and data processing
The genome-wide expression data of murine podocytes, mesangial, and endothelial cells were obtained from http://www.gudmap.org/. The identifications of data sets that have been utilized in our study are listed in Supplemental Table 5. The detailed protocol of data generation is described in a recently published paper by Brunskill et al. (2011) and can also be found in our Supplemental Methods. We preprocessed and normalized data as described in Microdissected Human Kidney Data. By comparing the expression level in podocytes versus the other two major cell types in glomeruli, we define a gene to be podocyte-specific if its expression in podocytes is 4.76-fold over mesangial cells and 4.65-fold over glomerular capillary endothelial cells. The cut-off values represent three standard deviations of the average difference between podocytes and mesangial/endothelial cell transcripts, respectively. By use of HomoloGene from NCBI Entrez (Maglott et al. 2011), 102 murine genes could be mapped to their Homo sapiens ortholog (Supplemental Table 6).
Evaluation of intrarenal protein localization in HPA
We evaluated the intrarenal localization pattern of protein products of predicted genes based on HPA 7.0-2010.11.15. Intraglomerular cell types are identified based on their localization pattern inside of the glomerular tuft by three investigators with expertise in renal histopathology independently in a blinded manner. Conflicts were resolved by a majority vote. The following staining patterns were considered inconclusive and excluded from the analysis: (1) proteins with negative staining or “data not available”; (2) proteins with only a single renal histology image available; and (3) proteins with a diffuse nonspecific staining pattern. If several antibodies were evaluated for a specific protein, the images from the antibody with the highest degree of specificity were used for evaluation. Tubular brush border staining was considered unspecific. In general, protein localization patterns were classified into three groups: (1) expressed exclusively in podocytes with no other cell types exhibiting staining in kidney section, referred to as “podocyte-specific in kidney” (i.e., nephrin [NPHS1] in Fig. 3A, I); (2) expressed specifically in podocytes within the glomerulus but with positive staining also observed in tubular-interstitial compartments, referred to as “podocyte-specific in glomerulus” (i.e., SYNPO and CD2-associated protein [CD2AP] in Fig. 3A, II and III, respectively); and (3) all remaining staining patterns as “other renal cell.” For clarity, we refer to category 1 and 2 in aggregate as “podocyte-specific” genes.
IHC staining of kidney biopsy tissues
Following a previously described protocol (Lorz et al. 2008), IHC studies were performed using a PCOLCE2 primary rabbit antibody (HPA013203, Sigma-Aldrich).
Association of RNA expression of cell-lineage–specific genes to kidney function
The expression of the top 136 candidate genes in a cohort of 139 patients with CKD was correlated to the square root of the GFR, calculated by MDRD equation (modified diet in renal disease formula) (Levey et al. 2006), using Pearson correlation. The correlations were compared against a randomized set by analyzing their correlation density plots using the sm R package (Bowman and Azzalini 2010). Randomization was performed by randomly reassigning expression values to GFR 100 times on the given data set, followed by recalculation of the correlation.
Manual tissue-of-origin sample annotation for the web server
Microarray experiments (Affymetrix GeneChip Human Genome U133 Plus 2.0) were manually annotated to the sample's tissue of origin using the controlled vocabulary in the Brenda Tissue Ontology. To ensure wide coverage of tissue types, a broad set of candidate samples for each tissue was identified with an initial term matching, corrected for linguistic variations with stemming, for each Brenda term (and its synonyms) on sample descriptions available in GEO. These term-to-experiment matches were then manually curated, verified, or corrected based on the corresponding sample descriptions. Only matches for terms across at least two independent data sets were reviewed. Only the tissue-of-origin information was considered in the manual evaluation, and so, tumor-adjacent normal breast biopsy samples were correctly annotated to “breast,” for example. We excluded tissue mixture samples, reference samples, and nonhuman samples. We also excluded samples with ambiguous descriptions, as well as cell line and cancer terms. Samples annotated to detailed terms in the controlled vocabulary were propagated up to organ-level annotations, based in the organ of origin. Terms with fewer than 10 annotated samples after propagation were excluded at this stage. This procedure resulted in a manually annotated compendium of 7539 samples from 28 tissues that we make available through the nanodissection web server for nanodissection analysis.
Data access
Normalized gene expression data files of microdissected human kidney biopsies have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/; Edgar et al. 2002) under accession number GSE47185.
Acknowledgments
We thank the members of the European Renal cDNA Bank-Kroener-Fresenius biopsy bank at the time of the expression profiling: Clemens David Cohen, Holger Schmid, Michael Fischereder, Lutz Weber, Matthias Kretzler, Detlef Schlöndorff (Munich/Zurich/AnnArbor/New York); Jean Daniel Sraer, Pierre Ronco (Paris); Maria Pia Rastaldi, Giuseppe D'Amico (Milano); Peter Doran, Hugh Brady (Dublin); Detlev Mönks, Christoph Wanner, (Würzburg); Andrew Rees (Aberdeen and Vienna); Frank Strutz, Gerhard Anton Müller (Göttingen); Peter Mertens, Jürgen Floege (Aachen); Norbert Braun, Teut Risler (Tübingen); Loreto Gesualdo, Francesco Paolo Schena (Bari); Jens Gerth, Gunter Wolf (Jena); Rainer Oberbauer, Dontscho Kerjaschki (Vienna); Bernhard Banas, Bernhard Krämer (Regensburg); Moin Saleem (Bristol); Rudolf Wüthrich (Zurich); Walter Samtleben (Munich); Harm Peters, Hans-Hellmut Neumayer (Berlin); Mohamed Daha (Leiden); Katrin Ivens, Bernd Grabensee (Düsseldorf); Francisco Mampaso (Madrid); Jun Oh, Franz Schaefer, Martin Zeier, Hermann-Joseph Gröne (Heidelberg); Peter Gross (Dresden); Giancarlo Tonolo (Sassari); Vladimir Tesar (Prague); Harald Rupprecht (Bayreuth); Hermann Pavenstädt (Münster); Hans-Peter Marti (Bern); and Peter Mertens (Magdeburg). This work was supported in part by R01 GM071966 to O.G.T. and M.K., by RO1 HG005998 and DBI0546275 to O.G.T., by P50 GM071508, and by R01 DK079912 and P30 DK081943 to M.K. O.G.T. is a Senior Fellow of the Canadian Institute for Advanced Research.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.155697.113.
References
- Beck LH, Bonegio RGB, Lambeau G, Beck DM, Powell DW, Cummins TD, Klein JB, Salant DJ 2009. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med 361: 11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MR, Czech KA, Arend LJ, Witte DP, Devarajan P, Potter SS 2007. Laser capture microdissection-microarray analysis of focal segmental glomerulosclerosis glomeruli. Nephron, Exp Nephrol 107: e30–e40 [DOI] [PubMed] [Google Scholar]
- Berthier CC, Bethunaickan R, Gonzalez-Rivera T, Nair V, Ramanujam M, Zhang W, Bottinger EP, Segerer S, Lindenmeyer M, Cohen CD, et al. 2012. Cross-species transcriptional network analysis defines shared inflammatory responses in murine and human lupus nephritis. J Immunol 189: 988–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman AW, Azzalini A 2010. R package sm: nonparametric smoothing methods (version 2.2-4)
- Brunskill EW, Georgas K, Rumballe B, Little MH, Potter SS 2011. Defining the molecular character of the developing and adult kidney podocyte. PLoS ONE 6: e24640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai JJ, Petrov DA 2010. Relaxed purifying selection and possibly high rate of adaptation in primate lineage-specific genes. Genome Biol Evol 2: 393–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Agati VD 2008. The spectrum of focal segmental glomerulosclerosis: New insights. Curr Opin Nephrol Hypertens 17: 271–281 [DOI] [PubMed] [Google Scholar]
- D'Agati VD, Kaskel FJ, Falk RJ 2011. Focal segmental glomerulosclerosis. N Engl J Med 365: 2398–2411 [DOI] [PubMed] [Google Scholar]
- Doublier S, Salvidio G, Lupia E, Ruotsalainen V, Verzola D, Deferrari G, Camussi G 2003. Nephrin expression is reduced in human diabetic nephropathy: Evidence for a distinct role for glycated albumin and angiotensin II. Diabetes 52: 1023–1030 [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endlich N, Sunohara M, Nietfeld W, Wolski EW, Schiwek D, Kränzlin B, Gretz N, Kriz W, Eickhoff H, Endlich K 2002. Analysis of differential gene expression in stretched podocytes: Osteopontin enhances adaptation of podocytes to mechanical stress. FAESB J 16: 1850–1852 [DOI] [PubMed] [Google Scholar]
- Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, Richardson C, Kopp JB, Kabir MG, Backx PH, et al. 2008. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 358: 1129–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda A, Wickman LT, Venkatareddy MP, Wang SQ, Chowdhury MA, Wiggins JE, Shedden KA, Wiggins RC 2012. Urine podocin:nephrin mRNA ratio (PNR) as a podocyte stress biomarker. Nephrol Dial Transplant 27: 4079–4087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadegbeku CA, Gipson DS, Holzman LB, Ojo AO, Song PXK, Barisoni L, Sampson MG, Kopp JB, Lemley KV, Nelson PJ, et al. 2013. Design of the Nephrotic Syndrome Study Network (NEPTUNE) to evaluate primary glomerular nephropathy by a multidisciplinary approach. Kidney Int 83: 749–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, et al. 2010. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329: 841–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein HC 2001. Albuminuria and risk of cardiovascular events, death, and heart failure in diabetic and nondiabetic individuals. JAMA 286: 421–426 [DOI] [PubMed] [Google Scholar]
- Groop P-H, Thomas MC, Moran JL, Wadèn J, Thorn LM, Mäkinen V-P, Rosengård-Bärlund M, Saraheimo M, Hietala K, Heikkilä O, et al. 2009. The presence and severity of chronic kidney disease predicts all-cause mortality in type 1 diabetes. Diabetes 58: 1651–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbritter J, Diaz K, Chaki M, Porath JD, Tarrier B, Fu C, Innis JL, Allen SJ, Lyons RH, Stefanidis CJ, et al. 2012. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J Med Genet 49: 756–767 [DOI] [PubMed] [Google Scholar]
- Henger A, Kretzler M, Doran P, Bonrouhi M, Schmid H, Kiss E, Cohen CD, Madden S, Porubsky S, Gröne EF, et al. 2004. Gene expression fingerprints in human tubulointerstitial inflammation and fibrosis as prognostic markers of disease progression. Kidney Int 65: 904–917 [DOI] [PubMed] [Google Scholar]
- Higgins JPT, Wang L, Kambham N, Montgomery K, Mason V, Vogelmann SU, Lemley KV, Brown PO, Brooks JD, van de Rijn M 2004. Gene expression in the normal adult human kidney assessed by complementary DNA microarray. Mol Biol Cell 15: 649–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkes B, Wiggins RC, Gbadegesin R, Vlangos CN, Seelow D, Nürnberg G, Garg P, Verma R, Chaib H, Hoskins BE, et al. 2006. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet 38: 1397–1405 [DOI] [PubMed] [Google Scholar]
- Hodgin JB, Borczuk AC, Nasr SH, Markowitz GS, Nair V, Martini S, Eichinger F, Vining C, Berthier CC, Kretzler M, et al. 2010. A molecular profile of focal segmental glomerulosclerosis from formalin-fixed, paraffin-embedded tissue. Am J Pathol 177: 1674–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenhower C, Schroeder M, Chikina MD, Troyanskaya OG 2008. The Sleipnir library for computational functional genomics. Bioinformatics 24: 1559–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelfinger JR 2011. MYO1E, focal segmental glomerulosclerosis, and the cytoskeleton. N Engl J Med 365: 368–369 [DOI] [PubMed] [Google Scholar]
- Jain S, De Petris L, Hoshi M, Akilesh S, Chatterjee R, Liapis H 2011. Expression profiles of podocytes exposed to high glucose reveal new insights into early diabetic glomerulopathy. Lab Invest 91: 488–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joachims T. 2006. Training linear SVMs in linear time. In Proceedings of the 12th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining–KDD '06, p. 217. ACM Press, New York, NY. [Google Scholar]
- Ju W, Eichinger F, Bitzer M, Oh J, McWeeney S, Berthier CC, Shedden K, Cohen CD, Henger A, Krick S, et al. 2009. Renal gene and protein expression signatures for prediction of kidney disease progression. Am J Pathol 174: 2073–2085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao WHL, Klag MJ, Meoni LA, Reich D, Berthier-Schaad Y, Li M, Coresh J, Patterson N, Tandon A, Powe NR, et al. 2008. MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat Genet 40: 1185–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A, et al. 2009. Human Protein Reference Database: 2009 update. Nucleic Acids Res 37: D767–D772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Wu H, Green G, Winkler CA, Kopp JB, Miner JH, Unanue ER, Shaw AS 2003. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science 300: 1298–1300 [DOI] [PubMed] [Google Scholar]
- Koop K, Eikmans M, Baelde HJ, Kawachi H, De Heer E, Paul LC, Bruijn JA 2003. Expression of podocyte-associated molecules in acquired human kidney diseases. J Am Soc Nephrol 14: 2063–2071 [DOI] [PubMed] [Google Scholar]
- Kopp JB, Smith MW, Nelson GW, Johnson RC, Freedman BI, Bowden DW, Oleksyk T, McKenzie LM, Kajiyama H, Ahuja TS, et al. 2008. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet 40: 1175–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, Friedman D, Briggs W, Dart R, Korbet S, et al. 2011. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol 22: 2129–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köttgen A, Pattaro C, Böger CA, Fuchsberger C, Olden M, Glazer NL, Parsa A, Gao X, Yang Q, Smith AV, et al. 2010. New loci associated with kidney function and chronic kidney disease. Nat Genet 42: 376–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriz W, Elger M, Nagata M, Kretzler M, Uiker S, Koeppen-Hageman I, Tenschert S, Lemley KV 1994. The role of podocytes in the development of glomerular sclerosis. Kidney Int Suppl 45: S64–S72 [PubMed] [Google Scholar]
- Kuhn A, Thu D, Waldvogel HJ, Faull RLM, Luthi-Carter R 2011. Population-specific expression analysis (PSEA) reveals molecular changes in diseased brain. Nat Methods 8: 945–947 [DOI] [PubMed] [Google Scholar]
- Levey AS, Coresh J, Greene T, Stevens LA, Zhang YL, Hendriksen S, Kusek JW, Van Lente F 2006. Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate. Ann Intern Med 145: 247–254 [DOI] [PubMed] [Google Scholar]
- Lindenmeyer MT, Eichinger F, Sen K, Anders H-J, Edenhofer I, Mattinzoli D, Kretzler M, Rastaldi MP, Cohen CD 2010. Systematic analysis of a novel human renal glomerulus-enriched gene expression dataset. PLoS ONE 5: e11545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorz C, Benito-Martín A, Boucherot A, Ucero AC, Rastaldi MP, Henger A, Armelloni S, Santamaría B, Berthier CC, Kretzler M, et al. 2008. The death ligand TRAIL in diabetic nephropathy. J Am Soc Nephrol 19: 904–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglott D, Ostell J, Pruitt KD, Tatusova T 2011. Entrez Gene: Gene-centered information at NCBI. Nucleic Acids Res 39: D52–D57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon AP, Aronow BJ, Davidson DR, Davies JA, Gaido KW, Grimmond S, Lessard JL, Little MH, Potter SS, Wilder EL, et al. 2008. GUDMAP: The genitourinary developmental molecular anatomy project. J Am Soc Nephrol 19: 667–671 [DOI] [PubMed] [Google Scholar]
- Mele C, Iatropoulos P, Donadelli R, Calabria A, Maranta R, Cassis P, Buelli S, Tomasoni S, Piras R, Krendel M, et al. 2011. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N Engl J Med 365: 295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewold TB 2011. Stabilizing the kidney's skeleton. Sci Translational Med 3: 95ec128 [Google Scholar]
- Pavenstädt H 2000. Roles of the podocyte in glomerular function. Am J Physiol Renal Physiol 278: F173–F179 [DOI] [PubMed] [Google Scholar]
- Reddy MM, Wilson R, Wilson J, Connell S, Gocke A, Hynan L, German D, Kodadek T 2011. Identification of candidate IgG biomarkers for Alzheimer's disease via combinatorial library screening. Cell 144: 132–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich HN, Tritchler D, Cattran DC, Herzenberg AM, Eichinger F, Boucherot A, Henger A, Berthier CC, Nair V, Cohen CD, et al. 2010. A molecular signature of proteinuria in glomerulonephritis. PLoS ONE 5: e13451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roselli S, Heidet L, Sich M, Henger A, Kretzler M, Gubler M-C, Antignac C 2004. Early glomerular filtration defect and severe renal disease in podocin-deficient mice. Mol Cell Biol 24: 550–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem MA, Zavadil J, Bailly M, McGee K, Witherden IR, Pavenstadt H, Hsu H, Sanday J, Satchell SC, Lennon R, et al. 2008. The molecular and functional phenotype of glomerular podocytes reveals key features of contractile smooth muscle cells. Am J Physiol Renal Physiol 295: F959–F970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Wharram BL, Lee SK, Wickman L, Goyal M, Venkatareddy M, Chang JW, Wiggins JE, Lienczewski C, Kretzler M, et al. 2009. Urine podocyte mRNAs mark progression of renal disease. J Am Soc Nephrol 20: 1041–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai K, Mukoyama M, Mori K, Yokoi H, Koshikawa M, Yoshioka T, Takeda R, Sugawara A, Kuwahara T, Saleem MA, et al. 2006. Redistribution of connexin43 expression in glomerular podocytes predicts poor renal prognosis in patients with type 2 diabetes and overt nephropathy. Nephrol Dial Transplant 21: 2472–2477 [DOI] [PubMed] [Google Scholar]
- Schmid H, Boucherot A, Yasuda Y, Henger A, Brunner B, Eichinger F, Nitsche A, Kiss E, Bleich M, Gröne H-J, et al. 2006. Modular activation of nuclear factor-κB transcriptional programs in human diabetic nephropathy. Diabetes 55: 2993–3003 [DOI] [PubMed] [Google Scholar]
- Shen-Orr SS, Tibshirani R, Khatri P, Bodian DL, Staedtler F, Perry NM, Hastie T, Sarwal MM, Davis MM, Butte AJ 2010. Cell type-specific gene expression differences in complex tissues. Nat Methods 7: 287–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sistani L, Dunér F, Udumala S, Hultenby K, Uhlen M, Betsholtz C, Tryggvason K, Wernerson A, Patrakka J 2011. Pdlim2 is a novel actin-regulating protein of podocyte foot processes. Kidney Int 80: 1045–1054 [DOI] [PubMed] [Google Scholar]
- Steiglitz BM, Keene DR, Greenspan DS 2002. PCOLCE2 encodes a functional procollagen C-proteinase enhancer (PCPE2) that is a collagen-binding protein differing in distribution of expression and post-translational modification from the previously described PCPE1. J Biol Chem 277: 49820–49830 [DOI] [PubMed] [Google Scholar]
- Tanaka M, Asada M, Higashi AY, Nakamura J, Oguchi A, Tomita M, Yamada S, Asada N, Takase M, Okuda T, et al. 2010. Loss of the BMP antagonist USAG-1 ameliorates disease in a mouse model of the progressive hereditary kidney disease Alport syndrome. J Clin Invest 120: 768–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G 2001. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci 98: 5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter EE, Goodstadt L, Ponting CP 2004. Elevated rates of protein secretion, evolution, and disease among tissue-specific genes. Genome Res 14: 54–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woroniecka KI, Park ASD, Mohtat D, Thomas DB, Pullman JM, Susztak K 2011. Transcriptome analysis of human diabetic kidney disease. Diabetes 60: 2354–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaoita E, Yao J, Yoshida Y, Morioka T, Nameta M, Takata T, Kamiie J, Fujinaka H, Oite T, Yamamoto T 2002. Up-regulation of connexin43 in glomerular podocytes in response to injury. Am J Pathol 161: 1597–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]